

Abstract
We have biochemically and kinetically characterized the polymerase and exonuclease activities of the third B-family polymerase (Dpo3) from the hyperthermophilic Crenarchaeon, Sulfolobus solfataricus (Sso). We have established through mutagenesis that despite incomplete sequence conservation; the polymerase and exonuclease active sites are functionally conserved in Dpo3. Using presteady-state kinetics, we can measure the fidelity of nucleotide incorporation by Dpo3 from the polymerase active site alone to be 103 to 104 at 37 °C. The functional exonuclease proofreading active site will increase fidelity by at least 102 making Dpo3 comparable to other DNA polymerases in this family. Additionally, Dpo3’s exonuclease activity is modulated by temperature, where a loss in promiscuous degradation activity can be attributed to a reorganization of the exonuclease domain when bound to primer template DNA at high temperatures. Unexpectedly, the DNA binding affinity is weak compared with other DNA polymerases of this family. A comparison of the fidelities, polymerization kinetics, and associated functional exonuclease domain with those previously reported for other Sso polymerases (Dpo1 and Dpo4) illustrates that Dpo3 is a potential player in the proper maintenance of the archaeal genome.
Keywords: DNA replication, polymerase, thermodynamics, Dpo3, kinetics, archaea, DNA binding, fidelity
INTRODUCTION
Over the past 20 years, many additional DNA polymerases have been discovered within the genome of most organisms. This raises the question of how multiple DNA polymerases are regulated with regards to their individual functions for the maintenance of the organism’s genetic material. DNA polymerases have been divided into six main families (A, B, C, D, X, and Y) (1, 2). It is clear that B-family DNA polymerases provide the bulk of DNA synthesis during replication, and that X and Y-family DNA polymerases only act under special circumstances in various DNA repair pathways (3). In addition to the twelve other DNA polymerases in humans, there are three B-family DNA polymerases that act both in the initiation and elongation phases of DNA replication (4).
DNA polymerases from the B-family are generally robust and accurate enzymes with inherent nucleotide specificities augmented further by exonuclease proofreading domains that give nucleotide fidelities greater than 108, or one error every 108 bases incorporated (5). They become processive for >10,000 bases after complexation with their respective processivity factors or clamps (6). The leading (ε) and lagging (δ) strand polymerases (7) as well as the pol-α primase complex (8) are the three B-family polymerases found in humans. There are few significant mechanistic differences in enzymatic activities between ε and δ except for their strand specificities which are not fully understood (9). Pol-α primase, on the other hand, is able to synthesize short oligoribonucleotide primers that can be extended further through incorporation of deoxyribonucleotides on both strands to initiate replication elongation (10, 11).
The DNA processing enzymes in archaea have been shown to be more similar to those of eukaryotes than bacteria and serve as a simplified model for understanding more complex eukaryotic activities (12–15). Of the two main phyla in archaea, Euryarchaea have both B-family and D-family (unique to this archaeal phyla) DNA polymerases (16), whereas Crenarchaea have B-family and Y-family DNA polymerases (17, 18). Crenarchaea have a more conserved arsenal of eukaryotic-like DNA polymerases; however the metal binding domain of eukaryotic polymerases (ε & δ) may have evolved from the Euryarchaeal D-family polymerases instead (19). The Crenarchaeon, Sulfolobus solfataricus (Sso), has within its genome three B-family polymerases (Dpo1, Dpo2, and Dpo3); similar in number to the main analogous DNA polymerases (α, δ, and ε) found in humans. Both the proposed DNA replication polymerase, Dpo1, and the lesion bypass polymerase, Dpo4, have been well characterized with regards to their nucleotide incorporation mechanism (20, 21), fidelities (22, 23), and structures (24, 25). Dpo2 and Dpo3, have been identified by sequence homologies (26, 27), and until very recently had no biochemical characterization of activities (28).
Here, we have characterized the mechanism and kinetics of polymerization, exonuclease proofreading, and DNA binding activities of the third B-family DNA polymerase from Sulfolobus solfataricus, Dpo3. We have identified and mutated conserved amino acids in both the polymerization and exonuclease domains to confirm conservation and quantify their respective activities. Surprisingly and unlike other DNA polymerases, Dpo3 binds weakly to DNA requiring higher concentrations of enzyme for efficient nucleotide incorporation. This weak binding can be attributed to a mutation in the universally conserved Pol I motif. We have also quantified the selectivity of nucleotide incorporation for all 16 possible combinations into undamaged DNA templates using presteady-state kinetics. Circular dichroism analyses suggest that Dpo3 is a stable enzyme with a Tm > 94 °C, although the thermostability of the specific exonuclease domain may be regulated by substrate binding and temperature. The nucleotide incorporation rates, fidelity values, and exonuclease results are compared to the more thoroughly characterized Sso DNA polymerases (Dpo1 and Dpo4) to gain a better understanding of the diversity of DNA polymerase functions in the cell.
MATERIALS AND METHODS
Materials
Oligonucleotide substrates were purchased from Integrated DNA Technologies (IDT (Coralville, IA) and are listed in Supplemental Table 1. DNA strands over 28 nucleotides were gel-purified utilizing denaturing acrylamide gel electrophoresis (29). Radioactive ATP [γ-32P] was purchased from MP Biomedicals (Solon, OH). Optikinase (USB, Cleveland, OH) was used for 5′-end labeling of DNA substrates according to manufacturer protocols. Radiolabeled primers were added to cold complementary template strands at a ratio of 1:1.2 to ensure proper annealing. Annealing was performed by heating the sample to 95 °C for five minutes, followed by slowly cooling to room temperature for at least two hours. Commercial enzymes were from NEB (Ipswich, MA). All other chemicals were analytical grade or better.
Dpo3 Protein Purification
Dpo3 was amplified from Sulfolobus solfataricus P2 genomic DNA (ATCC #35092) with Pfx50 polymerase (Invitrogen, Carlsbad, CA). The gene was initially subcloned into pGEM-T (Promega, Madison, WI), and then ligated into pET30a (EMD Chemicals, Gibbstown, NJ) using the restriction sites NdeI and XhoI included in the primer sequences (Supplemental Material) to allow for expression of a C-terminal 6X His tag. The DNA polymerase (D424A, D542A, and D424A/D542A) and exonuclease (D226A, D228A, D234A, D236A) mutant constructs were created using a standard QuikChange protocol (Agilent, Santa Clara, CA) with KAPA HiFi DNA polymerase (KAPA Biosystems, Woburn, MA).
BL21(DE3) Rosetta 2 cells containing the various pET30a-Dpo3 constructs were grown at 37 °C, and protein expression was autoinduced as described (30). Cell pellets were resuspended in 50 mM sodium phosphate buffer (pH 7.0), 50 mM NaCl, and 5 mM β-mercaptoethanol. The cells were lysed by the addition of lysozyme and sonicated. After centrifugation, the supernatant was heat treated at 70 °C for 20 minutes to precipitate host proteins and centrifuged again. The supernatant containing Dpo3 was purified using a Ni2+ column (Thermo Fisher, Waltham, MA) and eluted with a step gradient of 500 mM imidazole. Further purification was performed using an ATKA Prime FPLC with HiTrap DEAE and Heparin columns (GE Healthcare, Piscataway, NJ) and elution with a linear gradient to 1 M NaCl. Separation of a major degradation product can be performed using a pH gradient from 8.5 to 7.9 and a MonoQ column (GE Healthcare). Final cleanup and sizing was performed with a Superdex 200 26/60 (GE Healthcare). The extinction coefficient for Dpo3 was determined to be 117,893 M−1cm−1 (31). Typical yields of purified protein were greater than 3 mg/L of cells.
Polymerase Activity Assays
Standard assays were performed in polymerization buffer [20 mM Tris-acetate (pH 7.5), 100 mM potassium acetate, and 10 mM magnesium acetate] containing 36 nM primer template DNA (ptDNA), and various concentrations of Dpo3 as indicated. Prior to initiation by addition of dNTPs, the reaction components were incubated for five minutes at the reaction temperature. Experiments were conducted at temperatures, polymerase concentrations, dNTP concentrations, and times as indicated in each figure legend. Reactions were terminated by the addition of an equal volume of a formamide quench (100 mM EDTA, 0.1% SDS, 79% formamide). To examine the products for short templates denaturing gels [20% acrylamide, 8 M urea, and TBE buffer (45 mM Tris-borate, 1 mM EDTA)] were used. Phosphor screens were then exposed to the gels for a minimum of 4 hours, imaged by a Storm 820 Phosphorimager (GE Healthcare), and quantified using ImageQuant software (v. 5.0).
Divalent Cation Optimization
Optimization of divalent cations was performed in polymerase activity assay reaction conditions as detailed above, however, the concentration of divalent metal, M2+ (Mg2+ or Mn2+), was varied. Reactions were quenched and analyzed as above for standard polymerase reactions. Data obtained from M2+ titration reactions were fit to a Michaelis-Menten equation with a cooperativity (n) parameter:
| (1) |
where Vmax is the maximal rate, Km is the Michaelis constant, and [M2+] is the concentration of the divalent cation included in the reaction. For those reactions where inhibition was observed at higher concentrations of M2+, an inhibition reaction equation was used instead:
| (2) |
where Ki is the inhibition constant.
Polymerase Fidelity
Fidelity assays were performed as detailed above for polymerase reactions, however each pre-incubated reaction contained 9.6 nM DNA, 2 μM Dpo3 (D236A) and was initiated with varying concentrations of dNTPs. Samples were removed and placed 1:1 into quench at time points as indicated in each figure legend. The time course of product formation was fit to a single exponential equation for each concentration of dNTP:
| (3) |
where A represents the reaction amplitude, kobs is the observed polymerase rate, and t is time in seconds. The observed rates extracted from the time courses of product formation for each dNTP concentration were then plotted against their respective [dNTP] and fit to a hyperbolic equation:
| (4) |
where kp is the maximum rate of dNTP incorporation and Kd is the dissociation constant for the incoming nucleotide.
Fluorescent Anisotropy DNA Binding Assays
Anisotropy assays were performed in polymerization buffer, in the absence of dNTPs with 1 nM ssDNA or ptDNA, where a Cy3 fluorescent label was placed on the 5′ end of the single strand or template strand respectively, and varying concentrations of Dpo3 (D236A) as depicted. Anisotropy values were obtained using a Fluoromax-3 fluorimeter with automated polarizers (HORIBA Jobin Yvon – Edison, NJ.) Cy3 were excited at 550 nm during 1 second integration times and data points represent an average of five consecutive readings. The anisotropy values reported are the average and standard error from three independent titrations. The absolute fluorescence emission (565 nm) was unchanged during the course of the titration, eliminating the possibility that Dpo3 binds directly to Cy3. The fluorescence anisotropy (r) was calculated using the following equation:
| (5) |
where I is the polarization intensity and the subscripts, V and H, represent vertical or horizontal polarized light, respectively. G is a correction factor for any differences in intensity of horizontally and vertically polarized light and is calculated automatically by the included FluorEssence software (v2.5.2.0).
The change in anisotropy was fit to a single binding equation:
| (6) |
where A is the reaction amplitude, and Kd is the dissociation constant for the interaction between Dpo3 and DNA.
Exonuclease Assays
Standard exonuclease assays were performed in polymerization reaction buffer containing 18 nM ptDNA, and 2 μM Dpo3. Prior to initiation by addition of DNA, the reaction components were incubated for five minutes at 55 °C, unless otherwise indicated. Experiments were performed for times as indicated in each figure legend. Reactions were quenched and analyzed as described for polymerase reactions.
Circular Dichroism (CD) Denaturing Measurements
Circular dichroism (CD) experiments to monitor conformational changes of either polymerase alone or bound to DNA were performed using a DSM 17 (Olis Inc., Bogart, GA) using a 1 mm path length cell. The experiments were assembled either in the presence or absence of DNA hairpin (2 μM) and Dpo3 (4 μM). The molar ellipticity (θ) at 222 nm was monitored over a temperature range from 20 to 95 °C in 2 °C intervals controlled by a peltier. The spectra from at least three separate scans were averaged and analyzed as described (32).
Homology Modeling and Alignment
Local and global sequence alignments were performed using ClustalW2 analysis (http://www.ncbi.nlm.nih.gov/blast/bl2seq/wblast2.cgi). The homology model of Dpo3 was created by threading the global alignment of Dpo3 with Dpo1 onto the structure of Dpo1 (PDB ID: 1S5J)(25) using SWISS-MODEL (33). The DNA and incoming nucleotide were modeled into the active site of the homology model of Dpo3 by aligning the polymerase active site of the RB69 gp43/DNA/dTTP ternary structure (PDB ID: 1IG9) (34) with the homology model of Dpo3 using PyMol (http://www.pmol.org).
RESULTS
Conditions for optimal polymerase activity of Dpo3
After purifying wild-type (WT) Dpo3 to homogeneity (Figure 1A), we characterized the buffer conditions required for maximal polymerase activity in 100 mM NaCl at 60 °C. Purification with a gel-filtration column for size selection was required for maximal polymerase activity, although no significant shift in molecule size was noted in the chromatogram. Neither HEPES nor Tris buffers nor varying pH over a limited physiological range (6.0 – 8.5) yielded a significant change in the polymerase activity of Dpo3 (Supplemental Figure 1A). Maximal activity was observed with 20 mM Tris between pH (7 – 8.5), and so, pH 7.5 was used in all subsequent reactions. A comparison of the polymerization ability in our buffer compared with that used previously (28) showed no significant difference in the rate of incorporation (data not shown).
Figure 1.
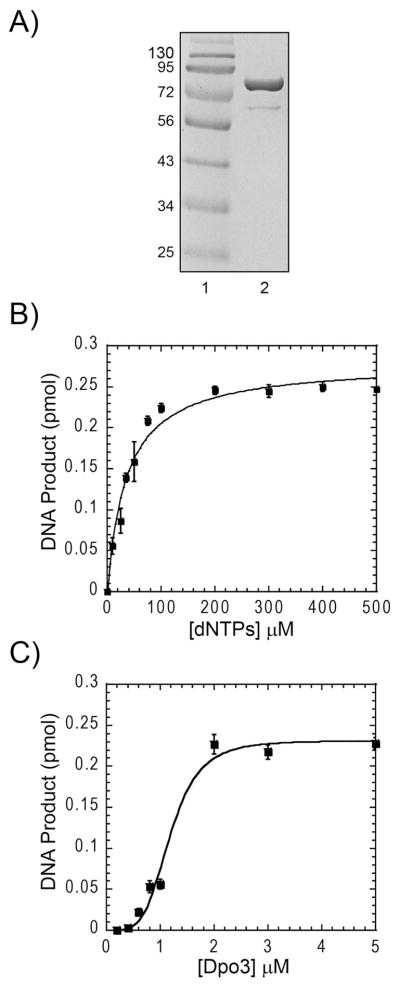
(A) Purified Dpo3 on a Coomassie-stained SDS-PAGE gel: lane 1, protein markers, and lane 2, Dpo3 (88 kDa). (B) Optimization of dNTP concentrations on total DNA product produced by 2 μM Dpo3 on ptDNA in Tris (pH = 7.5), 10 mM Mg2+ at 70 °C. The Km for dNTPs (36 ± 5 μM) was determined from the average of three independent experiments fit to Equation 1. (C) Effect of Dpo3 concentration on total DNA product produced using long ptDNA template. Reactions were performed in Tris (pH = 7.5), 200 μM dNTPs, 10 mM Mg2+ at 70 °C for 10 minutes. The apparent dissociation constant (K’d) for Dpo3 activity (1.2 ± 0.1 μM) was determined from the average of three independent experiments fit to Equation 1. The cooperativitiy parameter (n) was equal to 4.4 ± 1.1.
The importance of divalent cations in the nucleotide incorporation process has been well characterized, where the quantity of product extension is known to be dependent on metal concentration (35). We performed polymerase reactions while varying the concentration of Mg2+ (Supplemental Figure 1B) and fit the data to Equation 1 with an observed Km of 1.43 ± 0.08 mM. Identical experiments with Mn2+ showed an inhibition at concentrations greater than 0.5 mM. Based on these results, we chose to include 10 mM Mg2+ in the reaction buffer. Similarly, the optimal concentration of dNTPs was monitored (Figure 1B), where the Kd for incoming nucleotides was determined to be 36 ± 5 μM. 200 μM dNTPs were included in all subsequent polymerase reactions unless indicated otherwise. As a note, the exonuclease activity present in WT Dpo3 reduced the detected amount of fully extended product slightly from the theoretical maximum.
Having established appropriate reaction buffer conditions, we titrated Dpo3 to determine the optimal concentration for maximal activity (Figure 1C). Interestingly, very low activity levels were observed until a reaction concentration of 750 nM was reached. The data required a cooperativity coefficient for an appropriate fit to extract the apparent dissociation constant (K’d) equal to 1.2 ± 0.1 μM. The sigmoidal shape of the curve indicates that the concentration of the polymerase is critical for binding and associated activity. Further kinetic assays were performed at 2 μM Dpo3 determined to provide for maximal activity unless indicated otherwise.
Thermostability of Dpo3
The optimal reaction temperature was determined by examining total product synthesized from ptDNA at temperatures varying from 37 °C to 70 °C in a 10 minute reaction (Figure 2A). We observed maximal polymerase product formation at 65 °C. Reactions in the absence of dNTPs monitored the exonuclease activity and were maximal at 55 °C and decreased as temperature was increased further. The purification protocol included a 70 °C incubation step which typically eliminates any background nuclease activity. The exonuclease activity of any contaminating nucleases from E. coli should be maximal around 37 °C and decrease at temperatures greater than 45 °C. The parallel increases in both polymerase and exonuclease activities from 37 °C to 55 °C is evidence that the observed exonuclease activity is intrinsic to the polymerase.
Figure 2.
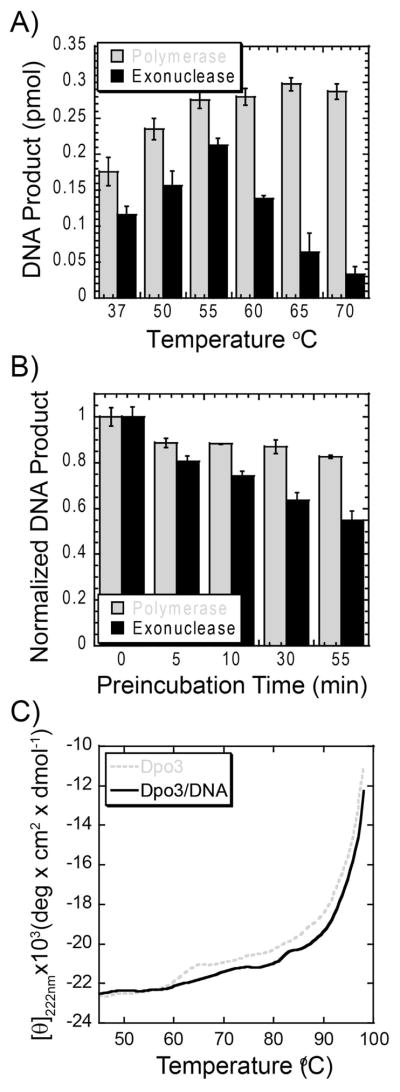
(A) Temperature dependence of 2 μM Dpo3 (WT) in 50 mM Tris (pH = 7.5), 10 mM Mg2+, 200 μM dNTPs on polymerase (grey) and exonuclease (black) activities on ptDNA (36 nM) in a 10 minute reaction. The reported values and errors are the average of three independent experiments. (B) Thermostability of 2μM Dpo3 (WT) after preincubation at 70 °C for the indicated time points. Quantification of product formation for polymerase activity (grey) after the addition of dNTPs and exonuclease activity (black) after the addition of ptDNA, in a 10 minute reaction at 70 °C (polymerase activity) or 55 °C (exonuclease activity). The error bars represent the standard error from at least three independent experiments. (C) Thermal melting of Dpo3 alone (grey, dashed) or bound (black, solid) to hairpin DNA monitored by circular dichroism at 222 nm (2 °C increments).
Although the polymerization activity of Dpo3 is maximal at temperatures greater than 65 °C, in order to assay the thermostability of the Dpo3 protein structure, we preincubated the wild-type polymerase at 70 °C for various times and then initiated the reaction by the addition of dNTPs or DNA to monitor polymerase or exonuclease activity, respectively (Figure 2B). Extension to full length product from primer template was used to evaluate any loss in polymerization activity. Even after incubation at 70 °C for 55 minutes, the loss in DNA polymerase activity was minimal. The reduction in exonuclease activity was slightly greater, but 55% of the activity remained after the 55 minute high temperature incubation period.
To get a more direct measure of protein structure thermostability, we used circular dichroism (CD) to monitor the change in molar ellipticity at 222 nm as a function of temperature in the absence and presence of hairpin DNA (Figure 2C). The protein structure of Dpo3 is highly thermostable with an estimated melting temperature (Tm) greater than 94 °C. The data could not be quantified accurately because the maximum of the denatured curve was not apparent at 100 °C. We also note a small but reproducible unfolding event that occurs at 63 °C. This may be localized unfolding of a small thermally unstable domain within Dpo3. In the presence of ptDNA, the Tm for Dpo3 shifts slightly higher (~96 °C) and the local unfolding event at 63 °C disappears.
Conserved Mutations in the Polymerase Active Site Disrupt Activity
There is a conserved aspartic acid residue (D424) within the polymerase six (Pol VI) domain and a single aspartic acid (D542) within the polymerase one (Pol I) domain of Dpo3 (Figure 3A). It has been previously shown that mutation of the conserved active site aspartates in homologous DNA polymerases abolishes nucleotide incorporation ability (36–39). Interestingly, Dpo3 lacks the first aspartate in the highly conserved DTD motif in Pol I domain (Figure 3A) known to be important in coordinating Mg2+ and contributing to translocation (37). To determine if the polymerase active site of Dpo3 is functionally conserved in in the absence of a conserved Pol I domain, we constructed three mutants; two single (D424A, D542A) and one double (D424A/D542A). We then examined the polymerase activity of the mutants compared to WT Dpo3 at 60 °C (Figure 3B). The polymerase activity for synthesis of full length products for the D424A mutant was reduced more than 30-fold from WT (0.010 ± 0.003 vs. 0.34 pmol sec−1, respectively), while the D542A mutant was reduced more than 50-fold (0.007 ± 0.002 pmol sec−1). The activity of the double mutant (D424A/D542A) was reduced to just above background levels and required much longer times to detect any product.
Figure 3.
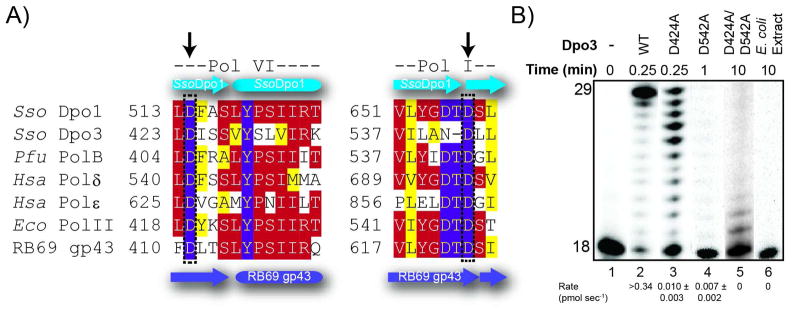
(A) Amino acid alignment of DNA polymerase domains VI and I using CLUSTAL W2 (http://www.ebi.ac.uk/Tools/clustalw2) for common members within the B-family of DNA replication polymerases. Color coding for slightly (yellow), mostly (red) and absolutely (purple) conserved residues are indicated. The secondary structure elements are derived from the crystal structure of Dpo1 (cyan) (PDBID: 1S5J) or RB69 gp43 (purple) (PDBID: 1CLQ). Species are each listed as a three letter code: Sso, Sulfolobus solfataricus, Pfu, Pyrococcus furiosus, Hsa, Homo sapien, and Eco, Escherichia coli. Arrows represent the residues in Dpo3 that were mutated and constitute the active site aspartates. (B) Effect of single (D424A or D542A) or double mutants (D424A/D542A) of Dpo3 on the extension of Template (T)G at 60 °C for indicated times in optimal buffer conditions. The average rates in pmol sec−1 from multiple independent experiments are listed below the corresponding lanes of the gel.
Conserved Mutations in the Exonuclease Active Site Disrupt Activity
The exonuclease domain of polymerases can be organized into three motifs (Exo I, Exo II, and Exo III). Aspartates within each domain have been implicated in the proofreading function in different polymerases. Dpo3 has a conserved aspartate (D172) in the exonuclease I domain shown to be important for E. coli Pol II (40) and pol ε (41), but mutation to alanine had no effect on the exonuclease activity (data not shown). Examination of the exonuclease II motif (Exo II) showed no universally conserved aspartate or glutamate (42–44) for Dpo3 but contained four potential catalytic aspartates (Figure 4A). We individually mutated each aspartate (D226A, D228A, D234A, and D236A) in Exo II and examined their effect on the exonuclease activity levels on three DNA substrates (ssDNA, ptDNA, dsDNA). As proof that D236 is involved in proofreading, the D236A mutant incorporates nucleotides fully to the end of the template unlike WT which stops one base prior to the end (−1) in this time frame (Figure 4B). D236A had the lowest exonuclease activity and therefore greatest perturbation (Figure 4C & D) of the four mutants. A double mutant D234A/D236A had no further reduction in exonuclease activity over the D236A single mutant (data not shown). The rate for the exonuclease product formation for WT was 0.031 ± 0.001 pmol sec−1, while the rate for D236A was reduced approximately seven fold to 0.0046 ± 0.0003 pmol sec−1, implicating this aspartate in the proofreading mechanism (Figure 4E).
Figure 4.

(A) Amino acid alignment of exonuclease domain II using CLUSTAL W2 (http://www.ebi.ac.uk/Tools/clustalw2) for common members within the B-family of DNA replication polymerases. Color coding for slightly (yellow), mostly (red) and absolutely (purple) conserved residues are indicated. The secondary structure elements are derived from the crystal structure of Dpo1 (cyan) (PDBID: 1S5J) or RB69 gp43 (purple) (PDBID: 1CLQ). Species are each listed as a three letter code: Sso, Sulfolobus solfataricus, Pfu, Pyrococcus furiosus, Hsa, Homo sapien and Eco, Escherichia coli. Arrows represent the residues in Dpo3 that were mutated. (B) Polymerase reactions comparing full length product formation from WT and D236A Dpo3 on Template T at 70 °C for three minutes, showing major products (29 base, blunt, 0) or (28 base, recessed, −1) for D236A or WT Dpo3, respectively. C) Exonuclease experiment on single strand (ssDNA), primer template (ptDNA), and double-strand (dsDNA) DNA for both WT and D236A Dpo3 at 55 °C for 10 minutes. (D) Quantification of exonuclease cleavage products for WT Dpo3 and each prospective exonuclease mutant (D226A, D228A, D234A, D236A), on all three DNA conformations: ssDNA (blue), ptDNA (red), or dsDNA (orange) at 55 °C for 10 minutes. (E) Quantification of the steady-state rate of exonuclease products produced from ptDNA by WT (0.031 ± 0.001 pmol s−1) or D236A (0.0046 ± 0.0003 pmol s−1) Dpo3 from at least two independent experiments. The error bars represent the standard error of the reaction.
DNA Binding Affinity of Dpo3 Monitored by Fluorescence Anisotropy
DNA binding of B-family polymerases is typically robust, and shows a preference for ptDNA over ssDNA (42). To determine if the relatively high concentrations of Dpo3 required for activity are a result of a weak binding affinity, we measured the DNA binding properties using fluorescence anisotropy. Fluorescence anisotropy measures the molecular rotational diffusion rates of molecules, where a decrease in these rates using fluorescently labeled DNA occurs upon formation of a protein DNA complex. The resulting increase in anisotropy (r) can be plotted to determine the binding coefficient (Kd). Both ssDNA and ptDNA were used as the substrate with a fluorescent dye, Cy3, located at the 5′ end of the template or single strand. Anisotropy values were measured as Dpo3 concentration was increased. The Kd for the interaction between Dpo3 and ssDNA was 1.10 ± 0.08 μM, while the Kd for the interaction between ptDNA was tighter at 0.81 ± 0.06 μM (Figure 5).
Figure 5.

Change in fluorescence anisotropy upon Dpo3 (D236A) binding to either Cy3-labeled ssDNA (Cy3DNA) (grey, dash) or ptDNA (black, solid). Data points were fit to Equation 4 to extract a Kd of binding for ssDNA (1.10 ± 0.08 μM) and ptDNA (0.81 ± 0.06 μM). The error bars represent the standard error from at least three independent experiments.
Correct Nucleotide Specificity
We analyzed the presteady-state dNTP incorporation rate of the Dpo3 exonuclease mutant (D236A) for correctly paired nucleotides across from all four different template bases. These reactions could be performed accurately by hand at 37 °C due to a decreased rate of synthesis at lower temperatures. Previously, Suo and coworkers established that fidelity reactions with Dpo1 and Dpo4 performed at lower temperatures did not have significantly different values than those performed at higher temperatures (22, 45). Single turnover reactions were performed where the enzyme concentration (2 μM) was held well above the DNA concentration (9.6 nM) as required based on the affinities and activities measured above. The quantified products for different concentrations of dCTP incorporated onto Template G were plotted as a function of the reaction times to obtain the apparent rate constants (kobs) according to Equation 3 (Figure 6A). These single-turnover rates were plotted as a function of the concentration of dCTP and fit to Equation 4 to yield the second order polymerization rate (kp), 0.045 ± 0.007 s−1 and dissociation constant for dCTP (Kd), 61 ± 25 μM (Figure 6B & Table 1). The measure of substrate specificity (kp/Kd) was calculated to be 7.4 × 10−3 mu;M−1s−1. If Dpo3 is able to bind nucleotides nonspecifically in the absence of DNA, high concentrations of enzyme could affect the Kd value measured if the concentration of enzyme was similar in magnitude. In our experiments, a large excess of enzyme was required to promote DNA binding, but this concentration is still at least 5 to 100 times less than the experimental concentration of dCTP.
Figure 6.

Concentration dependence of the pre-steady state rate of correct nucleotide incorporation. (A) A preincubated solution containing 2 μM Dpo3 and Template G (9.6 nM) was mixed with increasing concentrations of dCTP (10 μM – 200 μM) for the indicated time points. The data was fit to Equation 3 to determine the single-exponential rate (kobs). (B) kobs values were plotted as a function of dCTP concentration and fit with Equation 4 to yield kp (0.045 ± 0.008 s−1) and Kd (61 ± 26 μM).
Table 1.
Pre-Steady-State Kinetic Parameters of Dpo3
| dNTP | kp (s−1)1 | Kd (mM)1 | kp/Kd (μM−1 s−1) | Misincorporation Frequency2 |
|---|---|---|---|---|
| Template A | ||||
| dATP | 0.0010 ± 0.0002 | 0.23 ± 0.02 | 4.5 × 10−6 | 6.7 × 10−3 |
| dCTP | 0.0016 ± 0.0002 | 2.0 ± 0.6 | 7.9 × 10−7 | 1.2 × 10−3 |
| dGTP | 0.0017 ± 0.0004 | 1.7 ± 1.1 | 9.6 × 10−7 | 1.5 × 10−3 |
| dTTP | 0.038 ± 0.002 | 0.057 ± 0.008 | 6.6 × 10−4 | |
| Template C | ||||
| dATP | 0.00065 ± 0.00005 | 0.74 ± 0.20 | 8.9 × 10−7 | 3.0 × 10−4 |
| dCTP3 | - | - | - | - |
| dGTP | 0.069 ± 0.005 | 0.024 ± 0.007 | 2.9 × 10−3 | |
| dTTP | 0.0013 ± 0.0001 | 0.43 ± 0.16 | 3.1 × 10−6 | 1.1 × 10−3 |
| Template G | ||||
| dATP | 0.0025 ± 0.0005 | 1.9 ± 0.9 | 1.3 × 10−6 | 1.9 × 10−3 |
| dCTP | 0.045 ± 0.008 | 0.061 ± 0.025 | 7.4 × 10−4 | |
| dGTP4 | 0.0011 ± 0.0001 | 0.056 ± 0.017 | 2.0 × 10−5 | 2.8 × 10−2 |
| dTTP | 0.0016 ± 0.0001 | 0.30 ± 0.08 | 5.3 × 10−6 | 7.6 × 10−3 |
| Template (T)G | ||||
| dGTP | 0.0021 ± 0.0002 | 0.69 ± 0.20 | 3.1 × 10−6 | 4.4 × 10−3 |
| Template T | ||||
| dATP | 0.12 ± 0.01 | 0.018 ± 0.006 | 6.8 × 10−3 | |
| dCTP | 0.0009 ± 0.0001 | 0.79 ± 0.34 | 1.1 × 10−6 | 1.6 × 10−4 |
| dGTP5 | 0.0025 ± 0.0001 | 0.41 ± 0.03 | 6.0 × 10−6 | 8.7 × 10−4 |
| dTTP | 0.0015 ± 0.0001 | 0.39 ± 0.08 | 3.7 × 10−6 | 5.4 × 10−4 |
Calculated from the fit to Equation 4 of the second order plot.
Calculated as (kp/Kd)incorrect/(kp/Kd)correct.
Was not incorporated appreciably above background and unable to quantify.
Inhibition at [dGTP] > 1mM.
Inhibition at [dGTP] > 2 mM.
Single turnover experiments for other correct nucleotide incorporations (dTTP on Template A, dGTP on Template C, and dATP on Template T) were also performed similarly (Supplemental Figure 3). The kinetic parameters (kp, Kd, and kp/Kd) are reported in Table 1. Interesting observations from this analysis includes incorporation of dATP on Template T was roughly three-fold faster (0.12 ± 0.01 s−1 vs. 0.038 ± 0.002 s−1) than the reverse incorporation (dTTP on Template A). Also, the Kd for binding dATP was three-fold tighter than for dCTP or dTTP on their respective correctly base paired templates.
Incorrect Nucleotide Specificity
Presteady-state incorporation rates for incorrect nucleotides opposite all four templates were also analyzed similarly as described above. As an example, single nucleotide misincorporation of dTTP at various concentrations onto template T is shown in Figure 7A to obtain kobs. These observed rates were plotted against the dTTP concentration and fit to Equation 4 to give kp (0.0015 ± 0.0001 s−1), Kd (0.39 ± 0.08 mM), and kp/Kd (3.7 × 10−6 μM−1s−1) values (Figure 7B and Table 1). Misincorporation of dTTP on Template T was roughly 100-fold slower and with a 20-fold weaker Kd than for correct incorporation of dATP. This results in a misincorporation frequency for incorrect dNTPs of 5.4 × 10−4 (Table 1) or 1 error every 2000 bases with polymerase active site selection alone.
Figure 7.

Concentration dependence of the pre-steady state rate of incorrect nucleotide incorporation. (A) A preincubated solution containing 2 μM Dpo3 and Template T (9.6 nM) was mixed with increasing concentrations of dTTP (250 μM – 4 mM) for the indicated time points. The data was fit to Equation 3 to determine the single exponential rate (kobs). (B) kobs values were plotted as a function of dTTP concentration and fit with Equation 4 to yield kp (0.0015 ± 0.0001 s−1) and Kd (0.39 ± 0.08 mM).
In some cases, inhibition was noted at high concentrations of incorrect nucleotides. Incorporation of dGTP on Template T was inhibited at concentrations greater than 2 mM, and dGTP incorporation on Template G was inhibited at concentrations greater than 1 mM. Nucleotide selection seems to be the most discriminative for Template C and Template T provided by a concomitant decrease in both the polymerization rate and affinities.
For the most part, incorrect nucleotide incorporations are 20–100 times slower than for correct incorporations with Kd values 4–30 times weaker (Table 1). One exception seems to be that the Kd of dGTP and dCTP are identical on Template G. The 40-fold slower rate of incorporation for dGTP on that template is the only factor that provides for the selectivity in this case, leading to a weaker fidelity value of 2.77 × 10−2. For Template G, the preceding base (−1) in the template is cytosine which allows for the possibility that the strong binding of dGTP on Template G noted above is the result of a looping mechanism that base pairs the incoming nucleotide at the −1 position in the template. To test this, we changed the −1 base in the template strand to T to create Template (T)G. When single nucleotide misincorporation assays were performed with dGTP on this template, there was a significant increase in the Kd to 0.69 ± 0.20 mM (Table 1). This resulted in a decrease in kp/kd (3.1 × 10−6 μM−1 s−1) and misincorporation frequency (4.4 × 10−3) more consistent with the rest of Table 1. Except for dGTP on Template G, the error frequencies for Dpo3 range from 10−3 to 10−4 and are in line with other polymerases in this family (46, 47).
DISCUSSION
We have biochemically characterized both the DNA polymerization and exonuclease activities of the third B-family DNA polymerase in Sulfolobus solfataricus and verified that it is an active enzyme. Although Dpo3 is characterized as a B-family DNA polymerase, the Pol I motif in the active site is not absolutely conserved prompting speculation that it may be inactive (48). Dpo3 was proposed to have evolved thorough a gene duplication event (26), which eventually became the precursor for the human DNA polymerase ε implicated in leading strand DNA replication in eukaryotes(7, 19). In order to better understand the role of Dpo3 in chromosomal maintenance in archaea, we investigated the polymerase and exonuclease activities, fidelity, and thermostability of this polymerase.
Dpo3 is an accurate B-family DNA polymerase whose fidelity is increased further with the inclusion of an active exonuclease domain. Dpo3 has high thermostability that is increased slightly in the presence of DNA. Surprisingly, Dpo3 binds DNA weakly compared with other B-family polymerases (22, 23, 42) and the exonuclease and polymerase activities are maximal at different temperatures. While preparing this manuscript, a recent report has also characterized the in vitro activity of Dpo3 (28), and although there are some similarities such as weak DNA binding, we have measured significant differences in the kinetic and thermodynamic parameters as well as verified and compared the importance of the catalytic residues in the polymerase and exonuclease active sites. In spite of the nonconserved active site motifs, Dpo3 has been confirmed to be a B-family DNA polymerase with enzymatic activities that allow participation in coupled DNA replication or repair activities in archaea. The roles and activities of multiple DNA polymerases in this organism provide a model for understanding and characterizing DNA polymerase specificities in higher organisms.
Nonconserved Polymerase Motifs Reduce DNA Binding and Enzymatic Activity
Although Dpo3 is considered to be a B-family DNA polymerase, the most conserved Pol I domain contained in all DNA polymerases is mutated. This conserved YGDTD sequence is used to coordinate Mg2+ where the tyrosine and both aspartates are thought to be essential for polymerase activity (37, 38, 49). In Dpo3, the homologous sequence is LAN-D, where rather than a pair of aspartic acids residues coordinating Mg2+ and contributing to translocation, a single aspartic acid residue is important for the catalytic activity. Crystal structures for RB69 gp43 (34) and yeast pol δ (50) highlight the importance of the second aspartate (Pol I) in metal coordination and catalysis and is consistent with a similar role for D542 in Dpo3.
Conversely, the first aspartate in the Pol I motif is generally orientated away and towards the minor groove at the site of insertion (51, 52). Although this first aspartate is conserved, the actual role of this residue is elusive. Mutation of the first aspartate (Pol I) in Phi29 DNA polymerase disrupted the translocation step between catalysis steps and resulted in lower processivity, possibly representing a looser grip on DNA (53). Interestingly, high concentrations of Dpo3 (in excess of 750 nM) were required for stable binding and efficient DNA extension as also noted previously (28) and could be the consequence of the missing first aspartate. Mutation of the homologous first aspartate in human polymerase α required Mn2+ to restore the wild-type activity (38). Our metal dependent studies for Dpo3 may actually be similar in that at concentrations less than 1 mM metal, Mn2+ provides the optimal activity. At higher concentrations, Mn2+ is inhibitory and Mg2+ provides for optimal activity similar to the D1002N mutant of human pol α (38).
The other catalytic aspartate contained in the Pol VI motif and known to be involved in Mg2+ coordination is conserved in Dpo3 (D424) but residues adjacent to this residue have also been shown to affect catalysis. Previous work by Kennedy et al. implicated a conserved ALY motif within the Pol VI domain of the Pyrococcus furiosus B-family polymerase in a stacking interaction with the ribose of the incoming nucleotide (54). A408S and L409V mutations in PfuPol both result in a decrease in catalytic efficiency (kcat/Km). It seems that increased side chain volume interacting at the back of the incoming nucleotide negatively affect catalysis and fidelity. The homologous wild-type sequence in Dpo3 is 427-SVY and may also contribute to Dpo3’s slower incorporation rates and reduced fidelity.
To highlight the two active site aspartates primarily responsible for metal binding and catalysis, we created a ternary homology model of Dpo3 bound to DNA with an incoming dTTP (Figure 8A). In this model, it is clear that D424 and D542 are orientated properly for binding metals in the active site required for catalysis and incoming dNTP stabilization. Interestingly, N541 seems to adopt a similar conformational position as the first aspartate in the Pol I motif of other polymerases. The absence of a negative charge at this position in Dpo3 would disrupt native interactions with Mg2+ and the DNA template possibly explaining the reduced affinity of binding.
Figure 8.
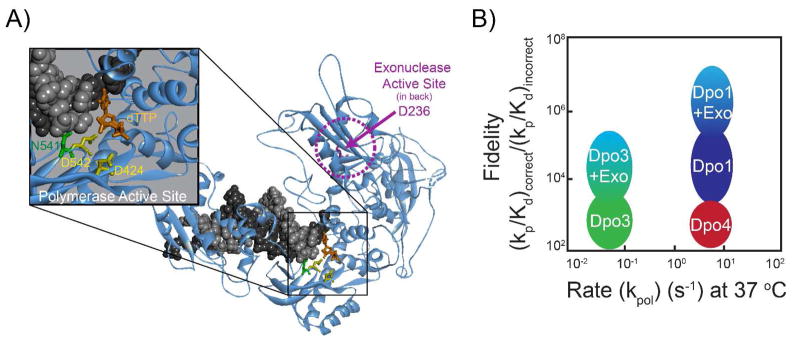
A) Homology model of the ternary complex of Dpo3 bound to DNA (black/grey) with incoming dTTP (orange) highlighting the aspartates in the polymerase (D424 and D542, yellow) and exonuclease (D236, pink) active sites. N541 (green) is also shown orientated towards the minor groove of the dsDNA template. B) Graphical representation of the fidelity [(kp/Kd)correct/(kp/Kd)incorrect] as a function of the rate (kpol) comparing Dpo1, Dpo3, and Dpo4.
Dpo3 has Moderate Polymerase Activity
We have shown through detailed kinetic analysis that the catalytic activity of Dpo3 is moderate, although the rate of nucleotide incorporation is significantly faster than previously published (28). During the purification of Dpo3, it became clear that inclusion of a gel filtration column for size selection and cleanup was required for maximal activity. A comparison of the reaction conditions used in our study with those from the previous report (28) noted no significant differences in extension rates using our purified protein. Polymerization reactions for Dpo3 at 37 °C or 50 °C converted more than 50–70% of the substrate to full length product in 10 minutes (Fig. 2A) compared with little full length product formed at these temperatures in a 30 minute reaction previously (28). The kinetic rate constants are difficult to compare between the studies, because we used presteady-state analysis monitoring the fastest rate of single nucleotide incorporation for Dpo3, while the previous study used steady-state analysis. Presteady-state analysis has been shown to be more accurate for measuring the kinetic and thermodynamic basis for fidelity of nucleotide incorporation (55). Moreover, we have verified that Dpo3 has an active exonuclease proofreading domain which would make steady-state analysis of polymerase fidelity with the wild-type enzyme difficult.
The fidelities of nucleotide incorporation for the Dpo3 polymerase active site alone are generally 102 – 103. One exception where the fidelity is less occurs when the incoming base is incorporated based on complementarity to the preceding −1 template base (dGTP on Template G). In this case, incorporation would require destabilization of the terminal base pair and looping out of the last base in the primer strand. Incorporation would then proceed opposite the −1 base in the template. An analogous looping out mechanism has been noted previously for Dpo4 (56, 57), although the looped-out base occurs in the template strand instead, in favor of nucleotide incorporation opposite the +1 position. We can rule out a similar mechanism for Dpo3 as incorporation dATP in Templates G (where the +1 base is T) has the weakest Kd value of the group.
In general, the presteady-state kinetics of Dpo3 are reduced compared with the other two well-characterized DNA polymerase (Dpo1 and Dpo4) in Sso (Table 2). We compared the nucleotide binding, kinetic incorporation rate, and the fidelity differences of Dpo3 with Dpo1 and Dpo4. Dpo3 possesses nucleotide selectivity (Kd difference) values more similar to the Y-family DNA polymerase, Dpo4. The ratio of the maximal rate of incorporation (kp) for correct versus incorrect nucleotides for Dpo3 is more similar to the selectivity provided by Dpo1, although the absolute rate of catalysis is roughly 100-fold slower. Therefore, the fidelity of Dpo3 is primarily driven by a reduced rate of incorporation for incorrect nucleotides rather than selectivity in nucleotide binding. The fidelity values for Dpo3 are comparable to those for Dpo4 but less than Dpo1 (Table 2). The exonuclease domain contained within Dpo3 will increase the fidelity value further; approaching that required for accurate DNA synthesis making this polymerase more accurate than Dpo4 (Figure 8B).
Table 2. Sulfolobus solfataricus.
DNA Polymerase Comparison
Variable Thermostabilities of the Polymerase and Exonuclease Domains
Interestingly, the temperatures in which the polymerase and exonuclease activities are maximal are different. The maximal exonuclease activity occurs at 55 °C which is below the physiological temperature while the maximal polymerase activity occurs above 65 °C. The thermostability of the entire Dpo3 structure is high with a TM of > 94 °C which is increased slightly in the presence of DNA. This is in direct contrast to a previous report which showed that preincubation for 20 minutes at 60 °C completely abolished activity (28). Conversely, we can show that preincubation at 70 °C for 55 minutes only minimally reduces polymerase activity and the exonuclease domain retains more than half the original activity. Thermostabilites for Dpo1 and Dpo4 have been measured previously (58, 59) and are similar to that found for Dpo3.
Looking closely at the CD spectra, we have noticed a reproducible but slight deviation around 63 °C suggesting that a local reversible unfolding event may be possible. This spectral change is not observed when DNA is included in the experiment suggesting that a more stable protein complex exists in the presence of DNA, possibly representing the closed conformation. Based on the decrease in promiscuous exonuclease activity above 55 °C and the CD spectra change at 63 °C, we conclude that the exonuclease domain is stabilized in a proofreading conformation in the presence of DNA at high temperatures.
When dNTPs are included, the role of the exonuclease activity is to increase the fidelity by monitoring correct base incorporations. Clearly, there is a cycle between nucleotide incorporation and exonuclease proofreading that occurs with the wild-type enzyme similar to other B-family DNA polymerases. The exonuclease activity is controlled by kinetics of the forward rate constant for nucleotide incorporation. When dNTPs are absent or in low abundance, the kinetic rate of incorporation is low and causes the slightly slower exonuclease rate to proceed instead. For Dpo3, the exonuclease activity is somewhat temperature dependent. The change in the CD spectrum coupled with slower kinetics at higher temperatures suggest that the DNA binding specificity (ssDNA or ptDNA) may be modulated by temperature. This may also help explain the lower exonuclease activity on ssDNA versus ptDNA at higher temperature in spite of somewhat similar affinities. Even so, the recognition or proofreading of misincorporated bases opposite damaged templates (hypoxanthine, 8-oxoG, and cyclobutane dimers) may be less restrictive in some cases dependent on the geometry as Dpo3 has been shown recently to be able to bypass these lesions (28).
Role of Multiple B-family DNA polymerases in Archaea
The DNA replication system from Sulfolobus is now an even more enticing model system with the discovery that there are multiple active B-family DNA polymerases along with a single Y-family lesion bypass polymerase. This is similar to other organisms which have multiple DNA replication polymerases at the replication fork (7, 60). The measured in vitro activity of Dpo3 is absolutely slower than the proposed DNA replication polymerase, Dpo1 (Figure 8B). Therefore, it seems that Dpo1 will provide the major replicative activity during replication. Of course, we cannot rule out an increase in activity for Dpo3 with additional accessory factors in vivo. The included active exonuclease proofreading domain of Dpo3 would suggest that it has a role in faithful DNA replication and not necessarily repair or lesion bypass. Of course, genetic knockouts of this polymerase could provide some further information on the proposed role in genomic maintenance.
DNA binding and recognition of the DNA template will be difficult for Dpo3 due to its weaker affinity. We have shown previously that Dpo1 can form a trimeric complex at a similar concentration range (42). It was noted recently, that the protein expression levels of Dpo3 are similar to Dpo1 and much greater than Dpo4 (28), suggesting that the cellular concentrations and thermodynamics may favor formation of a Dpo3/DNA complex. Therefore, the local concentrations of polymerases around the replication fork will strongly influence associations.
Based on the enzymatic properties described here, Dpo3 could be involved in initiating DNA replication, extension of Okazaki fragments on the lagging strand (61), or in a more directed role of synthesis across specific lesions (28). The initiation of DNA replication after RNA priming is performed by DNA polymerase α in humans. There is no direct homolog in archaea and the polymerase responsible for extending the RNA primer to initiate DNA replication has not been identified (62). The measured in vitro synthesis rate of Dpo3 extrapolated at 75 °C is roughly 150 s−1, based on the rate increase measured with temperature for Dpo1 (22). This is slightly faster than required (92 s−1) for bidirectional synthesis of the leading strand at three origins (63) on the Sso genome (2.99 × 106 bases) (64) over a 90 minute S-phase (65). It is equally possible that synthesis of the short 100–150 base Okazaki fragments (66) could be performed by Dpo3 in parallel to provide the necessary speed for genomic replication. Probably just as likely though, Dpo3 could be confined to a specialized role in DNA repair, although with the identification of an active exonuclease domain, the specific lesions that are processed remain to be determined. Identification of interacting protein partners with Dpo3 may be able to better reveal a potential role for this polymerase in vivo. Clearly, there are multiple kinetic and thermodynamic association events that occur with DNA in an organism with multiple DNA polymerases. The regulation of each DNA polymerase’s individual activity will be dynamically controlled by cellular concentrations and interactions with other accessory proteins that direct binding along with their individual kinetics to maintain the genome.
Supplementary Material
Acknowledgments
Funding was provided from startup funds from the University of Pittsburgh, Department of Chemistry (M.A.T.) and a Research Scholar Grant (RSG-11-049-01-DMC) to M.A.T. from the American Cancer Society.
We thank Hsiang-Kai Lin for performing the circular dichroism experiments and Brian Graham for critical reading of the manuscript.
ABBREVIATIONS
- Sso
Sulfolobus solfataricus
- Dpo1
Sso DNA polymerase I
- Dpo2
Sso DNA polymerase II
- Dpo3
Sso DNA polymerase III
- CD
circular dichroism
- ssDNA
single stranded DNA
- ptDNA
primer template DNA
- dsDNA
double stranded DNA
- Tm
Thermal melting temperature
- Kd
dissociation constant
- kp
presteady-state rate constant
- kobs
observed rate constant
- K’d
apparent dissociation constant
- WT
wild-type
Footnotes
SUPPORTING INFORMATION AVAILABLE
Table S1 (DNA substrates), Table S2 (DNA primers), Figure S1 (buffer and metal optimization), Figure S2 (example gels of time course of incorporation for polymerase active site mutants), Figure S3 (additional presteady-state correct incorporation second order plots) are available. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Filee J, Forterre P, Sen-Lin T, Laurent J. Evolution of DNA polymerase families: evidences for multiple gene exchange between cellular and viral proteins. J Mol Evol. 2002;54:763–773. doi: 10.1007/s00239-001-0078-x. [DOI] [PubMed] [Google Scholar]
- 2.Burgers PM, Koonin EV, Bruford E, Blanco L, Burtis KC, Christman MF, Copeland WC, Friedberg EC, Hanaoka F, Hinkle DC, Lawrence CW, Nakanishi M, Ohmori H, Prakash L, Prakash S, Reynaud CA, Sugino A, Todo T, Wang Z, Weill JC, Woodgate R. Eukaryotic DNA polymerases: Proposal for a revised nomenclature. J Biol Chem. 2001;276:43487–43490. doi: 10.1074/jbc.R100056200. [DOI] [PubMed] [Google Scholar]
- 3.Hubscher U, Maga G, Spadari S. Eukaryotic DNA polymerases. Annu Rev Biochem. 2002;71:133–163. doi: 10.1146/annurev.biochem.71.090501.150041. [DOI] [PubMed] [Google Scholar]
- 4.Bebenek K, Kunkel TA. Functions of DNA polymerases. Adv Protein Chem. 2004;69:137–165. doi: 10.1016/S0065-3233(04)69005-X. [DOI] [PubMed] [Google Scholar]
- 5.Kunkel TA, Bebenek K. DNA replication fidelity. Annu Rev Biochem. 2000;69:497–529. doi: 10.1146/annurev.biochem.69.1.497. [DOI] [PubMed] [Google Scholar]
- 6.Jeruzalmi D, O’Donnell M, Kuriyan J. Clamp loaders and sliding clamps. Curr Opin Struct Biol. 2002;12:217–224. doi: 10.1016/s0959-440x(02)00313-5. [DOI] [PubMed] [Google Scholar]
- 7.Nick McElhinny SA, Gordenin DA, Stith CM, Burgers PM, Kunkel TA. Division of labor at the eukaryotic replication fork. Mol Cell. 2008;30:137–144. doi: 10.1016/j.molcel.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beckman J, Kincaid K, Hocek M, Spratt T, Engels J, Cosstick R, Kuchta RD. Human DNA polymerase alpha uses a combination of positive and negative selectivity to polymerize purine dNTPs with high fidelity. Biochemistry. 2007;46:448–460. doi: 10.1021/bi061243s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burgers PM. Polymerase dynamics at the eukaryotic DNA replication fork. J Biol Chem. 2009;284:4041–4045. doi: 10.1074/jbc.R800062200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuchta RD, Stengel G. Mechanism and evolution of DNA primases. Biochim Biophys Acta. 2010;1804:1180–1189. doi: 10.1016/j.bbapap.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stillman B. DNA polymerases at the replication fork in eukaryotes. Mol Cell. 2008;30:259–260. doi: 10.1016/j.molcel.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grabowski B, Kelman Z. Archeal DNA replication: Eukaryal proteins in a bacterial context. Annu Rev Microbiol. 2003;57:487–516. doi: 10.1146/annurev.micro.57.030502.090709. [DOI] [PubMed] [Google Scholar]
- 13.Barry ER, Bell SD. DNA replication in the archaea. Microbiol Mol Biol Rev. 2006;70:876–887. doi: 10.1128/MMBR.00029-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Majernik AI, Jenkinson ER, Chong JP. DNA replication in thermophiles. Biochem Soc Trans. 2004;32:236–239. doi: 10.1042/bst0320236. [DOI] [PubMed] [Google Scholar]
- 15.Leipe DD, Aravind L, Koonin EV. Did DNA replication evolve twice independently? Nucleic Acids Res. 1999;27:3389–3401. doi: 10.1093/nar/27.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cann IKO, Komori K, Toh H, Kanai S, Ishino Y. A heterodimeric DNA polymerase: Evidence that members of Euryarchaeota possess a distinct DNA polymerase. Proc Natl Acad Sci U S A. 1998;95:14250–14255. doi: 10.1073/pnas.95.24.14250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kulaeva OI, Koonin EV, McDonald JP, Randall SK, Rabinovich N, Connaughton JF, Levine AS, Woodgate R. Identification of a DinB/UmuC homolog in the archeon Sulfolobus solfataricus. Mutat Res. 1996;357:245–253. doi: 10.1016/0027-5107(96)00164-9. [DOI] [PubMed] [Google Scholar]
- 18.Gruz P, Shimizu M, Pisani FM, De FM, Kanke Y, Nohmi T. Processing of DNA lesions by archaeal DNA polymerases from Sulfolobus solfataricus. Nucleic Acids Res. 2003;31:4024–4030. doi: 10.1093/nar/gkg447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tahirov TH, Makarova KS, Rogozin IB, Pavlov YI, Koonin EV. Evolution of DNA polymerases: an inactivated polymerase-exonuclease module in Pol epsilon and a chimeric origin of eukaryotic polymerases from two classes of archaeal ancestors. Biol Direct. 2009;4:11. doi: 10.1186/1745-6150-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brown JA, Suo Z. Elucidating the kinetic mechanism of DNA polymerization catalyzed by Sulfolobus solfataricus P2 DNA polymerase B1. Biochemistry. 2009;48:7502–7511. doi: 10.1021/bi9005336. [DOI] [PubMed] [Google Scholar]
- 21.Fiala KA, Suo Z. Mechanism of DNA polymerization catalyzed by Sulfolobus solfataricus P2 DNA polymerase IV. Biochemistry. 2004;43:2116–2125. doi: 10.1021/bi035746z. [DOI] [PubMed] [Google Scholar]
- 22.Zhang L, Brown JA, Newmister SA, Suo Z. Polymerization fidelity of a replicative DNA polymerase from the hyperthermophilic archaeon Sulfolobus solfataricus P2. Biochemistry. 2009;48:7492–7501. doi: 10.1021/bi900532w. [DOI] [PubMed] [Google Scholar]
- 23.Fiala KA, Suo Z. Pre-steady-state kinetic studies of the fidelity of Sulfolobus solfataricus P2 DNA polymerase IV. Biochemistry. 2004;43:2106–2115. doi: 10.1021/bi0357457. [DOI] [PubMed] [Google Scholar]
- 24.Ling H, Boudsocq F, Woodgate R, Yang W. Crystal structure of a Y-family DNA polymerase in action: a mechanism for error-prone and lesion-bypass replication. Cell. 2001;107:91–102. doi: 10.1016/s0092-8674(01)00515-3. [DOI] [PubMed] [Google Scholar]
- 25.Savino C, Federici L, Johnson KA, Vallone B, Nastopoulos V, Rossi M, Pisani FM, Tsernoglou D. Insights into DNA replication: the crystal structure of DNA polymerase B1 from the archaeon Sulfolobus solfataricus. Structure. 2004;12:2001–2008. doi: 10.1016/j.str.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 26.Edgell DR, Klenk HP, Doolittle WF. Gene duplications in evolution of archaeal family B DNA polymerases. J Bacteriol. 1997;179:2632–2640. doi: 10.1128/jb.179.8.2632-2640.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Prangishvili D, Klenk HP. Nucleotide sequence of the gene for a 74 kDa DNA polymerase from the archaeon Sulfolobus solfataricus. Nucleic Acids Res. 1993;21:2768. doi: 10.1093/nar/21.11.2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choi JY, Eoff RL, Pence MG, Wang J, Martin MV, Kim EJ, Folkmann LM, Guengerich FP. Roles of the four DNA polymerases of the crenarchaeon Sulfolobus solfataricus and accessory proteins in DNA replication. J Biol Chem. 2011;286:31180–31193. doi: 10.1074/jbc.M111.258038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sambrook JaDWR. Molecular Cloning: A Laboratory Manual. 1–3. CSHL Press; Cold Spring Harbor: 2001. [Google Scholar]
- 30.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 31.Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 32.Marky LA, Breslauer KJ. Calculating thermodynamic data for transitions of any molecularity from equilibrium melting curves. Biopolymers. 1987;26:1601–1620. doi: 10.1002/bip.360260911. [DOI] [PubMed] [Google Scholar]
- 33.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 34.Franklin MC, Wang J, Steitz TA. Structure of the replicating complex of a pol alpha family DNA polymerase. Cell. 2001;105:657–667. doi: 10.1016/s0092-8674(01)00367-1. [DOI] [PubMed] [Google Scholar]
- 35.Hillebrand GG, Beattie K. Template–dependent variation in the relative fidelity of DNA polymerase I of Escherichia coli in the presence of Mg2+ versus Mn2+ Nucleic Acids Res. 1984;12:3173–3183. doi: 10.1093/nar/12.7.3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Polesky AH, Steitz TA, Grindley ND, Joyce CM. Identification of residues critical for the polymerase activity of the Klenow fragment of DNA polymerase I from Escherichia coli. J Biol Chem. 1990;265:14579–14591. [PubMed] [Google Scholar]
- 37.Bernad A, Blanco L, Salas M. Site-directed mutagenesis of the YCDTDS amino acid motif of the phi 29 DNA polymerase. Gene. 1990;94:45–51. doi: 10.1016/0378-1119(90)90466-5. [DOI] [PubMed] [Google Scholar]
- 38.Copeland WC, Wang TS. Mutational analysis of the human DNA polymerase alpha. The most conserved region in alpha-like DNA polymerases is involved in metal-specific catalysis. J Biol Chem. 1993;268:11028–11040. [PubMed] [Google Scholar]
- 39.Dua R, Levy DL, Campbell JL. Analysis of the essential functions of the C-terminal protein/protein interaction domain of Saccharomyces cerevisiae pol epsilon and its unexpected ability to support growth in the absence of the DNA polymerase domain. J Biol Chem. 1999;274:22283–22288. doi: 10.1074/jbc.274.32.22283. [DOI] [PubMed] [Google Scholar]
- 40.Lowe LG, Guengerich FP. Steady-state and pre-steady-state kinetic analysis of dNTP insertion opposite 8-oxo-7,8-dihydroguanine by Escherichia coli polymerases I exo- and II exo. Biochemistry. 1996;35:9840–9849. doi: 10.1021/bi960485x. [DOI] [PubMed] [Google Scholar]
- 41.Korona DA, Lecompte KG, Pursell ZF. The high fidelity and unique error signature of human DNA polymerase epsilon. Nucleic Acids Res. 2011;39:1763–1773. doi: 10.1093/nar/gkq1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mikheikin AL, Lin HK, Mehta P, Jen-Jacobson L, Trakselis MA. A trimeric DNA polymerase complex increases the native replication processivity. Nucleic Acids Res. 2009;37:7194–7205. doi: 10.1093/nar/gkp767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bebenek A, Dressman HK, Carver GT, Ng S, Petrov V, Yang G, Konigsberg WH, Karam JD, Drake JW. Interacting fidelity defects in the replicative DNA polymerase of bacteriophage RB69. J Biol Chem. 2001;276:10387–10397. doi: 10.1074/jbc.M007707200. [DOI] [PubMed] [Google Scholar]
- 44.Fazlieva R, Spittle CS, Morrissey D, Hayashi H, Yan H, Matsumoto Y. Proofreading exonuclease activity of human DNA polymerase δ and its effects on lesion-bypass DNA synthesis. Nucleic Acids Research. 2009;37:2854–2866. doi: 10.1093/nar/gkp155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fiala KA, Sherrer SM, Brown JA, Suo Z. Mechanistic consequences of temperature on DNA polymerization catalyzed by a Y-family DNA polymerase. Nucleic Acids Res. 2008;36:1990–2001. doi: 10.1093/nar/gkn004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dieckman LM, Johnson RE, Prakash S, Washington MT. Pre-steady state kinetic studies of the fidelity of nucleotide incorporation by yeast DNA polymerase delta. Biochemistry. 2010;49:7344–7350. doi: 10.1021/bi100556m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhong X, Pedersen LC, Kunkel TA. Characterization of a replicative DNA polymerase mutant with reduced fidelity and increased translesion synthesis capacity. Nucleic Acids Res. 2008;36:3892–3904. doi: 10.1093/nar/gkn312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rogozin IB, Makarova KS, Pavlov YI, Koonin EV. A highly conserved family of inactivated archaeal B family DNA polymerases. Biol Direct. 2008;3:32. doi: 10.1186/1745-6150-3-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blasco MA, Bernad A, Blanco L, Salas M. Characterization and mapping of the pyrophosphorolytic activity of the phage phi 29 DNA polymerase. Involvement of amino acid motifs highly conserved in alpha-like DNA polymerases. J Biol Chem. 1991;266:7904–7909. [PubMed] [Google Scholar]
- 50.Swan MK, Johnson RE, Prakash L, Prakash S, Aggarwal AK. Structural basis of high-fidelity DNA synthesis by yeast DNA polymerase delta. Nat Struc Mol Biol. 2009;16:979–U107. doi: 10.1038/nsmb.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zahn KE, Tchesnokov EP, Gotte M, Doublie S. Phosphonoformic acid inhibits viral replication by trapping the closed form of the DNA polymerase. J Biol Chem. 2011;286:25246–25255. doi: 10.1074/jbc.M111.248864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xia S, Wang M, Blaha G, Konigsberg WH, Wang J. Structural insights into complete metal ion coordination from ternary complexes of B family RB69 DNA polymerase. Biochemistry. 2011;50:9114–9124. doi: 10.1021/bi201260h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saturno J, Lazaro JM, Blanco L, Salas M. Role of the first aspartate residue of the “YxDTDS” motif of phi29 DNA polymerase as a metal ligand during both TP-primed and DNA-primed DNA synthesis. J Mol Biol. 1998;283:633–642. doi: 10.1006/jmbi.1998.2121. [DOI] [PubMed] [Google Scholar]
- 54.Kennedy EM, Hergott C, Dewhurst S, Kim B. The mechanistic architecture of thermostable Pyrococcus furiosus family B DNA polymerase motif A and its interaction with the dNTP substrate. Biochemistry. 2009;48:11161–11168. doi: 10.1021/bi9010122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Johnson KA. Transient-State Kinetic Analysis of Enzyme Reaction Pathways. In: David SS, editor. The Enzymes. Academic Press; 1992. pp. 1–61. [Google Scholar]
- 56.Fiala KA, Suo Z. Sloppy bypass of an abasic lesion catalyzed by a Y-family DNA polymerase. J Biol Chem. 2007;282:8199–8206. doi: 10.1074/jbc.M610719200. [DOI] [PubMed] [Google Scholar]
- 57.Fiala KA, Hypes CD, Suo Z. Mechanism of abasic lesion bypass catalyzed by a Y-family DNA polymerase. J Biol Chem. 2007;282:8188–8198. doi: 10.1074/jbc.M610718200. [DOI] [PubMed] [Google Scholar]
- 58.Pisani FM, De FM, Rossi M. Amino acid residues involved in determining the processivity of the 3′-5′ exonuclease activity in a family B DNA polymerase from the thermoacidophilic archaeon Sulfolobus solfataricus. Biochemistry. 1998;37:15005–15012. doi: 10.1021/bi981127s. [DOI] [PubMed] [Google Scholar]
- 59.Boudsocq F, Iwai S, Hanaoka F, Woodgate R. Sulfolobus solfataricus P2 DNA polymerase IV (Dpo4): an archaeal DinB-like DNA polymerase with lesion-bypass properties akin to eukaryotic poleta. Nucleic Acids Res. 2001;29:4607–4616. doi: 10.1093/nar/29.22.4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McInerney P, Johnson A, Katz F, O’Donnell M. Characterization of a triple DNA polymerase replisome. Mol Cell. 2007;27:527–538. doi: 10.1016/j.molcel.2007.06.019. [DOI] [PubMed] [Google Scholar]
- 61.Duggin IG, Bell SD. The chromosome replication machinery of the archaeon Sulfolobus solfataricus. J Biol Chem. 2006;281:15029–15032. doi: 10.1074/jbc.R500029200. [DOI] [PubMed] [Google Scholar]
- 62.Zuo Z, Rodgers CJ, Mikheikin AL, Trakselis MA. Characterization of a functional DnaG-type primase in archaea: Implications for a dual-primase system. J Mol Biol. 2010;397:664–676. doi: 10.1016/j.jmb.2010.01.057. [DOI] [PubMed] [Google Scholar]
- 63.Robinson NP, Bell SD. Origins of DNA replication in the three domains of life. FEBS J. 2005;272:3757–3766. doi: 10.1111/j.1742-4658.2005.04768.x. [DOI] [PubMed] [Google Scholar]
- 64.She Q, Singh RK, Confalonieri F, Zivanovic Y, Allard G, Awayez MJ, Chan-Weiher CC, Clausen IG, Curtis BA, De MA, Erauso G, Fletcher C, Gordon PM, Heikamp-de JI, Jeffries AC, Kozera CJ, Medina N, Peng X, Thi-Ngoc HP, Redder P, Schenk ME, Theriault C, Tolstrup N, Charlebois RL, Doolittle WF, Duguet M, Gaasterland T, Garrett RA, Ragan MA, Sensen CW, Van der OJ. The complete genome of the crenarchaeon Sulfolobus solfataricus P2. Proc Natl Acad Sci U S A. 2001;98:7835–7840. doi: 10.1073/pnas.141222098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Duggin IG, McCallum SA, Bell SD. Chromosome replication dynamics in the archaeon Sulfolobus acidocaldarius. Proc Natl Acad Sci U S A. 2008;105:16737–16742. doi: 10.1073/pnas.0806414105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Matsunaga F, Norais C, Forterre P, Myllykallio H. Identification of short ‘eukaryotic’ Okazaki fragments synthesized from a prokaryotic replication origin. EMBO Rep. 2003;4:154–158. doi: 10.1038/sj.embor.embor732. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.