

Abstract
Approximately 30% of human tumors sequenced to date harbor mutations in the POLB gene that are not present in matched normal tissue. Many mutations give rise to enzymes that contain non-synonymous single amino acid substitutions, several of which have been found to have aberrant activity or fidelity and transform cells when expressed. The DNA Polymerase β (Pol β) variant Asp160Asn (D160N) was first identified in a gastric tumor. Expression of D160N in cells induces cellular transformation as measured by hyperproliferation, focus formation, anchorage-independent growth and invasion. Here, we show that D160N is an active mutator polymerase that induces complex mutations. Our data support the interpretation that complex mutagenesis is the underlying mechanism of the observed cellular phenotypes, all of which are linked to tumorigenesis or tumor progression.
Keywords: Base excision repair, Cancer, Polymerase beta
1. Introduction
Endogenous cellular DNA damage is estimated to occur upwards of 50,000 lesions per cell daily [1]. Left unrepaired, DNA lesions can give rise to genomic instability or mutagenesis. To guard against these consequences and maintain genomic integrity, cells have evolved various DNA repair mechanisms. The base excision repair (BER) pathway is responsible for resolving oxidative and alkylation damage, the predominant forms of endogenous DNA lesions [2]. BER is initiated by damage-specific glycosylases that recognize damaged bases, and the nature of the glycosylase determines which sub-pathway of BER the cell utilizes [3]. A monofunctional glycosylase will remove the damaged base, leaving an AP site that is then recognized by AP endonuclease (APE1). APE1 nicks the DNA backbone, leaving a 3′-OH and a 5′-deoxyribose phosphate (dRP) group. Pol β, having both polymerase and dRP lyase activity, binds this substrate, fills in the gap and removes the 5′-dRP group [4]. DNA ligase IIIα completes repair by sealing the nick [5]. Bifunctional glycosylases can remove the damaged base and nick the DNA backbone, resulting in a 3′-dRP group and a 5′-phosphate [6]. APE1 modifies the DNA by removing the 3′dRP group. BER again completes by Pol β filling in the gap and DNA ligase IIIα sealing the nick. The NEIL DNA glycosylases initiate an APE-independent BER sub-pathway, excising the damaged base and incising the DNA strand to leave 3′and 5′-phosphate groups [7]. Polynucleotide kinase modifies this gap by removing the 3′-phosphate. Pol β binds this substrate and catalyzes incorporation of the correct dNTP opposite the templating base, followed by DNA ligase IIIα sealing the nick to complete repair. Regardless of the type of damage or BER sub-pathway initiated, Pol β is the main polymerase that is responsible for gap-filling DNA synthesis.
If any component of this multi-step BER process is not functioning properly in cells, DNA lesions or BER intermediates may persist. This can result in genomic instability or mutagenesis, hallmarks of tumorigenesis and tumor progression. First proposed in 1974, the mutator phenotype hypothesis indicates that the mutation rate observed in tumors is too high to be accounted for by the somatic mutation rate alone [8]. Defects in DNA replication or repair are thought to underlie the elevated mutation rate observed in human tumors [9]. Such defective pathways have indeed been identified in human tumors, including mismatch repair, methylation reversal repair and BER [10]. Tumor-specific Pol β variants have been identified in 30% of 189 tumors characterized to date [11]. Several non-synonymous amino acid substitution variants have functional phenotypes that include polymerase deficiency, dRP lyase deficiency and subtle mutagenesis in vivo and in vitro [12–15]. Three of these variants, Glu295Lys, Ile260Met and Lys289Met, have also been shown to induce cellular transformation when expressed in cells [13,16]. These variants are found in the fingers sub-domain of Polβ, a region that contains residues that form the dNTP binding pocket and influence dNTP selectivity. The variant D160N was first identified in a small-scale screen that was focused upon sequencing POLB in 20 human gastric tumors [17]. Six gastric tumors were found to harbor a POLB mutation that would result in a unique non-synonymous single amino acid substitution. The previously described dRP-lyase domain variant Leu22Pro and fingers domain variant Glu295Lys were also identified in this screen. Residue D160 is found in the palm sub-domain, where the catalytic residues reside.
In this study, our goal was to determine if the gastric tumor-associated variant D160N exhibited altered biochemical properties and if cellular effects of D160N expression are linked to cancer etiology. In the work presented here, we provide evidence that D160N is an active polymerase that induces complex errors and that when expressed in cells, induces a variety of phenotypes related to cancer etiology. Taken together, our results are consistent with the interpretation that the D160N variant has a functional phenotype related to genomic instability and tumorigenesis.
2. Materials and methods
2.1. Plasmid DNA constructs and cloning
For protein purification, the wild-type (WT) Pol β cDNA was cloned into the pET28a vector containing an N-terminal 6X-histidine tag as previously described [18]. The D160N variant was generated using the QuickChange site-directed mutagenesis kit according to the manufacturer’s instructions (Stratagene) with the following DNA primers:
5′-GAGGAGATGCTGCAAATGCAGAACATTGTTCTTAATGAAGTTA-3′ and 5′-TAACTTCATTAAGAACAATGTTCTGCATTTGCAGCATCTCCTC-3′ (Invitrogen). Introduction of the desired mutation was confirmed by direct sequencing at the Keck DNA Sequencing Facility at the Yale University School of Medicine. For retroviral infection into mouse cells, the WT Pol β cDNA with a C-terminal hemagglutinin (HA) tag was cloned into the pRVYtet retroviral vector as previously described [16]. The pRVYtet vector contains a selectable hygromycin resistance gene driven by an internal SV40 promoter and drives expression of Pol β proteins in a Tetracycline (Tet)-repressible manner from the tetO/CMV promoter. The pVSVG vector (Clontech) expresses an env glycoprotein that is used to generate retrovirus using the GP2-293 packaging cell line system (Clontech).
2.2. Cell lines and cell culture
C127 cells are non-transformed epithelial cells generated from a murine mammary carcinoma of an RIII mouse and purchased from ATCC [19]. C127 cells were grown at 37 °C in a humidified 5% CO2 incubator and maintained in DME10 (Dulbecco Modified Eagle’s Medium supplemented with 10% fetal bovine serum, 1% penicillin–streptomycin). The mouse embryonic fibroblast (MEF) cell line deficient in Pol β (88Tag Pol β −/−) was a gift from Leona Samson (Massachusetts Institute of Technology). Pol β-deficient MEFs were maintained in DME10 supplemented with 1% L-glutamine (Invitrogen) and grown at 37 °C in a humidified 5% CO2 incubator. GP2-293 cells (Clontech) used for viral packaging were maintained in DME10 supplemented with 1% L-glutamine and 1 mM HEPES (Invitrogen) and grown at 37 °C in a humidified 5% CO2 incubator.
2.3. Transfection, infection and expression analysis
To generate retrovirus containing the D160N or WT Pol β construct, the GP2-293 packaging cell line was co-transfected with the pRVYtet and pVSVG plasmids using calcium phosphate [16]. GP2-293 cells were incubated for 72 h before virus was harvested. C127 cells were infected with retrovirus supplemented with 4 μg/mL polybrene (American Bioanalytical) as described [16]. Stable clones were selected with 300 μg/mL Hygromycin (Hyg, Invitrogen) and 2.5 μg/mL Tet to suppress expression. Exogenous expression was evaluated by Western blot as described [16]. Briefly, clones were split and grown in the presence or absence of Tet until they were 80% confluent. Cell lysates were harvested by scraping with heated SDS loading buffer (50 mM Tris pH 6.8, 100 mM DTT, 2% SDS and 10% glycerol) and were boiled for 10 min and resolved on a 10% SDS-PAGE gel. Proteins were transferred to a nitrocellulose membrane using a semi-dry transfer apparatus and probed with monoclonal anti-Pol β antibody (Abcam 1831) at 1:250 dilution. Following extensive washing in 1× PBS supplemented with 0.1% Tween, the membrane was incubated with horseradish peroxidase-conjugated secondary antibody at a 1:1000 dilution. Bands were developed using ECL plus (Amersham) and exposed to film. Only clones exhibiting approximately equal levels of exogenous and endogenous Pol β expression were selected for subsequent assays. Bands were quantified using ImageQuant software.
2.4. Focus formation assay
To determine if expression of D160N results in a loss of contact inhibition, C127-D160N clones were passaged as previously described [16]. Briefly, cells were passaged every 3–4 days in DME10 supplemented with 300 μg/mL Hyg with or without Tet where appropriate. Every fourth passage, 1 × 104 cells were seeded into T25 flasks and cells were fed every 3–4 days with DME10 supplemented with 300 μg/mL Hyg, with or without Tet. After 25 days flasks were stained with Giemsa to visualize foci. Resulting foci were counted by microscopic examination. Flasks containing greater than 350 foci were deemed to have too many foci to be accurately counted. To determine if the presence of foci is related to a heritable change, D160N expression was extinguished at later passages by adding Tet back to the growth media and continued monitoring of foci as described.
2.5. Anchorage-independent growth assay
D160N clones were evaluated for the ability to grow in soft agar as described [16]. Approximately 1 × 105 late-passage cells grown under inducing or non-inducing conditions were mixed with DME10 supplemented with 0.3% Difco Noble agar and Tet if appropriate. The resulting mixture was poured onto 60 mm dishes containing DME10 and 0.6% Difco Noble agar. Cells were fed twice a week with 1 mL of DME10 supplemented with 0.3% Difco Noble agar with or without Tet where appropriate. After 5 weeks, colonies were visualized by light microscopy, with 10 fields from each of 5 plates per treatment scored for colonies. The number of colonies per field (±SE) reported is the average across all plates and *p < 0.01 for the paired, two-tailed t-test.
2.6. Proliferation assay
To determine the effect of D160N or WT Pol β expression on cellular proliferation, late-passage cells were seeded at 2 × 104 cells per 60 mm dish with or without Tet as appropriate and allowed to attach overnight at 37 °C in a humidified 5% CO2 incubator. A replicate of each plating condition was trypsinized and counted every 24 h for four days using an automated cell counter (Nexelcom). Experiments were performed in triplicate and the data is reported as average change in cell number per 24-h period.
2.7. Chemoinvasion assay
To evaluate the invasive potential of cells expressing D160N or WT Pol β, late-passage C127 clones were used in a standard Boyden chamber invasion assay according to the manufacturer’s instructions (BD Biosciences). Cells were plated in DMEM on chambers containing a Matrigel membrane insert to evaluate invasion. Chambers containing a control membrane insert lacking Matrigel were used to evaluate migration. 2.5 × 104 cells were seeded per 24-well chamber in triplicate under inducing or non-inducing conditions in serum-free media. DMEM containing 10% FBS was used as a chemoattractant in the lower chamber. Chambers were incubated for 22 h at 37 °C in a humidified 5% CO2 incubator. Non-invading cells were removed from the inner chamber surface by scrubbing with a cotton swab. Cells remaining on the outer surface of the chamber were stained with the Diff-Quick staining kit according to the manufacturer’s instructions (BD Biosciences). Membranes were removed from the inserts and fixed on microscope slides. Five representative microscope fields were scored per replicate slide under 20× magnification. The percentage of migratory cells that are also invasive was determined for each cell line by dividing the average number of migratory cells by the average number of invading cells for each cell line. The invasion index for each clone is reported by taking the percentage of invasive cells under inducing conditions divided by the percentage of invasive cells under non-inducing conditions.
2.8. Protein expression and purification
Proteins were expressed and purified by FPLC as previously described [20].
2.9. DNA substrate preparation
The DNA substrates used in this study are listed in Table 1. The individual oligonucleotides were synthesized by the Keck Oligo Synthesis Facility at the Yale University School of Medicine and purified by PAGE chromatography (20% acrylamide, 8 M urea). Radiolabeled DNA substrates were generated as described [18].
Table 1.
DNA substrate sequences.
| Substrate | DNA sequence of annealed product |
|---|---|
| 1 bp-gap | 5′ CGCGCCAATCGAGCCATGTCGT GTCAACGACCCACCATTCAAGA |
| 3′ GCGCGGTTAGCTCGGTACAGCAGCAGTTGCTGGGTGGTAAGTTCT | |
| LPSD | 5′ CTGCAGCTGATGCGCUGTACGGATCCCCGGGTAC |
| 3′ GACGTCGACTACGCGGCATGCCTAGGGGCCCATG |
2.10. Pre-steady state burst kinetic analysis
To determine if D160N and WT follow the same general kinetic scheme, rapid chemical quench kinetics were performed using a KinTek apparatus [21] as previously described [20]. Two reaction mixtures at 2× concentrations (600 nM DNA and 200 nM enzyme and 200 μM correct dNTP and 20 mM MgCl2) were separately prepared in Reaction Buffer (50 mM Tris–Cl pH 8, 20 mM NaCl, 6 mM DTT, 10% glycerol) and loaded onto the KinTek apparatus thermostatted at 37 °C. Equal volumes of both mixtures were reacted on a millisecond timescale from 0.02 to 1 second and quenched with EDTA. Extended products were resolved on a 20% polyacrylamide gel containing 8 M urea, visualized using a Storm 860 Phosphorimager and quantified with ImageQuant software. Extended product was plotted as a function of time and fit by nonlinear regression to the full burst equation:
where [E]app is the apparent enzyme concentration, which is proportional to the burst amplitude, kobs is the rate of product formation and kss is the rate of product release.
2.11. Active site titration
To determine the DNA binding constant KD(DNA), a titration of DNA from 10 to 300 nM was performed in Reaction Buffer with a fixed 60 nM concentration of Pol β enzyme and 100 μM of correct dNTP for 0.3 s in a KinTek apparatus at 37 °C. This time interval was selected to allow maximum amplitude while minimizing multiple enzymatic turnovers. Reactions were analyzed as stated above and product formed was plotted as a function of DNA concentration and fit to the quadratic equation:
where [E]app is the apparent enzyme concentration, [D]0 is the initial concentration of the gapped DNA substrate, and KD(DNA) is the dissociation constant of the Pol β-DNA complex.
2.12. Whole cell extract base excision repair assay
To assess the ability of D160N to participate in BER, we prepared whole cell extract (WCE) from Pol β-deficient MEFs as described [22]. The DNA substrate LPSD was used for this assay (Table 1). The assay was performed as previously described in [15]. Briefly, 100 nM of DNA substrate was first treated with 10 U of uracil DNA glycosylase in UDG buffer (New England Biolabs) for 30 min at 37 °C then placed on ice. 5 nM of UDG-treated DNA was then reacted with 10 nM purified Pol β and 5 μg of WCE in BER buffer (45 mM HEPES, 70 mM KCl, 2 mM DTT, 7.5 mM MgCl2, 0.5 mM EDTA, 2 mM ATP and 10 μM dCTP) for 2, 5 and 20 min. Reactions were quenched with an equal volume of 90% formamide dye containing EDTA. Products were resolved on a 20% sequencing gel as described above and repaired products were visualized using a Storm 860 Phosphorimager and quantified using ImageQuant software.
2.13. Circular dichroism spectroscopy and thermal denaturation
Circular dichroism (CD) spectra were obtained for purified WT Pol β and D160N proteins as 1 μM solutions in 10 mM K2HPO4 at 23 °C measured in 1 nm steps from 190 nm to 260 nm. Scans were run using crystal cuvettes with 0.2 mm path length in a Chirascan™ Circular Dichroism Spectrometer (Applied Photophysics). The scan was performed in triplicate for each protein sample. Thermal denaturation profiles were also obtained on the Chirascan™ under the same conditions. Melting temperatures (Tm) were determined by estimating the midpoint of the transition phase of the denaturation curve from 5 °C to 65 °C measured at 208 nm.
2.14. Pull-down assays
To evaluate the ability of D160N to interact with known BER complex binding partners XRCC1 and DNA ligase IIIα, we performed pull-down assays using WCE prepared from Pol β-deficient MEFs. Pull-downs were performed similarly as described using 20 μg of purified His-tagged WT Pol β or D160N protein added to 100 μg of WCE [13]. The following protocol modifications were made. Following transfer, membranes were probed overnight with primary antibodies against either DNA ligase IIIα (1:250, BD Transduction Laboratories 611878) or XRCC1 (1:500 Abcam 9147). Following extensive washing with 1XPBS supplemented with 0.1% Tween20, membranes were incubated with horseradish peroxidase-conjugated secondary antibody (1:5000 Amersham) for 1 h at room temperature. Bands were visualized using an enhanced chemiluminescence kit (BioRad) according to the manufacture’s instructions and a super-cooled high resolution CCD camera on the BioRad ChemiDoc Imaging System.
2.15. HSV-tk forward mutation assay
Gapped duplex DNA substrates (vectors pSAStu2-5 and 2–6), encoding the HSV-tk gene and a [CA]5 or 6 tandem repeat were prepared as described [23]. In vitro polymerase reactions using D160N were performed in duplicate as described [23]. Completed synthesis across the gap during the polymerase reaction was confirmed by 0.6% agarose gel analysis. Completed reactions were used to transform the E. coli strain FT334 by electroporation and mutant selection was performed as described [24]. In brief, selection of HSV-tk mutant plasmids was accomplished by plating bacteria in the presence of 40 mM 5-fluoro-2′-dexoyuridine (FUdR) and 50 mg/mL chloramphenicol (Cm). The mutational specificity of D160N was obtained as described [24]. The mutation frequency (MFobs) was obtained by dividing the number of FUdRR + CmR colonies by the total number of CmR colonies. To generate the polymerase error frequency (EFest), we corrected for pre-existing mutations present within the DNA template (ssDNA frequency) as previously reported in [23], as well as the presence of multiple independent mutations and undetectable mutations [25]. For tk sequences that contained more than one mutation, we considered mutations separated by 15 bases or more to be independent errors and were counted separately for the error frequency calculation. Mutations present within 15 bases, referred to as tandem mutations, are considered to result from a single binding event, based on previous Pol β processivity studies [25]. Independent single mutations were only included in the EFest calculation if they were determined to be detectable, meaning that they resulted in a change in the amino acid sequence. Base substitutions that resulted in a non-synonymous amino acid change and frame shift mutations were considered detectable.
3. Results
3.1. Cells expressing D160N exhibit increased proliferation
Since D160N was isolated from a human gastric tumor, we evaluated the ability of D160N to induce cellular transformation. To examine phenotypes related to transformation, we used C127 mouse epithelial cell line because these cells are immortalized but not transformed. C127 cells were infected with retrovirus containing the pRVYtet vector harboring the HA-tagged D160N construct and clones were selected as described in Section 2. Western blot analysis was performed on lysates from clone pairs grown with or without Tet for a full passage. Clones were selected for subsequent assays based on a relatively equal level of expression of both endogenous and exogenous Pol β when Tet was removed, and no significant expression of exogenous Pol β in the presence of Tet (Fig. 1).
Fig. 1.
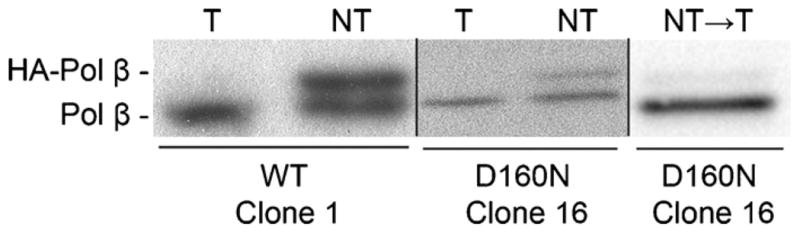
C127 cell lines inducibly express exogenous Pol β. Representative Western blots showing expression of WT Clone 1, D160N Clone 16. Exogenous Pol β is HA-tagged and runs higher than the untagged endogenous form. Blots were quantified using ImageQuant Software. Under inducing conditions (NT), WT clone 1 expresses at a ratio (exogenous:endogenous) of 0.97:1. D160N Clone 16 expresses at a ratio of 0.5:1. Adding Tet back to the media shuts off exogenous expression in D160N Clone 16 (shown for passage 22), as the ratio is reduced to 0.1:1.
Increased proliferation is a hallmark of tumor cells. We investigated whether D160N expression would impact proliferation capacity. For these studies we used clone 16 of D160N and clone 1 of WT in C127 cells and monitored the change in cell number per 24-h period for a total of four days. Expression of exogenous WT in this cell line does not significantly increase the rate of proliferation (ANOVA p = 0.66, Fig. 2A). Expression of exogenous D160N, however, does increase the cellular proliferation rate, a property of cellular transformation (Fig. 2B). The proliferation rate increases between 2 and 3-fold on days 4 and 5, compared to the non-induced (Tet) counterpart, a significant increase (ANOVA p ≤ 0.0001).
Fig. 2.

D160N expression induces cellular transformation. (A) Expression of exogenous WT Pol β does not affect the cellular proliferation rate, as compared to its matched, non-induced control (ANOVA p = 0.66). The solid line represents cells grown under inducing conditions (no Tet) and the dashed line represents cells grown under non-inducing conditions (Tet). (B) Exogenous expression of D160N (solid line) results in a significant increase (ANOVA p < 0.0001) in growth rate when compared to its matched, non-induced control (dashed line). (C) Focus formation of D160N clones; clone 6 (●), clone 16 (▼), clone 21 (■) Foci reported are from an average of two flasks. Solid lines represent cells assayed when D160N was expressed (no Tet) and dashed lines represent no expression (Tet included in medium). Foci counts over 350 were determined to be too numerous to count, indicated by the break in the y-axis. (D) Transformation persists in D160N-16 after expression has been extinguished. Tet was added at passage 22 to extinguish expression, indicated by the arrow. Data up to passage 22 is reproduced from panel C. (E) Expression of D160N results in anchorage-independent growth. D160N-16 was grown under inducing or non-inducing conditions before being plated in soft agar. When expression of D160N is induced, significantly more colonies grew in soft agar (*p < 0.01). (F) Expression of D160N increases cellular invasion capacity. The invasive index for cells expressing D160N is increased 4.5 fold compared to cells expressing WT.
3.2. Expression of D160N induces focus formation in C127 cells
Clones 6, 16 and 21 were serially passaged in the presence or absence of Tet. For all three clones evaluated, when expression of D160N is permitted by removal of Tet from the growth media, cells form foci by passage 12 (Fig. 2C). Cells of the same clone grown in Tet-containing media form significantly fewer foci, and only at much later passages. The same assay design using exogenous WT Pol β has previously shown that expression of WT Pol β does not result in focus formation [16]. Our data demonstrate that expression of the D160N variant induces focus formation.
To determine if expression of D160N is required for transformation to persist, we extinguished expression of exogenous Pol β in clone 16 by adding Tet back to the media at passage 22. We confirmed by Western blot that the addition of Tet turned off D160N expression (Fig. 1). We then assessed the ability of these cells to form foci as described above (Fig. 2D). The cells continue to form large numbers of foci after D160N expression is turned off, indicating that continuous D160N expression is not required to maintain the transformed phenotype over subsequent passages. This result suggests that D160N expression led to a heritable change in these cells resulting in focus formation.
3.3. D160N expression results in anchorage-independent growth
Another characteristic of transformed cells is anchorage-independent growth. We assessed the ability of cells expressing D160N to grow in soft agar, using D160N clone 16. When this clone was grown under non-inducing conditions, an average of 52 colonies were present per plate (Fig. 2E). Under inducing conditions, an average of 163 colonies were observed, a 3.1 fold increase. These results indicate that expression of D160N confers the ability to grow in an anchorage-independent manner.
3.4. Cells expressing D160N are invasive
Invasiveness as measured by the Boyden chamber chemoinvasion assay is a predictor of metastatic potential. We evaluated the chemoinvasion capacity in C127 clones expressing wild-type, clone 1, and D160N, clone 16 using a standard Boyden chamber assay as described in Section 2. For each clone, invasive index was calculated by comparing the percentage of invasive, migratory cells for both non-inducing and inducing conditions. The ratio of invasion percentage under inducing conditions to invasion percentage under non-inducing conditions is referred to as the invasive index. The invasive index in cells expressing wild type was determined to be 0.8, whereas the invasive index in cells expressing D160N was 3.6, a 4.5 fold increase (Fig. 2F). These results indicate that expression of D160N confers invasive potential in cells. Taken together, our data are consistent with the interpretation that D160N expression induces cellular transformation in this system.
3.5. D160N is an active polymerase
We have shown here that D160N expression results in cellular transformation that likely results from mutagenesis induced by expression of this protein. To determine if the transformed phenotype has a mutational basis we sought to test the hypothesis that D160N alone is a mutator polymerase. First, we characterized its biochemical properties.
CD spectroscopy was used to evaluate the effect of the single amino acid substitution D160N on global protein folding. The overall molar ellipticity profiles for WT Pol β and D160N are similar, indicating that the single substitution at position 160 does not affect the enzyme’s global helical structure (Fig. S1). D160N also has a similar thermal stability profile and melting temperature as WT Pol β (data not shown). These results indicate that the transformed phenotype does not result from aberrant folding or stability of the D160N enzyme.
When reacted with an excess of DNA on a millisecond timescale, WT Pol β exhibits biphasic burst kinetics with a kobs of 12 ± 2 s−1 representing the burst of product formation and a rate-limiting kss of 1.3 s−1 representing product release (Fig. 3). The WT burst rate is similar to previously reported results [14]. D160N exhibits a similar rapid burst of product formation with a kobs of 17 ± 4 s−1 and a kss of 1.4 s−1 (Fig. 3). These results indicate that both WT and D160N follow a similar kinetic mechanism for nucleotide incorporation, with the rate-limiting step occurring after chemistry.
Fig. 3.
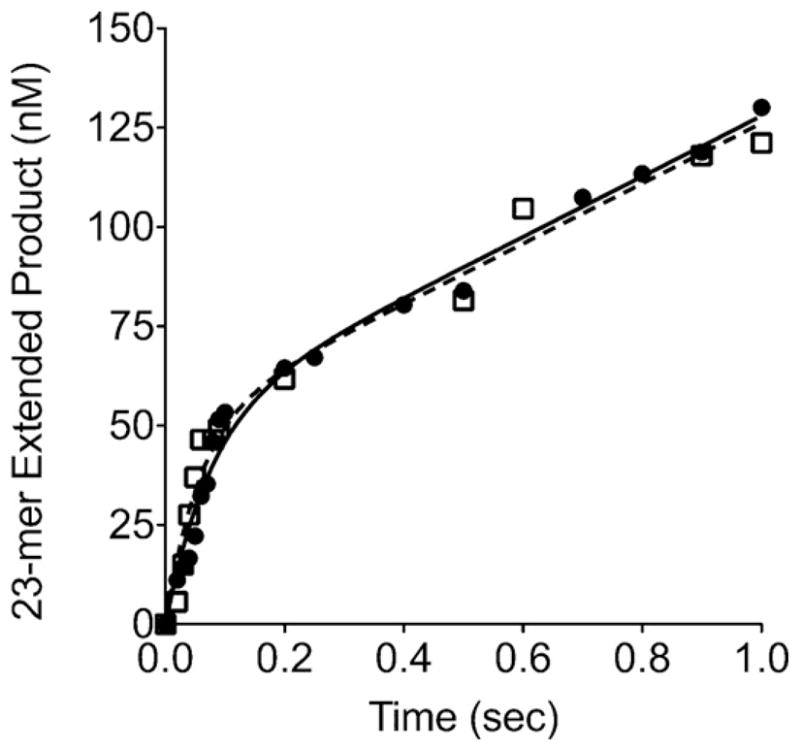
D160N exhibits biphasic pre-steady state burst kinetics. Insertion of dCTP opposite template G was evaluated on the 1 base pair-gapped DNA substrate. Reactions were carried out on a millisecond timescale using a KinTek apparatus as described in Section 2. Data for WT (●, solid line) was fit to the full burst equation with a kobs of 12 ± 2 s−−1 and a steady-state rate constant of 1.3 s−1. Data for D160N (△, dashed line) was fit to the full burst equation with a kobs of 17 ± 4 s−1 with a steady-state rate constant of 1.4 s−1.
3.6. D160N binds DNA with similar affinity as wild-type
By performing an active site titration as described in Section 2, we determined the equilibrium dissociation constant KD(DNA) for both WT and D160N. For single base pair gapped DNA substrate, the KD(DNA) values or WT and D160N are 16 ± 3 nM and 21 ± 4 nM respectively (data not shown). This suggests that the substitution of Asn for Asp at residue 160 does not affect the affinity of Pol β to bind DNA.
3.7. D160N is a mutator polymerase
Our biochemical characterization of D160N suggests that it binds to DNA and catalyzes DNA synthesis in a manner similar to WT. Next, we used the in vitro HSV-tk forward mutation assay to determine if D160N catalyzes DNA synthesis in an error-prone manner [23]. Using this assay, we evaluated the accuracy of Pol β synthesis through a target region containing the HSV-tk gene and either a [CA]5 and [CA]6 microsatellite sequence. We obtained mutation frequencies for D160N on HSV-tk templates containing the artificial microsatellites [CA]5 and [CA]6 (Table 2) and compared our results to previously reported WT data [23]. Data reported are an average of two independent polymerase reactions for each template. D160N exhibited an 8-fold and a 23-fold increased mutation frequency relative to WT on the [CA]6 and [CA]5 templates respectively. These results demonstrate that D160N is a mutator polymerase.
Table 2.
Observed HSV-tk mutant and estimated error frequencies of Pol β WT and D160N.
| Mutation classb | Frequency × 10−4 (no. sequenced)a
|
Ratio D160N/lβ-WT | |
|---|---|---|---|
| D160N | β-WT | ||
| (A) pSAStu2-5 template | |||
| MFobs | 320 (42) | 14 (24) | 23 |
| Single | 305 (40) | 14 (24) | 22 |
| Multiple | 15 (2) | 0 (0) | >26 |
| Pol EFest | 330 | 12 | 28 |
| [CA]5 | 46 (6) | 2.4 (5) | 19 |
| HSV-tk coding | 274 (36) | 9.3 (19) | 29 |
| Mutation class | Frequency × 10−4 (no. sequenced)a
|
Ratio D160N/β-WT | |
|---|---|---|---|
| D160N | β-WT | ||
| (B) pSAStu2-6 template | |||
| MFobs | 410 (45) | 54 (44) | 8 |
| Single | 391 (43) | 54 (44) | 7 |
| Multiple | 18 (2) | 0 (0) | >15 |
| Pol EFest | 430 | 52 | 8 |
| [CA]6 | 76 (8) | 11 (9) | 7 |
| HSV-tk coding | 353 (37) | 41 (35) | 9 |
The polymerase error frequency (Pol EFest) represents all independent errors generated by the polymerase that would result in a non-synonymous amino acid substitution. Mutations within the [CA]n repeat are indicated separately from the errors within the HSV-tk coding sequence.
To evaluate the types of mutations induced by D160N within the HSV-tk target sequence, we isolated plasmid DNA from mutant colonies and sequenced the target region as described in Section 2.
Mutation frequency (MFobs) is derived from the number of mutant colonies obtained. A mutant colony may harbor more than one mutation within the HSV-tk gene, as indicated by “multiple.”
3.8. D160N induces an increased frequency of mutations within microsatellites
To more directly compare errors made by the two enzymes, we calculated the estimated polymerase error frequency for distinct error classes. The observed distribution of D160N errors between the [CA] microsatellite and the HSV-tk coding region was not statistically different from that observed for WT (Table 2). WT and D160N also induce similar types of errors within the microsatellite region that consist mainly of deletions of CA repeats (Table S1). However, while WT exhibits a nearly 4-fold increase in error frequency between [CA]5 and [CA]6, D160N induces a similar error frequency on [CA]5 and [CA]6 DNA templates. Our results suggest that the WT and D160N enzymes interact with these DNA templates differently.
3.9. D160N induces tandem complex mutations
The mutation spectra for independent, non-tandem errors produced by D160N within the HSV-tk coding region are presented in Fig. S2 and are summarized in Table 3. We observed a significant change in the distribution of D160N errors among three mutation classes, relative to WT: base substitutions, frameshifts and tandems (X2 = 5.95; p = 0.05). The most striking distinction observed between D160N and WT in the context of this assay is the propensity to generate tandem mutations present within the target region. The proportion of tandem errors among all other coding region errors made by D160N is significantly different from that produced by WT (p = 0.0012, Fisher’s exact test, two-sided). Two mutations present within a 15 bp region are classified as tandem simple mutations. Tandem complex mutations are classified as a group of mutations (>2 mutations) present within a 15 bp region, considered to result from a single binding event. Tandem complex mutations identified in the HSV-tk coding region for both enzymes are shown in Fig. 4. On both DNA templates, 28% of mutations (25/90) made within the entire target by D160N were tandem complex, compared to only 3% (2/68) of WT mutations. For both DNA templates, the frequencies of tandem complex errors represent the most significantly elevated error class for D160N relative to WT.
Table 3.
Distribution and frequency of errors produced by WT and D160N Pol β within the HSV-tk coding sequence.
| Type of error | Frequency × 10−4 (no. events)
|
Ratio D160N/β-WT | |
|---|---|---|---|
| D160N | β-WT | ||
| Pol EFest (HSV-tk coding region) | 310 (90) | 25 (68) | 12 |
| Base substitutions (independent) | 55 (16) | 2 (6) | 28 |
| Frameshifts (independent) | 138 (40) | 16 (43) | 9 |
| Tandem errors | 117 (34) | 7 (19) | 17 |
| Simple | 31 (9) | 6 (17) | 5 |
| Complex | 86 (25) | 0.7 (2) | 123 |
The HSV-tk coding region Pol EF from Table 2 was averaged for both substrates, and the average used to calculate frequencies of specific error classes. Base substitutions and frameshifts are considered independent if they occur greater than 15 nucleotides apart. Tandem simple mutations refer to two single mutations that occur within 15 bases. Tandem complex mutations describe >2 mutations that occur within 15 bases.
Fig. 4.

Tandem complex mutations affecting the HSV-tk coding region generated by (A) D160N and (B) WT. The parent sequence is given on the top line and all mutant sequences are below. Mutations are highlighted in red and dotted lines indicate deleted bases. If a specific tandem mutation was identified in more than one mutant colony, the number of colonies is indicated to the right of the mutation sequence.
3.10. D160N interacts with XRCC1 and LigIIIα and participates in BER
To evaluate the ability of D160N to interact with other BER proteins, we performed pull-down assays using WCE from Pol β-deficient MEFs and purified his-tagged WT or D160N protein. As shown in Fig. 5, D160N maintains the ability to physically interact with both XRCC1 and LigIIIα.
Fig. 5.
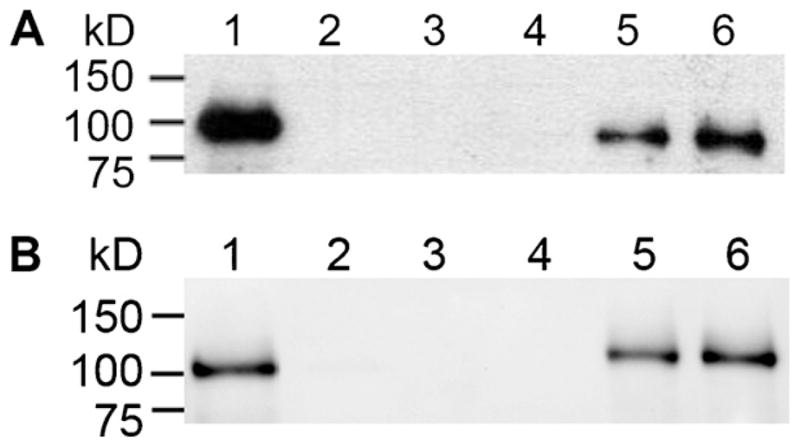
D160N maintains interactions with BER binding partners. A pull-down experiment was performed using WCE from Pol β-deficient MEFs and his-tagged purified Pol β enzyme as described in Section 2. (A) WT and D160N interact with XRCC1 (90 kDa). Lane 1, 25% input; Lane 2, WT with nickel beads alone; Lane 3, D160N with nickel beads alone; Lane 4, WCE with nickel beads alone; Lane 5, WT and WCE pull-down; Lane 6, D160N and WCE pulldown. (B) WT and D160N interact with LigaseIIIα (103 kDa). Lane 1, 25% input; Lane 2, WT with nickel beads alone; Lane 3, D160N with nickel beads alone; Lane 4, WCE with nickel beads alone; Lane 5, WT and WCE pulldown; Lane 6, D160N and WCE pulldown.
To determine if D160N has the ability to participate in the BER of uracil, a BER assay was performed using WCE from Pol β-deficient MEFs. An assessment of BER performed by either WT or D160N was carried out by adding purified Pol β to the DNA–WCE mixture and reacting at 37 °C on a minute timescale. Both WT and D160N are observed to form n + 1 product, which is then fully repaired by BER enzymes present in the WCE (Fig. 6). The overall percentage of repaired product in these reactions is calculated as the ratio of the repaired product intensity divided by the total intensity within the lane. Without addition of exogenous Pol β (Lane 3), the WCE could only repair 6% of the substrate; a polymerase other than Pol β likely functions in this repair. Addition of either WT or D160N result in at least 50% of the substrate repaired in 2 min and at least 86% repaired by 20 min. These results indicate that D160N interacts with partner BER proteins and participates in BER in a cellular context to a similar degree as WT Pol β.
Fig. 6.

D160N participates in base excision repair with similar efficiency as WT. WCE from Pol β-deficient MEFs was incubated with the UDG-treated LPSD DNA substrate. Addition of purified Pol β enzyme for 2, 5 or 20 min results in dCTP incorporation, followed by DNA ligation by WCE enzymes to form fully repaired product. After 2 min, WT and D160N repair 35% of the DNA substrate. By 20 min, WT repairs 86% and D160N repairs 87% of the DNA substrate. Lane 1, annealed DNA substrate; Lane 2, UDG-treated DNA substrate; Lane 3, UDG-treated DNA incubated with WCE; Lanes 4–6, UDG-treated DNA substrate incubated with WCE and WT Pol β for 2, 5 and 20 min respectively; Lanes 7–9, UDG-treated DNA substrate incubated with WCE and D160N for 2, 5 and 20 min respectively.
4. Discussion
In this study, we showed that the D160N gastric cancer-associated Pol β variant exhibits a functional phenotype linked to tumorigenesis and/or tumor progression. We also demonstrate that the mechanism of cellular transformation has a mutational basis because it does not require continuous expression of the D160N protein. Importantly, our in vitro experiments reveal that D160N is as active as WT Pol β, yet it is a strong mutator polymerase, supporting our hypothesis showing that cellular transformation stems from mutagenesis. D160N interacts with its protein partners and functions in BER similar to WT. Therefore, we conclude that D160N exhibits functional phenotypes that are associated with cancer.
4.1. D160N expression results in cancer hallmark phenotypes
Cellular transformation was assessed by four endpoints considered to be hallmarks of cancer [26]. Cells induced to express exogenous D160N proliferate ~3 fold faster than their non-induced counterpart as well as cells induced to express exogenous WT Pol β. Proliferation assays were performed after 25 passages under inducing and non-inducing conditions. Presumably, during this time, D160N expression lead to the induction of mutations within genes controlling growth, resulting in more rapid proliferation than that observed with cells expressing WT or cells grown under non-inducing conditions for D160N. An elevated proliferation rate in tumors, such as those expressing D160N, may lead to propagation of genomic instability [27]. Cells expressing D160N also exhibit anchorage-independent growth, as evidenced by the ability to grow in soft agar. The anchorage-independent growth phenotype is linked to tumor metastatic potential [28]. These cells also demonstrate a 4.5-fold increase in invasive index, as measured by a Boyden chamber chemoinvasion assay. These results indicate that D160N expression is linked to cellular transformation and could be one of the drivers of carcinogenesis in the gastric tumor from which it was isolated. Expression of D160N may also lead to metastasis and more aggressive disease compared to tumors expressing WT Pol β.
We have also shown that D160N expression in cells leads to a loss of contact inhibition, as measured by the ability to form foci. Each D160N clone formed large numbers of foci at different passages, consistent with the interpretation that the mechanism of transformation is stochastic in nature. These varying rates of transformation may result from different genes being mutated at random over time, along with selection. Continued expression of D160N was not required for the formation of foci to persist, indicating that expression of D160N resulted in a heritable change. These results are consistent with the interpretation that the mechanism underlying transformation is mutagenesis.
4.2. D160N is an active polymerase that participates in BER
Biochemical in vitro assays using purified D160N enzyme indicate that it has a WT-like burst rate and DNA-binding affinity. The substitution to Asn at position 160 does not affect the overall global folding structure or thermal stability of the enzyme. D160N retains the ability to interact with the BER enzymes XRCC1 and DNA LigaseIIIα, and participates in BER to a similar degree as WT. These results indicate that D160N would be expected to be active and function in BER like WT in a cellular environment.
4.3. D160N exhibits mutagenic properties that may contribute to cellular transformation
Using an in vitro forward mutation assay, we directly evaluated the mutagenic potential of the D160N variant on two DNA substrates. D160N is a mutator polymerase on both DNA substrates evaluated, with an EF increase of 8-fold for the CA6 substrate and a 28-fold increase for the CA5 substrate. While the overall proportion of mutations made that affected the microsatellite were similar to WT, the elevated overall EF of D160N translates into a marked increase in microsatellite mutation frequency: 19-fold for CA5 and 7-fold for CA6. Alterations within microsatellites, specifically within (CA)n repeat regions, have been reported in a significant proportion of human gastric tumors [29,30]. Our data suggest that gastric tumors expressing D160N may be at increased risk for mutagenesis within (CA)n microsatellites.
For D160N, the most enriched error class, in both proportion and frequency, was tandem complex errors. This error class is characterized by mutations that occur within a single binding event, with multiple errors (>2) present within the target. Tandem complex mutations generated by D160N occur predominantly within the tk coding region and range from base substitutions mixed with simple or large deletions, to a large insertion and a long-range complex mutation spanning 72 base pairs. Tandem complex mutations generated by WT were rarely seen, and only in the CA6 DNA substrate. The two WT tandem complex errors identified included large deletions and lacked any point mutations.
The nature of the base substitutions within tandem complex errors is of particular interest, as many of the tandem complex mutations contained consecutive regions of micro-homology to other nearby sequences. The AAGAA motif generated in six distinct tandem complex errors were found to replace a TTCTT motif, the exact complement of a downstream sequence. Since this sequence corresponds to the reverse complement of a nearby region, the mutation is likely to be templated in some manner, and not the result of individual base substitutions. A 4 bp insertion generated by D160N on the CA6 DNA substrate, TCTT, is found 8 bp downstream from the insertion site. A 5 bp region of base substitutions, GCCCG, on the CA6 DNA substrate corresponds to a duplication of the preceding 5 bases. D160N also produced a 49 bp insertion on the CA5 DNA substrate. The last 22 nucleotides of this inserted sequence are found immediately adjacent to the insertion. The remaining inserted bases were the complementary sequence of that nearby region. These data suggest that D160N is producing these errors by some type of templated mechanism and not by simple misincorporation. Our results indicate that the DNA may have altered positioning within the active site of D160N, allowing a more flexible orientation that permits a high degree of template movement.
Crystal structures of fellow X-family Polymerase member DNA Polymerase λ bound to a 2-nucleotide gap reveal that the templating strand can adopt a “scrunched” conformation [31]. This template scrunching allows the polymerase to simultaneously interact with the 3′-OH and 5′-dRP, while the extra templating base is extra-helical and found within a pocket that may help to stabilize this scrunched intermediate. Interactions with both sides of the DNA gap are maintained without resulting in significant conformational changes of the enzyme. Molecular modeling of DNA Polymerase λ indicated that longer gap sizes of up to 5 nucleotides would also be accommodated within the enzyme.
Single turnover kinetic studies using longer gaps up to 10 nucleotides with Pol λ and Pol β indicate that for both enzymes, dNTP misincorporation is based on the identity of the downstream position when the gap size is 2 or greater [32]. Pol β, however, was shown to have higher fidelity than Pol λ for all gap lengths and incorporations. This distinction was largely due to a dramatic increase in the binding constant for dNTP for Pol β. Interestingly, the catalytic efficiency of Pol β decreases with increasing gap size, but when the downstream oligonucleotide is absent, the efficiency improves.
Our results indicate that the Pol β variant D160N generates complex tandem mutations, many of which contain regions of microhomology present in the downstream sequence. We propose that the known ability of Pol β to use downstream sequence as a template is exacerbated by the mutation to Asn at position 160. Our data are consistent with the idea that when Asp is altered to Gln, the enzyme becomes more error prone and appears to catalyze some type of template-directed mutagenesis. The mechanism by which this is occurs is not known. The mutagenic capacity of D160N may be increased in long-patch BER, as our forward mutation assay uses a large gapped DNA substrate, facilitating processive Pol β synthesis. It is not clear how the Asn in position 160 would permit Pol β to perhaps “scrunch” more than a few bases of DNA. It is possible that this alteration could somehow increase the flexibility of the palm region, permitting additional DNA template bases to be scrunched.
We have previously observed mutator activity in three other distinct tumor-associated variants: Ile260Met, Lys289Met and Tyr265Cys [14,15,25]. All three variants are found in the fingers sub-domain, a region critical for dNTP binding and selectivity. Similar to D160N, Y265C increases overall mutation frequency by 31-fold in the context of the HSV-tk forward mutation assay [25]. Unlike D160N, Y265C exhibits general mutator activity and generates errors irrespective of DNA sequence context. Importantly, while D160N retains a wild type-like catalytic rate and DNA binding affinity, Y265C exhibits a 20-fold reduction in rate and a 5-fold reduction in DNA binding affinity [33]. Compared to Y265C, I260M and K289M are more subtly mutagenic. Both I260M and K289M have lower DNA binding affinity and exhibit biphasic burst kinetics with a slower catalytic rate than WT [14,15]. Unlike D160N or Y265C, in forward mutation assays, I260M and K289M generate modest, 3-fold increases in overall mutation frequency. These small increases in mutation frequency are primarily due to sequence context-dependent mutagenesis. Therefore, D160N is a unique Pol β mutator variant that retains WT-like catalytic efficiency.
The tandem complex mutations we have characterized here are found within the tk target region, downstream of a microsatellite sequence. McDonald and colleagues have used comparative population genomics to show that repeat DNA sequences increase the likelihood of base substitutions in the neighboring DNA sequence [34]. Their findings indicate that repeat sequences are linked to regions of sequence diversity that increase on an evolutionary time scale, from E. coli to humans. As evidenced by our forward mutation assay results, tumors containing the variant D160N may have increased sequence diversity at these regions compared to cells only expressing WT Pol β. Increased mutagenesis in these regions generated by D160N may contribute to tumor evolution and microenvironment adaptability, resulting in more aggressive disease.
Supplementary Material
{kind=link}
Acknowledgments
This work was supported by grants from the NIH Predoctoral Genetics Training Grant T32 GM007499 to K.A.D. and P01CA129186 to J.B.S. We thank members of the Sweasy lab, including D. Murphy, A. Nemec and S. Dalal, for insightful discussions regarding this work and G. Sun for technical assistance.
Abbreviations
- Pol β
DNA polymerase β
- BER
base excision repair
- AP
apurinic/apyrimidinic
- APE1
AP endonuclease 1
- dRP
deoxyribose phosphate
- NEIL
Nei-like
- WT
wild-type
- Tet
tetracycline
- MEF
mouse embryonic fibroblast
- Hyg
hygromycin
- SDS
sodium dodecyl sulfate
- PAGE
polyacrylamide gel electrophoresis
- DTT
dithiothreitol
- PBS
phosphate buffered saline
- FPLC
fast performance liquid chromatography
- dNTP
deoxynucleotide triphosphate
- kobs
observed rate constant
- kss
steady-state constant
- KD
equilibrium dissociation constant
- WCE
whole cell extract
- UDG
uracil DNA glycosylase
- CD
circular dichroism
- XRCC1
X-ray repair cross complementing 1
- FUdR
5-fluoro-2′-deoxyuridine
- Cm
chloram-phenicol
- HSV-tk
Herpes simplex virus-thymidine kinase
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.dnarep.2012.01.004.
Footnotes
Conflicts of interest
The authors declare that there are no conflicts of interest.
References
- 1.Lindahl T, Wood RD. Quality control by DNA repair. Science. 1999;286:1897–1905. doi: 10.1126/science.286.5446.1897. [DOI] [PubMed] [Google Scholar]
- 2.Sung JS, Demple B. Roles of base excision repair subpathways in correcting oxidized abasic sites in DNA. FEBS J. 2006;273:1620–1629. doi: 10.1111/j.1742-4658.2006.05192.x. [DOI] [PubMed] [Google Scholar]
- 3.Fortini P, Parlanti E, Sidorkina OM, Laval J, Dogliotti E. The type of DNA glycosylase determines the base excision repair pathway in mammalian cells. J Biol Chem. 1999;274:15230–15236. doi: 10.1074/jbc.274.21.15230. [DOI] [PubMed] [Google Scholar]
- 4.Prasad R, Beard WA, Strauss PR, Wilson SH. Human DNA polymerase beta deoxyribose phosphate lyase. Substrate specificity and catalytic mechanism. J Biol Chem. 1998;273:15263–15270. doi: 10.1074/jbc.273.24.15263. [DOI] [PubMed] [Google Scholar]
- 5.Srivastava DK, Berg BJ, Prasad R, Molina JT, Beard WA, Tomkinson AE, Wilson SH. Mammalian abasic site base excision repair. Identification of the reaction sequence and rate-determining steps. J Biol Chem. 1998;273:21203–21209. doi: 10.1074/jbc.273.33.21203. [DOI] [PubMed] [Google Scholar]
- 6.Svilar D, Goellner EM, Almeida KH, Sobol RW. Base excision repair and lesion-dependent subpathways for repair of oxidative DNA damage. Antioxid Redox Signal. 2011;12:2491–2507. doi: 10.1089/ars.2010.3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wiederhold L, Leppard JB, Kedar P, Karimi-Busheri F, Rasouli-Nia A, Weinfeld M, Tomkinson AE, Izumi T, Prasad R, Wilson SH, Mitra S, Hazra TK. AP endonuclease-independent DNA base excision repair in human cells. Mol Cell. 2004;15:209–220. doi: 10.1016/j.molcel.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 8.Loeb LA, Springgate CF, Battula N. Errors in DNA replication as a basis of malignant changes. Cancer Res. 1974;34:2311–2321. [PubMed] [Google Scholar]
- 9.Bielas JH, Loeb LA. Mutator phenotype in cancer: timing and perspectives. Environ Mol Mutagen. 2005;45:206–213. doi: 10.1002/em.20111. [DOI] [PubMed] [Google Scholar]
- 10.Jiricny J, Marra G. DNA repair defects in colon cancer. Curr Opin Genet Dev. 2003;13:61–69. doi: 10.1016/s0959-437x(03)00004-2. [DOI] [PubMed] [Google Scholar]
- 11.Starcevic D, Dalal S, Sweasy JB. Is there a link between DNA polymerase beta and cancer? Cell Cycle. 2004;3:998–1001. [PubMed] [Google Scholar]
- 12.Dalal S, Chikova A, Jaeger J, Sweasy JB. The Leu22Pro tumor-associated variant of DNA polymerase beta is dRP lyase deficient. Nucleic Acids Res. 2008;36:411–422. doi: 10.1093/nar/gkm1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lang T, Dalal S, Chikova A, DiMaio D, Sweasy JB. The E295K DNA polymerase beta gastric cancer-associated variant interferes with base excision repair and induces cellular transformation. Mol Cell Biol. 2007;27:5587–5596. doi: 10.1128/MCB.01883-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dalal S, Hile S, Eckert KA, Sun KW, Starcevic D, Sweasy JB. Prostate-cancer-associated I260M variant of DNA polymerase beta is a sequence-specific mutator. Biochemistry. 2005;44:15664–15673. doi: 10.1021/bi051179z. [DOI] [PubMed] [Google Scholar]
- 15.Lang T, Maitra M, Starcevic D, Li SX, Sweasy JB. A DNA polymerase beta mutant from colon cancer cells induces mutations. Proc Natl Acad Sci USA. 2004;101:6074–6079. doi: 10.1073/pnas.0308571101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sweasy JB, Lang T, Starcevic D, Sun KW, Lai CC, Dimaio D, Dalal S. Expression of DNA polymerase {beta} cancer-associated variants in mouse cells results in cellular transformation. Proc Natl Acad Sci USA. 2005;102:14350–14355. doi: 10.1073/pnas.0505166102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iwanaga A, Ouchida M, Miyazaki K, Hori K, Mukai T. Functional mutation of DNA polymerase beta found in human gastric cancer – inability of the base excision repair in vitro. Mutat Res. 1999;435:121–128. doi: 10.1016/s0921-8777(99)00036-1. [DOI] [PubMed] [Google Scholar]
- 18.Murphy DL, Kosa J, Jaeger J, Sweasy JB. The Asp285 variant of DNA polymerase beta extends mispaired primer termini via increased nucleotide binding. Biochemistry. 2008;47:8048–8057. doi: 10.1021/bi702104y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Law MF, Lowy DR, Dvoretzky I, Howley PM. Mouse cells transformed by bovine papillomavirus contain only extrachromosomal viral DNA sequences. Proc Natl Acad Sci USA. 1981;78:2727–2731. doi: 10.1073/pnas.78.5.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murphy DL, Jaeger J, Sweasy JB. A triad interaction in the fingers subdomain of DNA polymerase Beta controls polymerase activity. J Am Chem Soc. 2011;133:6279–6287. doi: 10.1021/ja111099b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson KA. Conformational coupling in DNA polymerase fidelity. Annu Rev Biochem. 1993;62:685–713. doi: 10.1146/annurev.bi.62.070193.003345. [DOI] [PubMed] [Google Scholar]
- 22.Biade S, Sobol RW, Wilson SH, Matsumoto Y. Impairment of proliferating cell nuclear antigen-dependent apurinic/apyrimidinic site repair on linear DNA. J Biol Chem. 1998;273:898–902. doi: 10.1074/jbc.273.2.898. [DOI] [PubMed] [Google Scholar]
- 23.Kelkar YD, Strubczewski N, Hile SE, Chiaromonte F, Eckert KA, Makova KD. What is a microsatellite: a computational and experimental definition based upon repeat mutational behavior at A/T and GT/AC repeats. Genome Biol Evol. 2010;2:620–635. doi: 10.1093/gbe/evq046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eckert KA, Hile SE, Vargo PL. Development and use of an in vitro HSV-tk forward mutation assay to study eukaryotic DNA polymerase processing of DNA alkyl lesions. Nucleic Acids Res. 1997;25:1450–1457. doi: 10.1093/nar/25.7.1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Opresko PL, Sweasy JB, Eckert KA. The mutator form of polymerase beta with amino acid substitution at tyrosine 265 in the hinge region displays an increase in both base substitution and frame shift errors. Biochemistry. 1998;37:2111–2119. doi: 10.1021/bi9722711. [DOI] [PubMed] [Google Scholar]
- 26.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 27.Preston-Martin S, Pike MC, Ross RK, Jones PA, Henderson BE. Increased cell division as a cause of human cancer. Cancer Res. 1990;50:7415–7421. [PubMed] [Google Scholar]
- 28.Mori S, Chang JT, Andrechek ER, Matsumura N, Baba T, Yao G, Kim JW, Gatza M, Murphy S, Nevins JR. Anchorage-independent cell growth signature identifies tumors with metastatic potential. Oncogene. 2009;28:2796–2805. doi: 10.1038/onc.2009.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mironov NM, Aguelon MA, Potapova GI, Omori Y, Gorbunov OV, Klimenkov AA, Yamasaki H. Alterations of (CA)n DNA repeats and tumor suppressor genes in human gastric cancer. Cancer Res. 1994;54:41–44. [PubMed] [Google Scholar]
- 30.Nakashima H, Honda M, Inoue H, Shibuta K, Arinaga S, Era S, Ueo H, Mori M, Akiyoshi T. Microsatellite instability in multiple gastric cancers. Int J Cancer. 1995;64:239–242. doi: 10.1002/ijc.2910640405. [DOI] [PubMed] [Google Scholar]
- 31.Garcia-Diaz M, Bebenek K, Larrea AA, Havener JM, Perera L, Krahn JM, Pedersen LC, Ramsden DA, Kunkel TA. Template strand scrunching during DNA gap repair synthesis by human polymerase lambda. Nat Struct Mol Biol. 2009;16:967–972. doi: 10.1038/nsmb.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brown JA, Pack LR, Sanman LE, Suo Z. Efficiency and fidelity of human DNA polymerases lambda and beta during gap-filling DNA synthesis. DNA Repair (Amst) 2011;10:24–33. doi: 10.1016/j.dnarep.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Washington SL, Yoon MS, Chagovetz AM, Li SX, Clairmont CA, Preston BD, Eckert KA, Sweasy JB. A genetic system to identify DNA polymerase beta mutator mutants. Proc Natl Acad Sci USA. 1997;94:1321–1326. doi: 10.1073/pnas.94.4.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McDonald MJ, Wang WC, Huang HD, Leu JY. Clusters of nucleotide substitutions and insertion/deletion mutations are associated with repeat sequences. PLoS Biol. 2011;9:e1000622. doi: 10.1371/journal.pbio.1000622. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.