

Abstract
Simplified model binding sites allow one to isolate entangled terms in molecular energy functions. Here, we investigate the effects on ligand recognition of the introduction of a histidine into a hydrophobic cavity in lysozyme. We docked 656040 molecules and tested 26 highly and nine poorly ranked. Twenty-one highly ranked molecules bound and five were false positives, while three poorly ranked molecules were false negatives. In the 16 X-ray complexes now known, the docking predictions overlaid well with the crystallographic results. Although ligand enrichment was high, the false negatives, the false positives, and the inability to rank order illuminated weaknesses in our scoring, particularly overweighed apolar and underweighted polar terms. Adjusting these led to new problems, reflecting the entangled nature of docking scoring functions. Changes in ligand affinity relative to other lysozyme cavities speak to the subtleties of molecular recognition even in these simple sites and to their relevance for testing different models of recognition.
Introduction
Molecular docking screens are widely used for ligand discovery.1−9 Whereas hit rates are often high enough for this method to be pragmatic, the technique retains many false positives and false negatives.10 Many of these may be attributed to approximations and errors in docking scoring functions.11 To screen millions of molecules rapidly, approximations in the individual terms that make up scoring functions, and their functional form, have been necessary. These approximations include the use of static atomic partial charges,12 an arbitrarily steep repulsive term, poor treatment of ligand internal energy,13 and crude minimization of docked orientations. Meanwhile, protein-binding sites are complicated: they display multiple, disparate functional groups in geometrically complicated arrangements, they involve ordered and often displaceable water molecules, and often have extensive interfaces with bulk solvent. Although much effort has been devoted to improving these terms in docking, these are typically only tested retrospectively.14,15
A common approach to entangled problems in biology and biophysics has been to simplify the challenge using model systems. In biology, this has included the use of organisms such as Cavia porcellus to study metabolism,16Danio rerio to study vertebrate development,17Tetrahymena to study nucleic acid biology,18Caenorhabditis elegans to study cell differentiation,19 and Drosophila melanogaster to study genetics.20 While in biophysics, simple model proteins like T4 lysozyme,21 staphylococcal nuclease,22 and barnase23 have been used to study interactions important for protein stability, chymotrypsin inhibitors, barstar, and model peptide trp-cages have been used to study protein folding,24 and barnase,25 lysozyme,26 and β-lactamase27 have been used to study trade-offs between stability and activity. In molecular docking, we and others have turned to small, engineered cavity sites28 (Figure 1), which are typically buried from solvent and dominated by single terms such as hydrophobicity, steric complementarity, hydrogen bonding, or ion pair interactions. Because they are small, on the order of 150 to 200 Å3, they bind small organic molecules; unlike normal binding sites, thousands of likely ligands are commercially available, making prospective testing against these sites straightforward. In this they resemble host–guest systems long studied in chemistry.29 Unlike many host–guest systems, however, these proteins are soluble in water, can be readily functionalized by mutagenesis, and can be readily overexpressed.
Figure 1.
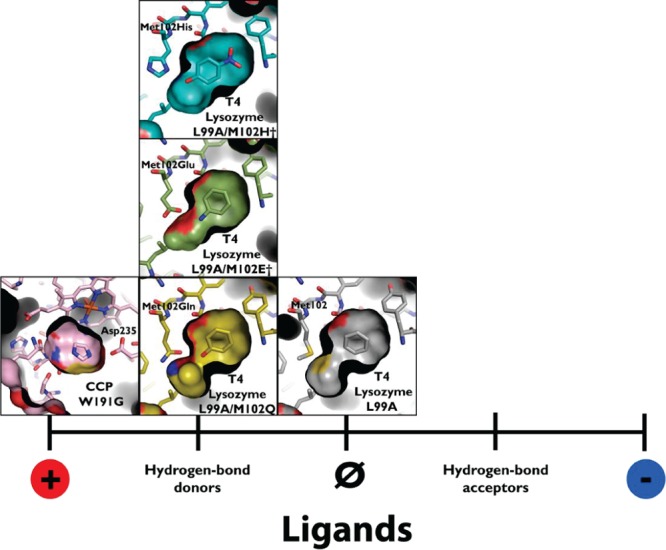
Model cavities to test molecular docking and protein–ligand interactions. These cavities range from the L99A cavity in T4 lysozyme (represented by PDB 181L(28)), which binds exclusively apolar, hydrophobic ligands, to the W191G cavity in cytochrome C peroxidase (CCP) (represented by PDB 1AES(73)). Intermediate to these are the more polar T4 lysozyme cavities L99A/M102Q (PDB 1LI2), which adds a polar glutamine residue to the cavity and L99A/M102E† (PDB 3GUN(55)), which adds a neutral glutamic acid, and L99A/M102H† (PDB 4EKQ(37)), the focus of this study, which adds a histidine residue to the cavity. These intermediate cavities bind both nonpolar and polar, hydrogen-bond donating ligands.
We have previously used two cavities introduced into the hydrophobic core of T4 lysozyme to isolate terms in docking: a hydrophobic site introduced by substituting Leu99→Ala (L99A mutant)28 and the same site with a single carbonyl group added to the cavity wall added by further substituting Met102→Gln (L99A/M102Q).30 Consistent with its hydrophobic nature, the over 70 ligands that have been characterized for the L99A site are largely apolar. Thus, whereas toluene binds to this cavity with an affinity of 102 μM,31 phenol does not measurably bind. Conversely, both phenol and toluene bind to the L99A/M102Q site, with affinities of 91 and 156 μM,30 respectively, and the 30 ligands verified for this site are largely apolar but can tolerate a singly hydrogen-bond donating group (Figure 1). In prospective screening and testing studies, we used these targets to identify weaknesses in docking scoring functions and subsequently optimize one of them.32
Here we introduce a histidine into this cavity, polarizing it still further, by making the substitution Met102→His in the L99A background of T4 lysozyme, L99A/M102H†. The introduction of the highly polar His into this apolar cavity destabilizes it to the point that expression required multiple stabilizing substitutions at sites distal from the cavity binding site—these distal substitutions are indicated by the dagger, † (Methods). Notwithstanding these substitutions, L99A/M102H† retains the features of the earlier cavities: the site is sequestered from solvent, dominated by a few simple terms,33 and thousands of likely ligands are commercially available and can be readily tested. We use this new cavity to investigate the following questions: How does the introduction of the highly polar His102 change the binding of what were ligands for the apolar and slightly polar cavities? Is the affinity of molecules like benzene and toluene reduced or those of molecules like phenol increased? Are new, perhaps more polar molecules accommodated by this site? Most importantly for us, can docking track these changes, which molecules that are mispredicted as false positives and false negatives, and can we isolate the terms that are most responsible for these failures? We investigate these questions using prospective molecular docking screens and testing predicted ligands using a combination of direct-binding assays and X-ray crystallography.
Results
Initial attempts to express and purify L99A/M102H were unsuccessful, likely owing to the stability insult34 resulting from introducing a highly polar histidine residue into an otherwise apolar, buried cavity in the hydrophobic core of T4 lysozyme. To express folded protein, it was necessary to add an N-terminal hexahistidine tag and introduce six substitutions previously shown to stabilize the protein: T21C, S38D, E108V, S117V, T142C, and N144D.21,26,35 This mutant protein, L99A/M102H†, was purified from Escherichia coli in yields of 5 mg/L. The protein was a typical member of the L99A family of cavities, with a buried site of ∼150 Å3. It is likely that this cavity exists in solution with ordered water molecules36 interacting with the buried His102, but we have only been able to crystallize it as a holocomplex, minimally with 2-mercaptoethanol (BME), a component of the crystallization buffer, and in these holostructures these waters are not observed. The BME complexed structure, which we had determined previously, along with complexes with benzisoxazole, nitrobenzene, 4-nitrophenol, and cyanophenol, provided a template structure for docking screens.37 Observation of hydrogen-bonds between donating groups from 4-nitrophenol, 2-mercaptoethanol, 2-cyanophenol, and benzisoxazole, and a hydrogen bond between the His102 Nε2 and the Sδ of Met106, supported a conformation of the histidine where its hydrogen-bond accepting Nδ1 nitrogen pointed into the pocket (Figure 1), available for ligand interaction; this conformation was preserved for the 11 new ligand–protein complex structures subsequently determined here (Figure 2). The complex between the cavity and 2-mercaptoethanol (PDB ID 4E97), determined to 1.30 Å resolution, was used in the docking screens.
Figure 2.

Crystallographic poses of the L99A/M102H† ligands newly reported here. In all cases, oxygens are represented in red, nitrogens blue, and sulfurs yellow. A cutaway of the crystallographically determined protein structure (and surface) is shown with cyan carbons, and the crystallographically determined ligand carbons are light yellow. The side of the surface occupied entirely by the protein is in black. His102 and the ligands are shown in a stick representation. Hydrogen bonds between the ligand and His102 are shown with red dashes. The Fo – Fc electron density map of the protein after refinement but before placement of the ligand is shown in black mesh at σ = 3.0. The figure was rendered with PyMol.70 (A) L99A/M102H† internal cavity showing the protein residues that make up the internal surface of the cavity in a thin stick reperesntation (other than His102). In all cases, the ligand binding cavity is completely physically isolated from bulk solvent. (B) L99A/M102H† with benzene bound, (C) L99A/M102H† with toluene bound, (D) L99A/M102H† with phenol bound (hydrogen bond distance = 2.86 Å), (E) L99A/M102H† with 2-allyl phenol bound (hydrogen bond distance = 3.00 Å), (F) L99A/M102H† with 1-phenyl-2-propyn-1-ol bound (hydrogen bond distance = 2.77 Å), (G) L99A/M102H† with 2-amino-5-chloro thiazole bound (hydrogen bond distance = 2.89 Å), (H) L99A/M102H† with 4-bromoimidazole bound (hydrogen bond distance = 2.93 Å), (I) L99A/M102H† with 4-trifluoromethyl imidazole bound (hydrogen bond distance =2.85 Å), (J) L99A/M102H† with 2-(pyrazolo-1-yl) ethanol bound (hydrogen bond distance = 2.77 Å), (K) L99A/M102H† with 3-trifluoromethyl-5-methyl pyrazole bound (hydrogen bond distance = 2.90 Å), (L) L99A/M102H† with 2-bromo-5-hydroxybenzaldehyde bound (hydrogen bond distance = 2.50 Å).
We screened a 650000 fragment subset of the ZINC database38 against this cavity (see Experimental Methods). Molecules were selected for testing based on four criteria: (1) ranking among the top 100 in the docking-prioritized list, (2) similarity to ligands from the apolar L99A or the slightly polar L99A/M102Q, (3) low topological similarity to such ligands but favorable ranks in the docking screen, or (4) low ranks in the screen but high topological similarities to ligands we ultimately discovered and tested for L99A/M102H† from the first three sets (these last might be docking false negatives) (Supporting Information Figure 1). Binding was measured by thermal denaturation (Tm) upshift, X-ray crystallography, and, for characteristic ligands, by isothermal titration calorimetry (ITC, Table 1). For 11 of the new ligands, X-ray crystal structures were determined in complex with the L99A/M102H† cavity, with resolutions ranging from 1.40 to 1.73 Å (Figure 2, Supporting Information Table 1).
Table 1. Binding Affinity by Isothermal Titration Calorimetry (ITC) to L99A/M102H†a.
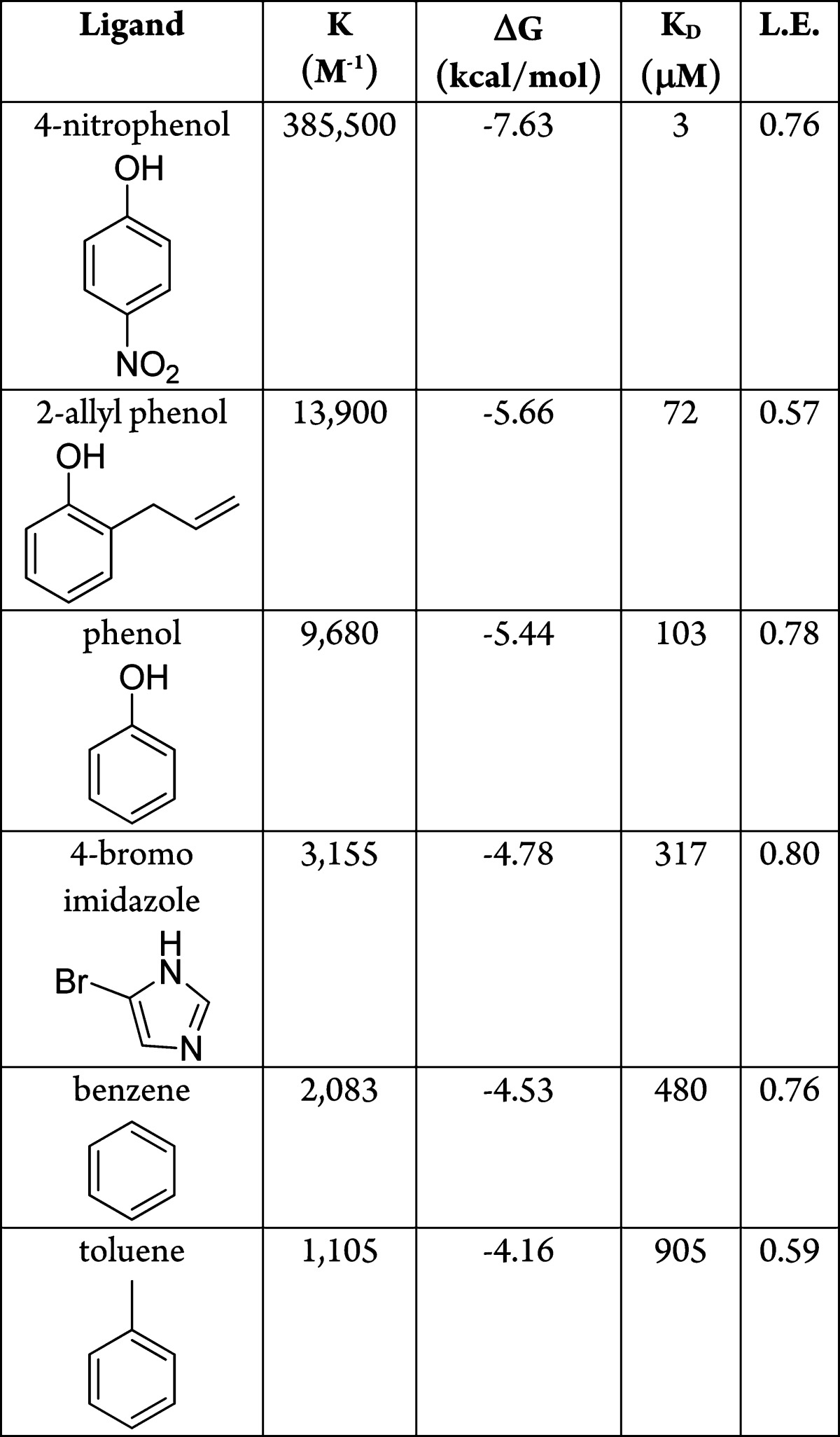
All values reflect the average of duplicate measurements from two separate ligand solutions. Nitrobenzene and 2-amino-5-chloro thiazole appear to have binding affinities of ≥1 mM by ITC measurement.
Of the 26 high ranking docked molecules that were tested, 21 were observed to bind either by melting temperature (Tm) upshift (at either pH values 3.01, 5.4, or 6.8 (values typically used in T4 lysozyme),30,31 and for ligand binding often determined by ligand pKa or by sensitivity requirments) or by X-ray crystallography. For five molecules, binding was not observed by either Tm upshift or by crystallography even at millimolar concentrations; we consider the latter to be docking decoys. Correspondingly, nine molecules with ranks worse than 32800 (5%), positive (unfavorable) docking energy scores, but high topological similarity to what were by now confirmed L99A/M102H† ligands were also tested. Six of these were not observed to bind, consistent with their low docking ranks, but three had measurable affinity; we consider these to be docking false negatives. Affinities measured by ITC tracked the magnitude of Tm upshift and the change in the folding stability of the protein (ΔΔG) on ligand binding (Supporting Information Figure 2), determined using the Schellman equation (ΔTm × ΔSm, Experimental Methods).39 Though ΔΔGmelt can only be used as a proxy for ligand affinity, the observation that it tracks the calorimetric affinity suggests that it is a good guide, at least in this system. Calorimetric binding constants ranged from Kd values of 3 μM for 4-nitrophenol to 103 μM for phenol to 905 μM for toluene. 4-Nitrophenol is the tightest binding ligand yet discovered for any of the lysozyme family of cavities, with a ligand efficiency of 0.76 kcal/mol/heavy-atom. This molecule binds over 40-fold more tightly to L99A/M102H† than it does to L99A/M102Q, consistent with the increased polarity of the histidine cavity. Conversely, benzene and toluene bind between 4-fold and 9-fold worse than they do in L99A/M102Q (Table 2), a point to which we will return.
Table 2. Comparison of Disassociation Constants for the T4 Lysozyme Cavities (All from ITC)a.
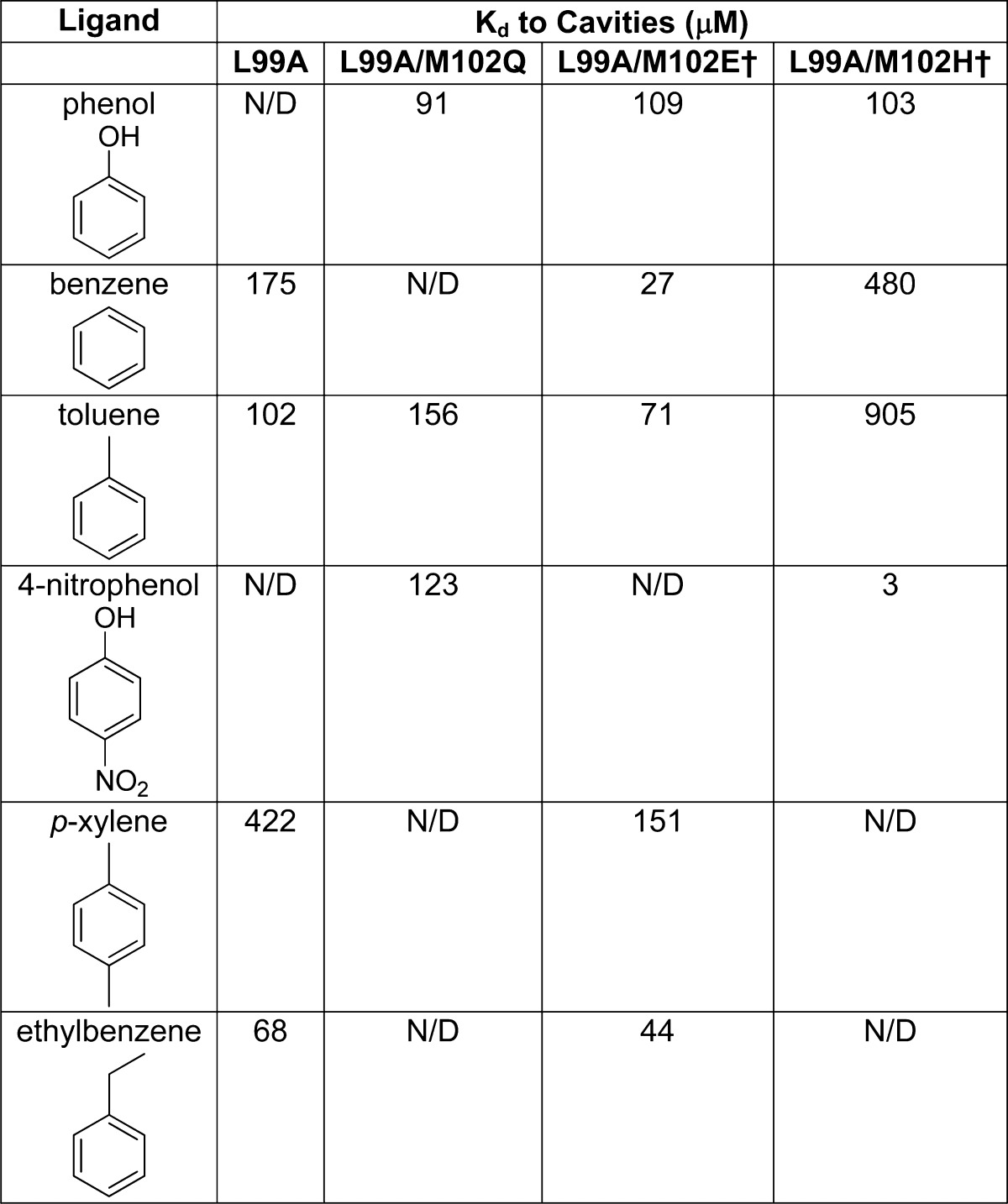
N/D indicates undetermined. L99A measurements performed as reverse titrations, 29 °C, pH 5.5.31 L99A/M102Q measurements performed at 10 °C, pH 6.8,30 except for the 4-nitrophenol measurement, which was performed identically to the L99A/M102H† measurements but at 10 °C. L99A/M102E† measurements performed at 4 °C, pH 4.7.55 L99A/M102H† measurements from this work, 25 °C, pH 5.0.
In this study, we consider molecules to be ligands that normally would not be considered so; some, like 2-mercaptoethanol, bind so weakly that they could not be observed except by crystallography, where they were present at tens of millimolar concentration. Meanwhile, ligands like cyanophenol appeared to bind in the low mM range and could only be detected by Tm upshift at the pH value where the temperature–stability curve for the protein is most sensitive to ligand binding. That said, much of the low binding of these molecules may be attributed to their small size, typically below even that explored in most fragment libraries, where molecules below 150 Da are rare. By ligand efficiency, many of the even weakest molecules still have values close to 0.4 kcal/mol/heavy-atom, and for many crystal structures of the bound complexes were determined, so there is little doubt as to their status as ligands. And from the perspective of illuminating docking problems (a main driver of this study, after all) many of the weak ligands seemed interesting to us. Thus, molecules like phenyl vinyl sulfide ranked at the top of the list but had very modest affinities, while molecules like 2-mercaptoethanol bound so weakly as to be only detectable by crystallography are considered docking false negatives.
By some criteria, the 81% (21 of 26) docking true positive and 33% (3 of 9) false negative rates were comforting, as was the overall enrichment curve (Figure 3). As was true in our earlier cavity docking studies,30,40,41 this hit-rate is far higher than what we have observed for soluble proteins recognizing larger ligands, consistent with the hypothesis that there is a far higher chance of finding ligands at this molecular weight than among larger molecules in screening libraries.42 On closer inspection, illuminating problems emerge. Taking the apparent change of stability on ligand binding as a proxy for affinity, there was no correlation between docking rank and this affinity-proxy even among the true positive ligands (Supporting Information Figure 3) (we use the Schellman equation39 of ΔTm × ΔS, comparing the ligand-bound to the free protein, Table 3). Thus, 4-nitrophenol, which appeared to stabilize the protein more than any other ligand (1.4 kcal/mol by Tm perturbation), did not even rank among the top 0.95% of the molecules screened although it has the best affinity of any ligand from all the lysozyme cavities43 (Table 2). Conversely, phenyl vinyl sulfide had the highest rank of all molecules tested, 10th out of over 656040, but it only induced a modest ΔTm (ΔGmelt = 0.25 kcal/mol), and 5-nitrothiophene-2-carbononitrile, the next highest ranked molecule tested at 19th overall, was not observed to bind at all (ΔTm < 0.4 °C). Phenol, with a rank of 2430 and a docking score of −14.6 kcal/mol, bound well (103 μM), but 2-cyanophenol, which scored only 0.4 kcal/mol worse, was barely detectable. On the other hand, 4-bromo-2-cyanophenol was scored poorly by DOCK (0.96 kcal/mol, rank 33,837) but had a substantial 2.2 °C ΔTm (ΔGmelt = 0.6 kcal/mol).
Figure 3.
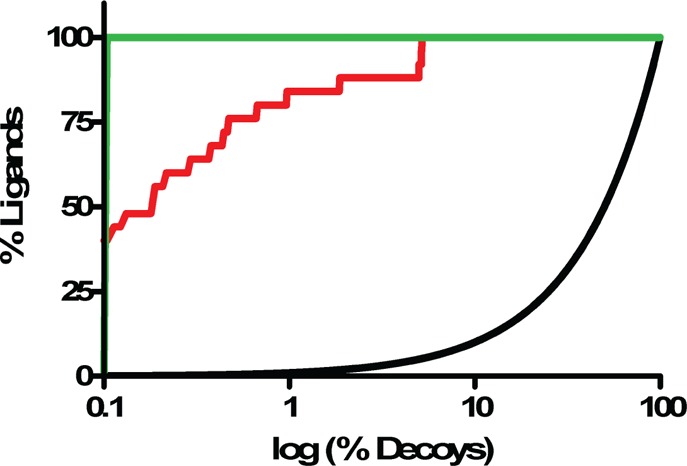
ROC plots for docking-predicted ligands in L99A/M102H† (red curve, logAUC = 70.14). All logAUC values are adjusted so that the random-selection curve (black) has a logAUC = 0, and a perfect enrichement curve (green) corresponds to a logAUC = 85.5.12
Table 3. The 37 Molecules Tested for Binding to L99A/M102H†a.
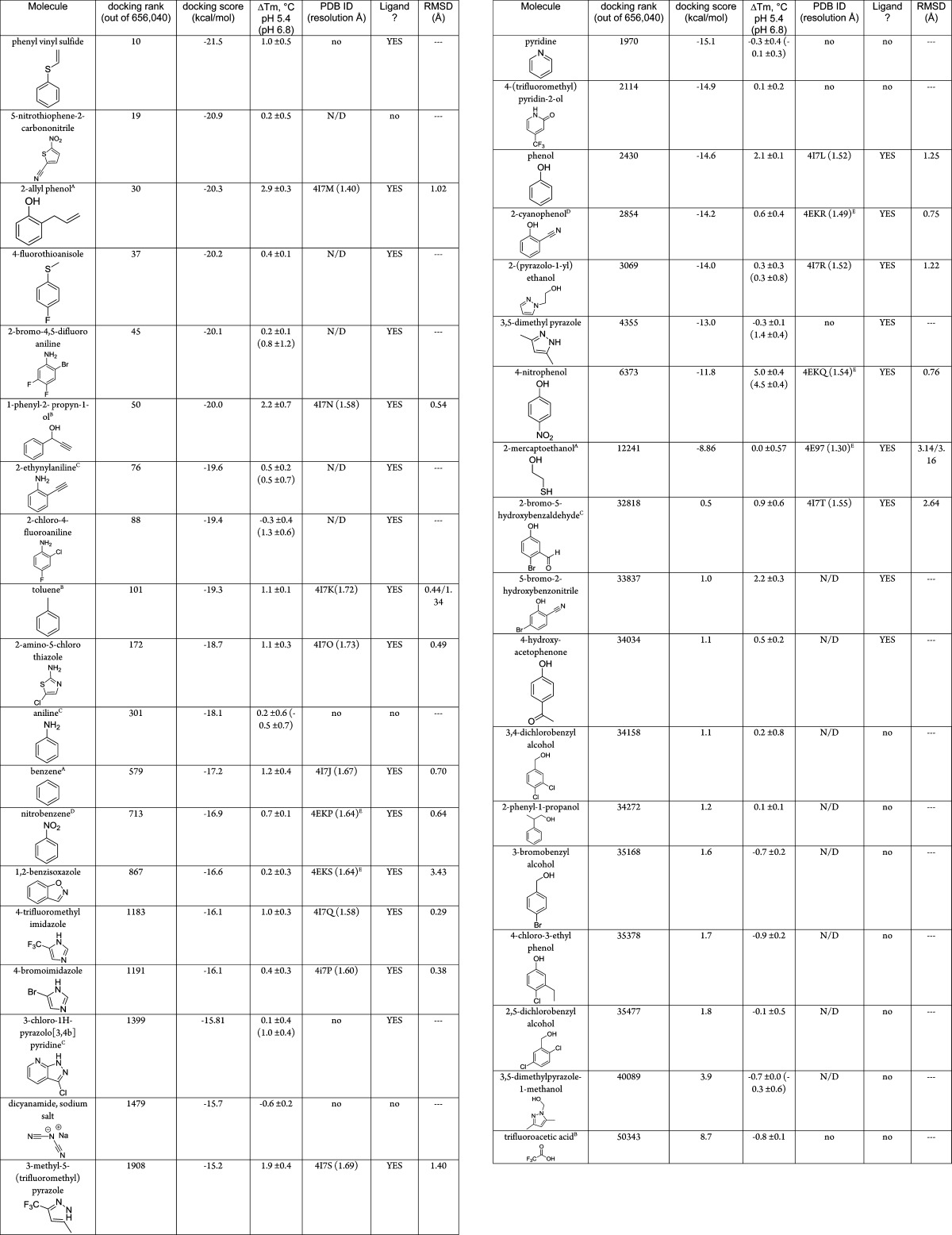
Molecules were identified as ligands if they showed a thermal unfolding upshift ΔTm of ≥0.4 °C in the presence of 1 mM ligand, unless otherwise noted, at pH 5.4, pH 6.8, or pH 3.05, or a co-crystal structure was successfully obtained (at pH 4.5). Those molecules which a crystal complex structure could not be obtained by soaking are indicated by “no” in the PDB ID column, those ligands that were not screened this way are reported as “N/D”. A more thorough treatment is included as Supporting Information Tables 2 and 3. The top 10000 molecules in the docking ranked list are included as Supporting Information Table 4. A = tested at 2 mM. B = tested at 5 mM. C = tested at 0.5 mM. D = measured at pH = 3.05. E = previously reported.37
We looked for patterns in the components of the overall docking energy score that might explain the poor rank ordering, the high ranks of the false positives, and the low ranks of the false negatives. The latter were the least informative, as these were larger molecules that simply could not be accommodated by the conformation of the cavity that we used in docking, as is represented by their poor van der Waals scores (Supporting Information Table 2). Likely the L99A/M102H† cavity expands on binding to these molecules, as seen with earlier cavities.32 Whereas conformational flexibility is certainly a key problem in docking, it is more of a sampling than a scoring problem and will not be further considered here. On the other hand, there was a clear pattern in the scores of the weak or nonbinding, high-ranking molecules: most lacked hydrogen-bond donating groups (Table 3, Supporting Information Figure 1). The docking scores of these molecules were characterized by favorable van der Waals and desolvation terms. By the same token, molecules that did hydrogen-bond to the cavity histidine in the docked poses, and had high apparent affinities, such as many of the hydroxyl-bearing ligands, were often ranked lower in the hit list. For these molecules, the electrostatic component of their docked energy was outweighed by their desolvation energies and, to a lesser extent, their diminished van der Waals interaction energy, itself likely reflecting trade-offs inflicted by the close approach to the histidine.
To investigate these problems at atomic resolution, the X-ray crystal structure of 11 newly discovered ligands with L99A/M102H† were determined, adding to the five previously reported,37 at resolutions better than 1.74 Å (Table 3, Supporting Information Table 1). Both refined (2Fo – Fc) and initial difference density features for the ligands were clear (Figure 2), allowing us to model the ligand geometries unambiguously. Overall, the docked poses corresponded well to the crystallographic results (Figure 4), with a mean RMSD of 1.20 Å over the 16 structures and a median RMSD of only 0.76 Å (Table 3) (the higher average owes to poor values of 3.14 Å for 2-mercaptoethanol (Figure 4D), 3.47 and 2.64 Å for the ring-flips in benzisoxazole (Figure 4E) and 2-bromo-5-hydroxybenzaldehyde (Figure 4P), respectively. Notwithstanding this good overall structural correspondence, problems were often observed with the His102 hydrogen bond to the ligand. For molecules like phenol, 2-mercaptoethanol, 4-hydroxyphenol, pyrazolo-ethanol, and 3-trifluoromethyl-5-methyl pyrazole (Figure 4C,D, G,J, and 4N,O), the discrepancies could be attributed mainly to imperfect donor–histidine interactions. In the crystal structures, the ligands adopt a geometry where the hydrogen-donating group is well positioned to hydrogen-bond with the histidine, while in the docking pose this geometry is offset, with the proton typically positioned out-of-line with the histidine acceptor (in DOCK3.6, there is no special term for hydrogen bonds, only overall electrostatic interaction energy). For the phenolic hydroxyls, this was compounded by the adoption of a conformation that placed the hydroxyl hydrogen out-of-plane of the aryl ring. This is likely a high-energy conformation relative to the in-plane geometry, the latter of which is consistent with the crystallographic poses.
Figure 4.

Comparison of the crystallographic (carbons light yellow) and docking-predicted poses (carbons magenta) of the L99A/M102H† ligands showing a cutaway of the surface of the cavity (otherwise colored as in Figure 1); His102 is shown. The figure was rendered with PyMol.70 In all cases, the cavity is buried from bulk solvent, and only small changes in the binding site were observed among the different structures. Values for RMSD between the docking poses and the crystallographic ligand geometries are given in Table 3. Shown are the complexes with (A) benzene, (B) toluene, (C) phenol, (D) 2-mercaptoethanol (PDB ID 4E97(37)), (E) 1,2-benzisoxazole (PDB ID 4EKS(37)), (F) nitrobenzene (PDB ID 4EKP(37)), (G) 4-nitrophenol (PDB ID 4EKQ(37)), (H) 2-hydroxy benzonitrile (PDB ID 4EKR(37)), (I)) 2-allyl phenol, (J) 1-phenyl-2-propyn-1-ol, (K) 2-amino-5-chloro thiazole, (L) 4-bromoimidazole, (M) 4-trifluoromethyl imidazole, (N) 2-(pyrazolo-1-yl) ethanol, (O) 3-trifluoromethyl-5-methyl pyrazole, (P) 2-bromo-5-hydroxybenzaldehyde.
We considered three strategies to overcome this apparent bias toward apolar molecules and imperfect hydrogen-bonding: increasing the local dipole on the cavity His102, down-weighting the contribution of the calculated van der Waals interaction to the overall energy score by 1/2, and increasing the sampling of phenolic hydrogens. The first strategy was accomplished by increasing the magnitude of the charge on histidine Nδ1 atom (pointing into the cavity), from −0.527 to −0.927 to −1.327 electrons, balancing that with an increased charge on the Nε2H, pointing away from the cavity, from 0.32 to 0.72 to 1.12. This is a strategy that we have often used to increase the polarity of docked ligands in prospective ligand discovery campaigns,44 but have never tested in a controlled manner. The second strategy was accomplished simply by multiplying the van der Waals term by 0.5 or by increasing the weighting of the electrostatics term by a factor of 2. The third strategy involved rescoring the crystallographic poses of the phenolic ligands with appropriate conformations of the phenolic hydrogen.
The effects of both increasing the local histidine dipole, and of reducing the weight on the van der Waals term, illustrate how changing scoring functions in a nonphysical way to achieve a particular goal, here, improving the rank of polar molecules that interact with the histidine, can have unintended consequences. Unexpectedly, scaling-down the van der Waals term actually reduced the number of top-ranking molecules that interacted with His102, from 39 in the standard calculation to 32. This reflects a lower stringency against bulkier molecules, which rise in the ranked list at the expense of those making polar interactions (Supporting Information Table 3). On the other hand, increasing the charge on the acceptor Nδ1 of His102 did increase the number of ligands that hydrogen bonded with this residue, from 39 of the top ranked 100 in the standard docking, to 65 when the magnitude of the partial atomic charge was increased by 0.4 electrons, to 93 when it was increased by 0.8 electrons. However, the ligands that rose in the list were almost exclusively amine-bearing molecules like anilines, imidazoles, guanidines, and pyrazoles, while phenolic-bearing ligands, which appear to bind more tightly, had at best mixed results.
The unexpected decline of the phenolic ligands relative to the amine-bearing ligands when the local charge on the His102 Nδ1 is increased may reflect a broader problem of undersampling, or even a mis-sampling, of the phenolic hydroxyl position in our ligand conformations. Unlike anilinic, pyrazolic, and imidazolic protons, the phenolic proton is rotatable and must be sampled in docking. In most of the ligands discovered here, the proton was sampled in positions largely out of the plane of the ring. Conversely, in the Cambridge Structural Database, most phenols orient their protons in the plane of the ring.45 When we insist that the docked molecules adopt this hydrogen conformation (Experimental Methods), their scores improve without cost to their desolvation energies, and their docked geometries improve. For instance, when we sample the phenolic hydrogens in the plane of the ring, the positional error in the docked pose improved by 0.05 to 0.97 Å rms (Table 4), illustrating the utility of this simple improvement.
Table 4. The Effect of Enforcing In-Plane Hydrogens on the Phenolic Ligandsa.
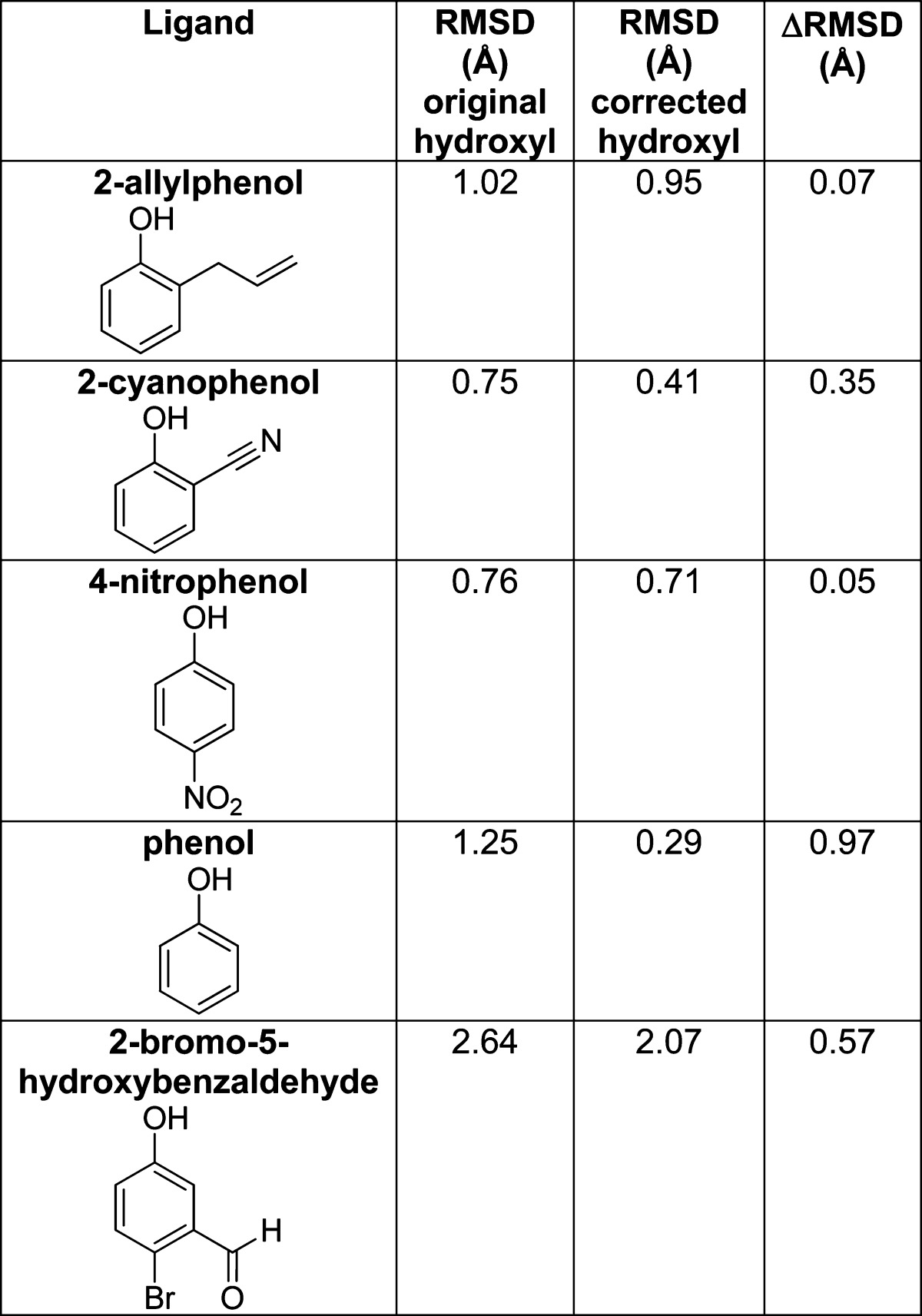
By forcing the phenolic hydrogen to adopt a physically realistic pose, the DOCK predicted pose becomes a better match to the crystallographically determined structure.
Discussion
Three principal observations emerge from the docking and ligand testing against the L99A/M102H† cavity. First, the molecules that ranked highly by docking, relative to a 650000 compound library, broadly tracked the increased polarity of the site. The observations that six of nine ligands chosen based on topological similarity alone often did not bind, and the observations of substantial changes in ligand affinities between this site and earlier apolar (L99A) and slightly polar (L99A/M102Q) cavities, suggests that the docking is capturing important aspects of ligand recognition. Second, and notwithstanding this high hit-rate, the inability to even monotonically rank order the hits, and the false positives that were observed, reflect difficulties in balancing electrostatic and nonpolar terms in docking. In screens against a large, diverse compound database, even in a model cavity, superficial changes to address these deficiencies can result in unexpected new problems. Third, the L99A/M102H† cavity contributes to what is now a spectrum of simple cavity sites characterized by small perturbations and dominated by one or two terms. These sites provide good templates for testing methods in docking, and other molecular recognition methods, and are available to the community toward that end.
By many of the criteria typically used to evaluate docking, the screen against the polar cavity was unusually successful. The 81% hit rate and ability to discriminate against topologically similar decoys supports the ability of the docking to capture gross features of the molecular recognition in this site, as does the mixture of different chemotypes46 found. The docking hits against L99A/M102H† tracked the changed polarity of the site relative to the more apolar L99A or L99A/M102Q cavities. Thus, there were fewer polar, hydrogen-bond donating molecules among the top 100 L99A docking hits than among the top L99A/M102H† hits (and those that were there typically interacted with the native Met102 Sδ sulfur). Many of the high-ranking polar molecules that were found to bind to L99A/M102H† ranked less well against the L99A cavity, while many of the apolar active ligands ranked better when docked against L99A (Table 2, Supporting Information Figure 4). For instance, benzene, toluene, and nitrobenzene are all ranked better in the L99A cavity screen than in the L99A/M102H† screen, while the polar 2-allyl phenol, 2-ethynylaniline, and 2-(pyrazolo-1-yl) ethanol all rank better in the newly polarized cavity (Supporting Information Table 3) This correspondence is further supported by the high fidelity of the docking poses to the subsequently determined X-ray crystallographic results, which superposed with an average RMSD of 1.2 Å to the initial docking poses (Figure 4, Table 3). Whereas we do not wish to overemphasize docking performance in these sites, as our goal here is more to interrogate docking weaknesses than to parade its strengths, it is perhaps worth mentioning that ligand discovery rose above what might be easily captured by physical features alone. For instance, categorizing the 37 molecules tested here according to whether they have fewer than 10 non-hydrogen atoms, at least one polar atom, an aromatic ring, and are neutral, leads to substantially worse performance in a truth table than the docking performance, as does simply categorizing ligands by polar surface area (e.g., docking had 21 true and five false positives, whereas polar surface area had 16 true and four false positives, with docking also performing better by the true and false negative criteria) (Supporting Information Figure 5). We cannot exclude the possibility that a more optimized QSAR might be devised to better categorize the ligands reported here, at least retrospectively.
From a molecular recognition standpoint, we were struck by the differential effects on polar and apolar ligand binding of the Gln102→His substitution in the binding site (Figure 1), especially for phenols and their apolar isosteres. The His substitution increased the affinities of phenol and 4-nitrophenol by 0.2 and 2.6 kcal/mol, respectively, relative to their affinities in the L99A/M102Q cavity, while toluene bound 0.8 kcal/mol better in the L99A/M102Q cavity (Table 2). Presumably, this reflects the greater polarity of the histadine cavity relative to the glutamine. Formal double perturbation analysis47,48 confirms this inference: using phenol and toluene, 4-nitrophenol and toluene, or 4-nitrophenol and phenol as the ligand pairs, and histadine and glutamine as the amino acid pairs, the interaction between phenol and His102 is 0.6 kcal/mol stronger than that between the Gln102, while that between 4-nitrophenol and His102 is 2.4 kcal/mol greater than the same interaction with Gln102 (e.g., for the strength of the phenol His102 interaction, one may subtract the ΔΔG of toluene binding to L99A/M102H versus toluene binding to L99A/M102Q from the ΔΔG of phenol binding to the same cavities, Table 2). This reflects the greater polarity conferred by the histidine, consistent with its destabilization of the protein26,37 and the polarization of the phenolic group by the para-nitro. Still, the strength of these effects was unexpected: Gln102→His only substitutes one hydrogen bond acceptor with another, and yet the differential effects on the ligand hydrogen bond were in the range of, or in the case of 4-nitrophenol substantially stronger than, an entire neutral hydrogen bond. This speaks to the strength of interactions in fully buried cavities and the ability to isolate these terms in such sites.49 Whereas this study was focused on tracking polarity changes in docking screens, and was not directed toward formal interaction energy analysis, undertaking double-perturbation studies in these cavities may merit further study.
If the virtual screen captured the elementary aspects of molecular recognition in this polarized cavity, it also illuminated liabilities not seen in the earlier cavities. The inability of docking to even rank-order compounds is well-known and has been attributed to several origins, including inadequate treatment of ligand conformational energies,13,50,51 receptor and ligand desolvation,12 protein and ligand flexibility,52 and treatment of ordered waters.53 In the L99A/M102H† cavity, most of these terms may be discounted: the ligands possess little flexibility, all desolvate the protein site and are desolvated by it to about the same extent, and over a dozen crystal structures reveal that when the cavity does change conformation, it is modest. What remains is an imbalance between the van der Waals and electrostatic energy, with the former apparently dominating the latter; relatively weak apolar ligands, like phenyl vinyl sulfide, rank well, whereas tight-binding polar ligands, like 4-nitrophenol, rank relatively poorly (Table 3). If it is easy to appreciate this imbalance, superficial solutions seemed inadequate to address it. Thus, down-weighting the van der Waals term in the scoring function, which is widely practiced,54 had the unintended consequence of actually reducing the number of high-ranking polar molecules in favor of more apolar molecules that could now be accommodated by a more permissive steric function. Correspondingly, increasing the weighting of the electrostatic term, or increasing the magnitudes of the partial atomic charges on His102, which served to favor local polar interactions, did increase the number of polar high-scoring molecules in the docking hit list, but this change favored the more rigid amine-based hydrogen-bond donors, like anilines, thiazoles, and pyrazoles, over hydroxyl-based donors; it is the latter of these that in reality bound better to the site. It is only when we addressed deficiencies in the sampling of the hydroxyl hydrogen conformational generation that this issue was partly addressed. When confronted by entangled physical terms, such as the role of electrostatics and van der Waals repulsion in polar interactions, it is tempting to adopt apparently straightforward “fixes”. It is perhaps a lesson of this study that, as sensible as these might seem, when confronted with a vast library of diverse chemicals, such as those in large-scale docking screens, these fixes often fall victim to chemotypes that exploit newly introduced liabilities in the scoring functions. Such unintended consequences are rarely apparent in wholly retrospective studies, which often fail to confront the full diversity of a large library screen. The model systems thus may teach that, to advance the field, both pragmatically and generally, we may have to confront the hard task of building more physically sophisticated models.
If the need to develop well-grounded physical methods may seem daunting, the field is well-positioned to test new algorithms in well-controlled systems. As in protein stability and folding, where simple model systems had so salutary effect on the field, docking as a field may be able to exploit the simple model cavity sites that we and others have introduced.28,55 Taken together, the L99A, L99A/M102Q, L99A/M102E†, and the L99A/M102H† cavities in lysozyme, and the W191G cavity in cytochrome C peroxidase, provide a scale of binding sites dominated, on one extreme, by nonpolar complementarity and hydrobophicity, to slight polarity with the introduction of three different hydrogen bond acceptors (the carbonyls of glutamine and glutamate and the Nδ1 nitrogen of histidine56), to a fully charged site in W191G at the other extreme (Figure 1). This polarity spectrum is reflected in the ligands that bind to each site, and the affinity with which they do so (Table 2). Apolar ligands bind less well to L99A/M102H† than they do to L99A, L99A/M102Q, and L99A/M102E†, while the more polar ligand, 4-nitrophenol, binds to L99A/M102H† with the greatest measured affinity for a ligand to any of the model cavities (3 μM). Even in these simple sites, there are, however, subtleties that reflect problems encountered in more complicated sites, as is appropriate for a model system.57,58 For instance, the affinities of benzene and toluene are essentially unaffected between L99A and L99A/M102Q, while the affinity of phenol is unchanged between L99A/M102Q and L99A/M102H†. Conversely, against L99A/M102H†, 4-nitrophenol is 40-fold more potent that for L99A/M102Q (Table 2), while the affinities of benzene and toluene drop by 3- and 9-fold, respectively, compared to L99A, and toluene drops by 6-fold compared to L99A/M102Q (Table 2). As simple as these cavities are, they still capture important aspects of the scoring problem in docking, and indeed simulation methods more broadly.32 Because they are small, there are thousands of likely ligands, still untested, in our libraries, enabling further prospective testing, which will be crucial to test new methods. The constructs to express these proteins are freely available to the community, as are small amounts of purified protein. We are, indeed, willing to test predictions, made by other methods and other investigators interested in investigating molecular recognition in well-controlled systems.59,60
Experimental Methods
Protein Expression
Unless otherwise noted, all compounds in this study were obtained from standard commercial sources. The gene containing T4 phage lysozyme L99A/M102H† in pET-28 (EMD Biosciences, Darmstadt) was transformed into E. coli BL21(DE3) cells and grown in Luria–Bertani media containing kanamycin to OD600 0.6–0.8 at 37 °C and then induced with 0.5 mM isopropyl β-d-1-thiogalactopyranoside overnight at 18 °C. Cells were lysed by microfluidizer, centrifuged at 18000g for 45 min, and then purified on a nickel-nitrilotriacetic acid column at 4 °C, imidazole buffer, pH 6.8, in the presence of 5 mM 2-mercaptoethanol and then dialyzed into 200 mM KCl, 5 mM 2-mercaptoethanol, 0.02% sodium azide, 50 mM phosphate buffer, pH 6.6 once and then two more times in the same buffer without 2-mercaptoethanol or azide, and finally concentrated to 10 mg/mL, aliquoted, and flash frozen in liquid nitrogen and stored at −80 °C until needed. Protein purity was ≥95% by SDS-PAGE. Upon thawing, the protein was centrifuged to remove precipitate and stored at 4 °C, further centrifugation as needed on subsequent days.
Ligand Binding
CD measurements were performed in at least triplicate at pH 5.4 as described31 at a final ligand concentration of 1 mM unless otherwise noted (Table 3). Briefly, the CD signal was observed at 228 nm with heating using a Jasco J-715 spectropolarimeter with a Jasco PTC-348WI Peltier, often using 1 mm Helma cuvette. The resulting thermal melt was fit using the program EXAM61 to yield Tm and ΔH values. Cuvettes were soaked overnight in a 5% Contrad solution to remove any residual protein before use. ΔCp was set to 10.5 kJ mol–1 K–1 for pH 5.4, 8.1 kJ mol–1 K–1 at pH 6.8, and 8.0 kJ mol–1 K–1 at pH 3.05. Ligands were dissolved in DMSO and added to the cuvette to a final concentration of 1 mM except as noted in Table 3. A ΔTm cutoff of 0.4 °C was used to differentiate binders from nonbinders. In all cases (including controls), a final DMSO concentration of 0.1% was used in the assay. Thermodynamic values from CD melts were calculated as ΔGmelt = ΔTm × ΔS, where ΔS = ΔH/Tm.39 ITC was performed using a MicroCal VP-ITC (GE Healthcare) at pH 5.0 using a 33 mM succinate, 44 mM imidazole, 44 mM Tris constant ionic strength buffer at 25 °C.62 Ligands were dissolved in the same buffer as the protein had been equilibrated in. All solutions were degassed immediately before use. The cell concentration of the protein was typically 10 μM. A initial set of injections of ligand into buffer without protein was used to account for the heat of dilution. Reported values are the average of two measurements.
Crystallography
Crystals were grown as previously described.37 Crystals were placed in the cryo solution containing enough of each ligand to make a 5 mM ligand solution and soaked overnight at 4 °C. An initial set of molecules expected to bind was used as an initial crystallographic screening test set (Supporting Information Figure 1). Data sets were obtained at the ALS beamline 8.3.1 and have been deposited in the PDB (Supporting Information Table 1). Reflections were integrated and scaled with XDS.63 Molecular replacement was performed with PHASER.64 Models were refined initially using simulated annealing using PHENIX65 and COOT66 and later were refined with TLS B-factors.67 Ligand parameters were generated using PRODRG.68 Complex structures were solved by molecular replacement using the structure of L99A/M102H† in complex with 2-mercaptoethanol.37 The models were checked with MolProbity.69 Figures were made with PyMOL.70 RMSD values were calculated between the crystal and DOCK poses manually; when discernible specific atoms were identified, although in cases of significant degeneracy (e.g., benzene) distances were measured between the nearest possibly correct atom.
Docking
Molecular docking to the structure of L99A/M102H† (PDB ID: 4E97) was performed using DOCK 3.6 using solvent-excluded volume (SEV) desolvation.12 The binding site His102 was protonated on Nε2, and the most occupied conformation was used in cases of multiple receptor conformations. We used the “model systems” subset in the ZINC database,38 which consists of commercially available molecules with molecular weights ≤250 Da. At the time of this study, this amounted to 656040 molecules; all were docked against L99A/M102H†. A set of molecules known to bind the other lysozyme L99A cavity ligands and those previously discovered to bind to the cavity were used as an initial set of “ligands” to calibrate the early docking runs. The ranks and DOCK scores (including the individual components of these scores) of the top 10000 molecules are included as Supporting Information Table 4. Expert examination was used to select a set of molecules from among the top 100 (0.02%) scoring molecules (high scoring set). This involved visual inspection of the DOCK generated pose for obvious errors, with some selection for diversity. Another set of molecules was chosen and screened by CD and later crystallography from the top 5000 molecules (0.8%) for dissimilarity to any of the ligands known to bind to any of the L99A, L99A/M102Q, or L99A/M102E† cavities (dissimilar set). Dissimilarity was defined as having a Tanimoto coefficient71 of <0.30 using the ECFP4 fingerprint72 to any of the published ligands of the other lysozyme cavity constructs.43 Expert examination of the ligands discovered at this point allowed the formalization of a ligand chemotype that was explicitly implemented with Pipeline Pilot (Accelrys, San Diego, CA) (≤10 heavy atoms, ≥1 hydrogen bond donor, ≥1 aromatic ring, and no atoms bearing formal charges). The DOCK list was then filtered for the molecules that fit the topological ligand description and above, and a final group was selected by filtering the molecules in the bottom 95% of the entire list that also fit the topological definition of ligands (although most of these molecules ended up being in the top 10% of the docking list). We then chose nine that were would nevertheless fit the 1D physical criteria (Supporting Information Figure 1); these were possible docking false negatives set. A graphical representation of the enrichment factor generated by the docking run considering only ligands that were experimentally verified to be binders is given as figure. The phenolic ligands were also rescored using the cocrystal structure after manually placing the phenolic hydrogen in plane with the ring of the ligand.
Acknowledgments
We thank Inbar Fish, Joel Karpiak, Gabriel Rocklin, and Drs. Trent E. Balius, Sarah Barelier, and Marcus Fischer for helpful comments on the manuscript, Dr. Oliv Eidam for help with crystallography, and Dr. John Chodera for help with ITC. This work was supported by US NIH grant GM59957.
Glossary
Abbreviations Used
- ZINC
ZINC is not commercial
- BME
2-mercaptoethanol
- PDB
Protein Data Bank
- Tm
melting temperature
- ITC
isothermal titration calorimetry
- Kd
binding constant
- RMSD
root-mean square deviation
- DMSO
dimethyl sulfoxide
- ALS
Advanced Light Source, Berkeley, CA
- TLS
translation libration screw
- SEV
solvent excluded volume
- CD
circular dichroism
- ECFP4
extended connectivity fingerprint (bond diameter 4)
- CCP
cytochrome C peroxidase
- ROC
receiver operating characteristic
- logAUC
area under the curve, log value
- LE
ligand efficiency
Supporting Information Available
The complete crystallographic table for protein–ligand complex structures reported, 2D images of the molecules tested and their docking scores, graphical representations of the relationship between ΔTm and binding affinity or docking rank, a truth table comparing the ability of molecular docking, a 1D chemotype and total polar surface area to predict ligands, as well as a list of the 10000 molecules in the top of the docking ranked list with scored separated into individual terms (van der Waals, electrostatic interactions, polar and nonpolar desolvation). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Kitchen D. B.; Decornez H.; Furr J. R.; Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nature Rev. Drug Discovery 2004, 311935–949. [DOI] [PubMed] [Google Scholar]
- Alvarez J. C. High-throughput docking as a source of novel drug leads. Curr. Opin. Chem. Biol. 2004, 84365–370. [DOI] [PubMed] [Google Scholar]
- Shoichet B. K.; Kobilka B. K. Structure-based drug screening for G-protein-coupled receptors. Trends Pharmacol. Sci. 2012, 335268–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doman T. N.; McGovern S. L.; Witherbee B. J.; Kasten T. P.; Kurumbail R.; Stallings W. C.; Connolly D. T.; Shoichet B. K. Molecular docking and high-throughput screening for novel inhibitors of protein tyrosine phosphatase-1B. J. Med. Chem. 2002, 45112213–2221. [DOI] [PubMed] [Google Scholar]
- Meiering S.; Inhoff O.; Mies J.; Vincek A.; Garcia G.; Kramer B.; Dormeyer M.; Krauth-Siegel R. L. Inhibitors of Trypanosoma cruzi trypanothione reductase revealed by virtual screening and parallel synthesis. J. Med. Chem. 2005, 48154793–4802. [DOI] [PubMed] [Google Scholar]
- Carlsson J.; Coleman R. G.; Setola V.; Irwin J. J.; Fan H.; Schlessinger A.; Sali A.; Roth B. L.; Shoichet B. K. Ligand discovery from a dopamine D-3 receptor homology model and crystal structure. Nature Chem. Biol. 2011, 711769–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mysinger M. M.; Weiss D. R.; Ziarek J. J.; Gravel S.; Doak A. K.; Karpiak J.; Heveker N.; Shoichet B. K.; Volkman B. F. Structure-based ligand discovery for the protein–protein interface of chemokine receptor CXCR4. Proc. Natl. Acad. Sci. U. S. A. 2012, 109145517–5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H.; Bower K. E.; Beuscher A. E.; Zhou B.; Bobkov A. A.; Olson A. J.; Vogt P. K. Stabilizers of the Max Homodimer Identified in Virtual Ligand Screening Inhibit Myc Function. Mol. Pharmacol. 2009, 763491–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utsintong M.; Talley T. T.; Taylor P. W.; Olson A. J.; Vajragupta O. Virtual Screening Against alpha-Cobratoxin. J. Biomol. Screening 2009, 1491109–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadhav A.; Ferreira R. S.; Klumpp C.; Mott B. T.; Austin C. P.; Inglese J.; Thomas C. J.; Maloney D. J.; Shoichet B. K.; Simeonov A. Quantitative Analyses of Aggregation, Autofluorescence, and Reactivity Artifacts in a Screen for Inhibitors of a Thiol Protease. J. Med. Chem. 2010, 53137–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perola E.; Walters W. P.; Charifson P. S. A detailed comparison of current docking and scoring methods on systems of pharmaceutical relevance. Proteins 2004, 562235–249. [DOI] [PubMed] [Google Scholar]
- Mysinger M. M.; Shoichet B. K. Rapid Context-Dependent Ligand Desolvation in Molecular Docking. J. Chem. Inf. Model. 2010, 5091561–1573. [DOI] [PubMed] [Google Scholar]
- Tirado-Rives J.; Jorgensen W. L. Contribution of conformer focusing to the uncertainty in predicting free energies for protein–ligand binding. J. Med. Chem. 2006, 49205880–5884. [DOI] [PubMed] [Google Scholar]
- Ferrara P.; Gohlke H.; Price D. J.; Klebe G.; Brooks C. L. Assessing scoring functions for protein–ligand interactions. J. Med. Chem. 2004, 47123032–3047. [DOI] [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30162785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunzl C.; Sachser N. Behavioural and endocrinological changes during the process of domestication in guinea pigs. Arch. Tierz. 2000, 43, 153–158. [Google Scholar]
- Laggner C.; Kokel D.; Setola V.; Tolia A.; Lin H.; Irwin J. J.; Keiser M. J.; Cheung C. Y. J.; Minor D. L.; Roth B. L.; Peterson R. T.; Shoichet B. K. Chemical informatics and target identification in a zebrafish phenotypic screen. Nature Chem. Biol. 2012, 82144–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodson S. A. Taming free energy landscapes with RNA chaperones. RNA Biol. 2010, 76677–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahringer J. Embryonic Tissue Differentiation in Caenorhabditis elegans Requires Dif-1, a Gene Homologous to Mitochondrial Solute Carriers. EMBO J. 1995, 14102307–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garza D.; Medhora M.; Koga A.; Hartl D. L. Introduction of the Transposable Element Mariner into the Germline of Drosophila melanogaster. Genetics 1991, 1282303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baase W. A.; Liu L. J.; Tronrud D. E.; Matthews B. W. Lessons from the lysozyme of phage T4. Protein Sci. 2010, 194631–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeker A. K.; GarciaMoreno B.; Shortle D. Contributions of the ionizable amino acids to the stability of staphylococcal nuclease. Biochemistry 1996, 35206443–6449. [DOI] [PubMed] [Google Scholar]
- Daggett V.; Fersht A. The present view of the mechanism of protein folding. Nature Rev. Mol. Cell Biol. 2003, 46497–502. [DOI] [PubMed] [Google Scholar]
- Simmerling C.; Strockbine B.; Roitberg A. E. All-atom structure prediction and folding simulations of a stable protein. J. Am. Chem. Soc. 2002, 1243811258–11259. [DOI] [PubMed] [Google Scholar]
- Schreiber C.; Buckle A. M.; Fersht A. R. Stability and Function: Two Constraints in the Evolution of Barstar and Other Proteins. Structure 1994, 210945–951. [DOI] [PubMed] [Google Scholar]
- Shoichet B. K.; Baase W. A.; Kuroki R.; Matthews B. W. A Relationship between Protein Stability and Protein Function. Proc. Natl. Acad. Sci. U. S. A. 1995, 922452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. J.; Minasov G.; Shoichet B. K. Evolution of an antibiotic resistance enzyme constrained by stability and activity trade-offs. J. Mol. Biol. 2002, 320185–95. [DOI] [PubMed] [Google Scholar]
- Eriksson A. E.; Baase W. A.; Wozniak J. A.; Matthews B. W. A Cavity-Containing Mutant of T4 Lysozyme Is Stabilized by Buried Benzene. Nature 1992, 3556358371–373. [DOI] [PubMed] [Google Scholar]
- Rekharsky M. V.; Mori T.; Yang C.; Ko Y. H.; Selvapalam N.; Kim H.; Sobransingh D.; Kaifer A. E.; Liu S. M.; Isaacs L.; Chen W.; Moghaddam S.; Gilson M. K.; Kim K. M.; Inoue Y. A synthetic host–guest system achieves avidin–biotin affinity by overcoming enthalpy–entropy compensation. Proc. Natl. Acad. Sci. U. S. A. 2007, 1045220737–20742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei B. Q. Q.; Baase W. A.; Weaver L. H.; Matthews B. W.; Shoichet B. K. A model binding site for testing scoring functions in molecular docking. J. Mol. Biol. 2002, 3222339–355. [DOI] [PubMed] [Google Scholar]
- Morton A.; Matthews B. W. Specificity of Ligand-Binding in a Buried Nonpolar Cavity of T4 Lysozyme—Linkage of Dynamics and Structural Plasticity. Biochemistry 1995, 34278576–8588. [DOI] [PubMed] [Google Scholar]
- Boyce S. E.; Mobley D. L.; Rocklin G. J.; Graves A. P.; Dill K. A.; Shoichet B. K. Predicting Ligand Binding Affinity with Alchemical Free Energy Methods in a Polar Model Binding Site. J. Mol. Biol. 2009, 3944747–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty D. A. Physical organic chemistry on the brain. J. Org. Chem. 2008, 73103667–3673. [DOI] [PubMed] [Google Scholar]
- Isom D. G.; Cannon B. R.; Castaneda C. A.; Robinson A.; Bertrand G. M. E. High tolerance for ionizable residues in the hydrophobic interior of proteins. Proc. Natl. Acad. Sci. U. S. A. 2008, 1054617784–17788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X. J.; Baase W. A.; Shoichet B. K.; Wilson K. P.; Matthews B. W. Enhancement of protein stability by the combination of point mutations in T4 lysozyme is additive. Protein Eng. 1995, 8101017–1022. [DOI] [PubMed] [Google Scholar]
- Prabhu N.; Sharp K. Protein–solvent interactions. Chem. Rev. 2006, 10651616–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merski M.; Shoichet B. K. Engineering a model protein cavity to catalyze the Kemp elimination. Proc. Natl. Acad. Sci. U. S. A. 2012, 1094016179–16183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin J. J.; Sterling T.; Mysinger M. M.; Bolstad E. S.; Coleman R. G. ZINC: A Free Tool to Discover Chemistry for Biology. J. Chem. Inf. Model. 2012, 5271757–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becktel W. J.; Schellman J. A. Protein Stability Curves. Biopolymers 1987, 26111859–1877. [DOI] [PubMed] [Google Scholar]
- Brenk R.; Vetter S. W.; Boyce S. E.; Goodin D. B.; Shoichet B. K. Probing molecular docking in a charged model binding site. J. Mol. Biol. 2006, 35751449–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari A. M.; Wei B. Q. Q.; Costantino L.; Shoichet B. K. Soft docking and multiple receptor conformations in virtual screening. J. Med. Chem. 2004, 47215076–5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hann M. M.; Leach A. R.; Harper G. Molecular complexity and its impact on the probability of finding leads for drug discovery. J. Chem. Inf. Comput. Sci. 2001, 413856–864. [DOI] [PubMed] [Google Scholar]
- Gaulton A.; Bellis L. J.; Bento A. P.; Chambers J.; Davies M.; Hersey A.; Light Y.; McGlinchey S.; Michalovich D.; Al-Lazikani B.; Overington J. P. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40D1D1100–D1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb P.; Rosenbaum D. M.; Irwin J. J.; Fung J. J.; Kobilka B. K.; Shoichet B. K. Structure-based discovery of beta(2)-adrenergic receptor ligands. Proc. Natl. Acad. Sci. U. S. A. 2009, 106166843–6848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen F. H. The Cambridge Structural Database: a quarter of a million crystal structures and rising. Acta Crystallogr., Sect. B:Struct. Sci. 2002, 58, 380–388. [DOI] [PubMed] [Google Scholar]
- Martin E. J.; Blaney J. M.; Siani M. A.; Spellmeyer D. C.; Wong A. K.; Moos W. H. Measuring diversity: experimental design of combinatorial libraries for drug discovery. J. Med. Chem. 1995, 3891431–1436. [DOI] [PubMed] [Google Scholar]
- Carter P. J.; Winter G.; Wilkinson A. J.; Fersht A. R. The Use of Double Mutants to Detect Structural Changes in the Active Site of the Tyrosyl-Transfer RNA-Synthetase (Bacillus Stearothermophilus). Cell 1984, 383835–840. [DOI] [PubMed] [Google Scholar]
- Otzen D. E.; Fersht A. R. Analysis of protein–protein interactions by mutagenesis: direct versus indirect effects. Protein Eng. 1999, 12141–45. [DOI] [PubMed] [Google Scholar]
- Deechongkit S.; Dawson P. E.; Kelly J. W. Toward assessing the position-dependent contributions of backbone hydrogen bonding to beta-sheet folding thermodynamics employing amide-to-ester perturbations. J. Am. Chem. Soc. 2004, 1265116762–16771. [DOI] [PubMed] [Google Scholar]
- Perola E.; Charifson P. S. Conformational analysis of drug-like molecules bound to proteins: an extensive study of ligand reorganization upon binding. J. Med. Chem. 2004, 47102499–2510. [DOI] [PubMed] [Google Scholar]
- Zhang L. Y.; Gallicchio E.; Friesner R. A.; Levy R. M. Solvent models for protein–ligand binding: comparison of implicit solvent Poisson and surface generalized Born models with explicit solvent simulations. J. Comput. Chem. 2001, 226591–607. [Google Scholar]
- Mukherjee S.; Balius T. E.; Rizzo R. C. Docking Validation Resources: Protein Family and Ligand Flexibility Experiments. J. Chem. Inf. Model. 2010, 50111986–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biela A.; Betz M.; Heine A.; Klebe G. Water makes the difference: rearrangement of water solvation layer triggers non-additivity of functional group contributions in protein–ligand binding. ChemMedChem 2012, 781423–1434. [DOI] [PubMed] [Google Scholar]
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; Shaw D. E.; Francis P.; Shenkin P. S. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 4771739–1749. [DOI] [PubMed] [Google Scholar]
- Liu L. J.; Baase W. A.; Michael M. M.; Matthews B. W. Use of Stabilizing Mutations To Engineer a Charged Group within a Ligand-Binding Hydrophobic Cavity in T4 Lysozyme. Biochemistry 2009, 48378842–8851. [DOI] [PubMed] [Google Scholar]
- Cashin A. L.; Torrice M. M.; McMenimen K. A.; Lester H. A.; Dougherty D. A. Chemical-scale studies on the role of a conserved aspartate in preorganizing the agonist binding site of the nicotinic acetylcholine receptor. Biochemistry 2007, 463630–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biela A.; Sielaff F.; Terwesten F.; Heine A.; Steinmetzer T.; Klebe G. Ligand binding stepwise disrupts water network in thrombin: enthalpic and entropic changes reveal classical hydrophobic effect. J. Med. Chem. 2012, 55136094–6110. [DOI] [PubMed] [Google Scholar]
- Brandt T.; Holzmann N.; Muley L.; Khayat M.; Wegscheid-Gerlach C.; Baum B.; Heine A.; Hangauer D.; Klebe G. Congeneric but still distinct: how closely related trypsin ligands exhibit different thermodynamic and structural properties. J. Mol. Biol. 2011, 40551170–1187. [DOI] [PubMed] [Google Scholar]
- Sharp K., Potential functions for virtual screening and ligand binding calculations: Some theoretical considerations. In Virtual Screening in Drug Discovery; Alvarez J., Shoichet B., Eds.; CRC Press: Boca Raton, FL, 2005. [Google Scholar]
- Gallivan J. P.; Dougherty D. A. A computational study of cation–pi interactions vs salt bridges in aqueous media: implications for protein engineering. J. Am. Chem. Soc. 2000, 1225870–874. [Google Scholar]
- Kirchhhoff W.EXAM; NIST: Gaithersburg, MD, 1993. [Google Scholar]
- Ellis K. J.; Morrison J. F. Buffers of Constant Ionic-Strength for Studying pH-Dependent Processes. Methods Enzymol. 1982, 87, 405–426. [DOI] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mccoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M. D.; Isupov M. N.; Murshudov G. N. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2001, 57, 122–133. [DOI] [PubMed] [Google Scholar]
- Schuttelkopf A. W.; van Aalten D. M. F. PRODRG: a tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 1355–1363. [DOI] [PubMed] [Google Scholar]
- Chen V. B.; Arendall W. B.; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano W. L.The PyMOL Molecular Graphics System, version 1.0; Schrödinger, LLC: San Carlos, CA, 2007. [Google Scholar]
- Willett P.; Barnard J. M.; Downs G. M. Chemical similarity searching. J. Chem. Inf. Comput. Sci. 1998, 386983–996. [Google Scholar]
- Rogers D.; Hahn M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 505742–754. [DOI] [PubMed] [Google Scholar]
- Fitzgerald M. M.; Musah R. A.; McRee D. E.; Goodin D. B. A ligand-gated, hinged loop rearrangement opens a channel to a buried artificial protein cavity. Nature Struct. Biol. 1996, 37626–631. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.