

Abstract
Fra-1 is aberrantly expressed in a large number of cancer cells and tissues, and emerging evidence suggests an important role for this Fos family protein in both oncogenesis and the progression or maintenance of many tumour types. Here, we show that the concentration of Fra-1 is high in invasive oestrogen receptor (ER)-negative (ER−) breast cancer cell lines, regardless of their Ras pathway status. All of the ER− cells express high levels of activated PKCθ, and the inhibition of PKCθ activity using RNA interference or the expression of a dominant-negative mutant results in a dramatic reduction in Fra-1 abundance. Conversely, the ectopic expression of constitutively active PKCθ leads to Fra-1 phosphorylation and accumulation in poorly invasive ER+ cells. This accumulation is due to the stabilisation of the Fra-1 protein through PKCθ signalling, whereas other members of the PKC family are ineffective. Both SPAK and ERK1/2, whose activities are up-regulated by PKCθ, participate in PKCθ-driven Fra-1 stabilisation. Interestingly, their relative contributions appear to be different depending on the cell line studied. ERK1/2 signalling has a major role in ER- MDA-MB-231 cells whereas Fra-1 accumulation occurs mainly through SPAK signalling in ER− BT549 cells. Fra-1 mutational analysis shows that the phosphorylation of S265, T223 and T230 is critical for PKCθ-driven Fra-1 stabilisation. Phosphorylation of the protein was confirmed using specific antisera against Fra-1 phosphorylated on T223 or S265. In addition, Fra-1 participates in PKCθ-induced cell invasion and is necessary for PKCθ-induced cell migration. In summary, we identified PKCθ signalling as an important regulator of Fra-1 accumulation in ER- breast cancer cells. Moreover, our results suggest that PKCθ could participate in progression of some breast cancers and could be a new therapeutic target.
Keywords: Breast Neoplasms; Cell Line, Tumor; Cell Movement; Female; Humans; Isoenzymes; genetics; metabolism; MAP Kinase Signaling System; Phosphorylation; Protein Kinase C; genetics; metabolism; Protein Processing, Post-Translational; Protein Stability; Protein-Serine-Threonine Kinases; metabolism; Proto-Oncogene Proteins c-fos; metabolism; Receptors, Estrogen; metabolism
INTRODUCTION
Activating protein 1 (AP-1) activity, which is induced by a vast number of extracellular stimuli, such as growth factors, cytokines, tumour promoters and environmental stresses, has diverse biological functions and plays critical roles in regulating cell growth, differentiation, apoptosis, development and tumourigenesis[1]. The AP-1 transcription factor is a dimeric protein complex comprising primarily Jun and Fos family members. While Jun proteins (c-Jun, JunB, and JunD) form homodimers or heterodimers with Fos proteins (c-Fos, Fra-1, Fra-2 and FosB), Fos proteins cannot associate with each other. Jun-Fos heterodimers interact more stably than Jun-Jun homodimers and therefore control transcription more efficiently. The combinatorial diversity of the dimers varies with the expression and activation levels of the individual components according to the cell type, the environmental situation and the phase of the cell cycle, suggesting that the various dimers display different properties and functions[2].
Emerging evidence suggests an important role for Fra-1 in oncogenesis and the progression or maintenance of many tumour types[3,4]. Fra-1 has been shown to be a mediator of the Ras-induced transformation of NIH3T3 cells and thyroid cells[5,6]. Whereas Fra-1 is constitutively expressed in a limited number of tissues, a high Fra-1 concentration is found in numerous cancer cell lines and tissues, including thyroid, breast, lung, brain, endometrial, prostate, bladder and colon carcinomas. Furthermore, in these different models, the manipulation of the Fra-1 concentration has indicated the active role of Fra-1 in the maintenance or acquisition of a more aggressive phenotype [7–12].
In breast cancer, the most invasive cell lines have high AP-1 DNA-binding activity that is mostly due to Fra-1-containing heterodimers[13]. Aberrant Fra-1 protein levels have been detected in cells expressing neither oestrogen α (ER) nor progesterone receptors (PR) and expressing a number of mesenchymal markers. Moreover, Fra-1 plays an active role in breast cancer cell growth, invasion, motility and the control of cell morphology[7,10]. Altogether, these data suggest that Fra-1 could be involved in breast cancer progression. In agreement with these studies, Fra-1 expression has been associated with hyperplastic and neoplastic proliferative breast disorders[14–17]. Moreover, the tumour cell-induced de novo overexpression of Fra-1 in macrophages has recently been suggested to play a role in the immunosuppressive mechanisms correlated with mammary tumour progression[18].
Fra-1 is regulated at the transcriptional level through numerous extracellular stimuli. However, Fra-1 is an intrinsically unstable protein and the regulation of its stability may be fundamental for its accumulation[4]. Fra-1 is among the most upregulated targets under Ras transformation conditions and its accumulation depends on both transcriptional auto-regulation and ERK-dependent post-translational stabilisation. Indeed, the Fra-1 half-life is increased upon ERK1/2 pathway activation in thyroid[19] and colon tumours[20,21]. The ERK1/2 pathway has been shown to lead to the phosphorylation of serines S252 and S265, thereby inhibiting Fra-1 degradation during both normal physiological induction and the constitutive activation of this cascade in human colon cancer cells expressing oncogenic forms of KRAS and BRAF, which both activate ERK. However, because Ras mutations are not frequent in breast cancer cells[22], we hypothesised that other kinases might play a role in the aberrant accumulation of hyperphosphorylated Fra-1 in these cells. Here, we tested the role of the PKCθ pathway.
PKCθ is a novel PKC that is activated by diacylglycerol but not by calcium[23]. This serine/threonine kinase is a critical component of the immune system, in which it controls T lymphocyte fate and function[24]. PKCθ is overexpressed in gastrointestinal stromal tumours[25] and has only recently been implicated in breast cancers. PKCθ has been reported to promote c-Rel-driven mammary tumourigenesis in mice by repressing ERα synthesis[26]. In addition, the PKCθ protein stimulates the proliferation and motility of breast cancer cells and is detectable and present in an active form only in ER-negative (ER−) breast cancer cells. Along the same line, ER− tumours in patients express an elevated level of PKCθ mRNA compared to ER+ tumours[27].
We report here that high PKCθ activity leads to a strong expression of Fra-1 in ER− invasive breast cancer cell lines. PKCθ acts through the activation of ERK1/2 and Ste20-related proline-alanine-rich kinase (SPAK) pathways and stabilises Fra-1 protein by inducing its phosphorylation on S265, T223 and T230. Moreover, the high accumulation of Fra-1 induced by the PKCθ pathway is critical to mediate the effect of this kinase on cell migration.
RESULTS
High PKCθ activity in ER-negative breast cancer cells leads to aberrant Fra-1 accumulation
We and others have previously reported that Fra-1 is aberrantly expressed in invasive ER-negative breast cancer cell lines and that the protein is mainly present in hyperphosphorylated forms, the migration of which appears retarded in SDS-PAGE analysis. As shown in Figure 1A, Fra-1 protein level was high even in ER− cells displaying modest ERK1/2 activity (P-ERK1/2). Interestingly, activated PKCθ (P-PKCθ) was expressed in all of these ER− cells, as assayed by analysing one of its activating phosphorylations (T538), leading us to hypothesise that this kinase might promote the strong accumulation of Fra-1 in these invasive cells.
Figure 1. High PKCθ activity in ER-negative breast cancer cell lines increases Fra-1 expression.
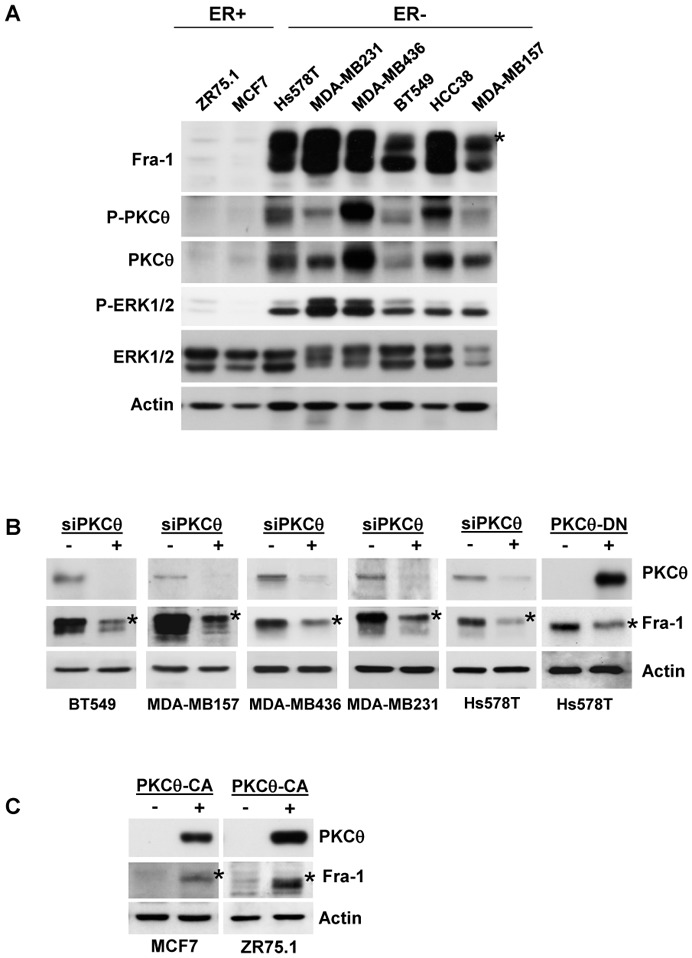
A. Whole-cell extracts (WCEs, 90 μg) from the indicated breast cancer cell lines were analysed using immunoblotting with antibodies against Fra-1, ERK1/2, phospho-ERK1/2 (T202/Y204), PKCθ, phospho-PKCθ (T538) and actin, which was used to confirm equal loading. B. The indicated ER− breast cancer cell lines were transfected with two different siRNAs (3.6 nM each) targeting the PKCθ mRNA. After two successive 48h-transfections, WCEs (90 μg) were subjected to immunoblotting for Fra-1, PKCθ, and actin, the latter being used to confirm equal loading. Alternatively, Hs578T cells were transiently transfected with 2 μg of PKCθ-DN expression vector (+) or empty vector (−) and, 48 h after transfection, WCEs (90 μg) were analysed using immunoblotting. C. The indicated ER+ breast cancer cell lines were transiently transfected with 2 μg of PKCθ-CA expression vector (+) or empty vector (−) and analysed as in B. In A, B and C, phosphorylated Fra-1 forms are indicated by an asterisk.
To verify our hypothesis, we inhibited activated PKCθ expression through the transfection of specific siRNA in ER− cells (Figure 1B). RNA interference resulted in a significant reduction in the Fra-1 levels in the ER− cell lines in which PKCθ silencing was efficient. In addition, the high transfection efficiency of plasmids in Hs578T cells allowed us to obtain a significant inhibition of Fra-1 expression after overexpression of a kinase-defective PKCθ variant (PKCθ-DN), which serves as a dominant negative mutant for PKCθ activity (Figure 1B). Conversely, the transient transfection of a constitutively active mutant of PKCθ (PKCθ-CA) in two ER+ breast cancer cell lines, MCF7 and ZR75.1, greatly increased endogenous Fra-1 content compared to control cells (Figure 1C). Altogether, these data indicate that the activation of PKCθ modulates the level of Fra-1 in breast cancer cells.
We then chose to pursue this study in the ER+ MCF7 and ZR75.1 cell lines and in the ER− MDA-MB-231 and BT549 cell lines, which express strong or more modest activated ERK1/2 level, respectively.
The PKCθ pathway increases Fra-1 protein stability and Fra-1 phosphorylation
To assess whether PKCθ signalling increased Fra-1 expression via a post-transcriptional mechanism, as the ERK1/2 pathway does, MCF7 and ZR75.1 cells were transfected with a pIRES2-EGFP construct allowing the expression of myc-tagged Fra-1 and of GFP from the same bicistronic mRNA[21]. The myc tag enabled us to discriminate ectopically expressed Fra-1 from endogenous Fra-1 and, due to its high stability, GFP served as an internal standard to assess Fra-1 expression level. As shown in Figure 2A, introduction of PKCθ-CA drastically increased the expression of the myc-tagged Fra-1 protein, indicating that the PKCθ pathway acted, at least in part, via a post-transcriptional mechanism. Conversely, the inhibition of PKCθ using siRNA significantly decreased Fra-1 protein expression in two ER-negative breast cancer cell lines. The effect of PKCθ on Fra-1 protein stabilisation was then tested in MCF7 cells transiently transfected with a Fra-1 expression vector in the presence or absence of PKCθ-CA followed by incubation with cycloheximide (an inhibitor of protein synthesis) for increasing periods of time (Figure 2B). In the absence of cycloheximide, PKCθ-CA increased Fra-1 expression level (note that a 10-fold higher concentration of total protein was analysed for control cells than for cells expressing the constitutively active kinase to allow for a more accurate comparison of Fra-1 levels). In addition, PKCθ-CA drastically reversed the ratio of the Fra-1 fastest- to slowest-migrating bands (the shift in migration was particularly drastic in this experiment due to a long electrophoretic migration). The most retarded band corresponded to hyperphosphorylated Fra-1, as established after treatment of cell extracts with alkaline phosphatase (not shown). Moreover, in the presence of cycloheximide, the Fra-1 protein was degraded more rapidly under control conditions than in cells expressing activated PKCθ, where Fra-1 was stable for at least 6 hours. We therefore concluded that PKCθ increased Fra-1 phosphorylation and stability.
Figure 2. PKCθ increases Fra-1 protein stability in breast cancer cells.
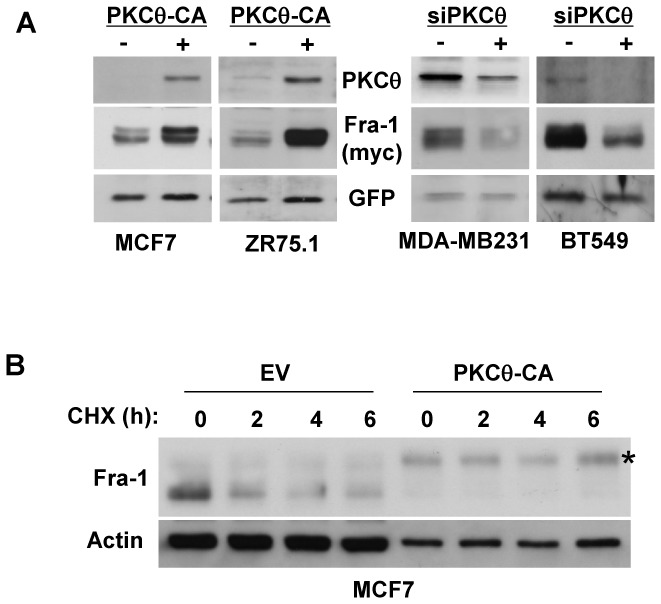
A. The indicated breast cancer cell lines were transiently transfected with 50 ng of pIRES2-EGFP vector expressing Fra-1 in the presence (+) or absence (−) of either the PKCθ-CA expression vector or siRNAs targeting the PKCθ gene as in Figure 1C and 1B, respectively. Then, WCEs (90 μg) were analysed by immunoblotting for the expression of PKCθ, Myc (detecting exogenous Fra-1) and GFP, which served as an internal standard for the normalisation of Myc-tagged Fra-1 expression. B. MCF7 cells were transiently transfected with 50 ng of pCI-Fra-1 expression vector in the presence (PKCθ-CA) or absence (EV) of the PKCθ-CA expression vector. Two days after transfection, cells were treated with 50 μg/ml of cycloheximide (CHX) for the indicated times. WCEs were then analysed using immunoblotting.
We then tested the effect of other members of the PKC family, specifically those that have been implicated in cancer (Figure 3). We performed the experiments in the ER− MDA-MB231 cells, in which Fra-1 is aberrantly expressed and hyperphosphorylated, to ensure that the kinases responsible for endogenous Fra-1 phosphorylation were active. Cells were transfected with Fra-1-pIRES2-EGFP and co-transfected with expression vectors expressing either PKCθ or other PKC proteins tagged with GFP. Although other PKCs, such as PKCα and PKCε, were preferentially expressed in ER− cells (not shown), all tested PKCs, except PKCθ, had little or no effect on Fra-1 protein stability in invasive MDA-MB231 cells (Figure 3). Similar data were obtained when the same experiment was performed in MCF7 cells (not shown). This result suggested that Fra-1 was a specific target of the PKCθ signalling pathway.
Figure 3. PKCθ stabilises Fra-1 protein, while other PKC subfamily members do not.
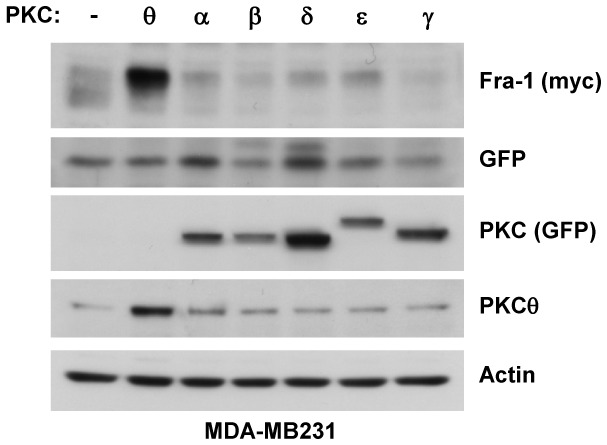
MDA-MB231 cells were transfected with 50 ng of the pIRES2-EGFP vector expressing Fra-1 together with 2 μg of vector expressing the indicated wild-type PKC subfamily members or empty vector (−). Two days later, WCEs were analysed using immunoblotting for the expression of PKCθ and GFP (detecting the ectopic expression of PKCα, α:, γ, δ, and ε on the top part of the blot), Myc (detecting exogenous Fra-1), GFP (detected at the bottom part of the blot and serving as an internal standard for the normalisation of Myc-tagged Fra-1 expression), and actin.
ERK1/2 and SPAK participate in Fra-1 regulation through the PKCθ pathway
We then addressed how PKCθ regulates Fra-1 stability. We observed that the overexpression of constitutively activated PKCθ in MCF7 cells increased the level of activated ERK1/2 (Figure 4A). However, the treatment of the cells with the pharmacological MEK/ERK inhibitor U0126, which efficiently inhibited ERK1/2 activation, only partially blocked PKCθ-induced Fra-1 expression, suggesting that ERK1/2 activation by PKCθ is only partly implicated in PKCθ-driven Fra-1 accumulation. Basbous et al. have reported that the ERK1/2 pathway leads to Fra-1 stabilisation via the phosphorylation of serines S252 and S265 in HeLa cells and colon cancer cell lines. To specify the role of ERK1/2 activity in PKCθ–driven Fra-1 stabilisation, we compared the effect of S252A and S265A mutations on the ability of ERK1/2 and PKCθ pathways to stabilise the Fra-1 protein. MCF7 cells were transfected with the bicistronic pIRES2-EGFP vector coding for either wild-type Fra-1 or a Fra-1 mutant in which the two serines were replaced by alanines. ERK1/2 and PKCθ signalling were activated through co-transfection of constitutively active MEK1 (S218D-S222D) or PKCθ-CA variants, respectively (Figure 4B). In agreement with published results, the S252A/S265A mutation strongly inhibited Fra-1 stabilisation induced by the ERK1/2 pathway. Interestingly, these two mutations were also sufficient to abrogate ERK5-driven Fra-1 expression in cells in which ERK5 signalling was activated after transfection with constitutively active MEK5 and wild-type ERK5. However, activated PKCθ still stabilised the mutant, although less efficiently than wild-type Fra-1, thus demonstrating that the activation of the ERK1/2 pathway by PKCθ is not sufficient to explain PKCθ-driven Fra-1 stabilisation in breast cancer cells.
Figure 4. The ERK1/2 pathway is partly involved in the stabilisation of Fra-1 protein through PKCθ signalling.
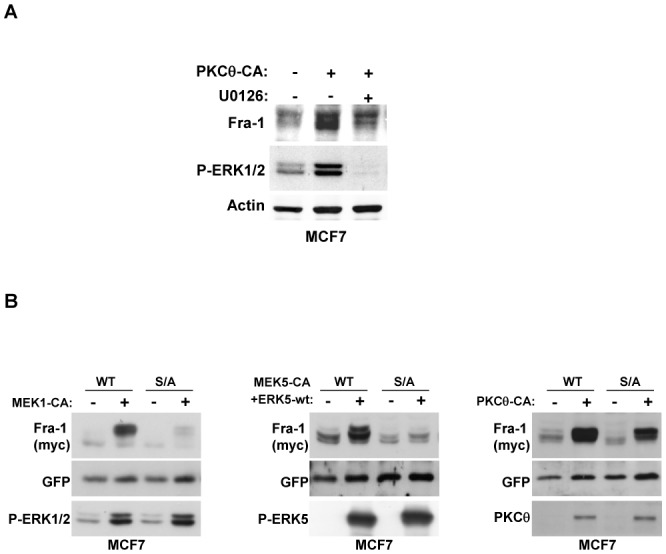
A. MCF7 cells were transfected with 2 μg of PKCθ-CA expression vector (+) or empty vector (−). One day later, cells were treated with 5 μM of the pharmacological inhibitor U0126 (+) or DMSO (−) for 16 h. WCEs were then analysed using immunoblotting for endogenous levels of activated ERK1/2, Fra-1, and actin. B. MCF7 cells were transiently transfected with 50 ng of the pIRES2-EGFP vector expressing either wild-type Fra-1 (WT) or mutated Fra-1 in which serines 252 and 265 were replaced by alanine (S/A). To activate the indicated pathways, cells were co-transfected with either 2 μg of the MEK1-CA expression vector, or 1 μg of MEK5-CA plus 1 μg of the ERK5-wt expression vector, or 2 μg of the PKCθ-CA expression vector, or their corresponding empty vector (−). Two days later, WCEs were analysed using immunoblotting with antibodies against Fra-1, GFP, phospho-ERK1/2 (T202/Y204), phospho-ERK5 (T218/Y220) and PKCθ as indicated.
Based on these data, we studied the potential role of the serine/threonine kinase SPAK, whose activity is stimulated by PKCθ in T lymphocytes[28]. The silencing of SPAK using RNA interference reduced the endogenous expression of Fra-1 in MDA-MB231 and BT549 cells (Figure 5A). This inhibition was also observed using another siRNA targeting the SPAK gene (not shown). Moreover, SPAK silencing decreased the endogenous Fra-1 accumulation induced by ectopic expression of activated PKCθ in MCF7 cells (Figure 5B, left panel). A similar experiment was performed by co-transfecting the bicistronic vector expressing wild-type Fra-1, and the data showed that PKCθ-induced stabilisation of exogenous Fra-1 was partially repressed after SPAK inhibition (Figure 5B, right panel). These results indicate that SPAK is involved in the stabilisation of Fra-1 upon PKCθ activation.
Figure 5. PKCθ stabilises Fra-1 protein through the cooperative activation of the SPAK and ERK1/2 pathways.
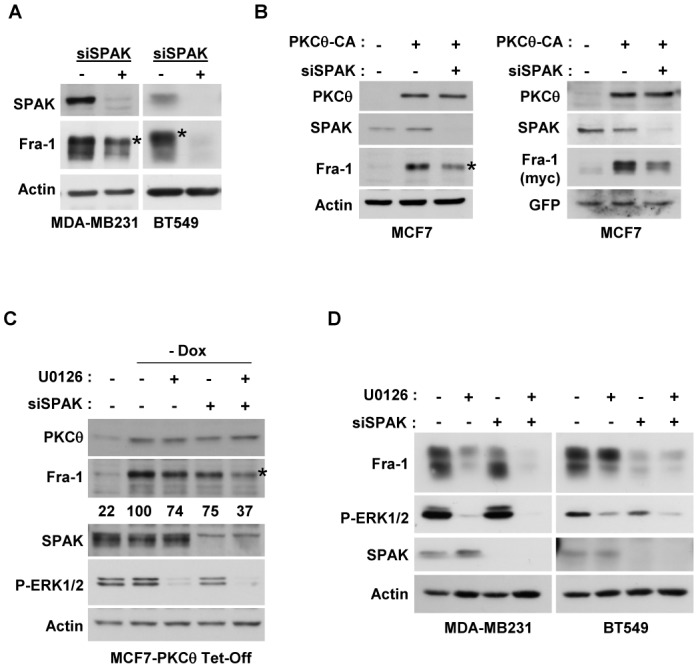
A. MDA-MB231 and BT549 cells were transfected with 3.6 nM of siRNA targeting the SPAK gene, and after two successive 48h-transfections, WCEs (90 μg) were subjected to immunoblotting for Fra-1, SPAK and actin. B. MCF7 cells were transfected with 3.6 nM of siRNA targeting the SPAK gene, and 24 h after two successive siRNA transfections, cells were transfected with 2 μg of the PKCθ-CA expression vector (+) or empty vector (−) in the absence (left panel) or presence of 50 ng of pIRES2-EGFP expressing wild-type Fra-1 (right panel). Two days after plasmid transfection, WCEs were analysed using immunoblotting. C. Stable MCF7 cells overexpressing PKCθ-CA under tetracycline control (MCF7-PKCθ Tet-Off) were transfected with 3.6 nM of siRNA targeting the SPAK gene in the presence (+) or absence (−) of doxycycline (Dox). One day after two successive siRNA transfections, cells were treated with 5 μM of the pharmacological inhibitor U0126 (+) or DMSO (−) for 16 h. Then, WCEs were analysed using immunoblotting. The blots were scanned, and the densitometry values of Fra-1 were normalised to actin. The values relative to the non-treated cells in the absence of Dox (set at 100%) are given below each lane. D. MDA-MB231 and BT549 cells were transfected with 3.6 nM of siRNA targeting the SPAK gene. One day after two successive siRNA transfections, cells were treated with 5 μM of the pharmacological inhibitor U0126 (+) or DMSO (−) for 16 h. Then, WCEs were analysed using immunoblotting. In A, B and C, phosphorylated Fra-1 forms are indicated by an asterisk.
The roles of SPAK and ERK1/2 were then investigated in MCF7 cells stably expressing constitutively active PKCθ under tetracycline control (Tet-off system) in which SPAK and ERK1/2 activities were inhibited using RNA interference or UO126, respectively (Figure 5C). Whereas each treatment alone slightly reduced PKCθ-induced Fra-1 expression, an inhibition of approximately 63% was observed when both the SPAK and ERK1/2 pathways were repressed. The relative efficiency of the two pathways in regulating endogenous Fra-1 expression was then tested in the invasive ER− breast cancer cell lines, MDA-MB231 and BT549, which express highly or moderately activated ERK1/2 levels, respectively (Figure 5D). Whereas Fra-1 stabilisation mainly occurred through the ERK1/2 pathway in MDA-MB231 cells, SPAK had an essential role in stabilising the Fra-1 protein in the BT549 cells.
The phosphorylation of serine 265 and threonines 223 and 230 is important for PKCθ-driven Fra-1 stabilisation
To determine whether the phosphorylation and the stabilisation of Fra-1 by the PKCθ pathway were directly linked, a series of Fra-1 mutants in which serines and threonines were replaced with alanine was constructed in the bicistronic pIRES2-EGFP expression vector.
The C-terminal portion of Fra-1 has been reported to be critical for its stabilisation[21] and we observed that the phosphorylation shift in SDS-PAGE analysis was due to phosphorylation on threonines rather than phosphorylation on serines. Therefore, we mutated all of the threonines (222, 223, 227, 230, 236, 240, and 266) present in the C-terminal part of Fra-1 and combined these mutations with the mutation of S265, which is the major phosphorylation site for ERK1/2-mediated stabilisation. The S265A mutation slightly reduced the stabilisation of Fra-1 by PKCθ (Figure 6A), while the mutation of all C-terminal threonines did not affect this stability (not shown). However, when we combined the S265A mutation with different threonine mutations, we found that the Fra-1 S265A-T223A-T230A triple mutant was no longer stabilised by activated PKCθ in ER-positive MCF7 and ZR75.1 cell lines (Figure 6A).
Figure 6. Serine 265 and threonines 223 and 230 are the major phosphorylation sites responsible for the stabilisation of Fra-1 by the PKCθ pathway.
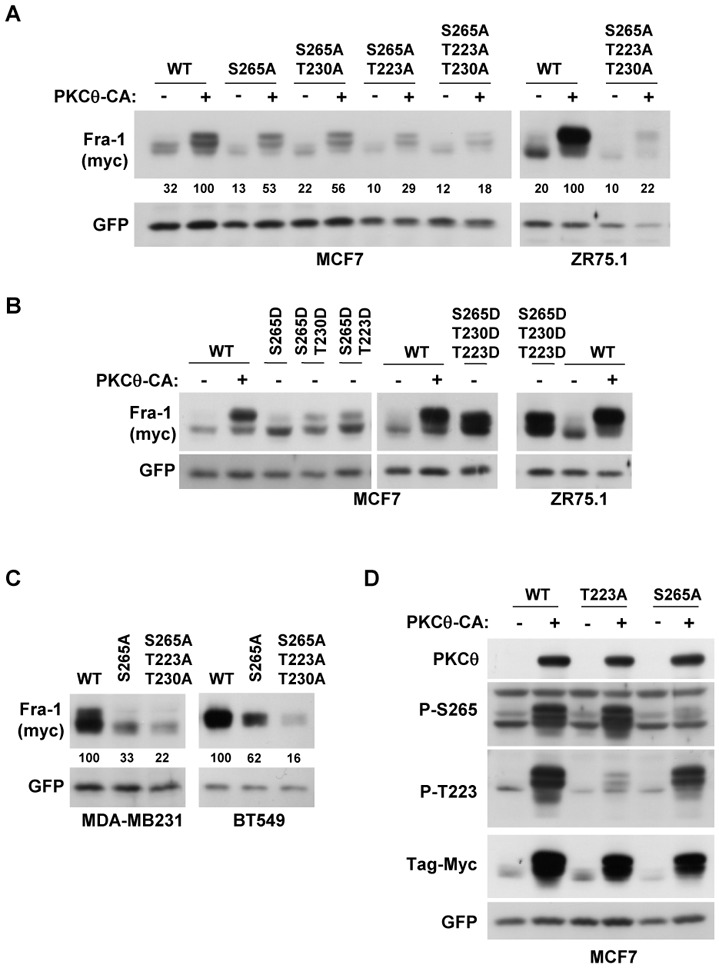
A. MCF7 and ZR75.1 cells were transiently transfected with 50 ng of pIRES2-EGFP vector expressing either wild-type Fra-1 (WT) or mutated Fra-1 in which the following amino acids were replaced by alanine as indicated: serine 265 (S265A), threonine 223 (T223A) or threonine 230 (T230A). To activate the PKCθ pathway, cells were co-transfected with either 2 μg of the PKCθ-CA expression vector (+) or the empty vector (−). Two days later, WCEs were analysed using immunoblotting as in Figure 2A. The blots were scanned, and the densitometry values of Fra-1 were normalised to GFP. The values relative to the cells transfected with the WT Fra-1 expression vector and PKCθ-CA (set at 100%) are given below each lane. B. MCF7 and ZR75.1 cells were transiently transfected with 50 ng of pIRES2-EGFP vector expressing either wild-type Fra-1 (WT) or mutated Fra-1 in which the following amino acids were replaced by aspartic acid as indicated: serine 265 (S265D), threonine 223 (T223D) or threonine 230 (T230D). To activate the PKCθ pathway, cells were co-transfected with 2 μg of either the PKCθ-CA expression vector (+) or the empty vector (−). Two days later, WCEs were analysed using immunoblotting as in Figure 2A. C. MDA-MB231 and BT549 cells were transiently transfected with 50 ng of pIRES2-EGFP vector expressing either wild-type Fra-1 (WT) or mutated Fra-1 in which either serine 265 or all three amino acids (serine 265 and threonines 223 and 230) were replaced by alanine. Two days later, WCEs were analysed using immunoblotting as in Figure 2A. The blots were scanned and the densitometry values of Fra-1 were normalised to GFP. The values relative to the cells transfected with the WT Fra-1 expression vector (set at 100%) are given below each lane. D. MCF7 cells were transiently transfected with 50 ng of pIRES2-EGFP vector expressing either wild-type Fra-1 (WT) or mutated Fra-1 in which either serine 265 (S265A) or threonine 223 (T223A) were replaced by alanine. To activate the PKCθ pathway, cells were co-transfected with 2 μg of either the PKCθ-CA expression vector (+) or the empty vector (−). Two days later, WCEs were analysed using immunoblotting with antibodies detecting Fra-1 only when phosphorylated at serine 265 (P265) or threonine 223 (P-223) and antibodies against PKCθ, Myc and GFP, as in Figure 2A.
We then mutated the same amino acids to aspartic acid to mimic phosphorylation. In MCF7 (Figure 6B), the single (S265D) and double mutants (S265D-T223D and S265D-T230D) were slightly stabilised compared to wild-type Fra-1 in the absence of PKCθ activation. However, in both MCF7 and ZR75.1 cells, the triple Fra-1 mutant S265D-T223D-T230D displayed an expression level comparable to that of wild-type Fra-1 in the presence of activated PKCθ. Conversely, compared to the wild-type Fra-1 protein, the expression of the Fra-1 S265A-T223A-T230A triple mutant was drastically reduced in the ER− MDA-MB231 and BT549 cells, which expressed endogenously activated PKCθ (Figure 6C). We therefore concluded that the phosphorylation of S265, T230 and T223 was crucial for Fra-1 stabilisation through PKCθ signalling. Moreover, in agreement with Figure 5D and the higher efficiency of ERK1/2 signalling in Fra-1 stabilisation in MDA-MB231 cells, the mutation of S265 alone was more efficient in destabilising Fra-1 in MDA-MB231 cells than in BT549 cells (Figure 6C).
Next, the use of specific antisera against Fra-1 phosphorylated on T223 or S265 allowed us to determine the potential interplay between ERK1/2-driven Fra-1 phosphorylation (PKCθ-dependent or PKCθ-independent) and other PKCθ-driven phosphorylation. In MCF7 cells co-transfected with expression vectors for PKCθ and either wild-type or mutated Fra-1 (S265A or T223A), these antisera specifically detected the corresponding phosphorylated protein in immunoblots (Fig. 6D). This result indicated that these residues were the actual targets of PKCθ signalling. Moreover, the phosphorylation of T223 and S265 was independent of each other.
Fra-1 is implicated in PKCθ-induced breast cancer cell motility and invasiveness
We have previously shown that both Fra-1 and PKCθ increase the invasion and motility of breast cancer cells in vitro [10, 26]. Therefore, we tested whether Fra-1 could mediate the effect of PKCθ on these biological responses. To address this question, we performed an in vitro invasion assay with an MCF7 cell clone overexpressing PKCθ-CA under tetracycline control (Figure 7A). As expected, the overexpression of activated PKCθ induced in the absence of doxycycline strongly increased cell invasion (Figure 7B). Interestingly, Fra-1 silencing partly repressed this stimulation. To further explore the role of Fra-1 in PKCθ-driven invasion, a similar experiment was performed in the absence of Matrigel. The activation of PKCθ led to a strong induction of cell motility (Figure 7C). Moreover, the inhibition of PKCθ-stimulated Fra-1 expression using siRNA completely abolished the migration induced by PKCθ. Therefore, our results demonstrate that Fra-1 plays a major role in the control of cell migration by PKCθ.
Figure 7. Fra-1 is involved in breast cancer cell migration and invasion induced by the PKCθ pathway.
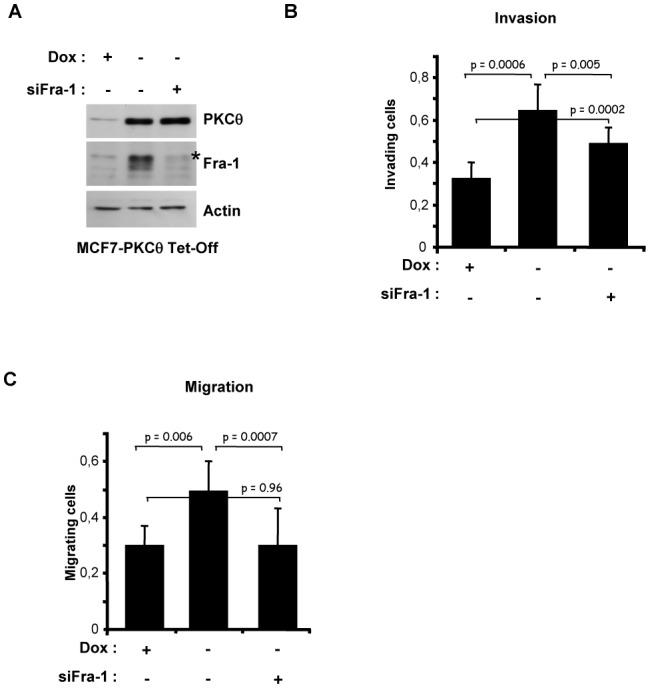
MCF7 cells stably overexpressing PKCθ-CA under tetracycline control (MCF7-PKCθ Tet-Off) were transfected with 3.6 nM of siRNA targeting the FOSL1 gene in the presence (+) or absence (−) of doxycycline (Dox). Three days later, cells were either plated in 12-well plates for immunoblotting analysis after 48 h of growth (A) or subjected, in triplicate, to an invasion (B) or migration (C) assay for 48 h. Cells that migrated to the lower side of the filter were quantified using spectrometric determination of the optical density at 540 nm and normalized by the OD540 obtained with the control cells as described in Materials and Methods. The values represent the mean (± SD) of three independent experiments performed in triplicate. The p-values were calculated using the paired Student’s t-test (two-tailed). In A, phosphorylated Fra-1 forms are indicated by an asterisk.
DISCUSSION
Here, we report that a high level of hyperphosphorylated Fra-1 can be found in invasive breast cancer cell lines independently of activated ERK1/2 level, indicating that other pathways are likely to be involved in Fra-1 phosphorylation and stabilisation. We addressed the role of PKCθ in Fra-1 accumulation, as it has recently been implicated in breast cancer[26] and is activated in invasive ER− breast cancer. We show that the PKCθ pathway participates in Fra-1 stabilisation through the activation of both ERK1/2 and SPAK pathways.
Although both pathways are activated by PKCθ in all of the breast cancer cells we tested, their relative contributions to the accumulation of Fra-1 appeared to be different depending on the cellular context (activation levels of PKCθ and ERK1/2). Fra-1 stabilisation by PKCθ in ER+ MCF7 cells requires equally the stimulation of both ERK1/2 and SPAK activities. However, in ER− MDA-MB231 cells, which display high ERK1/2 activity, the action of PKCθ mainly goes through this MAPK, while in ER− BT549 cells which show moderate ERK1/2 activity, its effect tends to be the result of SPAK activation. Interestingly, MDA-MB231 cells carry oncogenic constitutively active forms of K-Ras and B-Raf whereas BT549 cells express wild-type form of Ras and Raf[29,30]. Morevover, high Fra-1 levels were detected in ER− breast cancer cells harbouring (Hs578T and MDA-MB231) or not (MDA-MB436, BT549, HCC38 and MDA-MB157) Ras pathway activating mutations. Our data suggest that the strong Fra-1 expression results not only from the stimulation of Ras-ERK1/2 pathway, but also from the activation of PKCθ-ERK1/2 and PKCθ-SPAK pathways in a cell-dependent manner.
We show that the phosphorylation of S265, T223 and T230 is crucial for Fra-1 stabilisation by the PKCθ pathway. S265 is the main residue responsible for ERK1/2-driven Fra-1 stabilisation and, by analogy with c-Fos, is likely a target for ERK1 and/or ERK2 (ref. [21]). Interestingly, SPAK has been reported to be crucial for the induction of PKCθ-mediated AP-1 activity in T lymphocytes[28], but the effector(s) downstream of SPAK is unknown. It is therefore tempting to speculate that this factor is a Fra-1-containing AP-1 dimer. We do not know yet whether Fra-1 is a direct substrate of SPAK, and other kinases may act downstream of SPAK. Fra-1 does not possess the consensus motif [S/G/V]RFx[V/I]xx[V/I/T/S]xx[31] shown to mediate the binding of SPAK to its known substrates, such as members of the cation chloride co-transporters superfamily[32] and the TNF receptor RELT[33]. In addition, we cannot exclude a direct effect of PKCθ on Fra-1, and other kinases could act downstream of PKCθ. Only a few PKCθ substrates or potential candidate substrates, such as moesin, SPAK and CARMA1 (ref. [23]), are known, and there is no described consensus sequence of phosphorylation by PKCθ.
While both Fra-1 and PKCθ expressions are inversely correlated with the ERα status of breast cancer cells, activated PKCθ level does not systematically follow Fra-1 level among the different cell lines (Figure 1A). PKCθ is a strong inducer of Fra-1 expression; however according to the literature, it is not the only one[3] and PKCθ-independent regulators of FOSL1 mRNA synthesis and Fra-1 protein stability may be differently expressed depending on the ER− cell lines. Moreover, PKCθ activation in T lymphocytes requires diacylglycerol and at least phosphorylation of T538 and Y9034. In our study, PKCθ activation was measured by T538 phosphorylation and it would be interesting to evaluate the phosphorylation level of Y90 to determine whether it could correlate better with Fra-1 expression. Currently, PKCθ activation is not well understood and other activating phosphorylation sites not yet discovered could be critical in breast cancer. The 3-phosphoinositide-dependent kinase 1 (PDK1) and the src family protein tyrosine kinase Lck have been proposed to phosphorylate PKCθ on residues 538 and 90, respectively[34]. Recently, the GCK-like kinase (MAP4K3) has been shown to directly phosphorylate PKCθ on T538 (ref. [35]). While expression of GLK has not been studied in breast cancer, both LCK[36] and PDK1 (ref. [37]) have been reported to be overexpressed, especially in ER− compared to ER+ tumours for the tyrosine kinase. Therefore, further studies in breast cancer field are required to discover the factors regulating PKCθ activation, including the enzymes responsible for the accumulation of diacylglycerol and/or the kinase(s) phosphorylating PKCθ.
Fra-1 is an important target of the PKCθ pathway in breast cancer cells because PKCθ-driven Fra-1 expression mediates PKCθ effects on cell migration and invasion. Interestingly, we found that Fra-2, which also enhances cell invasion, was also regulated by the PKCθ pathway (not shown). However, Fra-2-induced invasion occurs through a different mechanism involving de novo RelB synthesis[38]. Fra-2 could therefore also participate in the enhancement of cell invasion induced by the kinase.
It is noteworthy that Fra-1 has recently been implicated in the epithelial-mesenchymal transition (EMT) of mammary cells by regulating slug expression[39], ZEB1/2 expression[40] or the neo-synthesis of microRNAs miR-221 and miR-222, which are associated with the basal-like subtype of breast cancer[41]. Fra-1 is a critical and necessary downstream target of ERK for the induction of EMT and the acquisition of a motile and invasive profile in non-tumourigenic epithelial cells[40,42]. Conversely, Fra-1 extinction increases the expression of epithelial genes and decreases the levels of mesenchymal markers in the Ras-mutated MDA-MB231 cell line[39]. We could therefore speculate that activated PKCθ may also induce EMT in cells in which it is aberrantly expressed.
In summary, our findings identify PKCθ as an important regulator of Fra-1 accumulation in ER− basal-like breast cancer cells and suggest that PKCθ may participate in progression of some breast cancers. As a consequence, it may be important to assess the potential effect of PKCθ in the progression of other cancers in which Fra-1 has been associated with a more aggressive phenotype.
MATERIALS AND METHODS
Cell culture
ER− and ER+ breast cancer cell lines were purchased from the American Type Culture Collection (ATCC, Manassas, VA) and maintained in standard culture medium as recommended by the ATCC. To generate stable cell lines expressing constitutively active PKCθ in a tetracycline-controlled expression system, MCF7 cells were infected with the pRevTet-off retroviral vector (Clontech Laboratories, Palo Alto, CA), which encodes the tetracycline-repressible transactivator tTA and a neomycin resistance gene. The cells were co-infected with pRev-TRE (Clontech), which harbours a hygromycin resistance gene and the entire human PKCθ-CA gene. Neomycin- and hygromycin-resistant cellular clones were isolated and maintained in 200 μg/ml neomycin and 20 μg/ml hygromycin B. The inhibition of PKCθ-CA expression was induced by treatment with 0.01 μg/ml doxycycline for 48 h when indicated.
Plasmids and siRNAs
The human wild-type full-length Fra-1 was cloned in pCI[13] and in the bicistronic pIRES2-EGFP, as previously reported[21]. Mutagenesis was performed using the Quick Change II site-directed mutagenesis kit from Stratagene (Agilent Technology France, Massy), and mutants were verified using nucleotide sequencing. Constitutively active PKCθ (PKCθ-CA), kinase-defective PKCθ (PKCθ-DN) and SPAK, subcloned in the pEF4/His expression vector, were generously provided by Amnon Altman (San Diego, CA)[43,28]. Constitutively active MEK1 and constitutively active MEK5 and wild-type ERK5 were gifts from Jacques Pouyssegur (Nice, France) and Robert A. Hipskind (Montpellier, France), respectively[44,45]. The sequence of the Fra-1 siRNA duplex has been described previously (Belguise et al., 2005). For PKCθ inhibition experiments, two different siRNAs were used and targeted 5′-GACCATCTCTGCAGATTAA-3′ and 5′-ACCACCGTGGAGCTCTACT-3′ sequences (sense strand). For SPAK knockdown experiments, the siRNA targeted the 5′-ATTCAAGCCATGAGTCAGT-3′ sequence. The control siRNA had the following sequence: 5′-AGGTAGTGTAATCGCCTTG-3′.
Transient transfections
For plasmid transfections, cells were plated in 6-well plates, and 24 h later, cells were transfected with 2 μg of DNA using the Jet PEI transfection reagent. When necessary, the pSP64 plasmid was used to obtain 2 μg of DNA. For siRNA transfections, duplexes (3.6 nM final) were introduced into the cells using reverse transfection using the Interferin transfection reagent (Ozyme) according to the manufacturer’s protocol. When two successive siRNA transfection rounds were performed, cells were re-plated 48 h after the first transfection (cell dilution 1:3).
Invasion and migration assays
Doxycycline was removed at the same time as Fra-1 siRNA was transfected in MCF7-PKCθ Tet-Off cells. Three days after transfection, a suspension of 400,000 cells was seeded in the upper compartment of a Transwell insert (Costar, Cambridge, MA) on an 8-mm diameter polycarbonate filter (8 μm pore size) and incubated at 37°C for 48 h. In parallel, 20,000 cells were plated in 12 well-plates either as a control of proliferation for the invasion/migration assay or for immunoblotting analysis. For invasion assays, filters were pre-coated with 30 μg Matrigel as previously described[10]. Cells that migrated to the lower side of the filter were quantified by the mitochondrial dehydrogenase enzymatic assay using 3(4,5-dimethyl-thiazol-2-yl)2,5-diphenol tetrazolium bromide (MTT) and OD540 determination. The same assay was used to quantify the cells plated in 12-well plates as a control of proliferation. Then, OD540 obtained for the migrating cells were normalized by the OD540 measured for the control cells.
Cell extracts and immunoblots
Cell extracts were prepared and subjected to immunoblotting as previously described[10]. Anti-human Fra-1 (R-20), anti-Myc and anti-GFP were purchased from Santa Cruz (Santa Cruz, CA, USA). Antibodies against PKCθ, phosphorylated PKCθ (T538), SPAK, ERK1/2, phosphorylated ERK1/2 (T202/Y204) and phosphorylated ERK5 (T218/Y220) were purchased from Cell Signalling Technology. Anti-actin was purchased from Sigma-Aldrich (L’Isle d’Abeau Chesnes, France). The anti-phosphoserine 265 antibody has previously been described[21]. The anti-phosphothreonine 223 antibody was obtained after the immunisation of rabbits with the PTLMTpTPSLTPFC peptide coupled to keyhole limpet haemocyanin. Both antibodies were produced and purified by Eurogentec (Belgium) as described[21].
Acknowledgments
We are grateful to A. Altman, J. Pouyssegur and A. Hipskind for providing plasmids and I. Jariel-Encontre for critical reading of the manuscript. This work was supported by the « Institut National de la Santé et de la Recherche Médicale », the University of Montpellier 1, the « Ligue Nationale Contre le Cancer-comite de l’Herault », the « Association pour la Recherche sur le Cancer » (fellowship to K. Belguise) and the French “Ministère de la Recherche et de l’Enseignement Supérieur» (fellowship to S. Milord). M. Piechaczyk’s laboratory was supported as an « Equipe Labellisée » of the « Ligue Nationale Contre le Cancer ».
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- 1.Shaulian E, Karin M. AP-1 in cell proliferation and survival. Oncogene. 2001;20:2390–2400. doi: 10.1038/sj.onc.1204383. [DOI] [PubMed] [Google Scholar]
- 2.Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer. 2003;3:859–868. doi: 10.1038/nrc1209. [DOI] [PubMed] [Google Scholar]
- 3.Young MR, Colburn NH. Fra-1 a target for cancer prevention or intervention. Gene. 2006;379:1–11. doi: 10.1016/j.gene.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 4.Verde P, Casalino L, Talotta F, Yaniv M, Weitzman JB. Deciphering AP-1 function in tumorigenesis: fra-ternizing on target promoters. Cell Cycle. 2007;6:2633–2639. doi: 10.4161/cc.6.21.4850. [DOI] [PubMed] [Google Scholar]
- 5.Mechta F, Lallemand D, Pfarr CM, Yaniv M. Transformation by ras modifies AP1 composition and activity. Oncogene. 1997;14:837–847. doi: 10.1038/sj.onc.1200900. [DOI] [PubMed] [Google Scholar]
- 6.Vallone D, Battista S, Pierantoni GM, Fedele M, Casalino L, Santoro M, et al. Neoplastic transformation of rat thyroid cells requires the junB and fra-1 gene induction which is dependent on the HMGI-C gene product. EMBO J. 1997;16:5310–5321. doi: 10.1093/emboj/16.17.5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kustikova O, Kramerov D, Grigorian M, Berezin V, Bock E, Lukanidin E, et al. Fra-1 induces morphological transformation and increases in vitro invasiveness and motility of epithelioid adenocarcinoma cells. Mol Cell Biol. 1998;18:7095–7105. doi: 10.1128/mcb.18.12.7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Risse-Hackl G, Adamkiewicz J, Wimmel A, Schuermann M. Transition from SCLC to NSCLC phenotype is accompanied by an increased TRE-binding activity and recruitment of specific AP-1 proteins. Oncogene. 1998;16:3057–3068. doi: 10.1038/sj.onc.1201845. [DOI] [PubMed] [Google Scholar]
- 9.Vial E, Sahai E, Marshall CJ. ERK-MAPK signalling coordinately regulates activity of Rac1 and RhoA for tumor cell motility. Cancer Cell. 2003;4:67–79. doi: 10.1016/s1535-6108(03)00162-4. [DOI] [PubMed] [Google Scholar]
- 10.Belguise K, Kersual N, Galtier F, Chalbos D. FRA-1 expression level regulates proliferation and invasiveness of breast cancer cells. Oncogene. 2005;24:1434–1444. doi: 10.1038/sj.onc.1208312. [DOI] [PubMed] [Google Scholar]
- 11.Debinski W, Gibo DM. Fos-related antigen 1 modulates malignant features of glioma cells. Mol Cancer Res. 2005;3:237–249. doi: 10.1158/1541-7786.MCR-05-0004. [DOI] [PubMed] [Google Scholar]
- 12.Sayan AE, Stanford R, Vickery R, Grigorenko E, Diesch J, Kulbicki K, et al. Fra-1 controls motility of bladder cancer cells via transcriptional upregulation of the receptor tyrosine kinase AXL. Oncogene doi:10.1038, in press. Oncogene. 2011 doi: 10.1038/onc.2011.375. e-pub ahead of print 29 August 2011. [DOI] [PubMed] [Google Scholar]
- 13.Philips A, Teyssier C, Galtier F, Rivier-Covas C, Rey JM, Rochefort H, et al. FRA-1 expression level modulates regulation of activator protein-1 activity by estradiol in breast cancer cells. Mol Endocrinol. 1998;12:973–985. doi: 10.1210/mend.12.7.0133. [DOI] [PubMed] [Google Scholar]
- 14.Milde-Langosch K, Kappes H, Riethdorf S, Loning T, Bamberger AM. FosB is highly expressed in normal mammary epithelia, but down-regulated in poorly differentiated breast carcinomas. Breast Cancer Res Treat. 2003;77:265–275. doi: 10.1023/a:1021887100216. [DOI] [PubMed] [Google Scholar]
- 15.Song Y, Song S, Zhang D, Zhang Y, Chen L, Qian L, et al. An association of a simultaneous nuclear and cytoplasmic localization of Fra-1 with breast malignancy. BMC Cancer. 2006;6:298–304. doi: 10.1186/1471-2407-6-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chiappetta G, Ferraro A, Botti G, Monaco M, Pasquinelli R, Vuttariello E, et al. FRA-1 protein overexpression is a feature of hyperplastic and neoplastic breast disorders. BMC Cancer. 2007;7:17–28. doi: 10.1186/1471-2407-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Logullo AF, Stiepcich MM, Osorio CA, Nonogaki S, Pasini FS, Rocha RM, et al. Role of Fos-related antigen 1 in the progression and prognosis of ductal breast carcinoma. Histopathology. 2011;58:617–625. doi: 10.1111/j.1365-2559.2011.03785.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Q, Ni H, Lan L, Wei X, Xiang R, Wang Y. Fra-1 protooncogene regulates IL-6 expression in macrophages and promotes the generation of M2d macrophages. Cell Res. 2010;20:701–712. doi: 10.1038/cr.2010.52. [DOI] [PubMed] [Google Scholar]
- 19.Casalino L, De Cesare D, Verde P. Accumulation of Fra-1 in ras-transformed cells depends on both transcriptional autoregulation and MEK-dependent posttranslational stabilisation. Mol Cell Biol. 2003;23:4401–4415. doi: 10.1128/MCB.23.12.4401-4415.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vial E, Marshall CJ. Elevated ERK-MAP kinase activity protects the FOS family member FRA-1 against proteasomal degradation in colon carcinoma cells. J Cell Sci. 2003;116:4957–4963. doi: 10.1242/jcs.00812. [DOI] [PubMed] [Google Scholar]
- 21.Basbous J, Chalbos D, Hipskind R, Jariel-Encontre I, Piechaczyk M. Ubiquitin-independent proteasomal degradation of Fra-1 is antagonized by Erk1/2 pathway-mediated phosphorylation of a unique C-terminal destabiliser. Mol Cell Biol. 2007;27:3936–3950. doi: 10.1128/MCB.01776-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harris TJ, McCormick F. The molecular pathology of cancer. Nat Rev Clin Oncol. 2010;7:251–265. doi: 10.1038/nrclinonc.2010.41. [DOI] [PubMed] [Google Scholar]
- 23.Hayashi K, Altman A. Protein kinase C theta (PKCtheta): a key player in T cell life and death. Pharmacol Res. 2007;55:537–544. doi: 10.1016/j.phrs.2007.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marsland BJ, Kopf M. T-cell fate and function: PKC-theta and beyond. Trends Immunol. 2008;29:179–185. doi: 10.1016/j.it.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 25.Blay P, Astudillo A, Buesa JM, Campo E, Abad M, Garcia-Garcia J, et al. Protein kinase C theta is highly expressed in gastrointestinal stromal tumors but not in other mesenchymal neoplasias. Clin Cancer Res. 2004;10:4089–4095. doi: 10.1158/1078-0432.CCR-04-0630. [DOI] [PubMed] [Google Scholar]
- 26.Belguise K, Sonenshein GE. PKCtheta promotes c-Rel-driven mammary tumorigenesis in mice and humans by repressing estrogen receptor alpha synthesis. J Clin Invest. 2007;117:4009–4021. doi: 10.1172/JCI32424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Klijn JG, Zhang Y, Sieuwerts AM, Look MP, Yang F, et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005;365:671–679. doi: 10.1016/S0140-6736(05)17947-1. [DOI] [PubMed] [Google Scholar]
- 28.Li Y, Hu J, Vita R, Sun B, Tabata H, Altman A. SPAK kinase is a substrate and target of PKCtheta in T-cell receptor-induced AP-1 activation pathway. EMBO J. 2004;23:1112–1122. doi: 10.1038/sj.emboj.7600125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hollestelle A, Elstrodt F, Nagel JH, Kallemeijn WW, Schutte M. (Phosphatidylinositol-3-OH kinase or RAS pathway mutations in human breast cancer cell lines. Mol Cancer Res. 2007;5:195–201. doi: 10.1158/1541-7786.MCR-06-0263. [DOI] [PubMed] [Google Scholar]
- 30.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 31.Delpire E, Gagnon KB. Genome-wide analysis of SPAK/OSR1 binding motifs. Physiol Genomics. 2007;28:223–231. doi: 10.1152/physiolgenomics.00173.2006. [DOI] [PubMed] [Google Scholar]
- 32.Piechotta K, Lu J, Delpire E. Cation chloride cotransporters interact with the stress-related kinases Ste20-related proline-alanine-rich kinase (SPAK) and oxidative stress response 1 (OSR1) J Biol Chem. 2002;277:50812–50819. doi: 10.1074/jbc.M208108200. [DOI] [PubMed] [Google Scholar]
- 33.Polek TC, Talpaz M, Spivak-Kroizman T. The TNF receptor, RELT, binds SPAK and uses it to mediate p38 and JNK activation. Biochem Biophys Res Commun. 2006;343:125–134. doi: 10.1016/j.bbrc.2006.02.125. [DOI] [PubMed] [Google Scholar]
- 34.Manicassamy S, Gupta S, Sun Z. Selective function of PKC-theta in T cells. Cell Mol Immunol. 2006;3:263–270. [PubMed] [Google Scholar]
- 35.Chuang HC, Lan JL, Chen DY, Yang CY, Chen YM, Li JP, et al. The kinase GLK controls autoimmunity and NF-κB signaling by activating the kinase PKC-θ in T cells. Nat Immunol. 2011;12:1113–1118. doi: 10.1038/ni.2121. [DOI] [PubMed] [Google Scholar]
- 36.Elsberger B, Fullerton R, Zino S, Jordan F, Mitchell TJ, Brunton VG, et al. Breast cancer patients’ clinical outcome measures are associated with Src kinase family member expression. Br J Cancer. 2010;103:899–909. doi: 10.1038/sj.bjc.6605829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maurer M, Su T, Saal LH, Koujak S, Hopkins BD, Barkley CR, et al. 3-Phosphoinositide-dependent kinase 1 potentiates upstream lesions on the phosphatidylinositol 3-kinase pathway in breast carcinoma. Cancer Res. 2009;69:6299–6306. doi: 10.1158/0008-5472.CAN-09-0820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang X, Belguise K, Kersual N, Kirsch KH, Mineva ND, Galtier F, et al. Oestrogen signalling inhibits invasive phenotype by repressing RelB and its target BCL2. Nat Cell Biol. 2007;9:470–478. doi: 10.1038/ncb1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen H, Zhu G, Li Y, Padia RN, Dong Z, Pan ZK, et al. Extracellular signal-regulated kinase signalling pathway regulates breast cancer cell migration by maintaining slug expression. Cancer Res. 2009;69:9228–9235. doi: 10.1158/0008-5472.CAN-09-1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shin S, Dimitri CA, Yoon SO, Dowdle W, Blenis J. ERK2 but not ERK1 induces epithelial-to-mesenchymal transformation via DEF motif-dependent signalling events. Mol Cell. 2010;38:114–127. doi: 10.1016/j.molcel.2010.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stinson S, Lackner MR, Adai AT, Yu N, Kim HJ, O’Brien C, et al. TRPS1 Targeting by miR-221/222 Promotes the Epithelial-to-Mesenchymal Transition in Breast Cancer. Sci Signal. 2011;4:ra41. doi: 10.1126/scisignal.2001538. [DOI] [PubMed] [Google Scholar]
- 42.Doehn U, Hauge C, Frank SR, Jensen CJ, Duda K, Nielsen JV, et al. RSK is a principal effector of the RAS-ERK pathway for eliciting a coordinate promotile/invasive gene program and phenotype in epithelial cells. Mol Cell. 2009;35:511–522. doi: 10.1016/j.molcel.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baier-Bitterlich G, Uberall F, Bauer B, Fresser F, Wachter H, Grunicke H, et al. Protein kinase C-theta isoenzyme selective stimulation of the transcription factor complex AP-1 in T lymphocytes. Mol Cell Biol. 1996;16:1842–1850. doi: 10.1128/mcb.16.4.1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brunet A, Pages G, Pouyssegur J. Constitutively active mutants of MAP kinase kinase (MEK1) induce growth factor-relaxation and oncogenicity when expressed in fibroblasts. Oncogene. 1994;9:3379–3387. [PubMed] [Google Scholar]
- 45.Mulloy R, Salinas S, Philips A, Hipskind RA. Activation of cyclin D1 expression by the ERK5 cascade. Oncogene. 2003;22:5387–5398. doi: 10.1038/sj.onc.1206839. [DOI] [PubMed] [Google Scholar]