

Abstract
The cellular responses to infection are many, and include programmed cell death to inhibit microbial dissemination and the production and secretion of interferons (IFNs), which confer resistance to uninfected cells. In addition to the antimicrobial effects of IFNs, these cytokines have been used clinically for the treatment of various neoplasias to inhibit proliferation and stimulate apoptosis. However, the precise mechanisms of action of IFNs remain to be completely understood. One of the primary response genes induced after an infection or treatment with type I or III IFN is known as IFN-stimulated gene 54 (ISG54) or IFN-induced gene with tetratricopeptide repeats 2 (IFIT2). ISG54/IFIT2 is a member of a family of IFN-induced genes related in the sequence and structure. Expression of this protein has been found to promote cellular apoptosis by a mitochondrial pathway dependent on the action of Bcl2 proteins. ISG54/IFIT2 does not function as a monomer, and it forms complexes with itself and with the related ISG56/IFIT1 and ISG60/IFIT3 proteins to elicit complex cellular responses. The apoptotic response to ISG54/IFIT2 may contribute to other functions that have been reported, including translational regulation, inhibition of tumor colonization, and protection against a lethal viral infection.
Introduction
The biological effects of interferons (IFNs) are complex, and reflect the impact of these cytokines on hundreds of cellular genes (de Veer and others 2001; Gomez and Reich 2003; Borden and others 2007). Initially discovered for their ability to inhibit viral replication, they are known to influence activation, differentiation, proliferation, and survival of various cell types. They have been used clinically as antiviral agents, for the treatment of immune diseases such as multiple sclerosis, and for the inhibition of various neoplasias. IFNs bind to the receptors in the plasma membrane and initiate a signal transduction pathway that leads to transcriptional induction of nuclear genes. The concerted and balanced effects of the induced genes, commonly referred to as IFN-stimulated genes (ISGs), are responsible for the unique physiological responses to IFN. Only by understanding the actions of individual ISGs will the biological effects of IFNs be understood. The ISG54 gene codes for a protein of 54 kDa (472 a.a.) with 9 tetratricopeptide repeats (TPR) (Levy and others 1986; Wathelet and others 1986; Stawowczyk and others 2011; Yang and others 2012). TPR motifs are protein–protein interaction modules that are known to play a role in the formation of multiprotein complexes (Blatch and Lassle 1999; D'Andrea and Regan 2003). For this reason, ISG54 has also been designated IFN-induced protein with tetratricopeptide repeat 2 (IFIT2). It is 1 of 4 related human ISGs (ISG54/IFIT2, ISG56/IFIT1, ISG58/IFIT5, and ISG60/IFIT3), and 3 murine ISGs (ISG54/IFIT2, ISG56/IFIT1, and ISG49/IFIT3) (Fensterl and Sen 2011). Human ISG54/IFIT2 will be the focus of this article, as it has the ability to promote cell death (Stawowczyk and others 2011). The proapoptotic effects of ISG54/IFIT2 likely contribute to both antiviral and antiproliferative actions of IFN.
There are 3 families of IFN cytokines (type I, II, and III) that bind to 3 distinct cell surface receptors associated with Janus tyrosine kinases (JAKs) (de Weerd and Nguyen 2012). JAKs are activated after IFN binding and subsequently phosphorylate multiple signaling substrates; of particular significance are the signal transducers and activators of transcription (STATs) (Levy and Darnell 2002; Platanias 2005; Schindler and others 2007; Stark and Darnell 2012). Tyrosine phosphorylation of the STATs promotes the reciprocal interaction of STAT monomers via their phosphotyrosine and SH2 domains to form dimers with the ability to recognize specific DNA target sites in the responsive genes. Type I IFNs (including α- and βIFNs) and type III IFNs (λIFNs) stimulate the formation of STAT1 tyrosine-phosphorylated dimers, and as well the unique multimeric transcription factor complex known as the IFN-stimulated gene factor 3 (ISGF3) (Fu and others 1990). ISGF3 is composed of a tyrosine-phosphorylated STAT1 and tyrosine-phosphorylated STAT2 that is associated with the IFN regulatory factor 9 (IRF9) (Martinez-Moczygemba and others 1997; Ozato and others 2007). ISGF3 binds specifically to an inducible enhancer sequence corresponding to a tandem repeat, designated as the IFN-stimulated response element (ISRE) (Levy and others 1988). In contrast, the tyrosine-phosphorylated STAT1 dimer and other STAT dimers recognize a DNA sequence with an inverted repeat, the γIFN-activated site, originally described in the type II IFN (γIFN) pathway (Decker and others 1991). The promoter of the ISG54/IFIT2 gene has an ISRE that binds ISGF3, and it is robustly induced in response to type I and III IFNs (Fig. 1).
FIG. 1.

Response of the human IFN-stimulated gene 54 (ISG54)/the IFN-induced protein with tetratricopeptide repeats 2 (IFIT2) gene to interferons (IFNs) and pattern recognition receptor (PRR) activation. A simple diagram is used to illustrate the IFN-stimulated response element (ISRE) in the promoter of the ISG54/IFIT2 gene, recognized by the tyrosine phosphorylated (pY) ISGF3 transcription factor activated by type I or III IFNs, and by the serine phosphorylated (pS) IFN regulatory factor 3 (IRF3) or IRF7 transcription factors activated by PRR signaling.
Although originally discovered as a direct response to type I IFN, the ISG54/IFIT2 gene is also induced as a primary stress response to infection, independent of IFNs (Daly and Reich 1993, 1995). Transmembrane and cytosolic pattern recognition receptors (PRRs) that recognize the microbial nucleic acids and other macromolecules activate the signal pathways that induce IFNs, but a subset of the ISGs are also induced before the action of IFNs. PRRs, including Toll-like receptors (TLRs), retinoic acid-inducible gene I-like receptors, and DNA-dependent activator of IRFs, bind microbial ligands and initiate a signaling cascade that leads to the serine phosphorylation and activation of the transcription factors IRF3 and/or IRF7 (Weaver and others 1998; Yoneyama and others 1998; Hiscott 2007; Uematsu and Akira 2007; Yoneyama and Fujita 2010; Theofilopoulos and others 2011). Phosphorylated IRF3 not only contributes to the induction of the IFN genes, it also directly recognizes the ISRE within a subset of ISGs and induces their transcription (Daly and Reich 1993, 1995; Andersen and others 2008). ISG54 is one of the genes induced directly in response to activated IRF3 (Fig. 1).
The rapid action of the innate immune system is of paramount importance to survive pathogenic infections. Apoptosis is one of the innate cellular defense responses. Although apoptosis results in the demise of the infected cell, it prevents the dissemination of replicating pathogens. In fact, many DNA viruses have evolved strategies to control the cellular apoptotic response (Benedict and others 2002; Best 2008; Galluzzi and others 2008). IFN signaling and activation of the IRF3 transcription factor are both known to promote apoptosis (Heylbroeck and others 2000; Weaver and others 2001; Thyrell and others 2002; Chawla-Sarkar and others 2003; Clemens 2003). The clinical efficacy of IFNs for the treatment of both pathogenic infections and various human neoplasias may depend on its apoptotic effects. Some of the ISGs known to play a role in apoptosis include promyelocytic leukemia, protein kinase R, IRF-1, RNAse L, and tumor necrosis factor (TNF)-related apoptosis-inducing ligand (Tamura and others 1995; Zhou and others 1997; Wang and others 1998; Jagus and others 1999; Chawla-Sarkar and others 2003). ISG54/IFIT2 is a newly identified mediator of mitochondrial cell death in the direct primary response to IFNs and PRRs (Stawowczyk and others 2011).
ISG54/IFIT2 Promotes Apoptosis
To evaluate the function of ISG54/IFIT2, it was expressed in HeLa-transformed cells without IFN stimulation. ISG54 was tagged with monomeric green fluorescence protein (mGFP), and the effect of mGFP or ISG54-mGFP expression was evaluated in cell cultures by flow cytometry. There was a dramatic decrease with time in the cell population expressing ISG54-mGFP, indicating that ISG54 had a negative effect on cell viability. This was confirmed by measuring propidium iodide (PI) uptake with flow cytometry in cells expressing ISG54-mGFP. PI is a fluorescent molecule excluded from viable cells, but enters the cells with permeabilized membranes and subsequently intercalates nuclear DNA. The loss of viability in cells expressing ISG54 was evident by 48 h (Stawowczyk and others 2011). To determine if cell death was mediated by apoptosis, cells were stained with allophycocyanin–annexin-V. Annexin-V binds to the phosphatidylserine groups that become exposed on the surface of apoptotic cells. Cells expressing ISG54-mGFP displayed a significant increase in annexin-V staining, indicating that cell death was mediated by apoptosis. Ectopic protein levels of ISG54-mGFP were comparatively similar to the levels of ISG54 in cells treated with IFN, indicating a physiologically relevant expression level of ISG54-mGFP. Transient expression of a tetracycline-inducible ISG54-mGFP elicited a similar apoptotic death response, and the response was demonstrated in various human transformed cell lines.
Since IFNs have a documented antiproliferative and apoptotic effect on transformed cells, we tested the prediction that ISG54/IFIT2 contributes to apoptosis stimulated in response to IFN. Stable cell lines were generated that knocked down endogenous ISG54/IFIT2 mRNA by expressing shRNA. Cells were evaluated for the level of ISG54 protein induced in response to IFNα, and the apoptosis assays were performed in the cells expressing normal levels or reduced levels of ISG54. Measurement of annexin-V staining by flow cytometry demonstrated reduced apoptosis in the knocked down cell line, indicating that ISG54 plays a significant role in the biological context of IFN-induced apoptosis (Stawowczyk and others 2011).
ISG54/IFIT2 Apoptosis Executed via the Mitochondrial Pathway
The hallmark of apoptosis is activation of caspases that are responsible for morphological cellular changes that include shrinkage, blebbing, and nuclear fragmentation (Earnshaw and others 1999; Strasser and others 2000). These changes were evident in the cells expressing ISG54-mGFP. Caspases are produced as catalytically inactive zymogens and are activated by proteolytic cleavage. Immunofluorescence studies with antibodies reactive to activated caspase-3, a primary executioner enzyme, showed a significant correlation with ISG54-mGFP expression (Stawowczyk and others 2011). Pathways that lead to caspase activation have been described as extrinsic, triggered by ligands that bind cell surface death receptors, or intrinsic, triggered by intracellular events. ISG54 does not appear to stimulate the extrinsic pathway, since a dominant-negative Fas-associated protein with a death domain and a caspase-8 cell-permeable inhibitor do not block apoptosis in response to ISG54.
Intrinsic apoptosis is triggered by a shift in the delicate balance of antiapoptotic and proapoptotic factors that regulate mitochondrial permeability (Adams and Cory 2007; Youle and Strasser 2008; Llambi and others 2011). When mitochondrial outer membrane permeabilization (MOMP) occurs in response to apoptotic factors, cytochrome c is released and forms a complex with Apaf-1 and caspase-9 (apoptosome). The apoptosome subsequently activates the effector caspases, caspase-3 and caspase-7. MOMP is tightly controlled by the interactions between members of a family of B-cell lymphoma-2 (Bcl2) proteins that have either proapoptotic or antiapoptotic effects (Fig. 2). The antiapoptotic proteins (Bcl2, Bclxl, Bclw, and Mcl-1) are associated with the outer membrane of mitochondria via their transmembrane domain. The activators of apoptosis (Bim, Bmf, Bid, Bik, Bad, Hrk, Noxa, and Puma) function when they are released from a sequestered cellular location, or cleaved from a larger protein, or are newly synthesized. They bind to the effectors of apoptosis (Bax, Bak, and less-studied Bok) to change their conformation. The conformational change promotes Bax, Bak effector binding to mitochondria, and MOMP. Current models indicate that the prosurvival Bcl2 proteins bind to and inhibit both the activators and effectors, but discerning the mechanisms that regulate their interactions and functions is an active field of study.
FIG. 2.
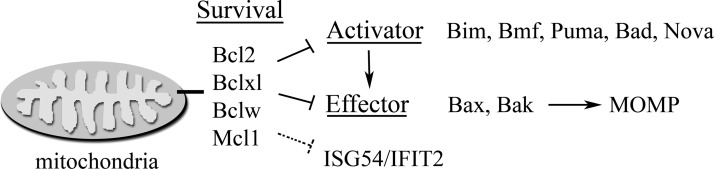
Conceptual diagram of B-cell lymphoma (Bcl) protein family interactions, as they impact mitochondrial outer membrane permeabilization (MOMP). Bcl survival proteins block apoptosis induced by ISG54/IFIT2, but the mechanism remains to be determined (dashed line).
To investigate the role of the Bcl2 family in the apoptotic effects of ISG54, Bclxl was ectopically expressed with ISG54 (Stawowczyk and others 2011). Overexpression of Bclxl blocked ISG54-induced cell death, indicating that ISG54 promotes a mitochondrial pathway of apoptosis. In a converse approach, the response of cells that lack the proapoptotic Bax and Bak genes was evaluated. The bax−/−;bak−/− double-knockout cells are resistant to diverse stimuli that trigger mitochondrial apoptosis. These cells and corresponding wild-type cells were transfected with ISG54-mGFP. There was no increase in cell death in bax−/−;bak−/− cells even after 72 h of ISG54 expression, indicating that Bax and Bak are required for apoptosis in response to ISG54-mGFP. Although ISG54/IFIT2 promotes mitochondrial apoptosis, live-cell imaging studies with ISG54-mGFP indicate that it does not enter the mitochondria. It does, however, enter the endoplasmic reticulum (ER), although ER stress is not measurably activated. The mechanism by which ISG54 impacts the Bcl family of proteins to promote apoptosis remains to be determined.
Protein Interactions Reveal ISG54/IFIT2 Complexes
The biological impact of any cellular protein is dependent on its functional interactions with other proteins. To investigate the potential binding partners, a proteomic approach was used to identify the proteins that could associate with ISG54/IFIT2 (Stawowczyk and others 2011). T7-epitope-tagged ISG54 was expressed in cells, and endogenous proteins bound to tagged ISG54 were identified by mass spectrometry as endogenous ISG54/IFIT2, ISG56/IFIT1, and ISG60/IFIT3 (Fig. 3A). The ISG58/IFIT5 member of the family did not associate with ISG54. The interaction of ISG54 with these 3 related family members was confirmed by coimmunoprecipitation after expression of V5-tagged ISG54 with FLAG-tagged partners. Glycerol-gradient analyses indicated that ISG54 does not exist as a monomer, but is part of a larger ISG/IFIT interactome (Stawowczyk and others 2011). Expression of ISG60 with ISG54 has the effect of inhibiting apoptosis promoted by ISG54, indicating that different homomeric or heteromeric complexes of ISG54 may elicit distinct biological responses.
FIG. 3.

ISG54/IFIT2 forms complexes with itself and ISG56/IFIT1 and ISG60/IFIT3. (A) ISG54 protein is able to dimerize with itself and/or bind ISG56 and ISG60. (B) Ribbon diagram of the crystal structure of the ISG54 homodimer (Protein Data Bank ID:PO9913) (Yang and others 2012). One monomer is highlighted in black and the other in gray with respective N-termini and C-termini indicated.
A different proteomic approach was also used to identify the interaction of these 3 ISG/IFIT proteins (Pichlmair and others 2011). A survey was performed to identify the proteins able to associate with RNA containing 5′-triphosphate, and ISG56/IFIT2 and ISG58/IFIT5 were identified. Subsequent analyses with ISG56/IFIT1 determined that it could form complexes with ISG54/IFIT2 and ISG60/IFIT3.
The crystal structure of ISG54/IFIT2 was solved recently, providing an insight to the molecular interactions of ISG54 with itself and potentially with the family members (Yang and others 2012). The structure reveals ISG54 as a tightly associated homodimer (Fig. 3B). The monomer consists of 22 tandem antiparallel α-helices; most are part of 9 TPR-like motifs grouped into 3 regions: N-terminal (helices 1–6); middle (helices 7–9); and C-terminal (helices 10–22), linked by long loops. The 3 regions of the monomer form a triangle with a space in the center. ISG54 does not exist as a monomer in solution; it forms a homodimer by interaction of the N-terminal and middle regions. The middle region of 1 monomer inserts itself into the empty space of the other monomer, forming a swapped dimer. Future studies are needed to determine if the molecular interactions between ISG54 and ISG56 or ISG60 generate similar swapped dimers. It is possible that trimers, tetramers, or larger complexes also form with ISGs or other binding partners. The functional outcomes of ISG54, ISG56, and ISG60 as homodimers or heterodimers may differ.
Other Functional Properties and Binding Partners Reported for ISG54/IFIT2
Translation
Expression of ISG54/IFIT2 has been reported to elicit several inhibitory effects, most notably on the initiation of translation. Terenzi and others (2005, 2006) demonstrated that the recombinant ISG54 or ISG56 protein added to an in vitro rabbit reticulocyte translation system was able to significantly reduce protein synthesis. They showed coimmunoprecipitation of epitope-tagged ISG54 and the translation initiation factor subunits eIF3c and eIF3e. The binding of ISG54 to the eIF3 subunits was proposed, thereby to inhibit translation. In contrast, studies by Pichlmair and others (2011) found the minimal effects of ISG54 protein added to a standard in vitro rabbit reticulocyte translation system. They provided evidence that the related ISG56 had an inhibitory effect on in vitro translation, and this was due to the ability of ISG56 to bind and sequester RNA with 5′-triphosphate groups, the common template used in the in vitro studies.
Since translational inhibition could trigger apoptosis, the in vivo effect of ISG54 on translation was investigated. Cells expressing ISG54-mGFP or control mGFP were transfected with either 5′-m7G-capped polyadenylated luciferase mRNA or polyadenylated luciferase mRNA regulated by the hepatitis C virus IRES (internal ribosome entry site). GFP-expressing cells were isolated by flow cytometry hours later and evaluated for luciferase protein. ISG54-mGFP did not demonstrate any negative effect on translation of the either capped mRNA or IRES mRNA in vivo (Stawowczyk and others 2011). Therefore, translational inhibition does not appear to be a primary means by which ISG54 promotes cellular apoptosis. This may be different during viral infection in the presence of viral RNA with 5′-triphosphate groups and the induced ISG54-binding partner, ISG56. In this case, there may be a concerted effect on translation. However, in the absence of an infection or an IFN response, a direct negative effect of ISG54 on translation of cellular mRNAs in vivo was not apparent.
Tumor colonization
Studies by Lai and others (2012) with an oral squamous cell carcinoma (OSCC) line indicate that ISG54/IFIT2 can inhibit cell migration in vitro and tumor colonization in vivo. Two independent cell lines were generated that knocked down endogenous ISG54 mRNA by introducing shRNA with a lentiviral expression system. Control shRNA cells and the ISG54 knockdown cell lines were compared for various parameters, including cell migration in vitro. An in vitro wound-healing assay was performed, in which a scrape was made in a confluent tissue culture monolayer with a pipette tip. The extent of cell migration to close the wound was measured after 24 h. The ISG54-knocked down cell lines showed a significantly increased migration that closed the wound. The enhanced migration of the knockdown cells was also apparent in the transwell invasion assays. These used a modified Boyden chamber assay in which cells were placed in a serum-free medium in the upper chamber separated from the medium with serum in the lower chamber by a membrane precoated with Matrigel. The knockdown of ISG54 correlated with increased migration. For this reason, the knockdown cells were evaluated for the markers associated with the epithelial–mesenchymal transition (EMT), a process associated with migration and invasion in vivo. These studies indicated that the patterns of protein expression in the ISG54 knockdown cells were correlative with the EMT.
The OSCC control shRNA cells and ISG54/IFIT2 shRNA knockdown cells were also evaluated for the establishment of tumors in NOD/SCID mice (Lai and others 2012). Cells were injected into the tail veins of mice, and 8 weeks later, tumor colonization was determined. Control cells expressing ISG54 established the tumors that were primarily localized to the lung. However, knockdown cell lines established tumors in the lungs, heart, head, and neck, peritoneum, and retroperitoneal cavity, and in some mice, lymph node, bone, and muscle colonization. The results indicated that ISG54 expression inhibited dissemination and a widespread establishment of tumor colonization.
Microbial pathogenesis
IFNs are unique among the cytokines in their ability to establish an antiviral response and restrict the pathogenic effects of both lytic viruses and tumor viruses. For this reason, studies of ISG54/IFIT2 have focused on its potential role in cellular defense to viral infection. Viruses, however, have evolved strategies to block the effectiveness of the IFN system. DNA viruses, in particular, have evolved mechanisms to inhibit cellular apoptosis. In fact, the adenoviral E1B 19K protein, known to function as a viral homolog of Bcl2, and the KSHV (Kaposi's sarcoma-associated herpesvirus) viral Bcl2 homolog can block apoptosis induced by ISG54 (Stawowczyk and others 2011) (M. Stawowczyk and N. Reich, unpublished).
Recent targeted gene disruption studies have been used to evaluate the impact of ISG54/IFIT2 on viral pathogenesis. Fensterl and others (2012) demonstrated that mice with an ISG54/IFIT2 gene knockout succumb to intranasal infection by vesicular stomatitis virus (VSV), a negative-sense ssRNA rhabdovirus. Neuropathogenesis and death resulted together with increased titers of VSV in the brains of knockout mice in comparison to WT animals. The basis of the ISG54 protective effect in the brain remains to be elucidated, and it is possible that ISG54-mediated apoptosis plays a defensive role in VSV neuropathogenesis. A group reported a protective role for ISG56/IFIT2 with a significant decrease in survival of VSV infection in ISG56/IFIT2 knockout mice (Pichlmair and others 2011). Other studies with ISG56/IFIT1 knockout mice have been performed with defective flavivirus or coronavirus that lack the viral 2′-O-methyltransferase activity (Daffis and others 2010; Zust and others 2011; Szretter and others 2012). These mice had decreased survival after infection by 2′-O-methylation-deficient viruses, but not by WT virus. The results indicate that 2′-O-methylation of the 5′-viral RNA cap can overcome an inhibitory impact of ISG56.
Loss-of-function studies with a genetic knockout may not provide a strong phenotype if there is a partial functional redundancy with other ISGs. For this reason, the gain of function by ectopic expression in the absence of other ISGs has also been used to evaluate the impact of individual proteins on viral replication. Most studies with ectopic expression of ISG54/IFIT2 and other members of the ISG/IFIT family have reported the inhibitory effects on infection by RNA viruses in tissue culture (Wang and others 2003; Daffis and others 2010; Schmeisser and others 2010; Liu and others 2011; Perwitasari and others 2011; Yang and others 2012). Notably, ISG56/IFIT1 was found to inhibit replication of a DNA virus, HPV (human papilloma virus), a virus linked to intraepithelial neoplasia (Terenzi and others 2008; Saikia and others 2010). ISG56 binds and inhibits the HPV E1 protein, which is essential for viral replication.
Several DNA and RNA viruses are associated with human cancers, and these include HPV, hepatitis B virus, Epstein-Barr virus, KSHV, human T-lymphotrophic virus, and hepatitis C virus. Since ISG54 is induced by viruses in response to activation of viral PRRs or IFNs, the proapoptotic effect of ISG54 may inhibit the establishment of neoplasia by cancer viruses. IFNs are used clinically in the treatment of chronic infection by some of these cancer viruses, and the proapoptotic role of ISG54 may contribute to suppression of neoplastic progression.
One of the cellular binding partners identified for both ISG54/IFIT2 and ISG56/IFIT1 is STING (stimulator of the IFN genes) (also known as MITA/MPYS/ERIS) (Li and others 2009; Barber 2011). STING mediates induction of type I IFNs in response to microbial dsDNA. Overexpression of ISG54 and ISG56 was reported to reduce the function of STING and IFN production: an effect that at first seemed counterintuitive for a defensive role of these ISGs. The authors propose that by modulating STING, ISG54 and ISG56 act as negative feedback regulators of IFN induction. The ability of ISG54 and ISG56 to promote apoptosis was not addressed in this study, and could be a significant influence.
RNA binding
Several studies indicate that ISG56/IFIT1 and ISG54/IFIT2 can bind to RNA. Since a subset of PRRs are activated during viral infection by binding dsRNA or uncapped 5′-triphosphorylated (5′ppp) RNA (TLR3, RIG-I, and MDA5), Pichlmair and others (2011) used affinity proteomics to search for proteins that bind 5′ppp RNA. They identified ISG56/IFIT1 and ISG58/IFIT5. Their subsequent studies indicated that ISG56/IFIT1 antagonized viral replication by physically sequestering viral 5′ppp RNA generated during infection. Another group provided evidence that the ISG54/IFIT2 protein can bind AU-rich RNA (Yang and others 2012). This work derived from the crystal structure of ISG54 that revealed an area of basic residues along an inner channel of its C-terminus. The positively charged region prompted investigations to evaluate the ability of ISG54 to bind nucleic acid. Following a series of in vitro binding assays, the preferential binding of ISG54 to AU-rich dsRNA and ssRNA was demonstrated. Studies by a different group indicated a negative impact of ISG54 expression on the stability of TNFα mRNA after gene induction by bacterial lipopolysaccharide (Berchtold and others 2008). The effect was dependent on the 3′-untranslated region of the TNFα mRNA and a region, including a constitutive decay element, but it did not involve the AU-rich region.
The potential biological effects resulting from ISG54 binding to RNA remain to be explored more thoroughly. Efforts to date have focused on protein-coding mRNAs; however, it is known that the protein-coding genes account for only a small amount of the human genome (Birney and others 2007; Djebali and others 2012). The majority of transcripts are noncoding RNAs that include long and short noncoding RNAs. For this reason, it is feasible that ISG54 binds and inhibits the function of one or more noncoding regulatory RNAs. A major class of short noncoding RNAs is micro-RNA (miRNA). miRNAs are complementary to their target mRNAs and decrease gene expression by translational inhibition and/or mRNA degradation, and more significantly, miRNAs have been shown to function as either oncogenes or tumor suppressors (Iorio and Croce 2012; Lujambio and Lowe 2012). Although speculative, ISG54 may affect gene expression and biological responses by binding AU-rich elements in mRNA or inactivating target noncoding regulatory RNAs.
Conclusions
This article is part of a special issue of the journal that highlights the antineoplastic effects of IFNs. To understand the mechanism of action of IFNs, it is necessary to determine the functional contribution of each individual ISGs. Ectopic expression of ISG54/IFIT2 was found to promote cell death of transformed cells, and thereby was the focus of this review. ISG54 expression promotes apoptosis via a mitochondrial pathway, and overexpression of antiapoptotic Bcl2 proteins or antiapoptotic viral proteins can block that apoptosis. In light of the ability of ISG54 to promote apoptosis, it remains to be determined how this response contributes to the reported effects of ISG54 on tumor colonization and antiviral responses (Fig. 4). The antiviral and anticancer effects of IFNs result from a concerted function of many ISGs, and ISG54 is one of the factors responsible for IFN-stimulated cell death. The ISG54 gene is normally induced in response to type I and III IFNs, but it is also induced directly in response to PRR activation of IRF3 during microbial infection. In the context of these responses, ISG54 does not exist as a monomer, but as a complex with itself and/or with 2 other related ISGs, ISG56/IFIT2 and ISG60/IFIT3. The functions ascribed to ISG54 may differ depending on the composition of this ISGosome and the potentially larger ISG interactome network. Likewise, the noted effects of the ISG56 and ISG60 proteins may differ depending on their interaction with ISG54. Understanding the mechanisms of action of ISG54 and its interactive partners is expected to support the development of future directed therapies for infectious disease and cancer.
FIG. 4.
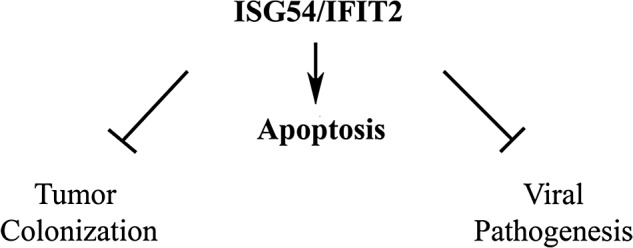
Potential impact of ISG54/IFIT2 on cellular processes.
Acknowledgments
Many thanks to Dr. Marcin Stawowczyk, who led the studies on the ISG54/IFIT2 apoptotic effects, described by our group and the laboratory members Dr. Hui Chen Foreman and Ha Youn Shin. Thanks also extended to my colleague, Dr. Laurie Krug, for stimulating and helpful discussions. Studies were supported by the National Institutes of Health.
Author Disclosure Statement
The author has no competing financial interests.
References
- Adams JM. Cory S. Bcl-2-regulated apoptosis: mechanism and therapeutic potential. Curr Opin Immunol. 2007;19:488–496. doi: 10.1016/j.coi.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen J. VanScoy S. Cheng TF. Gomez D. Reich NC. IRF-3-dependent and augmented target genes during viral infection. Genes Immun. 2008;9:168–175. doi: 10.1038/sj.gene.6364449. [DOI] [PubMed] [Google Scholar]
- Barber GN. Innate immune DNA sensing pathways: STING, AIMII and the regulation of interferon production and inflammatory responses. Curr Opin Immunol. 2011;23:10–20. doi: 10.1016/j.coi.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedict CA. Norris PS. Ware CF. To kill or be killed: viral evasion of apoptosis. Nat Immunol. 2002;3:1013–1018. doi: 10.1038/ni1102-1013. [DOI] [PubMed] [Google Scholar]
- Berchtold S. Manncke B. Klenk J. Geisel J. Autenrieth IB. Bohn E. Forced IFIT-2 expression represses LPS induced TNF-alpha expression at posttranscriptional levels. BMC Immunol. 2008;9:75. doi: 10.1186/1471-2172-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best SM. Viral subversion of apoptotic enzymes: escape from death row. Annu Rev Microbiol. 2008;62:171–192. doi: 10.1146/annurev.micro.62.081307.163009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birney E. Stamatoyannopoulos JA. Dutta A. Guigo R. Gingeras TR. Margulies EH. Weng Z. Snyder M. Dermitzakis ET. Thurman RE, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blatch GL. Lassle M. The tetratricopeptide repeat: a structural motif mediating protein-protein interactions. Bioessays. 1999;21:932–939. doi: 10.1002/(SICI)1521-1878(199911)21:11<932::AID-BIES5>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Borden EC. Sen GC. Uze G. Silverman RH. Ransohoff RM. Foster GR. Stark GR. Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov. 2007;6:975–990. doi: 10.1038/nrd2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla-Sarkar M. Lindner DJ. Liu YF. Williams BR. Sen GC. Silverman RH. Borden EC. Apoptosis and interferons: role of interferon-stimulated genes as mediators of apoptosis. Apoptosis. 2003;8:237–249. doi: 10.1023/a:1023668705040. [DOI] [PubMed] [Google Scholar]
- Clemens MJ. Interferons and apoptosis. J Interferon Cytokine Res. 2003;23:277–292. doi: 10.1089/107999003766628124. [DOI] [PubMed] [Google Scholar]
- D'Andrea LD. Regan L. TPR proteins: the versatile helix. Trends Biochem Sci. 2003;28:655–662. doi: 10.1016/j.tibs.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Daffis S. Szretter KJ. Schriewer J. Li J. Youn S. Errett J. Lin TY. Schneller S. Zust R. Dong H, et al. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature. 2010;468:452–456. doi: 10.1038/nature09489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly C. Reich NC. Double-stranded RNA activates novel factors that bind to the interferon-stimulated response element. Mol Cell Biol. 1993;13:3756–3764. doi: 10.1128/mcb.13.6.3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly C. Reich NC. Characterization of specific DNA-binding factors activated by double-stranded RNA as positive regulators of interferon alpha/beta-stimulated genes. J Biol Chem. 1995;270:23739–23746. doi: 10.1074/jbc.270.40.23739. [DOI] [PubMed] [Google Scholar]
- de Veer MJ. Holko M. Frevel M. Walker E. Der S. Paranjape JM. Silverman RH. Williams BR. Functional classification of interferon-stimulated genes identified using microarrays. J Leukoc Biol. 2001;69:912–920. [PubMed] [Google Scholar]
- de Weerd NA. Nguyen T. The interferons and their receptors—distribution and regulation. Immunol Cell Biol. 2012;90:483–491. doi: 10.1038/icb.2012.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker T. Lew DJ. Mirkovitch J. Darnell JE., Jr Cytoplasmic activation of GAF, an IFN-gamma-regulated DNA-binding factor. EMBO J. 1991;10:927–932. doi: 10.1002/j.1460-2075.1991.tb08026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djebali S. Davis CA. Merkel A. Dobin A. Lassmann T. Mortazavi A. Tanzer A. Lagarde J. Lin W. Schlesinger F, et al. Landscape of transcription in human cells. Nature. 2012;489:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earnshaw WC. Martins LM. Kaufmann SH. Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu Rev Biochem. 1999;68:383–424. doi: 10.1146/annurev.biochem.68.1.383. [DOI] [PubMed] [Google Scholar]
- Fensterl V. Sen GC. The ISG56/IFIT1 gene family. J Interferon Cytokine Res. 2011;31:71–78. doi: 10.1089/jir.2010.0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fensterl V. Wetzel JL. Ramachandran S. Ogino T. Stohlman SA. Bergmann CC. Diamond MS. Virgin HW. Sen GC. Interferon-Induced Ifit2/ISG54 Protects Mice from Lethal VSV Neuropathogenesis. PLoS Pathog. 2012;8:e1002712. doi: 10.1371/journal.ppat.1002712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu XY. Kessler DS. Veals SA. Levy DE. Darnell JE., Jr ISGF3, the transcriptional activator induced by interferon alpha, consists of multiple interacting polypeptide chains. Proc Natl Acad Sci U S A. 1990;87:8555–8559. doi: 10.1073/pnas.87.21.8555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L. Brenner C. Morselli E. Touat Z. Kroemer G. Viral control of mitochondrial apoptosis. PLoS Pathog. 2008;4:e1000018. doi: 10.1371/journal.ppat.1000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez D. Reich NC. Stimulation of primary human endothelial cell proliferation by IFN. J Immunol. 2003;170:5373–5381. doi: 10.4049/jimmunol.170.11.5373. [DOI] [PubMed] [Google Scholar]
- Heylbroeck C. Balachandran S. Servant MJ. DeLuca C. Barber GN. Lin R. Hiscott J. The IRF-3 transcription factor mediates Sendai virus-induced apoptosis. J Virol. 2000;74:3781–3792. doi: 10.1128/jvi.74.8.3781-3792.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiscott J. Triggering the innate antiviral response through IRF-3 activation. J Biol Chem. 2007;282:15325–15329. doi: 10.1074/jbc.R700002200. [DOI] [PubMed] [Google Scholar]
- Iorio MV. Croce CM. microRNA involvement in human cancer. Carcinogenesis. 2012;33:1126–1133. doi: 10.1093/carcin/bgs140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagus R. Joshi B. Barber GN. PKR, apoptosis and cancer. Int J Biochem Cell Biol. 1999;31:123–138. doi: 10.1016/s1357-2725(98)00136-8. [DOI] [PubMed] [Google Scholar]
- Lai KC. Liu CJ. Chang KW. Lee TC. Depleting IFIT2 mediates atypical PKC signaling to enhance the migration and metastatic activity of oral squamous cell carcinoma cells. Oncogene. 2012 doi: 10.1038/onc.2012.384. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Levy D. Larner A. Chaudhuri A. Babiss LE. Darnell JE., Jr Interferon-stimulated transcription: isolation of an inducible gene and identification of its regulatory region. Proc Natl Acad Sci U S A. 1986;83:8929–8933. doi: 10.1073/pnas.83.23.8929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D. Reich N. Kessler D. Pine R. Darnell JE., Jr Transcriptional regulation of interferon-stimulated genes: a DNA response element and induced proteins that recognize it. Cold Spring Harb Symp Quant Biol. 1988;53(Pt 2):799–802. doi: 10.1101/sqb.1988.053.01.090. [DOI] [PubMed] [Google Scholar]
- Levy DE. Darnell JE., Jr Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- Li Y. Li C. Xue P. Zhong B. Mao AP. Ran Y. Chen H. Wang YY. Yang F. Shu HB. ISG56 is a negative-feedback regulator of virus-triggered signaling and cellular antiviral response. Proc Natl Acad Sci U S A. 2009;106:7945–7950. doi: 10.1073/pnas.0900818106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XY. Chen W. Wei B. Shan YF. Wang C. IFN-induced TPR protein IFIT3 potentiates antiviral signaling by bridging MAVS and TBK1. J Immunol. 2011;187:2559–2568. doi: 10.4049/jimmunol.1100963. [DOI] [PubMed] [Google Scholar]
- Llambi F. Moldoveanu T. Tait SW. Bouchier-Hayes L. Temirov J. McCormick LL. Dillon CP. Green DR. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol Cell. 2011;44:517–531. doi: 10.1016/j.molcel.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujambio A. Lowe SW. The microcosmos of cancer. Nature. 2012;482:347–355. doi: 10.1038/nature10888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Moczygemba M. Gutch MJ. French DL. Reich NC. Distinct STAT structure promotes interaction of STAT2 with the p48 subunit of the interferon-alpha-stimulated transcription factor ISGF3. J Biol Chem. 1997;272:20070–20076. doi: 10.1074/jbc.272.32.20070. [DOI] [PubMed] [Google Scholar]
- Ozato K. Tailor P. Kubota T. The interferon regulatory factor family in host defense: mechanism of action. J Biol Chem. 2007;282:20065–20069. doi: 10.1074/jbc.R700003200. [DOI] [PubMed] [Google Scholar]
- Perwitasari O. Cho H. Diamond MS. Gale M., Jr Inhibitor of kappaB kinase epsilon (IKK(epsilon)), STAT1, and IFIT2 proteins define novel innate immune effector pathway against West Nile virus infection. J Biol Chem. 2011;286:44412–44423. doi: 10.1074/jbc.M111.285205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichlmair A. Lassnig C. Eberle CA. Gorna MW. Baumann CL. Burkard TR. Burckstummer T. Stefanovic A. Krieger S. Bennett KL, et al. IFIT1 is an antiviral protein that recognizes 5′-triphosphate RNA. Nat Immunol. 2011;12:624–630. doi: 10.1038/ni.2048. [DOI] [PubMed] [Google Scholar]
- Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005;5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- Saikia P. Fensterl V. Sen GC. The inhibitory action of P56 on select functions of E1 mediates interferon's effect on human papillomavirus DNA replication. J Virol. 2010;84:13036–13039. doi: 10.1128/JVI.01194-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler C. Levy DE. Decker T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem. 2007;282:20059–20063. doi: 10.1074/jbc.R700016200. [DOI] [PubMed] [Google Scholar]
- Schmeisser H. Mejido J. Balinsky CA. Morrow AN. Clark CR. Zhao T. Zoon KC. Identification of alpha interferon-induced genes associated with antiviral activity in Daudi cells and characterization of IFIT3 as a novel antiviral gene. J Virol. 2010;84:10671–10680. doi: 10.1128/JVI.00818-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark GR. Darnell JE., Jr The JAK-STAT pathway at twenty. Immunity. 2012;36:503–514. doi: 10.1016/j.immuni.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stawowczyk M. Van Scoy S. Kumar KP. Reich NC. The interferon stimulated gene 54 promotes apoptosis. J Biol Chem. 2011;286:7257–7266. doi: 10.1074/jbc.M110.207068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser A. O'Connor L. Dixit VM. Apoptosis signaling. Annu Rev Biochem. 2000;69:217–245. doi: 10.1146/annurev.biochem.69.1.217. [DOI] [PubMed] [Google Scholar]
- Szretter KJ. Daniels BP. Cho H. Gainey MD. Yokoyama WM. Gale M., Jr. Virgin HW. Klein RS. Sen GC. Diamond MS. 2′-O methylation of the viral mRNA cap by West Nile virus evades ifit1-dependent and -independent mechanisms of host restriction in vivo. PLoS Pathog. 2012;8:e1002698. doi: 10.1371/journal.ppat.1002698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura T. Ishihara M. Lamphier MS. Tanaka N. Oishi I. Aizawa S. Matsuyama T. Mak TW. Taki S. Taniguchi T. An IRF-1-dependent pathway of DNA damage-induced apoptosis in mitogen-activated T lymphocytes. Nature. 1995;376:596–599. doi: 10.1038/376596a0. [DOI] [PubMed] [Google Scholar]
- Terenzi F. Hui DJ. Merrick WC. Sen GC. Distinct induction patterns and functions of two closely related interferon-inducible human genes, ISG54 and ISG56. J Biol Chem. 2006;281:34064–34071. doi: 10.1074/jbc.M605771200. [DOI] [PubMed] [Google Scholar]
- Terenzi F. Pal S. Sen GC. Induction and mode of action of the viral stress-inducible murine proteins, P56 and P54. Virology. 2005;340:116–124. doi: 10.1016/j.virol.2005.06.011. [DOI] [PubMed] [Google Scholar]
- Terenzi F. Saikia P. Sen GC. Interferon-inducible protein, P56, inhibits HPV DNA replication by binding to the viral protein E1. EMBO J. 2008;27:3311–3321. doi: 10.1038/emboj.2008.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theofilopoulos AN. Kono DH. Beutler B. Baccala R. Intracellular nucleic acid sensors and autoimmunity. J Interferon Cytokine Res. 2011;31:867–886. doi: 10.1089/jir.2011.0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thyrell L. Erickson S. Zhivotovsky B. Pokrovskaja K. Sangfelt O. Castro J. Einhorn S. Grander D. Mechanisms of Interferon-alpha induced apoptosis in malignant cells. Oncogene. 2002;21:1251–1262. doi: 10.1038/sj.onc.1205179. [DOI] [PubMed] [Google Scholar]
- Uematsu S. Akira S. Toll-like receptors and Type I interferons. J Biol Chem. 2007;282:15319–15323. doi: 10.1074/jbc.R700009200. [DOI] [PubMed] [Google Scholar]
- Wang C. Pflugheber J. Sumpter R., Jr. Sodora DL. Hui D. Sen GC. Gale M., Jr Alpha interferon induces distinct translational control programs to suppress hepatitis C virus RNA replication. J Virol. 2003;77:3898–3912. doi: 10.1128/JVI.77.7.3898-3912.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZG. Ruggero D. Ronchetti S. Zhong S. Gaboli M. Rivi R. Pandolfi PP. PML is essential for multiple apoptotic pathways. Nat Genet. 1998;20:266–272. doi: 10.1038/3073. [DOI] [PubMed] [Google Scholar]
- Wathelet M. Moutschen S. Defilippi P. Cravador A. Collet M. Huez G. Content J. Molecular cloning, full-length sequence and preliminary characterization of a 56-kDa protein induced by human interferons. Eur J Biochem. 1986;155:11–17. doi: 10.1111/j.1432-1033.1986.tb09452.x. [DOI] [PubMed] [Google Scholar]
- Weaver BK. Ando O. Kumar KP. Reich NC. Apoptosis is promoted by the dsRNA-activated factor (DRAF1) during viral infection independent of the action of interferon or p53. FASEB J. 2001;15:501–515. doi: 10.1096/fj.00-0222com. [DOI] [PubMed] [Google Scholar]
- Weaver BK. Kumar KP. Reich NC. Interferon regulatory factor 3 and CREB-binding protein/p300 are subunits of double-stranded RNA-activated transcription factor DRAF1. Mol Cell Biol. 1998;18:1359–1368. doi: 10.1128/mcb.18.3.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. Liang H. Zhou Q. Li Y. Chen H. Ye W. Chen D. Fleming J. Shu H. Liu Y. Crystal structure of ISG54 reveals a novel RNA binding structure and potential functional mechanisms. Cell Res. 2012;22:1328–1338. doi: 10.1038/cr.2012.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneyama M. Fujita T. Recognition of viral nucleic acids in innate immunity. Rev Med Virol. 2010;20:4–22. doi: 10.1002/rmv.633. [DOI] [PubMed] [Google Scholar]
- Yoneyama M. Suhara W. Fukuhara Y. Fukuda M. Nishida E. Fujita T. Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J. 1998;17:1087–1095. doi: 10.1093/emboj/17.4.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ. Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- Zhou A. Paranjape J. Brown TL. Nie H. Naik S. Dong B. Chang A. Trapp B. Fairchild R. Colmenares C, et al. Interferon action and apoptosis are defective in mice devoid of 2′,5′-oligoadenylate-dependent RNase L. EMBO J. 1997;16:6355–6363. doi: 10.1093/emboj/16.21.6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zust R. Cervantes-Barragan L. Habjan M. Maier R. Neuman BW. Ziebuhr J. Szretter KJ. Baker SC. Barchet W. Diamond MS, et al. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat Immunol. 2011;12:137–143. doi: 10.1038/ni.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]