

Abstract
Spironucleus muris is a protozoan that can colonize the intestinal tract of many rodent species. Although its effects on animal health and research are debated, S. muris is often included on exclusion lists for rodent facilities. Common diagnostic tests for S. muris are insensitive and typically are performed at postmortem examination. We sought to develop a PCR-based diagnostic test with sufficient sensitivity and specificity for use on fecal samples from live rodents. We designed and optimized a PCR assay that targeted the 16S-like rRNA gene of S. muris. The assay was highly specific, given that samples from mice contaminated with S. muris were PCR positive, whereas samples from mice contaminated with other protozoa were negative. The assay also was highly sensitive, detecting as few as 5 template copies per microliter diluent. All mice positive for S. muris on postmortem exams also were positive by fecal PCR. Moreover, S. muris was detected by PCR in mice negative by postmortem examination but from colonies known to be contaminated as well as in rats and hamsters. To assess protozoal loads in mice of differing ages, the PCR assay was adapted to a quantitative format. Fecal loads of S. muris were highest in 4-wk-old mice and declined with age. The PCR assay developed promises to be a highly specific antemortem diagnostic assay with higher sensitivity than that of existing postmortem tests.
Spironucleus muris (formerly Hexamita muris) is considered an opportunistic pathogen of several rodent species including mice, hamsters, and rats.18 This flagellated protozoan inhabits the small intestine after the cysts are ingested.9,10 Reports suggest that during the life cycle of S. muris, trophozoites are found predominately throughout the small intestine in young rodents. In contrast, trophozoites are sequestered in the crypts of the pylorus and are fewer in adult rodents, very often to the point of not being detectable by standard diagnostic techniques.3,7
Infections in adult mice are usually subclinical.4,12 However, in young and immunocompromised mice, clinical signs can include enteritis, poor growth, hunched posture, distended abdomen, and death.1 In addition to being associated with increased morbidity and mortality, S. muris infection results in perturbations of the immune system that may confound research.14 Some studies have shown changes in macrophage activity in mice infected with S. muris.6,8 In the words of one researcher, “[mice infected with S. muris] are unsuitable for immunological studies.”16 In many studies of S. muris, the presence of other pathogens is often unknown or unreported, so the extent to which clinical signs or effects on research can be attributable to S. muris is uncertain.
Common methods for diagnosing S. muris in laboratory rodents include postmortem observation of trophozoites on wet mounts of fresh intestinal contents or in histologic sections of small intestine.9 Although these methods are specific, they require that the animal be euthanized to obtain the needed samples. In addition, these postmortem methods lack sensitivity, allowing subclinical infections to go undetected.
The most recent assessment of S. muris prevalence (in 2009) showed that it was detected in less than 1% of samples from laboratory mice in North America.13 However, this assessment was based on the use of wet mounts of intestinal contents, a test that may lack sensitivity. Therefore, the actual prevalence may be higher. Moreover, with a reported low prevalence, a test with low sensitivity also would have a low positive predictive value. Collectively these findings highlight the need for diagnostic assays for S. muris that have high sensitivity and specificity.
To this end, we developed a PCR assay for S. muris that would be as specific as and more sensitive than current diagnostic methods. PCR assays for enteric pathogens can be used on feces collected from live animals, precluding the need to euthanize animals to detect these agents.2
Materials and Methods
Sample and animal sources.
Mouse feces and tissue samples for PCR development and optimization, along with rat and hamster feces were obtained from submissions to IDEXX-RADIL (Columbia, MO) for diagnostic services. SENCARC/PtJ and CRL:CD1 mice were obtained from Harlan Laboratories (Indianapolis, IN) and Charles River Labs (Wilmington, MA), respectively, and were used in accordance with approved IACUC protocols.
PCR development and optimization.
We developed several primer pairs by using the S. muris 16S-like rRNA gene sequence (GenBank accession no. X84231) and DS Gene (Accelrys, San Diego, CA). These primer pairs covered different regions of the gene and were screened with NCBI BLAST to exclude those that would amplify mouse DNA or other genetic material that might be found in the intestine of mice. We chose 3 pairs of primers for testing (Table 1).
Table 1.
Sequence and predicted product size of primer pairs developed and tested.
| Primer set | Forward primer | Reverse primer | Product size (bp) |
| 1 | TCTGCCGCATCATCTAGAAG | CTCAGCCCGTAAGACAAAG | 196 |
| 2 | ACTTTGTCTTACGGGCTGAGC | TGCGATTCGCATGAACCGTC | 186 |
| 3 | TTCATGCGATCGCACAGG | AACGGTCTCTAATCGTC | 195 |
For PCR optimization and assessment, candidate primers were used on feces from mice diagnosed as positive for S. muris via wet mount of fresh intestinal contents. PCR products of the expected size were isolated and sequenced to confirm they were from S. muris; and positive control plasmids were generated for each primer set by using a TopoTA cloning kit (Invitrogen, Carlsbad, CA). The plasmids were isolated by using a plasmid miniprep kit (Eppendorf, Hauppauge, NY). Plasmid concentrations were obtained by using a Nanodrop 1100 spectrophotometer (Thermo Scientific, Wilmington, DE), and template copy number was estimated as:
where n the mass of template used (in nanograms) and l is the length of the template (in basepairs).17
DNA processing and PCR.
Samples were processed in a robotic system (model M48, GenoVision Extraction Robot, Qiagen, Germantown, MD), and the extracted DNA was stored at −20 °C. The PCR assay master mix was: 26.75 μL water, 5.00 μL 10× buffer (Roche Diagnostics, Indianapolis, IN) , 8.00 μL 5 mM dNTPs, 2.50 μL forward primer and 2.50 μL reverse primer (primer stock concentrations were 20 ng/µL), and 0.25 μL Faststart Taq polymerase (Roche Diagnostics). The thermocycling parameters used for all primer pairs were an initial melting step at 95 °C for 4 min, followed by 40 cycles of 94 °C for 30 s, 62 °C for 30 s, and 72 °C for 30 s. The completed reaction remained at 4 °C prior to gel analysis. PCR products were electrophoresed through 3% agarose gels containing ethidium bromide (BioRad Laboratories, Hercules, CA) and visualized under UV light. The gel image was captured by using a gel documentation system (InGenius system, Syngene, Frederick, MD).
PCR specificity.
To assess specificity, feces from mice that were positive for S. muris trophozoites by wet-mount exams of intestinal contents at the time of necropsy were examined by PCR using all 3 primer sets. In addition, 13 mice that were negative on wet mounts for S. muris but positive for other intestinal protozoa (Entamoeba muris, Chilomastix bettencourti, various trichomonads, nonspeciated flagellates) were tested. PCR bands of the expected product size (Table 1) from several mice were collected and sequenced (University of Missouri DNA Core lab or SeqWright [Houston, TX)]) to confirm specificity.
Sensitivity.
To assess sensitivity, the positive-control plasmids described earlier were serially diluted in water, and PCR was performed on sample dilutions ranging from 1 to 1.0 × 106 copies per microliter. To evaluate whether potential polymerase inhibitors present in feces affected the sensitivity of this assay, serial dilutions were performed by using fecal DNA extract from S. muris negative mice as diluents.11
Comparison of PCR assays.
PCR testing was performed on fecal samples from 74 mice (SENCARC/PtJ: n = 14; age, 16 to 17 wk; CRL:CD1: n = 45; age, 12 to 13 wk; and 15 mice of unknown age and background). To assess whether the PCR detected S. muris in other species, samples from 5 hamsters and 6 pet rats from sources historically positive for S. muris also were tested. Results of wet-mount examinations of the 74 mice were compared with those from our PCR assay.
In addition, feces from mice and hamsters were tested by using a previously described S. muris PCR assay.5 The assay was performed according to the reaction conditions as published as well as with the following modified conditions: HotStart buffer; 5 µM of each primer; initial denaturation step of 95 °C for 4 min; and 40 cycles of melting at 94 °C for 30 s, annealing at 62 °C for 30 s, and extension at 72 °C for 30 s.
Quantitative PCR.
A previous report suggests that S. muris loads decline with age.3 To confirm or refute this premise by using a more sensitive molecular assay, we modified the PCR we developed to a SYBR Green-based quantitative assay. The master mix for the quantitative PCR was as follows: H2O (46 µL), 3mM MgCl (4 µL), 158F primer (5 µL), 158R primer (5 µL), and Quantitect (Qiagen, Germantown, MD) mix (100 µL). The master mix was vortexed and pulse-centrifuged, and 16-µL aliquots were combined with 4 µL template DNA. The tubes were centrifuged at 845 × g for 5 s, capped, and placed in a LightCycler (Roche Applied Science, Indianapolis, IN). The LightCycler protocol started with the reaction being brought to 95 °C at a rate of 20 °C/s and then held for 15 min, followed by 40 cycles of 94 °C for 15 s, 60 °C for 20 s, and 72 °C for 30 s. The last of these 3-step cycles was followed by a melting curve analysis, with the reaction being brought to 95 °C, allowed to cool to 70 °C for 15 s, and returned to 95 °C at a rate of 0.1 °C/s. Absolute quantification, a standard curve, and a melting curve were calculated by using LightCycler software version 4.0 (Roche). Six standards consisting of logarithmic serial dilutions of plasmid from 1.0 × 105 to 1 copy were used with each quantitative PCR reaction. These standards produced a standard curve with a correlation coefficient of greater than 0.99, with the standards and samples forming concentric melting peaks, indicating that quantitative data were from single amplification products (that is, no primer dimers).
To assess changes, in fecal load, feces were collected from 10 female SENCAR mice at weekly intervals beginning when the mice were 3 wk old and continuing for 5 wk. At the end of 5 wk, the mice were euthanized via CO2 inhalation followed by cervical dislocation. Fecal pellets were collected from the colon for PCR, and wet-mount preparations were prepared from ileal contents. DNA was extracted from the feces as described earlier and analyzed by using the quantitative assay.
Results
Specificity of PCR primer sets.
Samples from mice known to be positive for S. muris were PCR-analyzed by using all 3 sets of primers and produced bands of predicted sizes. Amplicons from all 3 PCR assays were sequenced and found to be 100% homologous to the 16S-like rRNA gene from S. muris. The assay using primer set 1 did not produce any bands in samples from mice known to be negative for S. muris. In addition, mice positive for other intestinal parasites, such as Chilomastix or trichomonads, tested negative by PCR with primer set 1, whereas faint spurious bands were present when primer sets 2 and 3 were used to test these samples (Figure 1). These data show that, among the 3 sets tested, primer set 1 was the most specific for S. muris.
Figure 1.
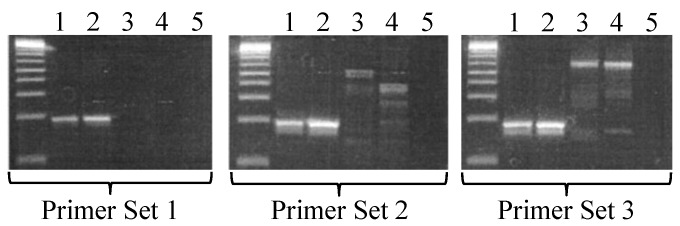
Specificity of PCR primers to S. muris. In each gel image, the far-left lane contains size markers (1-kb ladder). Lanes 1 and 2 contain DNA extracted from the feces of mice diagnosed as positive for S. muris by wet mount. Lane 3 contains DNA extracted from the feces of a mouse diagnosed with nonS. muris protozoa; lane 4 contains DNA extracted from the feces of a mouse diagnosed as negative for S. muris. Lane 5 contains a no-template control.
Sensitivity of PCR primer sets.
The PCR primer sets first were tested by using serial dilutions of a cloned plasmid. Primer set 1 produced a clearly observable band at 1 copy template per microliter, whereas primer sets 2 and 3 yielded only faint bands when tested at this dilution (Figure 2 A). When DNA extracted from the feces of mice free of S. muris was used as diluent for serial dilutions of plasmid, the sensitivity of the primers decreased by 1 log (Figure 2 B). We decided to use primer set 1 in additional testing because of its superior sensitivity and specificity compared with those of primer sets 2 and 3.
Figure 2.

Serial dilutions of plasmid with PCR target insert. (A) Water as diluent. (B) DNA extracted from the feces of mice negative for S. muris and used as diluent. Lane 1 contains 1000 copies per μL diluent; lane 2 contains 100 copies per μL diluent; lane 3 contains 10 copies per μL diluent; lane 4 contains 1 copy per μL diluent; and lane 5 contains a no-template control.
To compare the sensitivity of our PCR with that of a common method of protozoan diagnosis, intestinal content wet mount, 74 mice were screened by both wet mounts and fecal PCR. PCR testing identified all 10 mice that were positive by wet mount, as well as 10 additional mice that were negative by wet mount; the remaining 54 mice were negative by both assays. We then tested primer set 1 on DNA extracted from the feces of hamsters and rats. All 5 hamsters and 1 of the 6 juvenile rats tested positive for S. muris. PCR products from the positive rat and from one of the hamsters were sequenced and found to be 100% homologous to S. muris by BLAST analysis.
Comparison with existing PCR assays.
We then used mouse and hamster fecal DNA samples to compare the sensitivity of our PCR assay using primer set 1 with a previously described PCR assay.5 When we used the published reaction conditions for the previously described assay, we obtained no positive results (Figure 3). Modification of the reaction conditions improved sensitivity of the published assay, but not to the level of the PCR assay we developed. In addition, our assay identified several hamsters as positive for S. muris, whereas the published assay yielded only a single equivocal result among these samples (Figure 3).
Figure 3.

Comparison of (A) the S. muris PCR developed herein, (B) a previously published assay,5 and (C) the previously published assay run with modified conditions (see Methods). Lane 1, 1-kb DNA ladder; lane 2, no-template control; lane 3, DNA from a mouse that was negative for S. muris by direct smear; lane 4, DNA from a mouse that was positive for S. muris by direct smear; lanes 5–11, 10-fold dilutions of the sample in lane 4; lanes 12 and 13, DNA from additional mice identified as positive for S. muris by direct smear; lanes 15–20, DNA from hamsters.
Spironucleus muris loads during the course of infection.
Quantitative PCR was performed on weekly fecal samples from mice obtained from a colony historically positive for S. muris, to evaluate changes in parasite load over the course of infection. At the time of necropsy, 6 of the 10 SENCAR mice were diagnosed as negative for S. muris via wet mount; all 6 of those mice that were negative by wet mount tested positive for S. muris by qualitative PCR. Quantitative PCR of samples from preceding time points showed an initial increase in shedding, from an average of approximately 1000 copies at 3 wk of age to nearly 3000 copies at 4 wk of age. There was considerable variation in the amount of shedding, with several 4-wk-old mice shedding fewer than 1000 copies and some shedding more than 10,000. Shedding dropped dramatically by 5 wk of age and fluctuated at a nearly undetectable level throughout the remainder of the study (Figure 4). These data indicate that parasite loads decrease with age and, in young adult mice, may not be detectable by commonly used diagnostic methods.
Figure 4.
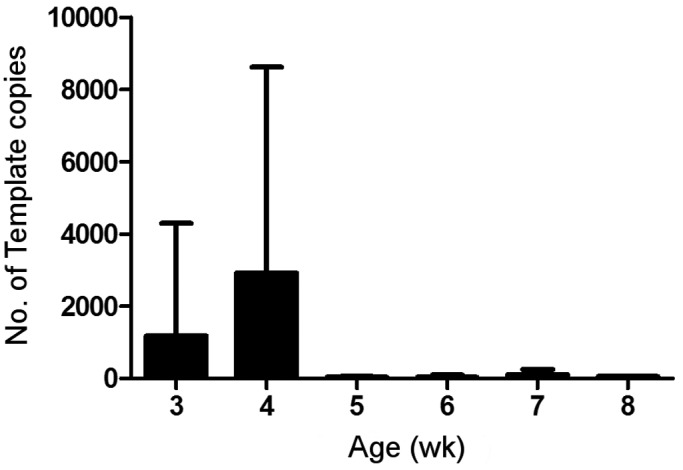
Shedding of S. muris in feces from female SENCAR mice. The y-axis indicates the average number of template copies detected by SYBR green qPCR. Error bars indicate standard deviation.
Discussion
S. muris is a potentially pathogenic organism of rodents,15 and as such, testing for this protozoa typically is included in health monitoring programs. The prevalence of S. muris in research animal colonies is unknown but believed to be low. However, current diagnostic tests, including postmortem exam of intestinal contents via wet-mount slides, histologic exam of small intestines, and fecal flotation, may lack sensitivity. Therefore the true prevalence of S. muris may be underestimated. Here we developed a PCR assay aimed at increasing the sensitivity of S. muris detection while maintaining high specificity. In addition to increasing sensitivity, PCR analysis supports antemortem testing of animals, thus increasing the feasibility of testing colonies.
Our studies show that the PCR assay we developed is specific for S. muris, in that it amplified products from rodents infected with S. muris but not those positive for other protozoa. The assay was not tested against all other ‘important’ protozoa—for example, we were unable to find Giardia-positive samples that were negative for Spironucleus. Therefore we cannot rule out the possibility of crossreactivity with some protozoa. However, sequencing of PCR amplicons confirmed the product as originating from a single organism, S. muris; no Giardia sequences were identified.
Evaluation of sensitivity showed that primer set 1 was sufficiently sensitive to detect fewer than 10 copies of the targeted gene per μL diluent. Quantitative PCR corroborated this finding, identifying as few as 1 copy per μL fecal DNA extract (data not shown). In addition, animals that were negative for S. muris by standard wet-mount testing, but that were from known positive colonies, were found to be positive for the organism by PCR. Collectively these findings indicate that our PCR assay has superior sensitivity when compared with the existing standard method of diagnosing S. muris.
Comparison of our PCR assay with one previously described5 showed that the previous test was less sensitive for S. muris, even when reaction conditions were optimized, than was the test we developed. Our assay also identified hamsters that were positive for S. muris but were not detected by the published assay. The reasons for the discrepancies between these 2 assays are unknown but may be related to the genes targeted by each. The previously developed S. muris PCR assay targets the elongation factor 1α gene,5 whereas our assay targets the 16S-like rRNA gene. It is conceivable that these different targets may affect sensitivity or that host-specific isolates of S. muris exist. For example, the 16S-like rRNA gene may be more conserved than is the elongation factor 1α gene between the S. muris strains in mice compared with hamsters. Further studies are necessary to evaluate whether such differences exist.
A previous study has shown that the number of trophozoites and cysts, which are detected by direct examination of intestinal contents, declines over a short period of time.3 In the current study, we developed a quantitative PCR assay and confirmed that shedding of S. muris decreases rapidly during the first several weeks of age in SENCAR mice with endemic infections. There was wide variation in fecal gene levels, which could be due to the age at which the mice were infected, infectious dose, differences in intestinal microbiota, and genetic variation of the outbred mice. Regardless of the variation in levels of shedding, the same pattern was seen in all mice, with a peak output at 4 wk of age which quickly declined within 1 wk. Although levels declined, S. muris remained detectable throughout the study. Of note, the common wet-mount method of S. muris detection failed to detect S. muris in PCR-positive animals at the end of the 9-wk study. Therefore, the use of the standard wet-mount technique may result in underestimation of the prevalence of S. muris.
Collectively the current study shows that PCR amplification is an excellent assay for the detection of S. muris. The assay is highly sensitive, allowing for detection of this protozoan even after parasitic loads have declined. Moreover, PCR testing can be performed on fecal samples from live rodents, precluding the need for euthanasia. This benefit allows for the assessment of colony animals in the face of a suspected outbreak without having to euthanize study animals.
Our PCR assay is more sensitive for detecting S. muris in mice than are current testing methods. This assay detects carriers and yields a better estimate of the prevalence of S. muris in colonies of mice and potentially hamsters and rats. In addition, our S. muris PCR assay may facilitate studies of the true pathogenicity of this parasite and its potential effects on research.
References
- 1.Baker DG. 1998. Natural pathogens of laboratory mice, rats, and rabbits and their effects on research. Clin Microbiol Rev 11:231–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beckwith CS, Franklin CL, Hook RR, Jr, Besch-Williford CL, Riley LK. 1997. Fecal PCR assay for diagnosis of Helicobacter infection in laboratory rodents. J Clin Microbiol 35:1620–1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brett SJ, Cox FE. 1982. Immunological aspects of Giardia muris and Spironucleus muris infections in inbred and outbred strains of laboratory mice: a comparative study. Parasitology 85:85–99 [DOI] [PubMed] [Google Scholar]
- 4.Brett SJ, Cox FE. 1982. Interactions between the intestinal flagellates Giardia muris and Spironucleus muris and the blood parasites Babesia microti, Plasmodium yoelii, and Plasmodium berghei in mice. Parasitology 85:101–110 [DOI] [PubMed] [Google Scholar]
- 5.Fain MA, Karjala Z, Perdue KA, Copeland MK, Cheng LI, Elkins WR. 2008. Detection of Spironucleus muris in unpreserved mouse tissue and fecal samples by using a PCR assay. J Am Assoc Lab Anim Sci 47:39–43 [PMC free article] [PubMed] [Google Scholar]
- 6.Goodrum KJ, Guzman GS, Lindsey JR, Silberman M, Spitznagel JK. 1984. Peritoneal macrophages of pathogen-free rats but not of conventional rats secrete elastolytic activity. J Leukoc Biol 36:161–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Institute for Laboratory Animal Science 1991. Natural infectious diseases of mice and rats. Washington (DC): National Academies Press [Google Scholar]
- 8.Keast D, Chesterman FC. 1972. Changes in macrophage metabolism in mice heavily infected with Hexamita muris. Lab Anim 6:33–39 [DOI] [PubMed] [Google Scholar]
- 9.Kunstyr I. 1977. Infectious form of Spironucleus (Hexamita) muris: banded cysts. Lab Anim 11:185–188 [DOI] [PubMed] [Google Scholar]
- 10.Kunstyr I, Ammerpohl E. 1978. Resistance of faecal cysts of Spironucleus muris to some physical factors and chemical substances. Lab Anim 12:95–97 [DOI] [PubMed] [Google Scholar]
- 11.Lou Q, Chong SK, Fitzgerald JF, Siders JA, Allen SD, Lee CH. 1997. Rapid and effective method for preparation of fecal specimens for PCR assays. J Clin Microbiol 35:281–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perdue KA, Copeland MK, Karjala Z, Cheng LI, Ward JM, Elkins WR. 2008. Suboptimal ability of dirty-bedding sentinels to detect Spironucleus muris in a colony of mice with genetic manipulations of the adaptive immune system. J Am Assoc Lab Anim Sci 47:10–17 [PMC free article] [PubMed] [Google Scholar]
- 13.Pritchett-Corning KR, Cosentino J, Clifford CB. 2009. Contemporary prevalence of infectious agents in laboratory mice and rats. Lab Anim 43:165–173 [DOI] [PubMed] [Google Scholar]
- 14.Ruitenberg EJ, Kruyt BC. 1975. Effect of intestinal flagellates on immune response in mice. Parasitology 71:30 [Google Scholar]
- 15.Schagemann G, Bohnet W, Kunstyr I, Friedhoff KT. 1990. Host specificity of cloned Spironucleus muris in laboratory rodents. Lab Anim 24:234–239 [DOI] [PubMed] [Google Scholar]
- 16.Sebesteny A. 1974. The transmission of intestinal flagellates between mice and rats. Lab Anim 8:79–81 [DOI] [PubMed] [Google Scholar]
- 17.Staroscik A. [Internet]. 2004. URI Genomics and Sequencing Center: calculator for determining the number of copies of a template. [Cited 27 December 2007]. Available at: http://www.uri.edu/research/gsc/resources/cndna.html.
- 18.Wagner JE, Doyle RE, Ronald NC, Garrison RG, Schmitz JA. 1974. Hexamitiasis in laboratory mice, hamsters, and rats. Lab Anim Sci 24:349–354 [PubMed] [Google Scholar]