

Abstract
Regulation of endothelial cell apoptosis is a critical modulator of normal and pathological angiogenesis. In this study, we examined the role of the protein kinase Akt/PKB in endothelial cell survival in response to growth factor and matrix attachment signals. Vascular endothelial growth factor(VEGF)-induced cytoprotection of endothelial cell monolayers correlated with the wortmannin-sensitive induction of Akt activity. Transfection of an adenovirus expressing a dominant-negative Akt mutant decreased endothelial cell viability in the presence of VEGF. Conversely, adenoviral transduction of wild-type Akt facilitated the cell survival effects of VEGF, whereas transduction of constitutively active Akt conferred endothelial cell survival in the absence of VEGF. Constitutively active Akt also conferred survival to endothelial cells in suspension culture, whereas stimulation with VEGF did not. In suspension cultures, VEGF stimulation was unable to activate Akt, and Akt protein levels were repressed in cells undergoing anoikis. These data suggest that cross-talk between growth factor- and anchorage-dependent signaling pathways are essential for Akt activation and endothelial cell survival.
The survival of endothelial cells is critical for angiogenesis and the maintenance of blood vessel integrity. VEGF1 is an endothelial cell-specific mitogen that functions to induce blood vessel formation during normal development and various pathological processes (1). VEGF biological activity is mediated through its binding to at least two endothelial cell-specific receptors, fms-like tyrosine kinase (flt-1) and fetal liver kinase (flk-1) (2, 3). Heterozygous deletion of the VEGF gene results in the embryonic lethality due to the abnormal blood vessel development (4, 5). VEGF also functions in adult organisms to promote angiogenesis in ischemic tissue (6) and during wound healing (7). Augmented expression of VEGF by gene transfer has been shown to promote the formation of collateral arteries in patients with peripheral artery disease (8), and VEGF is also associated with pathological angiogenesis such as in tumors that express high levels of this factor (9, 10). VEGF activates receptor tyrosine kinases leading to the activation of various signal transducers including phosphoinositide 3-kinase (PI3-kinase) (11, 12). In addition to its mitogenic effects, VEGF acts to promote endothelial cell survival both in vitro and in vivo (13-15). Consistent with its function as a survival factor, VEGF withdrawal is associated with vascular regression in both developing retina and tumors (13, 16, 17).
Endothelial cells are also dependent on adhesion to extracellular matrix for their survival (18). Cell adhesion to matrix is mediated by integrins and disruption of proper integrin-ligand interactions leads to a phenomenon termed anoikis, apoptosis induced by the disruption of cell-matrix interactions (19). Endothelial cell adhesion to matrix is mediated, at least in part, by integrin αvβ3, which transmits an “outside-in” survival signal that is essential for angiogenesis (20). Therefore, agents that induce endothelial cell apoptosis, either by antagonizing integrin binding or perturbing adhesion plaque formation, are currently being considered for cancer therapy by virtue of their ability to inhibit tumor neovascularization (21-23).
The serine/threonine protein kinase Akt/PKB is recognized increasingly as a key regulator of cell viability in a number of systems (24-27). Activation of Akt occurs through the direct binding of the phosphoinositide products of the PI3-kinase reaction to its pleckstrin homology domain (28). Phosphoinositol lipids also activate a protein kinase that phosphorylates Akt, resulting in further activation (29, 30). Here, we examined the involvement of Akt in the survival of cultured human umbilical vein endothelial cells (HUVEC). VEGF-mediated HUVEC survival correlated with the wortmannin-sensitive induction of Akt activity. Adenovirus-mediated expression of a dominant-negative mutant of Akt reduced HUVEC viability in the presence of VEGF. Forced expression of wild-type Akt promoted cell survival in response to low doses of VEGF, whereas constitutive overexpression of Akt was sufficient for endothelial cell survival in the absence of VEGF. VEGF did not promote Akt activation in suspension culture nor did it inhibit anoikis. However, expression of constitutively active Akt was sufficient for survival in the absence of attachment. These data suggest that growth factor and attachment-mediated signaling pathways converge on Akt to control endothelial cell survival under conditions of angiogenesis and vessel regression.
MATERIALS AND METHODS
Reagents, Plasmids, and Cell Culture
Anti-Akt antibody was purchased from Santa-Cruz. Wortmannin was purchased from Sigma. Akt plasmids were a gift from Dr. J. Testa. Recombinant human VEGF was the 165 amino acid isoform. HUVECs were cultured in monolayers in Dulbecco’s modified Eagle medium (DMEM) under conditions of serum deprivation or stimulation with VEGF as described by Spyridopoulos et al. (14). For suspension cultures, HUVEC monolayers were washed with phosphate-buffered saline (PBS) twice and incubated with PBS containing 0.5% EDTA. Cells were collected with centrifugation (2,000 rpm for 5 min) and washed with serum-depleted media. For suspension culture, bacterial culture dishes were precoated with 20 mg/ml bovine serum albumin overnight to prevent the cells from attaching to the substratum.
Akt Kinase Assay
Cells were washed twice with PBS and lysed with cell lysis buffer (1% Nonidet P-40, 10% glycerol, 137 mm NaCl, 20 mm Tris-HCl, pH 7.4, 20 mm NaF, 2 μg/ml leupeptin, 1 mm phenylmethyl-sulfonyl fluoride). Lysates were precleaned with protein G-agarose for 30 min at 4 °C, and immunoprecipitated for 2 h with anti-Akt antibodies in the presence of 2 mg/ml bovine serum albumin with or without 16 μg/ml competitor peptides (Santa Cruz). Immunoprecipitates were washed twice with cell lysis buffer, once with water, and once with kinase buffer (20 mm HEPES, pH 7.2, 10 mm MgCl2, 10 mm MnCl2). Immunoprecipitated proteins were incubated in 50 μl of kinase buffer containing 2 μg of histone H2B (Roche Molecular Biochemicals) and [γ-32P]ATP (5 μm, 10 μCi) for 30 min at room temperature. Kinase reactions were stopped by the addition of SDS sample buffer and subjected to SDS-polyacrylamide gel electrophoresis on 15% polyacrylamide gels before autoradiography.
Cell Viability Assay
Cells were plated on 24-well dishes at a density of 4 × 104 cells/well. After overnight incubation, the media was changed to DMEM containing 20% fetal bovine serum or the indicated concentrations of VEGF for 21 h. Cells were then washed with PBS, harvested by trypsinization, and viability was determined by the trypan blue exclusion assay (31, 32). Alternatively cells were analyzed for the appearance of pyknotic nuclei. After removal of culture media, 3.7% formaldehyde in PBS was added to the culture. After fixation for 20 min, cells were washed in PBS twice and stained with Hoechst 33342. Cells were examined with a Nikon Diaphot microscope. Cell viability was also assessed by the MTS assay. Cells were cultured in 96-well dish pre-coated with bovine serum albumin in the presence or absence of VEGF (100 ng/ml) for 21 h. MTS assay was performed according to the manufacturer’s protocol (Promega). Cells were incubated with MTS reaction solution for 2 h.
Adenoviral Constructs and Transfection
Replication-defective adenovirus vectors expressing mouse Akt proteins fused in-frame to the hemagglutinin (HA) epitope under the control of the cytomegalovirus (CMV) promoter were constructed according to the method of Becker (33). In brief, HA-Akt fragment from pcDNA3-HA-Akt plasmid was inserted into the multicloning site of pACCMVpLpA plasmid. pACCM-VpLpA-HA-Akt was co-transfected with pJM17 plasmid into 293 cells to allow for homologous recombination. The mutant Akt and constitutively active Akt adenoviral vectors were constructed by the same protocol. The dominant-negative mutant Akt (T308A,S473A) protein cannot be activated by phosphorylation (34) and it functions in a dominant-negative fashion (35). The constitutively active Akt construct has the c-src myristoylation sequence fused in-frame to the N terminus of the HA-Akt (wild-type) coding sequence. Adenoviral Akt constructs were amplified in 293 cells and purified by ultracentrifugation through a CsCl gradient. Adeno-β-gal expresses the bacterial β-galactosidase gene from the CMV promoter (36). HUVEC cultures were infected for 24 h with Ad-Akt or Ad-β-galactosidase at a multiplicity of infection of 50, achieving a 90% transduction efficiency (data not shown).
Electrophoretic Gel Mobility Analysis of DNA Fragmentation
Cells were cultured under the indicated conditions. Floating and attached cells were collected and resuspended in 200 μl of PBS containing proteinase K (0.5 mg/ml), RNase A (0.5 mg/ml), and 1% SDS. After 30 min incubation at 37 °C, 300 μl of NaI solution (6 m NaI, 13 mm EDTA, 0.5% SDS, 10 mg/ml glycogen, 26 nm Tris-HCl, pH 8.0) was added. Cell lysates were incubated at 60 °C for 15 min and 500 μl of isopropanol was added. DNA was precipitated by centrifugation at 15,000 rpm for 15 min. After washing once with 50% isopropanol and once with 100% isopropanol, the DNA was dried. DNA (15 μg/lane) was examined following electrophoresis on a 1.5% agarose gel.
Western Immunoblot Analyses
Cells were treated as described in the figure legends, and cell lysates were prepared as described for the Akt kinase assays. 20 μg of protein was separated on SDS-polyacrylamide gel electrophoresis gel and transferred onto a polyvinylidene difluoride membrane (Millipore). After blocking with T-PBS (PBS containing 0.2% Tween 20) containing 5% milk for 1 h, the membrane was incubated with anti-Bcl-2 antibody (Transduction Labs), anti-Akt antibody (Santa Cruz), phospho-specific Akt antibody (New England Biolabs), or tubulin (Calbiochem). ECL (Amersham) was used for detection.
RESULTS
VEGF Promotes HUVEC Survival
Consistent with previous reports (15, 37, 38), serum deprivation induces endothelial cell death (Fig. 1). Serum deprivation for 21 h resulted in 50–80% decreases in HUVEC viability depending on the preparation and passage number, where higher passage cells (passage 4 or 5) were more sensitive to death than lower passage cells (not shown). It has been reported that VEGF inhibits HUVEC apoptosis induced by serum deprivation (15). A dose dependence of VEGF in preventing HUVEC death is shown in Fig. 1A. Under the conditions of our assays, 100 ng/ml VEGF reversed cell loss induced by serum deprivation. Consistent with an anti-apoptotic effect, treatment with VEGF also diminished the frequency of endothelial cells exhibiting pyknotic nuclei (Fig. 1B). Inclusion of 100 ng/ml VEGF diminished the frequency of pyknotic nuclei from 19.6 ± 2.6 to 10.7 ± 0.8 in cultures that were incubated in the absence of serum for 21 h.
Fig. 1. VEGF protects HUVEC cultures against cell death induced by the serum depletion.

A, dose dependence of VEGF cytoprotection. Cells were plated in 24-well dishes overnight and then cultured in DMEM containing 20% fetal bovine serum (open bar) or the indicated concentrations of VEGF (closed bars). After 21 h in culture, cell viability was determined by the trypan blue exclusion assay (31, 32) performed in quadruplicate. B, VEGF reduces the frequencies of pyknotic nuclei in HUVECs cultured under serum depletion conditions. HU-VECs were incubated in the presence (+) or absence (−) of VEGF (100 ng/ml) for 21 h. Cells were stained with Hoechst 33342 as described under “Materials and Methods.”
The cell survival effects of VEGF were blocked by the PI3-kinase inhibitor wortmannin. Inhibition of VEGF-mediated cell viability by wortmannin was dose-dependent (Fig. 2A). Partial inhibition of VEGF-mediated HUVEC survival occurred when cultures were incubated with 10 or 50 nm wortmannin, whereas 200 nm wortmannin completely abrogated the survival effects of VEGF. 200 nm wortmannin also abrogated the VEGF-mediated diminution of pyknotic nuclei in the serum-deprived cultures (Fig. 2B).
Fig. 2. Wortmannin abrogates the cytoprotection of VEGF.

Cells were plated overnight and then cultured in DMEM containing the indicated concentrations of wortmannin in the presence (closed bars) or absence (open bar) of VEGF (100 ng/ml). A, after a 21-h culture, viability was determined by the trypan blue exclusion assay performed in quadruplicate. Data are shown as the mean ± S.E. (n = 4). B, effects of wortmannin on VEGF-mediated reduction in the frequency of HUVECs with pyknotic nuclei. Data are shown as the mean ± S.E. (n = 4). Cells were cultured in the presence or absence of VEGF with or without wortmannin for 21 h. Cells were stained with Hoechst 33342, and cells with pyknotic nuclei were counted.
VEGF Activates Akt in HUVEC Cultures
To determine whether VEGF regulates the activity of Akt family proteins in endothelial cells, HUVEC cultures were incubated in serum-free DMEM with or without VEGF (100 ng/ml). The kinase activity of Akt was determined in lysates immunoprecipitated with specific anti-Akt antibody. As shown in Fig. 3, VEGF activated Akt kinase activity in mitogen-deprived HUVEC cultures. Because Akt is regulated by PI3-kinase in other cell types (39), we investigated the effects of wortmannin on Akt activation by VEGF. A 30-min preincubation period with 200 nm wortmannin blocked VEGF-induced Akt activity (Fig. 3). Incubation with anti-Akt antibody competitor peptide diminished the H2B phosphorylation signal to levels seen in cultures incubated with wortmannin. Collectively, these data show that HUVEC survival correlates with Akt activity.
Fig. 3. VEGF activates Akt in HUVEC cultures in a wortmannin-dependent manner.
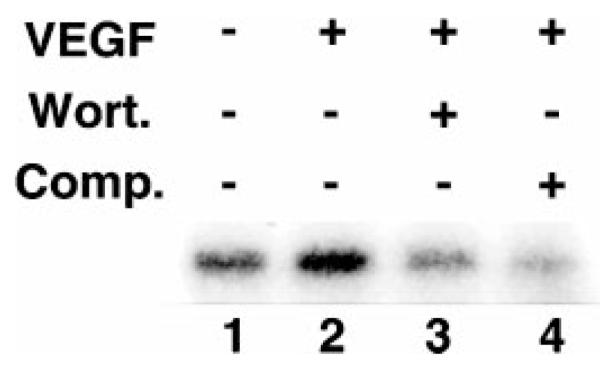
HUVEC cultures were preincubated in serum-free media in the presence (lane 3) or absence (lanes 1, 2, and 4) of wortmannin for 30 min. Cells were then stimulated with VEGF (100 ng/ml) (lanes 2, 3, and 4) for 15 min. Cell lysates were prepared and immunoprecipitated with anti-Akt antibody with (lane 4) or without (lanes 1, 2, and 3) competitor peptide. Kinase activity was measured with histone H2B as a substrate.
Properties of Adenoviral Transgenes Expressing Wild-type, Dominant-Negative, and Constitutively Active Akt
To explore the functional significance of VEGF-induced Akt activity in endothelial cell survival, replication-defective adenoviral vectors expressing wild-type (Adeno-wtAkt), dominant-negative (Adeno-dnAkt), or constitutively active Akt (Adeno-myrAkt) were constructed (Fig. 4A). All adenovirus-encoded transgenes were fused with the HA epitope to distinguish exogenous from endogenous Akt. Control cultures were infected with an adenoviral vector expressing β-galactosidase (Ad-β-gal), which does not affect endothelial cell viability under the conditions of our assay (not shown). Western blot analyses, using anti-Akt antibodies on anti-HA immunoprecipitated material, revealed that Adeno-wtAkt, Adeno-dnAkt, and Adeno-myrAkt expressed comparable levels of protein (Fig. 4B). Akt protein kinase activity was detected in the anti-HA immunoprecipitates from cells infected with Adeno-wtAkt in the presence, but not the absence, of VEGF (Fig. 4B) In contrast, no kinase activity was detected in the cultures infected with the Adeno-dnAkt vector in the presence or absence of VEGF. Cultures infected with Adeno-myrAkt contained high levels of HA-associated Akt activity that was not influenced by VEGF stimulation.
Fig. 4. Adenovirus constructs expressing wild-type Akt and its mutants.

A, structures of adenovirus vectors expressing wild-type, dominant-negative, and constitutively active Akt are indicated. B, HUVECs were infected with adenovirus vector expressing Akt or β-galactosidase at multiplicity of infection 50 as described under “Materials and Methods.” After 30 min of serum starvation, cells were stimulated with VEGF (100 ng/ml) for 15 min. Cell lysates were immunoprecipitated with anti-HA antibody. Kinase activity was measured with histone H2B as a substrate (top panel). Anti-HA-immunoprecipitated protein was Western blotted with anti-Akt antibody (bottom panel).
Akt Is Essential for VEGF-mediated Cell Survival
HUVEC cultures were infected with Adeno-dnAkt to test whether Akt is essential for VEGF-mediated survival. In the absence of growth factor, the dominant-negative Akt construct did not affect endothelial cell survival, but it inhibited the cytoprotective action of VEGF (Fig. 5A). DNA prepared from the serum-deprived HUVEC cultures displayed the typical nucleosome spacing ladder upon agarose gel electrophoresis that is indicative of apoptosis (Fig. 5B). The DNA ladder was diminished by the inclusion of VEGF in the culture media. Consistent with the data from the trypan blue exclusion assays, infection with Adeno-dnAkt blocked the ability of VEGF to inhibit DNA fragmentation.
Fig. 5. Dominant-negative Akt abrogates VEGF-mediated cell survival.

HU-VEC cultures were infected with adenovirus expressing β-galactosidase (β-gal) or dominant-negative Akt (dnAkt). Cells were incubated with (+) or without (−) VEGF (100 ng/ml) for 21 h (A). Viable cells were counted using a trypan blue exclusion assay. Data are shown as the mean ± S.E. (n = 4) (B). Floating and attached cells were collected, and DNA fragmentation was analyzed as described under “Materials and Methods.”
Akt Promotes Endothelial Cell Survival
Adenoviral transduction of wild-type Akt markedly augmented VEGF-induced endothelial cell survival (Fig. 6A). Cultures infected with Ad-Akt displayed 75 and 65% less cell death than control cultures at 1 and 10 ng/ml, respectively, whereas no decrease in cell death was detected in the cultures exposed to serum-free media in the absence of VEGF. These data show that forced expression of wild-type can enhance the sensitivity of endothelial cells to VEGF survival signals at subsaturating levels of VEGF.
Fig. 6. Akt promotes endothelial cell survival.

A, HUVEC cultures were transfected with adenovirus expressing β-galactosidase (β-gal) or wild-type Akt (wtAkt) at a multiplicity of infection of 50. Cells were cultured in the indicated concentrations of VEGF for 21 h. Viable cells were determined by trypan blue exclusion assay. Data are shown as the mean ± S.E. (n = 4). B, HUVEC cultures were transfected with adenovirus expressing β-galactosidase (β-gal) or constitutively active Akt (myrAkt). Cells were cultured in the serum-depleted condition for 21 h. Cells were fixed and stained with Hoechst 33342. The percentage of cells with pyknotic nuclei is demonstrated. Data are shown as the mean ± S.E. (n = 4).
In contrast to infection with Adeno-wtAkt, infection of cultures with Adeno-myrAkt promoted endothelial cell survival in the absence of growth factor. Analyses of serum-deprived HUVEC cultures revealed that infection with Adeno-myrAkt significantly decreases the frequency of pyknotic nuclei (Fig. 6B) to levels similar to that observed in the presence of VEGF (Fig. 2B).
Akt Activation Is Essential but not Sufficient for VEGF-mediated Induction of Bcl-2
A previous study showed that VEGF induces the anti-apoptotic protein Bcl-2 in HUVECs and that forced expression of Bcl-2 is sufficient to prevent apoptosis in the absence of VEGF (15). Thus, we used the adenoviral Akt vectors to test for the potential involvement of Akt activation in VEGF-mediated Bcl-2 induction. As shown in Fig. 7, VEGF stimulation led to a modest induction of Bcl-2. This up-regulation was blocked by infection with Adeno-dnAkt, demonstrating that Akt is essential for VEGF-mediated induction of Bcl-2. However, infection with Adeno-myrAkt was not sufficient to induce Bcl-2, suggesting that the endothelial cell survival conferred by Akt under these conditions involves other apoptosis-regulatory proteins.
Fig. 7. Akt activation is essential but not sufficient for VEGF-mediated induction of Bcl-2.

HUVECs were transfected with adenovirus expressing β-galactosidase (β-gal), dominant-negative Akt (dnAkt) or constitutively active Akt (myrAkt). Cells were cultured in media containing 2% fetal calf serum in the presence or absence of VEGF (100 ng/ml) for 24 h. Cells were harvested, and cell lysates were analyzed by Western blot with anti-Bcl-2 or anti-α-tubulin antibody.
Akt-mediated Anchorage-dependent Survival Signals
To examine the role of Akt in anchorage-dependent endothelial cell survival, HUVECs were cultured in suspension under serum-deprivation conditions in nonadhesive bacteriological dishes. Under these conditions, endothelial cells rapidly underwent anoikis (19) as indicated by cell shrinkage and membrane blebbling, characteristic of the apoptotic morphology (Fig. 8A). Inclusion of VEGF in the suspension culture media had no effect on cell morphology. However, cells pre-infected with the adenoviral construct expressing constitutively active Akt appeared viable in suspension culture (Fig. 8A). To confirm that constitutively active Akt construct promotes endothelial cell viability under these conditions, MTS assays were performed on cells cultured under these conditions (Fig. 8B). Consistent with observations of cell morphology, infection with Adeno-myrAkt preserved mitochondrial function, an indicator of cellular viability (40). These data show that constitutive Akt activity, but not stimulation with VEGF, is sufficient to confer survival in endothelial cells in the absence of matrix attachment.
Fig. 8. Constitutively active Akt confers resistance to apoptosis induced by cell detachment.

Cells were transfected with adenovirus expressing β-galactosidase (β-gal) or constitutively active Akt (myrAkt). Cells were collected with EDTA treatment and cultured in a suspension system with (+) or without (−) VEGF (100 ng/ml) as described under “Materials and Methods.” A, cells were cultured for 12 h in suspension system and examined with a phase contrast microscope. B, cells were cultured in suspension system for 21 h and then MTS assays were performed. Data are shown as the mean ± S.E. (n = 5).
To further study the role of Akt in anchorage-dependent endothelial cell survival, activated Akt levels were assessed in HUVEC monolayer and suspension cultures using an antibody that is specific for Akt phosphorylated at residue 473 and indicative of the status of Akt activation (34). Consistent with measurements of Akt-associated histone H2B-kinase activity (Fig. 3A), VEGF stimulation of HUVEC monolayers increased the level of phosphorylated (activated) Akt with no detectable change in the total level of Akt protein (Fig. 9A). However, VEGF stimulation had no effect on the level of Akt phosphorylation within 1 h of incubation in suspension culture (Fig. 9A). Moreover, these suspension culture cells (<1 h) displayed lower levels of basal Akt phosphorylation though levels of Akt protein were comparable between attached cells and cells in suspension. VEGF stimulation of HUVECs in suspension for 21 h also did not induce Akt phosphorylation. At this time, HUVEC death is prevalent (Fig. 8) and Akt protein levels are reduced relative to tubulin (Fig. 9B).
Fig. 9. VEGF cannot activate Akt in suspension culture.

Cells were cultured in either monolayer (M) or suspension (S) conditions for 15 min (A) or 21 h (B). After 45 min of serum deprivation, cells were cultured with (+) or without (−) VEGF (100 ng/ml) for 15 min (A) or 21 h (B). Cell lysates were prepared and immunoblotted with anti-phosphospecific Akt (p-Akt), anti-Akt (Akt), or anti-tubulin antibody.
DISCUSSION
Exposure of endothelial cells to survival factors and proper cell-matrix attachments are required for the formation of new blood vessels as well as the maintenance of existing vessels. Consistent with its function as a survival factor, VEGF withdrawal is associated with vascular regression in both developing retina and tumors (13, 16, 17, 41). In this study, we explored the role of the Akt protein kinase in mediating the survival functions of VEGF and matrix attachment in endothelial cells. We have shown that VEGF-induced survival in monolayer cultures correlates with the wortmannin-sensitive activation of the Akt protein. The functional significance of Akt activation is indicated by the finding that adenovirus-mediated expression of a dominant-negative Akt mutant inhibited the cell survival effect of VEGF. We also showed that forced expression of constitutively active Akt is sufficient to confer survival to serum-deprived endothelial cells. Following the initial submission of this paper, Gerber et al. (42) reported that VEGF activates Akt and that plasmid-mediated overexpression of constitutively active Akt protects endothelial cells from apoptosis, whereas a dominant-negative Akt construct inhibits the cytoprotection conferred by VEGF. The combined results of these two studies demonstrate that Akt activation is essential for VEGF-induced cytoprotection and that plasmid- or adenovirus-mediated gene transfer of constitutively active Akt can suffice for VEGF with regard to cell survival. Here, we have also shown that adenovirus-mediated transfer of wild-type Akt does not promote endothelial cell survival in the absence of VEGF, but it could potentiate the survival effects of subsaturating levels of VEGF. Thus, these data indicate that mitogen-induced activation of Akt can be a limiting event in the signaling cascade that controls endothelial cell viability.
Though growth factors initiate angiogenic process, it has been proposed that proper matrix associations are essential for endothelial cell survival during neovascularization as cells migrate toward the angiogenic source (20). Thus, it was of interest to test whether Akt was also involved in attachment-mediated endothelial cell survival and, if so, how VEGF and matrix survival signals were coordinated with regard to Akt. Consistent with reports in endothelial cells and other cell types (18), HUVECs rapidly underwent apoptosis when incubated in suspension culture. Though VEGF stimulation did not inhibit apoptosis under these conditions, transduction of constitutively active Akt was sufficient to promote cell viability in the absence of matrix attachment. Immediately following cell detachment, VEGF was unable to stimulate Akt activation via phosphorylation of serine 473 suggesting that matrix attachment is essential for signal transduction events upstream of Akt. At later time points in suspension culture, Akt protein was selectively lost suggesting that reductions in Akt protein might also contribute to this reduction in cell viability. However, adenovirus-mediated overexpression of wild-type Akt in either the presence or absence of VEGF stimulation had no effect on endothelial cell viability in suspension culture (data not shown). Therefore, it appears that the loss of Akt protein is a consequence of the apoptotic process, as has been found in Jurkat and U937 cell death (43), and that apoptosis initiates from a failure of VEGF to promote Akt activation in the absence of matrix attachment.
As noted above, attachment appears to be required for VEGF-initiated signal transduction events upstream of Akt. It has recently been shown that αvβ3 occupancy on smooth muscle cells stimulates insulin-like growth factor-1 signal transduction by enhancing the tyrosine kinase activity of the receptor (44), and similar mechanisms may be operating in endothelial cells with regard to VEGF activation of the Flk-1 receptor. An alternative hypothesis is that VEGF may function by modulating attachment-mediated survival signals. Of note, in the absence of VEGF stimulation, cell attachment has a marked influence on the basal level of Akt activation (see Fig. 9A). These data suggest that growth factor-independent mechanisms of Akt activation exist within endothelial cells, and it is possible that VEGF stimulation serves to facilitate these anti-apoptotic signals from integrins and the extracellular matrix, the production of which are also regulated by VEGF (14).
In endothelial cells, VEGF promotes tyrosine phosphorylation of various signal transducers, including PI3-kinase, Ras GTPase-activating protein, and phospholipase C-γ (11). Here, we have shown that VEGF-induced Akt activation and cytoprotection in endothelial cells was suppressed by wortmannin, a PI3-kinase inhibitor, indicating that Akt acts downstream of PI3-kinase in the VEGF signaling pathway. In contrast, VEGF mitogenic functions are transduced through protein kinase C, which is not inhibited by wortmannin (12). Taken together, these data suggest that the cytoprotective and mitogenic signals of VEGF are transduced by independent pathways in endothelial cells.
In summary, the findings of this study show that Akt is essential in regulating both VEGF- and attachment-mediated survival signals in endothelial cells. In particular, these data document the importance of cross-talk between these two signaling pathways in promoting Akt activation with consequences on endothelial cell survival. Because endothelial cell viability is essential for the maintenance of new blood vessels, further analysis of Akt may provide new insights about the regulatory control of angiogenesis during normal development and tumorigenesis.
Acknowledgments
We thank Tomono Takahashi for helpful advice.
Footnotes
This work was supported by National Institutes of Health Grants AR40197 and AG15052 (to K. W.).
The abbreviations used are: VEGF, vascular endothelial growth factor; PI3-kinase, phosphoinositide 3-kinase; HUVEC, human umbilical vein endothelial cell; DMEM, Dulbecco’s modified Eagle’s medium; PBS, phosphate-buffered saline; HA, hemagglutinin; CMV, cytomegalovirus.
REFERENCES
- 1.Risau W. Nature. 1997;386:671–674. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- 2.de Vries C, Escobedo JA, Ueno H, Houck KA, Ferrara N, Williams LT. Science. 1992;255:989–991. doi: 10.1126/science.1312256. [DOI] [PubMed] [Google Scholar]
- 3.Millauer B, Wizigmann-Voos S, Schnurch H, Martinez R, Moller NPH, Risau W, Ulrich A. Cell. 1993;72:835–846. doi: 10.1016/0092-8674(93)90573-9. [DOI] [PubMed] [Google Scholar]
- 4.Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, Declercq C, Pawling J, Moons L, Collen D, Risau W, Nagy A. Nature. 1996;380:435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- 5.Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O’Shea K, Powell-Braxton L, Hillan KJ, Moore MW. Nature. 1996;380:439–442. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- 6.Banai S, Jaklitsch MT, Shou M, Lazarous DF, Scheinowitz M, Biro S, Epstein SE, Unger EF. Circulation. 1994;89:2183–2189. doi: 10.1161/01.cir.89.5.2183. [DOI] [PubMed] [Google Scholar]
- 7.Brown LF, Yeo KT, Berse B, Yeo TK, Senger DR, Dvorak HF, Van De Water L. J. Exp. Med. 1992;176:1375–1379. doi: 10.1084/jem.176.5.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baumgartner I, Pieczek A, Manor O, Blair R, Kearney M, Walsh K, Isner JM. Circulation. 1998;97:1114–1123. doi: 10.1161/01.cir.97.12.1114. [DOI] [PubMed] [Google Scholar]
- 9.Plate KH, Breier G, Weich HA, Risau W. Nature. 1992;359:845–848. doi: 10.1038/359845a0. [DOI] [PubMed] [Google Scholar]
- 10.Kim KJ, Li B, Winer J, Armanini M, Billett N, Phillips HS, Ferrara N. Nature. 1993;362:841–844. doi: 10.1038/362841a0. [DOI] [PubMed] [Google Scholar]
- 11.Guo D, Jia Q, Song H-Y, Warren RS, Donner DB. J. Biol. Chem. 1995;270:6729–6733. doi: 10.1074/jbc.270.12.6729. [DOI] [PubMed] [Google Scholar]
- 12.Xia P, Aiello LP, Ishii H, Jiang ZY, Park DJ, Robinson GS, Takagi H, Newsome WP, Jirousek MR, King GL. J. Clin. Invest. 1996;98:2018–2026. doi: 10.1172/JCI119006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alon T, Hemo I, Itin A, Pe’er J, Stone J, Keshet E. Nat. Med. 1995;1:1024–1028. doi: 10.1038/nm1095-1024. [DOI] [PubMed] [Google Scholar]
- 14.Spyridopoulos I, Brogi E, Kearney M, Sullivan AB, Cetrulo C, Isner JM, Losordo DW. J. Mol. Cell. Cardiol. 1997;29:1321–1330. doi: 10.1006/jmcc.1996.0365. [DOI] [PubMed] [Google Scholar]
- 15.Gerber H-P, Dixit V, Ferrara N. J. Biol. Chem. 1998;273:13313–13316. doi: 10.1074/jbc.273.21.13313. [DOI] [PubMed] [Google Scholar]
- 16.Yuan F, Chen Y, Dellian M, Safabakhsh N, Ferrara N, Jain RK. Proc. Natl. Acad. Sci. U. S. A. 1996;93:14765–14770. doi: 10.1073/pnas.93.25.14765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Benjamin LE, Keshet E. Proc. Natl. Acad. Sci. U. S. A. 1997;94:8761–8766. doi: 10.1073/pnas.94.16.8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meredith JE, Fazeli B, Schwartz MA. Mol. Biol. Cell. 1993;4:953–961. doi: 10.1091/mbc.4.9.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frisch SM, Francis H. J. Cell Biol. 1994;124:619–626. doi: 10.1083/jcb.124.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brooks PC, Montgomery AMP, Rosenfeld M, Reisfeld RA, Hu T, Klier G, Cheresh DA. Cell. 1994;79:1157–1164. doi: 10.1016/0092-8674(94)90007-8. [DOI] [PubMed] [Google Scholar]
- 21.Claesson-Welsh L, Welsh M, Ito N, Anand-Apte B, Soker S, Zetter B, O’Reilly M, Folkman J. Proc. Natl. Acad. Sci. U. S. A. 1998;95:5579–5583. doi: 10.1073/pnas.95.10.5579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gasparini G, Brooks PC, Biganzoli E, Vermeulen PB, Bonoldi E, Dirix LY, Ranieri G, Miceli R, Cheresh DA. Clin. Cancer Res. 1998;4:2625–2634. [PubMed] [Google Scholar]
- 23.Nicolaou KC, Trujillo JI, Jandeleit B, Chibale K, Rosenfeld M, Diefenbach B, Cheresh DA, Goodman SL. Bioorg. Med. Chem. 1998;6:1185–1208. doi: 10.1016/s0968-0896(98)00090-x. [DOI] [PubMed] [Google Scholar]
- 24.Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- 25.Kauffmann-Zeh A, Rodriguez-Viciana P, Ulrich E, Gilbert C, Coffer P, Downward J, Evan G. Nature. 1997;385:544–548. doi: 10.1038/385544a0. [DOI] [PubMed] [Google Scholar]
- 26.Kennedy SG, Wagner AJ, Conzen SD, Jordan J, Bellacosa A, Tsichlis PN, Hay N. Genes Dev. 1997;11:710–713. doi: 10.1101/gad.11.6.701. [DOI] [PubMed] [Google Scholar]
- 27.Kulik G, Klippel A, Weber MJ. Mol. Cell. Biol. 1997;17:1595–1606. doi: 10.1128/mcb.17.3.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Franke TF, Kaplan DR, Cantley LC. Cell. 1997;88:435–437. doi: 10.1016/s0092-8674(00)81883-8. [DOI] [PubMed] [Google Scholar]
- 29.Kohn AD, Takeuchi F, Roth RA. J. Biol. Chem. 1996;271:21920–21926. doi: 10.1074/jbc.271.36.21920. [DOI] [PubMed] [Google Scholar]
- 30.Stokoe D, Stephens LR, Copeland T, Gaffney PRJ, Reese CB, Painter GF, Holmes AB, McCormick F, Hawkins PT. Science. 1997;277:567–570. doi: 10.1126/science.277.5325.567. [DOI] [PubMed] [Google Scholar]
- 31.Mesner PW, Winters TR, Green SH. J. Cell Biol. 1992;119:1669–1680. doi: 10.1083/jcb.119.6.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou S, Baltimore D, Cantley LC, Kaplan DR, Franke TF. Proc. Natl. Acad. Sci. U. S. A. 1997;94:11345–11350. doi: 10.1073/pnas.94.21.11345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Becker TC, Noel RJ, Coats WS, Gomez-Foix AM, Alam T, Gerard RD, Newgard CB. Methods in Cell Biology. Vol. 43. Academic Press; New York, NY: 1994. pp. 161–189. [DOI] [PubMed] [Google Scholar]
- 34.Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. EMBO J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- 35.Kitamura T, Ogawa W, Sakaue H, Hino Y, Kuroda S, Takata M, Matsumoto M, Maeda T, Konishi H, Kikkawa U, Kasuga M. Mol. Cell. Biol. 1998;18:3708–3717. doi: 10.1128/mcb.18.7.3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith RC, Branellec D, Gorski DH, Guo K, Perlman H, Dedieu J-F, Pastore C, Mahfoudi A, Denèfle P, Isner JM, Walsh K. Genes Dev. 1997;11:1674–1689. doi: 10.1101/gad.11.13.1674. [DOI] [PubMed] [Google Scholar]
- 37.Levkau B, Koyama H, Raines EW, Clurman BR, Herren B, Orth K, Roberts JM, Ross R. Mol. Cell. 1998;1:553–563. doi: 10.1016/s1097-2765(00)80055-6. [DOI] [PubMed] [Google Scholar]
- 38.Scatena M, Almeida M, Chaisson ML, Fausto N, Nicosia RF, Giachelli CM. J. Cell Biol. 1998;141:1083–1093. doi: 10.1083/jcb.141.4.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Franke TF, Yang S-I, Chan TO, Datta K, Kazlauskas A, Morrison DK, Kaplan DR, Tsichlis PN. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- 40.Tanaka M, Suda T, Haze K, Nakamura N, Sato K, Kimura F, Motoyoshi K, Mizuki M, Tagawa S, Ohga S, Hatake K, Drummond AH, Nagata S. Nat. Med. 1996;2:317–322. doi: 10.1038/nm0396-317. [DOI] [PubMed] [Google Scholar]
- 41.Benjamin LE, Golijanin D, Itin A, Pode D, Keshet E. J. Clin. Invest. 1999;103:159–165. doi: 10.1172/JCI5028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gerber H-P, McMurtrey A, Kowalski J, Yan M, Key BA, Dixit V, Ferrara N. J. Biol. Chem. 1998;273:30336–30343. doi: 10.1074/jbc.273.46.30336. [DOI] [PubMed] [Google Scholar]
- 43.Widmann C, Gibson S, Johnson GL. J. Biol. Chem. 1998;273:7141–7147. doi: 10.1074/jbc.273.12.7141. [DOI] [PubMed] [Google Scholar]
- 44.Zheng B, Clemmons DR. Proc. Natl. Acad. Sci. U. S. A. 1998;95:11217–11222. doi: 10.1073/pnas.95.19.11217. [DOI] [PMC free article] [PubMed] [Google Scholar]