

Abstract
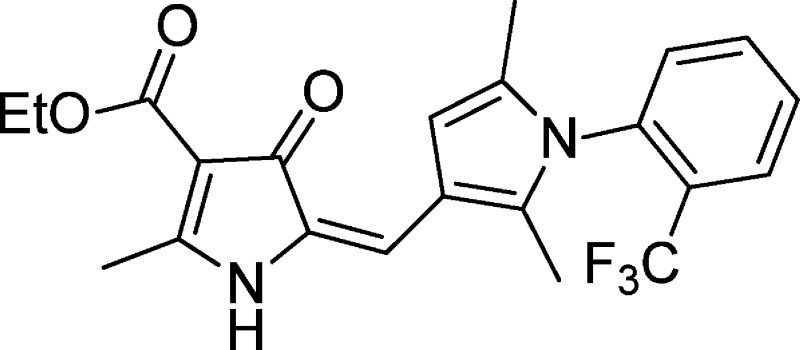
In the pursuit of new antimalarial leads, a phenotypic screening of various commercially sourced compound libraries was undertaken by the World Health Organisation Programme for Research and Training in Tropical Diseases (WHO-TDR). We report here the detailed characterization of one of the hits from this process, TDR32750 (8a), which showed potent activity against Plasmodium falciparum K1 (EC50 ∼ 9 nM), good selectivity (>2000-fold) compared to a mammalian cell line (L6), and significant activity against a rodent model of malaria when administered intraperitoneally. Structure–activity relationship studies have indicated ways in which the molecule could be optimized. This compound represents an exciting start point for a drug discovery program for the development of a novel antimalarial.
Introduction
In recent years, the emergence of drug-resistant pathogens has led to treatment failures for many infectious diseases, such as malaria. Resistance to current antimalarials is a major problem, e.g., chloroquine, sulfadoxine-pyrimethamine, and in some areas, mefloquine.1 To reduce the spread of resistance, the World Health Organisation (WHO) recommends treatment for malaria to be artemisinin combination therapy; although worryingly there are now reports of increased parasite clearance times with the artemisinins2−7 which may herald resistance. Thus, there is an urgent need for novel classes of compounds that are effective against these re-emerging infections. With the elimination of malaria now considered as a goal, it would be useful for scientists and policy makers to have an expanded arsenal of antimalarials that could help make this goal a reality.8,9
As part of a program for the discovery of new starting points for drug discovery programmes, the World Health Organisation Programme for Research and Training in Tropical Diseases (WHO-TDR) phenotypically screened various compound libraries. One of these was a 5000 strong structurally diverse compound collection from ChemDiv, which was screened against Plasmodium falciparum, the causative organism for the most pathogenic form of malaria. This led to the identification of pyrrolone (8a) shown in Figure 1. This compound had potent activity against P. falciparum K1, with an EC50 ∼9 nM, and was efficacious in a P. berghei mouse model when given intraperitoneally (ip), but was relatively inactive orally.
Figure 1.

Initial SAR of lead 8a.
The screening of several hundred commercially available compounds related to 8a containing the pyrrolone motif gave some indication of structure–activity relationships (SAR) (Figure 1). Here we report the further evaluation of 8a as a novel antimalarial lead, with further systematic SAR studies, in parallel with studies to assess drug metabolism and pharmacokinetics (DMPK), with the initial aim of achieving oral efficacy in the P. berghei mouse model, to establish the potential for further development of the pyrrolones as antimalarials.
Figure 2.

Strategy for elaboration of SAR around 8a.
Results and Discussion
Chemistry
An efficient synthesis of 8a and congeners was developed (Scheme 1), through acylation of ethyl-3-aminocrotonate (1) with chloroacetyl chloride10 and subsequent cyclization to the pyrrolone 3,10 which was not stored, but used immediately, due to its relatively poor stability. The condensation of 3 with 3-formyl pyrroles (7a–x) in the presence of potassium hydrogen sulfate11,12 generated the (E)-isomer predominantly.a This three-step sequence could be carried out in yields of up to ∼60%, and only required chromatography at step 1. The 3-formylpyrroles (7a–x) were obtained through condensation of the appropriate aniline with 2,5-hexandione (4) (Paal–Knorr pyrrole synthesis) and subsequent Vilsmeier–Haack formylation.13,14 The use of silica-supported p-toluenesulfonic acid in the condensation of anilines with 4, in a solvent-free procedure using microwave heating, provided a considerable rate advantage over classical procedures (i.e., reaction complete within 15–20 min compared to over 12 h with external heating); even relatively non-nucleophilic amines were condensed smoothly in the absence of Lewis acid catalysts.
Scheme 1. General Synthetic Approach to Pyrrolones.

(a) Chloroacetyl chloride, pyridine, 0 °C, 30 min, 75%; (b) KOH, EtOH, 95%; (c) p-toluenesulfonic acid bound with silica gel, 15–20 min, microwave, 80–90%, or p-toluenesulfonic acid, toluene, 90 °C, 3 h Dean–Stark apparatus; (d) POCl3, DMF, 100 °C, 3 h, 80–95% ; (e) KHSO4, EtOH, 3 h, reflux, 80–95%; R and R1 as defined in Tables 1 and 3.
Ring A
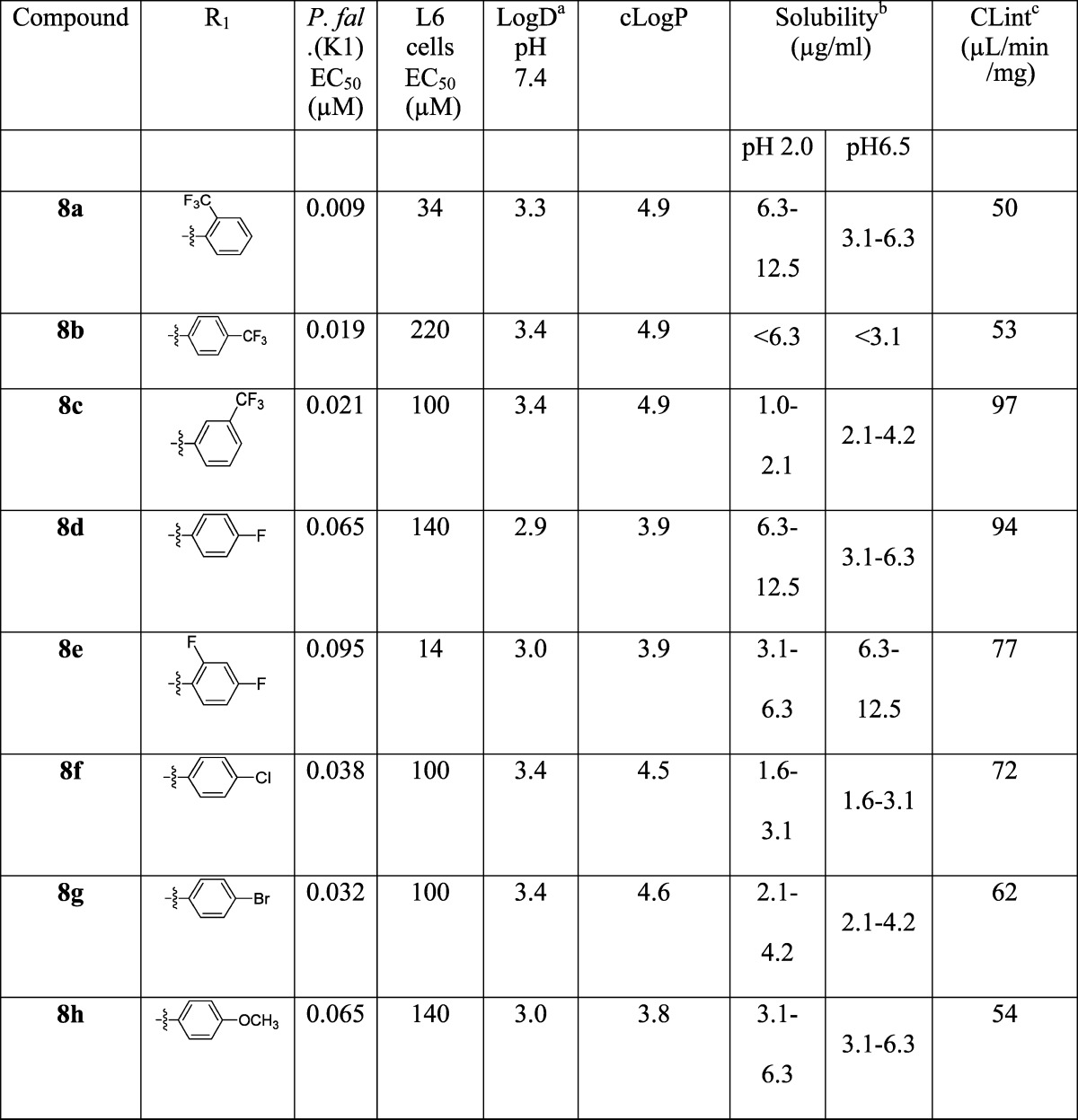
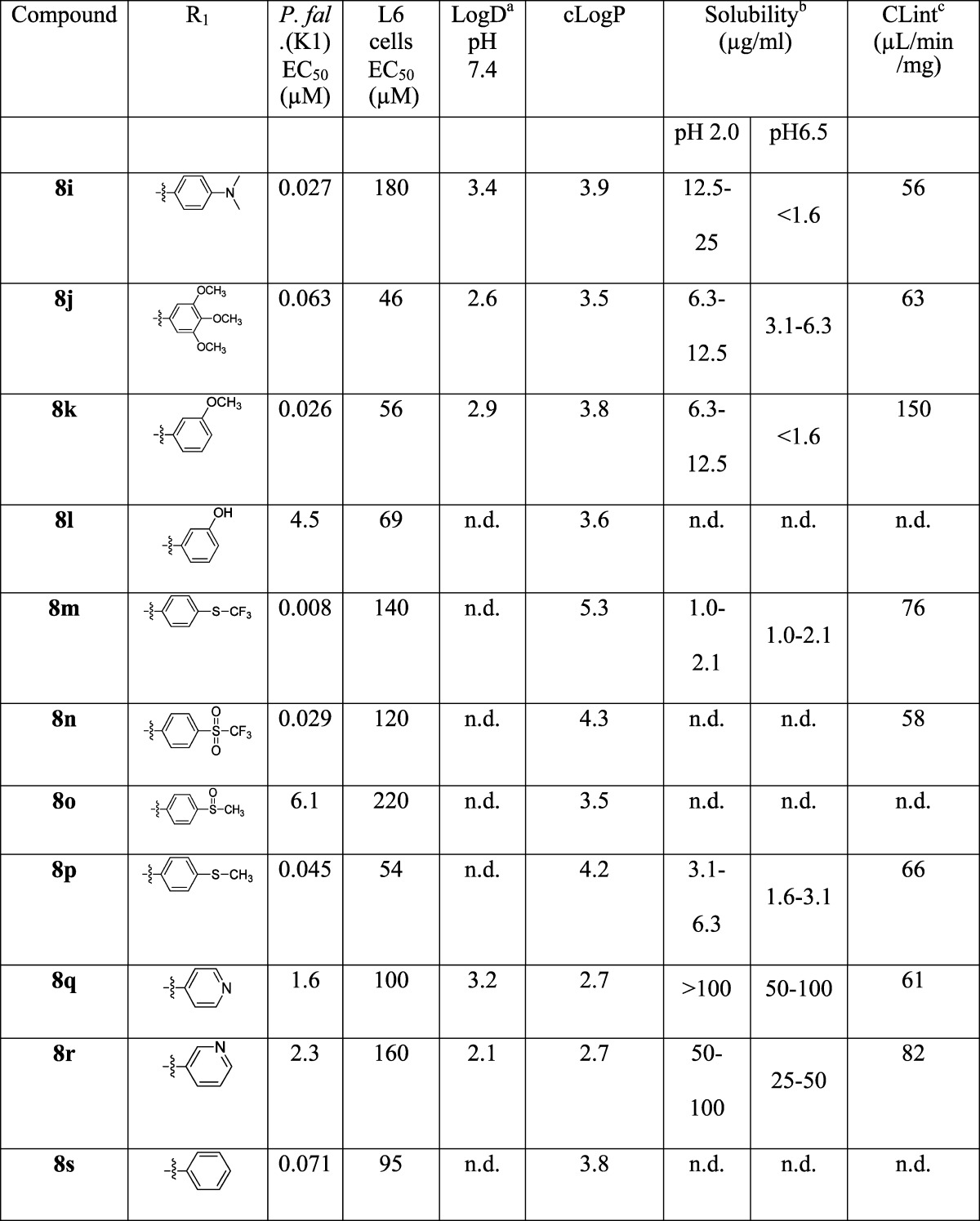
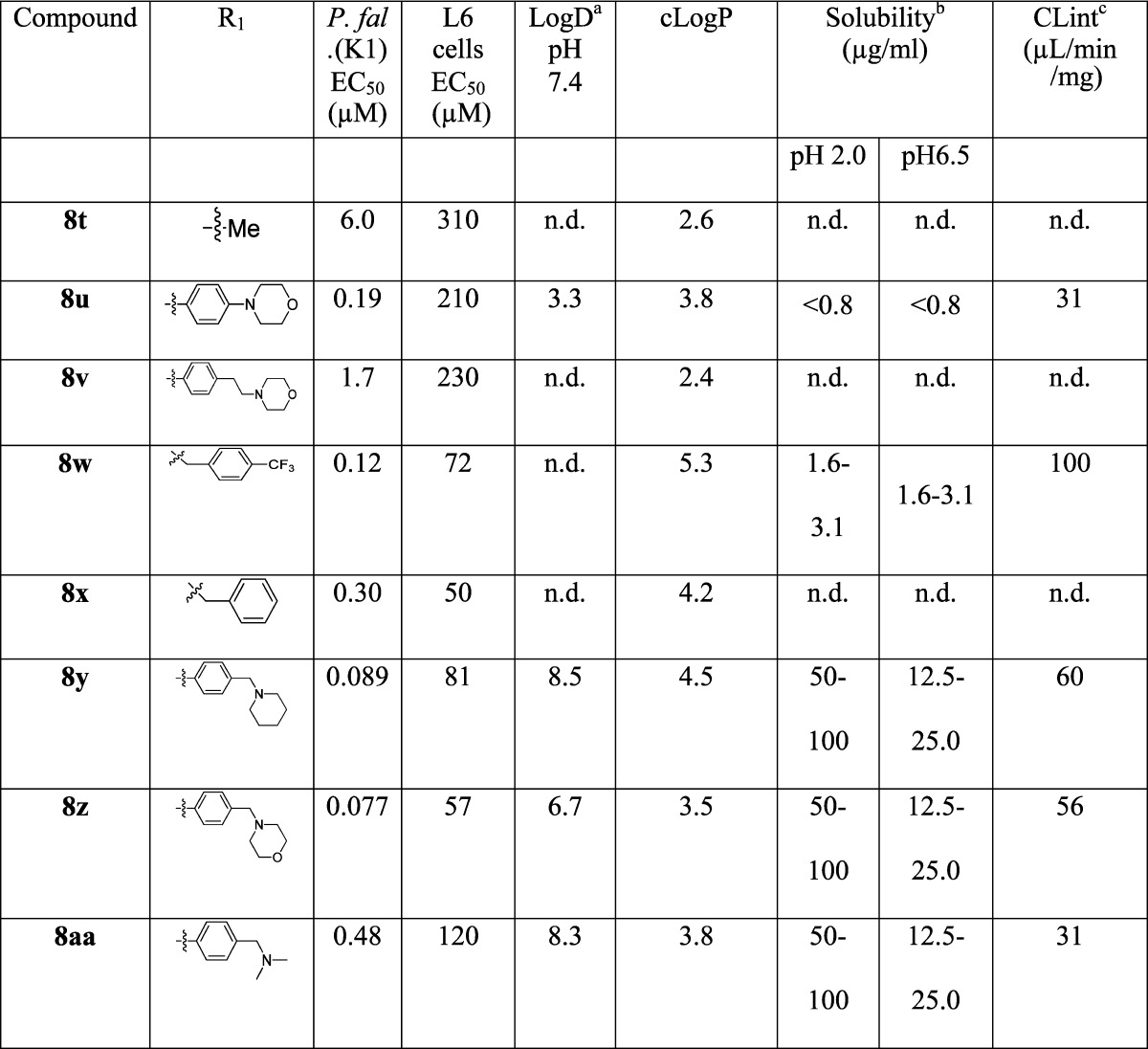
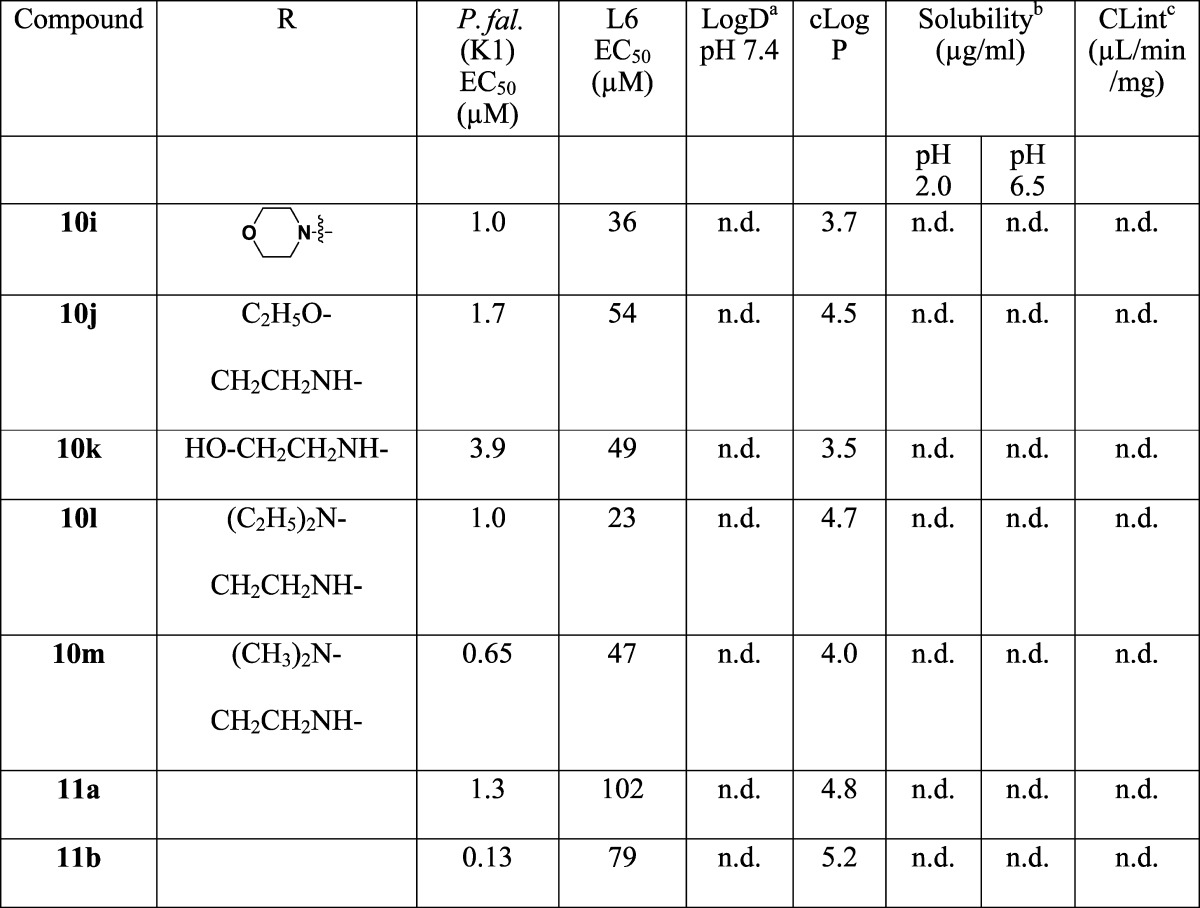
Using the approach outlined in Scheme 1, a number of analogues at the A ring (Table 1) were prepared by varying the aniline or amine 5a–x, with a particular aim of increasing the solubility. This provided a means of incorporating solubilizing groups either within the aromatic ring (e.g., pyridine analogues 8q and 8r), or as pendant amines (8y and 8aa), morpholines (8u), or sulfones (8n). The aryl ring was also replaced with a simple alkyl group (8t).
Table 1. LogD/LogP, Kinetic Solubility, in Vitro Intrinsic Clearance and Activity against P. falciparum for Ring A Variantsd.

Value measured using a chromatographic gLogD technique.
Estimated using nephelometry.
In vitro intrinsic clearance determined in mouse hepatic microsomes.
Chloroquine, EC50P. falciparum K1 0.095–0.172 μM; podophyllotoxin EC50 L6 cells 0.009–0.022 μM. The EC50 values are the means of two independent assays and varies less than ±50%. n.d = not determined.
Ring B
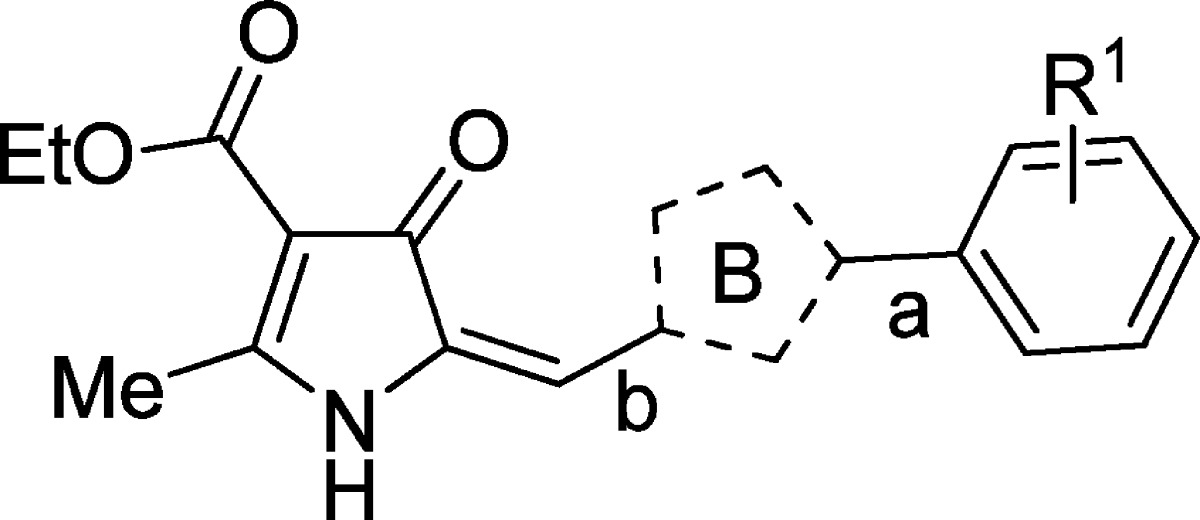
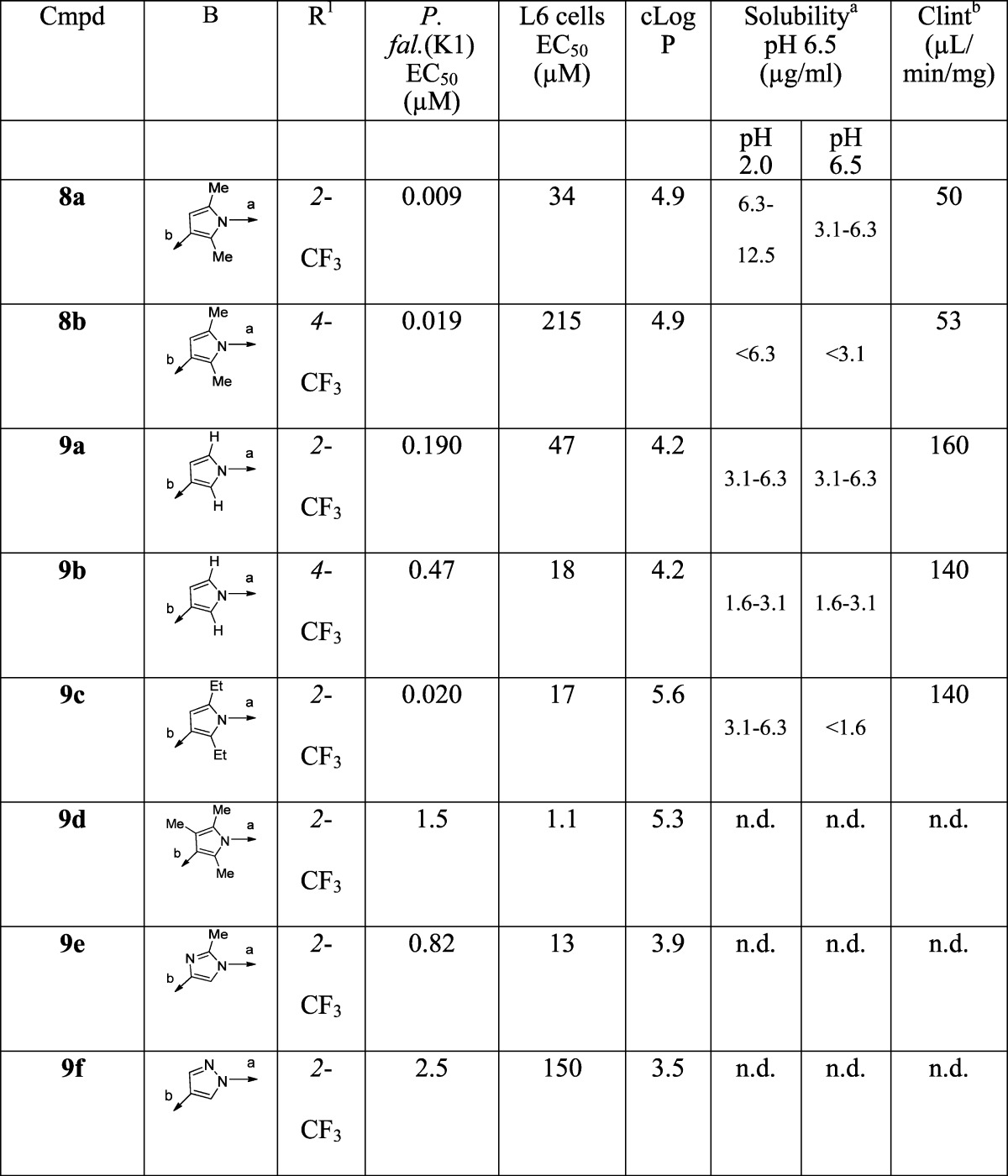
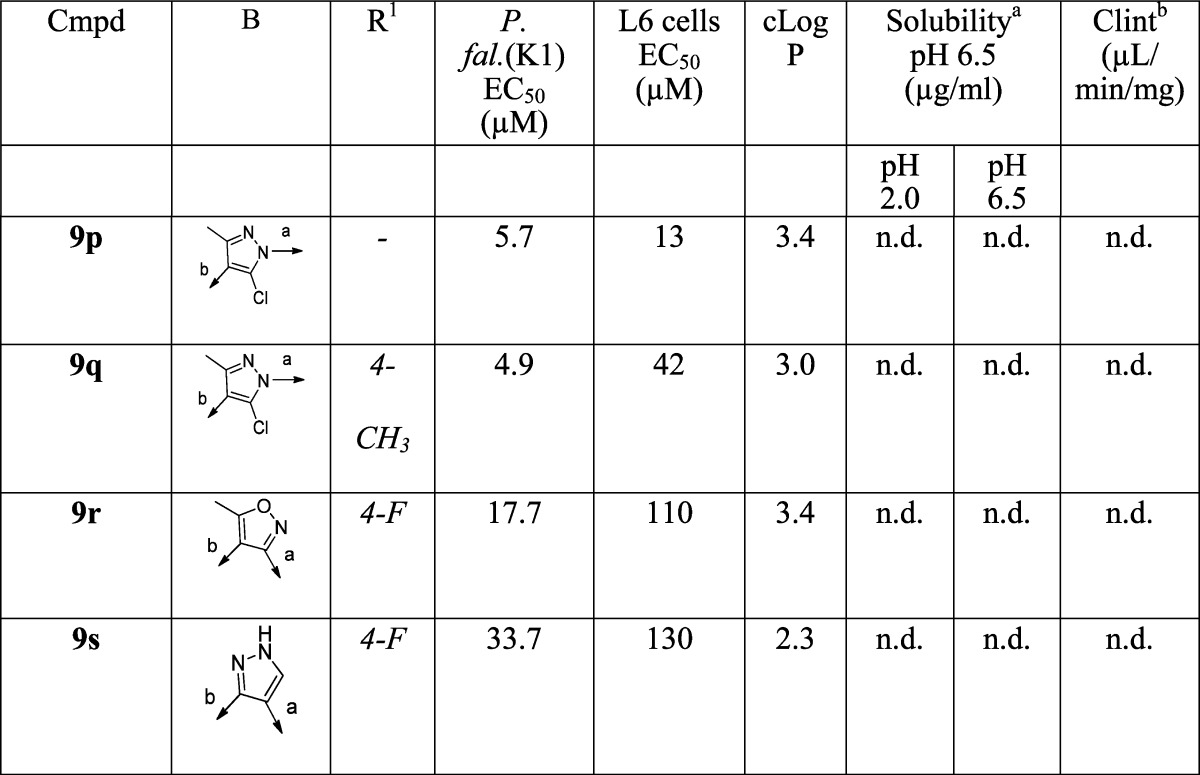
The methyl groups on the pyrrole were predicted computationally to be likely sites of metabolic vulnerability, and it was therefore of interest to investigate the effect of modifications of the pyrrole, including removing the methyl groups (9a–b) and replacing them with ethyl groups (9c). The 3-formyl pyrrole intermediates were prepared according to Scheme 2. Variants in which the B-ring pyrrole was replaced with other 5-ring heterocycles (imidazole, pyrazole, triazole, or thiazole) were synthesized as in Scheme 3. For the furan (9n–o) and chloro pyrazole (9p) derivatives, the requisite aldehydes were commercially available.
Scheme 2. Synthesis of Pyrrole B-ring Variants.

(a) Acetic acid, 90 °C, 3 h; (b) KHSO4, EtOH, reflux, 3 h; (c) 2-trifluoromethylaniline, p-toluenesulfonic acid bound with silica gel, 90 °C, reflux, 3 h; (d) POCl3, DMF, 100 °C, 3 h; (e) MeLi (3 M) in DME, THF, 0 °C – rt, 23 h, 75 °C for 17 h.
Scheme 3. Synthesis of Formyl Pyrazole, Thiazole, Imidazole, and Triazole Intermediates.

(a) aq EtOH, HCl 90 °C, 3 h; (b) POCl3, DMF, 100 °C, 3 h; (c) acetylacetone, 90 °C, 3 h; (d) ethyl-2-chloroacetoacetate, EtOH, reflux; (e) LiAlH4, THF, 0 °C; (f) PCC, CH2Cl2, rt; (g) PCl5 in CHCl3, reflux, 3 h; (h) tetrachloroethylene, 5-aminopyrimidine, few drops of POCl3; (i) t-BuONO/TMSN3, propargyl alcohol, MeCN; (j) Dess–Martin Reagent, CH2Cl2.
A number of B-ring variants in which an aryl ring replaces the pyrrole (9j–m) were prepared using a Suzuki coupling to make the requisite aldehyde intermediates (Scheme 4, Table 2).
Scheme 4. Synthesis of Aldehyde Intermediates for B-Ring Aryl Derivatives: (a) Tetrabutyl Ammonium Bromide (TBAB), K2CO3, Pd(OAc)2, Dioxane/Water (1:1).
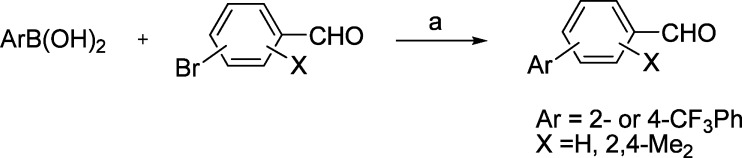
Table 2. cLogP, Kinetic Solubility, in Vitro Intrinsic Clearance and in Vitro Activity against P. falciparum for Ring B Variants.

Estimated using nephelometry.
In vitro intrinsic clearance determined in mouse hepatic microsomes, n.d. = not determined.
Ring C
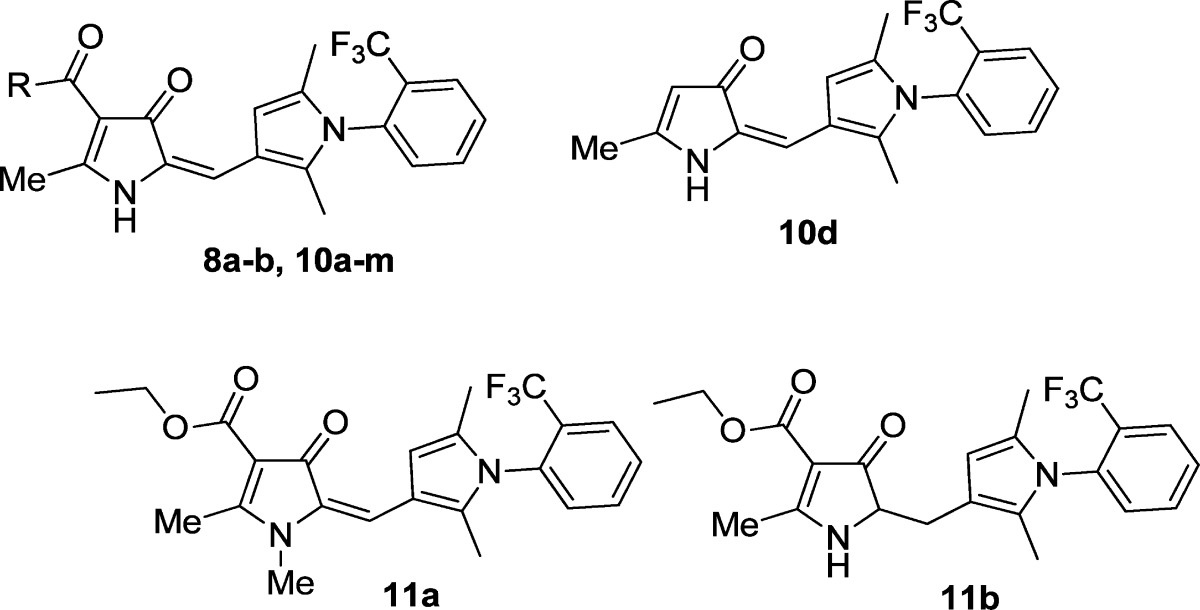
Concern that hydrolysis of the ethyl ester in 8a might adversely affect oral bioavailability and the elimination half-life led to an investigation of potentially more stable ester (Scheme 1) and amide derivatives, accessible from the carboxylic acid 10c (Scheme 5, Table 3). Base catalyzed hydrolysis of the ethyl ester 8a to generate 10c proved problematic due to decarboxylation to 10d under the conditions required. Decarboxylation could be reduced to a minimum (∼10%) by utilizing acid-catalyzed deprotection of the tert-butyl ester derivative 10a, with 10d readily removed by chromatography. The effect of methylating the pyrrolone NH was also investigated.
Scheme 5. Synthetic Approach to Amide Derivatives.

(a) HCl (6 M), 3 h, reflux, 80–95% (ratio 10c/10d ∼ 90:10); (b) N,N-carbonyldiimidazole, THF, reflux, excess amine in MeOH, 0° C, 18-24 h; (c) TBTU, DIPEA, secondary amine, DMF, 0 °C; 18–24 h.
Table 3. LogD/LogP, Kinetic Solubility, in Vitro Intrinsic Clearance and in Vitro Activity against P. falciparum for Ring C Variants.

Value measured using a chromatographic gLogD technique.
Estimated using nephelometry.
In vitro intrinsic clearance determined in mouse hepatic microsomes. n.d. = not measured.
In Vitro Biological Activity
Compounds were evaluated against P. falciparum (K1) strain and counter-screened for cytotoxicity against mammalian L6 cells (Tables 1–3). Compound 8a retained activity across a variety of drug-sensitive and -resistant strains (Table 4) and showed a high degree of selectivity for P. falciparum relative to L6.
Table 4. In Vitro Antiplasmodial and Cytotoxic Activities of 8a.
|
P.
falciparum EC50 (nM) |
cytotoxicity EC50 (nM) |
||||||
|---|---|---|---|---|---|---|---|
| K1a | D6b | W2c | TM90C2Bd | TM91C235e | L6 | ||
| 8a | 2–28 (n = 7) | 2.2–3.8 | 8.4 | 11–14 | 8.6–9.3 | 15000 | |
| chloroquine | 63–430 | 14 | 260 | 250 | 140 | ||
| mefloquine | 11 | 4.9 | 9.8 | 27 | |||
| artemesinin | 6.0 | ||||||
K1 = chloroquine and pyrimethamine resistant (STI).
D6 = chloroquine sensitive.
W2 = chloroquine resistant (WRAIR).
TM90C2B = atovaquone resistant (WRAIR).
TM91C235 = multidrug-resistant + atovaquone sensitive (WRAIR).
The activities of the compounds varied from 0.008 μM to 12 μM; the most active 8a–c, 8m and 9c had P. falciparum (K1) EC50 values of 0.008–0.021 μM, superior to chloroquine and the majority of the compounds showed good selectivity (>1000-fold) for P. falciparum compared to L6 mammalian cells. From the variations across rings A–C the following SAR was noted:
Ring A (Table 1)
(1) Replacing the phenyl ring with a methyl group (8t) led to a significant loss of activity (600-fold), suggesting that a hydrophobic group was required. Replacing the phenyl (8s) with a benzyl (8x) resulted in a small drop in activity (4-fold), indicating that the phenyl ring did not need to be attached directly to the pyrrole.
(2) In varying the position of the CF3 on the aryl ring there seemed to be relatively little difference in activity comparing the ortho (8a), meta (8c), or para (8b) derivatives. An ortho CF3 would be expected to reduce the conformational flexibility of ring A by hindering coplanarity of the pyrrole and phenyl rings, but this did not seem to have an effect on either activity or solubility (see below). Compounds 8a–c were only ∼5–10-fold more active than the unsubstituted derivative 8s, consistent with the idea that the lipophilicity contributed by ring A is the most significant variable in this part of the pyrrolones affecting in vitro activity.
(3) Further variation of substitution on the aromatic ring focused on the para position, as this is likely to be the most susceptible aryl position to cytochrome P450 (CYP)-mediated oxidation. In general, a wide variety of substituents were tolerated with relatively slight variation in potency, with the notable exception of the sulfoxide (8o), which was ∼100-fold less active than the corresponding sulphide (8p) and sulfone (8n). Similarly, the meta-substituted hydroxyl derivative (8l) was not tolerated, while the meta-substituted methoxy (8k) was. Solubilizing substituents were tolerated to a degree: although the pyridine derivatives (8q–r) and dimethylamine (8aa) derivatives lost significant activity, the sulphone (8n) retained activity, while the piperidine (8y) and the morpholine (8z) were only ∼5–10-fold less active than 8a.
Ring B (Table 2)
The methyl substituents on the pyrrole were identified as potential points of CYP-mediated metabolism.
1. Removal of the methyl substituents (9a, 9b) resulted in a significant (∼20–25-fold) loss in activity.
2. Replacing both methyls with ethyl (9c) did not significantly affect activity.
3. Replacement of the pyrrole with imidazole (9e), pyrazole (9f, 9g, 9p, 9q, and 9s), triazole (9h), thiazole (9i), aryl (9j–m), furan (9n–o) or isoxazole (9q) gave a significant loss of activity (∼20–1000-fold).
Ring C (Table 3)
1. Methylation of the NH (11a) resulted in a significant reduction of potency (>100-fold) relative to 8a, consequently no further N-substituted derivatives were investigated.
2. The potential for degradation of 8a through cleavage of the ester by esterases was of concern, although 8a was fairly resistant to chemical hydrolysis. Both the free acid 10c and the product from decarboxylation (10d) were significantly less active (∼50–150-fold) than 8a.
3. All modifications of the ester in 8a, including amides 10f–m, resulted in considerably reduced activity, although with the benzyl ester 10b the reduction in activity was relatively moderate (∼5-fold). The amides were inactive, which may be due to them adopting different conformations.
4. Reduction of the double bond linking the pyrrolone and pyrrole rings would be expected to improve solubility by reducing planarity. However the reduced derivative 11b proved significantly less active (>10-fold) than 8a; furthermore 11b was less stable chemically.
Thus there appears to be scope for modification of the aryl A-ring, with less room for maneuver around the 2,5-dimethyl pyrrole in ring B. As only fairly conservative modifications to ring C have been investigated there may be further potential in modifying the pyrrolone.
In Vivo Efficacy Studies in P. berghei Mouse Model
Compounds 8a and 8b showed good activity in the mouse model of P. berghei when dosed ip at 100 mg/kg once daily for 4 days, with >99% reduction in parasitaemia, comparable to chloroquine at 10 mg/kg; no dose-ranging experiments were undertaken. Chloroquine was dosed ip to give comparison to a known drug by the same route. Both 8a and 8b were relatively inactive when given orally (Table 5). Although full PK studies were not undertaken in mice, the oral exposure of 8a was evaluated in mice at a single dose of 50 mg/kg (formulated as a suspension in PEG400). This confirmed that relatively low plasma levels of 8a were achieved (<0.01 μM after ∼5 h), which is consistent with the lack of oral efficacy in the mouse P. berghei model.
Table 5. In Vivo Antimalarial Activity of 8a in Mice Infected with P. berghei.
| % reduction
parasitaemia |
||||||
|---|---|---|---|---|---|---|
| dose (mg/kg/day × 4) | route | day 4 | day 5 | day 6 | MSDa | |
| Experiment 1 | ||||||
| 8a | 100 | ip | 97.72 | 98.35 | 99.6 | 11 |
| 8a | 100 | po | 25.51 | 5.3 | 0 | 7.7 |
| chloroquine | 10 | ip | 99.9 | 11 | ||
| control | 5.7 | |||||
| Experiment 2 | ||||||
| 8a | 100 | ip | 99.93 | 10.7 | ||
| 8b | 100 | ip | 99.91 | 11.7 | ||
| 8b | 100 | po | 37.5 | 7 | ||
| chloroquine | 10 | ip | 99.97 | 20 | ||
| chloroquine | 100 | po | 99.91 | >30 | ||
| control | 7 | |||||
MSD = mean survival time (days).
Physicochemical Properties and in Vitro DMPK
The molecules generally showed reasonable physicochemical properties (MW in the region of 350–450, 1 HBD, 5–7 HBA, PSA in the range 60–100 Å2), compatible with good membrane permeability, but the aqueous solubility of most compounds was generally very low (Tables 1–3). The low solubility is likely a consequence of the conjugated planar nature of the molecules, and in some cases the lipophilicity. For some compounds, particularly (8i, 8k, 8r) and (9c), there was a reduction in the solubility between pH 2.0 and pH 6.5. In the case of (8i) and (8r), this is probably due to protonation at pH 2.0, but not at pH 6.5; although the reason for the high pH dependence on solubility is not clear for (8k) and (9c). Similarly compounds with a pendant amine also showed greater solubility (8y, 8z, 8aa) as did those with a pyridine replacement for the phenyl ring (8q, 8r).
Attempts to improve the solubility were made as follows:
-
•
Addition of solubilizing groups such as morpholine (8u, 8z) and piperidine (8y) on ring A.
-
•
Conversion of ester to amide linkages with various solublizing groups such as methyl amine, piperidine, morpholine, ethanolamine, dimethylethane-1,2-diamine, diethylethane-1,2-diamine, 2-ethoxyethanamine.
Compound 8a is predicted to be uncharged at physiological pH as are those compounds where amines are appended directly to the phenyl ring C (8i, 8u) or the pyridine analogues (8q, 8r). The more basic derivatives, where the amine is appended via a methylene linker (8y, 8z, 8aa), are predicted to have varying degrees of ionization, which correlates with their significantly higher aqueous solubility.
Compounds showed moderate to high rates of degradation when incubated with mouse liver microsomes (see Supporting Information for methodology). The main exceptions to this were (8u, 8aa) and (10d), which appeared to be slightly more stable. Putative metabolites having molecular weight consistent with the products of mono-oxygenation (P+16 for 8a, 8d, 8e, 8h, 8m, 8p, 8q, 9b, 9c, 10a, 10f), bis-oxygenation (P+32 for 8q, 10a), O-demethylation (P-14 for 8i), and/or morpholine ring cleavage (P-12 for 8u), were detected for a number of analogues (see Supporting Information). Compound 10c showed an increased rate of degradation in microsomes containing the dual cofactors NADPH and uridine-5′-diphospho-glucuronic acid (UDPGA) (the cofactors for CYP450-mediated metabolism and glucuronidation, respectively) relative to NADPH alone, suggesting that this compound is susceptible to primary glucuronidation in the microsomal test system. A putative glucuronide metabolite (P+176) was also detected for 10c (see Supporting Information).
It was predicted computationally that the methyl groups on the pyrrole (ring B) were likely to be metabolically unstable. Hence, these were removed (9a, 9b) or replaced with an ethyl group (9c), but these compounds were found to have even lower metabolic stability. All attempts to replace the pyrrole with other heterocycles resulted in much reduced antimalarial activity.
In addition to investigating their stability with mouse liver microsomes, key compounds (8a and 8b) were investigated in rat and human liver microsomes, in order to obtain an idea of species variability in metabolic degradation (Table 6). These results suggested only marginal differences in microsomal stability between species.
Table 6. Metabolic Stability Parameters for 8a and 8b Based on NADPH-Dependent Degradation Profiles in Human/Rat/Mouse Liver Microsomes.
| compound | species | T1/2a (min) | CLintb (μL/min/mg) | EH | metabolites detected |
|---|---|---|---|---|---|
| 8a | human | 58 | 30 | 0.62 | none |
| rat | 44 | 40 | 0.56 | P+16 | |
| mouse | 35 | 50 | 0.68 | none | |
| 8b | human | 35 | 49 | 0.73 | none |
| rat | 56 | 31 | 0.50 | none | |
| mouse | 32.9 | 53 | 0.70 | none |
Half-life.
In vitro intrinsic clearance.
Protein binding was investigated for selected compounds (Supporting Information). With the exception of the pyridine containing compounds (8q) (80% bound) and (8r) (79% bound), compounds were found to have moderate to high plasma protein binding (93.7–98.8% bound).
Compound 8a showed insignificant reactivity on incubation with reduced glutathione ethyl ester, indicating that it does not act as a Michael acceptor (which could adversely affect stability in vivo and might give rise to toxicity), and no adducts resulting from conjugation with glutathione were detected in the urine of a rat dosed intravenously with 8a.
The ester in both 8a and 8b is a potential point of metabolism through cleavage by esterases (Figure 3). In rat blood and plasma, ∼25% of 8a was hydrolyzed to the acid 10c over 4 h at 37 °C. Compound 10c was accompanied by traces of the decarboxylated product 10d, consistent with the observed chemical instability of 10c (although the formation of 10d during the extraction and/or analytical procedure cannot be ruled out).
Figure 3.

Degradation of 8ain vivo through ester hydrolysis.
In Vivo DMPK Studies
In order to better understand the efficacy data, PK studies for compounds 8a and 8b were conducted in rats, with iv, ip, and po administration (Table 7; Figure 4; see Supporting Information for Methodology). No sign of toxicity was observed when 8a or 8b were administered orally or ip at doses up to ∼20 mg/kg. There was a slight degree of hemolysis when the compounds were dosed iv, most likely arising from the vehicle required for formulation. Both compounds had terminal half-lives of at least 8 h, likely due to the high volumes of distribution. Compound 8a exhibited high plasma clearance (73 mL/min/kg), which in vitro studies suggest is due to a combination of moderate hepatic metabolic clearance and blood-mediated degradation, but not renal elimination given that there was minimal recovery of 8a excreted unchanged in urine. While plasma/blood stability studies were not conducted with 8b, similar degradation would be expected given the structural similarity to 8a.
Table 7. Pharmacokinetic Properties of 8a and 8b in Sprague Dawley Ratsa.
|
8a |
8b |
||||
|---|---|---|---|---|---|
| oralb | ivc | ipd | oralb | ivc | |
| dose (mg/kg) | 20 | 4.5 | 19 | 19 | 1.0 |
| apparent t1/2 (h) | 7.9 | 12 | 9.1 | c.n.c. | 87 |
| % dose in urine | 0.08 | 0.05 | 0.01 | n.d. | n.d. |
| Cmax (μM) | 0.11 | 3.3 | 0.19 | ||
| Tmax (min) | 200 | 15 | 100 | ||
| oral bioavailability F% | 6–7 | 45.7 | 4–5 | ||
| Vdss (L/kg) | 13 | 19 | |||
| plasma clearance (mL/min/kg) | 73 | 22 | |||
n.d. = not determined. c.n.c = could not calculate.
Oral suspension formulated with aqueous HPMC.
iv solution formulated in aqueous vehicle with 40% (v/v) propylene glycol.
ip suspension formulated in aqueous vehicle with 10% DMSO.
Figure 4.

Plasma concentrations of 8a (top) and 8b (bottom) following iv (filled circles), oral (open triangles), and ip (open squares, 8a only) administration to male Sprague–Dawley rats at nominal iv doses of 5 mg/kg (8a) and 1 mg/kg (8b), and oral and ip doses of 20 mg/kg. Each profile represents the average of n = 2 rats.
Both 8a and 8b had low oral bioavailability (7 and 5%, respectively) which is likely due to the combined effect of high first pass clearance and poor absorption resulting from the low aqueous solubility. There is also the potential for hydrolysis of the ester linkage in the gut, and during passage through the enterocytes and the liver. Despite the low oral bioavailability, the Cmax of both compounds when dosed at 20 mg/kg po was about 10-fold higher than the EC50 against parasites in vitro.
Conclusions
From phenotypic screening a series of pyrrolone derivatives have been identified with good in vitro activity against P. falciparum combined with good selectivity relative to the L6 mammalian cell line. A lack of cross resistance with standard antimalarials suggests they may have a novel mode of action, although this has not been investigated. While some of the pyrrolones have shown good in vivo activity in a P. berghei mouse model when administered by the intraperitoneal route, oral activity has so far proved elusive, likely resulting from a combination of poor absorption due to the low aqueous solubility and rapid first-pass metabolism through cytochrome P450-mediated oxidation and/or esterase cleavage. The lack of oral activity is a key hurdle that needs to be overcome in establishing the potential for further development of this series. SAR studies in conjunction with in vitro microsomal stability studies have indicated that there is scope for modification at several points on the lead compounds.
Experimental Section
Parasitology and DMPK methods are described in the Supporting Information.
Profiling Software
StarDrop (www.optibrium.com) was used to predict the sites of metabolism of the compounds.
Chemistry
All commercially available reagents, solvents, and starting materials were purchased from Aldrich Chemical Co. (UK). Where necessary a Biotage FLASH 25+ column chromatography system was used to purify mixtures; reagent-grade solvents used for chromatography were purchased from Fisher Scientific (UK) and flash column chromatography silica cartridges were obtained from Biotage (UK). Analytical thin-layer chromatography (TLC) was performed on precoated TLC plates (layer 0.20 mm silica gel 60 with fluorescent indicator UV254, from Merck). Developed plates were air-dried and analyzed under a UV lamp (UV 254/365 nm). Microwave irradiation was conducted using a BIOTAGE INITIATOR unit. The machine consists of a continuous focused microwave power delivery system with operator-selectable power output (0–400 W at 2.45 GHz). All 1H and 13C NMR spectra were recorded on a Bruker ARX-500 spectrometer (500 and 125 MHz for 1H and 13C NMR, respectively). Chemical shifts (δ) are reported in ppm relative to the residual solvent peak or internal standard (tetramethylsilane), and coupling constants (J) are reported in hertz (Hz). Data are reported as follows: chemical shift, multiplicity (br = broad, s = singlet, d = doublet, t = triplet, m = multiplet), integration. LC–MS analyses were performed with either an Agilent HPLC 1100 series connected to a Bruker Daltonics MicrOTOF or an Agilent Technologies 1200 series HPLC connected to an Agilent Technologies 6130 quadrupole spectrometer, where both instruments were connected to an Agilent diode array detector. LC–MS chromatographic separations were conducted with a Waters X bridge C18 column, 50 mm × 2.1 mm, 3.5 μm particle size; mobile phase, water/acetonitrile +0.1% HCOOH, or water/acetonitrile +0.1% NH3; linear gradient from 80:20 to 5:95 over 3.5 min and then held for 1.5 min; flow rate of 0.5 mL min–1. All assay compounds had a measured purity of ≥95% (by TLC and UV) as determined using this analytical LC–MS system. High resolution electrospray measurements were performed on a Bruker Daltonics MicrOTOF mass spectrometer.
Ethyl (E)-3-Amino-2-(2-chloroacetyl)but-2-enoate (2)
A solution of ethyl-3-aminocrotonate (2.0 g, 0.015 mol, 1.0 equiv) and pyridine (1.2 g, 0.015 mol, 1.0 equiv) in diethylether (10 mL) was cooled to 0 °C, and a solution of chloroacetylchloride (4.1 g, 0.037 mol, 2.4 equiv) in diethylether (5 mL) was added dropwise over 30 min, maintaining the temperature at 0 °C. After the mixture was stirred for a further 3 h at 0 °C the solvent was removed in vacuo. The resultant solid was washed with cold water to yield 2 as a cream yellow powder (2.7 g, 87% yield), mp 131–132 °C. 1H NMR (500 MHz; DMSO-d6): δ 5.92 (br s, 2H, NH2), 4.57 (s, 2H, −CH2Cl), 4.27 (q, 2H,-OCH2, J = 7.2 Hz), 2.36 (s, 3H, CH3), 1.36 (t, 3H, OCH2CH3, J = 7.2 Hz); 13C NMR (125 MHz, DMSO-d6): δ 190.6, 169.9, 168.3, 100.8, 60.5, 49.6, 24.6, 14.3; MS (ESI) m/z 206.1 [M + H]+ 100%.
Ethyl 5-Methyl-3-oxo-1,2-dihydropyrrole-4-carboxylate (3)
Ethyl (E)-3-amino-2-(2-chloroacetyl)but-2-enoate (2) (2.0 g, 0.0097 mol) was dissolved in absolute ethanol (5 mL) and cooled to 0 °C. Potassium hydroxide (1.09 g, 0.019 mol) was added, and the mixture was stirred for 3 h at 0 °C and then acidified to pH 2.0 using 2 M HCl to afford a yellow precipitate, which was washed with cold water to yield 3 as a yellow solid (1.6 g, 99% yield), mp 215 °C. 1H NMR (500 MHz; DMSO-d6): δ 10.7 (br s, 1H, -OH), 9.4 (br s, 1H,-NH), 7.64 (s, 1H, -NH), 6.05 (d, 1H, J = 2.4 Hz), 4.27 (q, 2H,-OCH2, J = 7.1 Hz), 4.08 (m, 2H, -OCH2), 3.80 (d, 2H), 2.4 (t, 3H, J = 1.6 Hz), 2.3(s, 3H), 1.8 (t, 3H, J = 7.1 Hz), 1.2 (t, 3H, J = 7.0 Hz, OCH2CH3); 13C NMR (125 MHz, DMSO-d6): δ 189.2, 171.5, 159.9, 99.3, 86.1, 60.4, 28.02, 14.2; MS (ESI) m/z 170.14 [M + H]+ 100%.
General Procedure for the Microwave-Accelerated Synthesis of 2,5-Dimethyl-1-aryl-1H-pyrroles (6a-6aa)
A mixture of 2,5-hexandione (4) (1 mmol), the appropriate aniline (5a-5aa) (1.2 mmol) and p-toluenesulfonic acid bound with silica gel (0.4 equiv) was stirred in an oven-dried pressure vial fitted with a magnetic stir bar. The vial was then placed in a microwave oven and heated twice (180 °C, 5 min) under microwave irradiation (0–400 W at 2.45 GHz). After being stirred for 15 min at room temperature, the mixture was filtered and the residual silica was washed with DCM (10 mL). The solvent was removed under reduced pressure, affording the pyrrole 6a-6aa (purity >95%, 80–90% yield), which was used without further purification.
General Procedure for the Synthesis of 2,5-Dimethyl-1-aryl-3-formylpyrroles (7a-7aa)
Phosphorus oxychloride (6 mmol) was added dropwise to ice-cooled DMF (12 mL) stirred under a N2 atmosphere. The mixture was kept at room temperature for 15 min, and then a solution of the requisite pyrrole 6a-6aa (1 mmol) in DMF (5 mL) was added and the mixture was heated at 100 °C for 3 h. After cooling, 30% NaOH was added dropwise to adjust to pH 10. The resultant precipitate was filtered and washed with water, affording the 2,5-dimethyl-1-aryl-3-formylpyrroles 7a-7aa (80–95% yield), which were used without further purification.
General Procedure for the Synthesis of Ethyl (5E)-5-[[2,5-dimethyl-1-[Substituted phenyl]pyrrol-3-yl]methylene]-2-methyl-4-oxo-1H-pyrrole-3-carboxylates (8a-8aa, 9a-9s)
To a solution of ethyl 5-methyl-3-oxo-1,2-dihydropyrrole-4-carboxylate 3 (1.0 equiv/mol) in absolute ethanol (3 mL) was added the requisite 2,5-dimethyl-1-aryl-3-formylpyrrole 7a-7aa (1.0 equiv/mol) and potassium hydrogen sulfate (0.2 equiv/mol). The mixture was heated at 70–80 °C for 3 h resulting in the formation of a yellow precipitate. The mixture was poured onto ice and filtered to afford the ethyl (5E)-5-[[2,5-dimethyl-1-[Substituted phenyl]pyrrol-3-yl]methylene]-2-methyl-4-oxo-1H-pyrrole-3-carboxylate 8a-8aa as a yellow powder (80–95% yield). In general no further purification by column chromatography was required.
Ethyl (5E)-5-[[2,5-dimethyl-1-[2-(trifluoromethyl)phenyl]pyrrol-3-yl]methylene]-2-methyl-4-oxo-1H-pyrrole-3-carboxylate (8a)
Yellow powder (0.22 g, 89%); mp 230–235 °C. 1H NMR (500 MHz, DMSO-d6): δ 10.3 (s, 1H, -NH), 8.01 (t, 1H, J = 7.7 Hz) 7.91 (t, 1H), 7.8 (t, 1H, J = 7.7 Hz), 7.51 (t, 1H, J = 7.7 Hz), 6.71 (s, 1H), 6.69 (s, 1H), 4.13–4.09 (q, 2H, J = 6.9 Hz), 2.58 (s, 3H), 1.99 (s, 3H), 1.90 (s, 3H), 1.23 (t, 3H, J = 7.1 Hz); 13C NMR (125 MHz, DMSO-d6): δ 180.5, 170.2, 163.3, 135.9, 134.36 (2C), 131.6, 131.6, 130.43 (2C), 129.17 (2C), 127.5, 113.5, 169.7, 105.9, 102.4, 58.2, 15.7, 14.4, 11.9, 10.3. IR (KBr) ν 3500–2000 (max at 3176.47, 2926.66, and 2340.11 N–H, and C–H st), 1661.80 and 1583.65 (C=O, ar–C-C and ar–C-N st) cm–1. LCMS m/z 419.15 (M + 1); HRMS m/z calculated (C22H22N2O3F3): 419.1577 (M + H)+; found: 419.1566. (Delta PPM: 2.40 ppm; acquired on Microtof; mass resolution: 10000 (fwhm)).
Ethyl (5E)-5-[[2,5-Dimethyl-1-[4-(trifluoromethyl)phenyl]pyrrol-3-yl]methylene]-2-methyl-4-oxo-1H-pyrrole-3-carboxylate (8b)
Yellow powder, mp 252–255 °C; 1H NMR (500 MHz, DMSO-d6): δ 10.27 (s, 1H, -NH), 7.96 (d, 2H) 7.65 (d, 2H), 6.75 (s, 1H), 6.70 (s, 1H), 4.16 (q, 2H, J = 5.4 Hz), 2.59 (s, 3H), 2.13 (s, 3H), 2.04 (s, 3H), 1.23 (t, 3H, J = 1.0, 1.1 Hz); 13C NMR (125 MHz; DMSO-d6): δ 180.4, 170.2, 163.2, 140.6, 134.5, 130.4, 129.2, 128.9, 128.6, 126.67, 126.64, 124.9, 122.8, 113.9, 109.4, 106.6, 102.4, 58.1, 15.8, 14.4, 12.6, 10.7; LCMS m/z 419.1527 (M + 1); HRMS m/z 419.1579 ([M + H]+; calcd for C22H21F3N2O3+ 419.1577.
Ethyl (5E)-5-[[2,5-Diethyl-1-[2-(trifluoromethyl) phenyl] pyrrol-3-yl]methylene]-2-methyl-4-oxo-1H-pyrrole-3-carboxylate (9c)
Yellow powder, mp 245–250 °C; 1H NMR (500 MHz, DMSO-d6): δ 10.31 (s, 1H, -NH), 8.01 (d, 1H), 7.92 (t, 1H), 7.83 (t, 1H), 7.60 (d, 1H), 6.71 (s, 1H), 6.69 (s, 1H), 4.13 (m, 2H, J = 5.6, 1.1 Hz), 2.61 (br s, 2H), 2.50 (s, 2H), 2.16 (m, 3H, J = 6.2, 7.6 Hz), 1.23 (t, 3H); 13C NMR (125 MHz; DMSO-d6): δ 187.7, 180.5, 170.2, 163.3, 141.6, 137.7, 134.4, 134.0, 132.0, 132.0, 129.1, 127.6, 126.9, 112.8, 109.3, 104.5, 102.4, 58.2, 19.3, 17.6, 15.8, 15.3, 14.4, 12.4; IR (KBr) ν 3500–2000 (max at 3159.66, 2926.40, and 2358.21 N–H, and C–H st), 1661.05 and 1582.35 (C=O, ar–C-C and ar–C-N st) cm–1; LCMS m/z 447.18 [M + H]+; HRMS m/z 447.1882 ([M + H]+; calcd for C24H26F3N2O3+ 447.1890).
tert-Butyl-(5E)-5-[[2,5-dimethyl-1-[2-(trifluoromethyl)phenyl]pyrrol-3-yl]methylene]-2-methyl-4-oxo-1H-pyrrole-3-carboxylate (10a)
Yellow powder (99% yield), mp 201–205 °C. 1H NMR (500 MHz; DMSO-d6): δ 10.13 (br s, 1H, -NH), 8.02 (d, 2H, J = 7.0 Hz), 7.93 (t, 1H, J = 7.6), 7.83 (t, 1H, J = 7.7 Hz), 6.7 (s, 1H), 6.63 (s, 1H), 4.3 (m, 2H, J = 5.0 Hz), 1.98 (s, 3H), 1.9 (s, 3H), 1.4 (s, 9H), 1.07 (t, 3H, J = 7.0 Hz); 13C NMR (125 MHz, DMSO-d6): δ 181.6, 180.8, 168.6, 169.6, 162.4, 135.5, 131.7, 129.2, 127.6, 123.8, 113.6, 112.1, 108.9, 106.0, 78.1, 55.9, 28.2(3C), 18.5, 16.6, 12.0,(2C) 10.3; LCMS m/z 447.18 (M + 1); HRMS m/z 447.1888 ([M + H]+; calcd for C24H26F3N2O3+ 447.1890).
Benzyl (5E)-5-[[2,5-Dimethyl-1-[2-(trifluoromethyl)phenyl]pyrrol-3-yl]methylene]-2-methyl-4-oxo-1H-pyrrole-3-carboxylate (10b)
Yellow powder (95% yield); mp 233–236 °C. 1H NMR (500 MHz; DMSO-d6): δ 10.36 (br s, 1H, -NH), 8.02 (d, 2H, J = 7.7 Hz), 7.9 (t, 1H, J = 7.0 Hz), 7.8 (t, 1H, J = 7.5 Hz), 7.54 (d, 2H, J = 7.5 Hz), 7.47 (d, 1H, J = 7.2 Hz), 7.39 (t, 1H, J = 7.2 Hz), 7.32 (t, 1H, J = 7.3 Hz), 6.72 (s, 1H), 6.70 (s, 1H), 5.19 (br s, 2H, CH2Ph), 2.5 (s, 3H), 2.0 (s, 3H), 1.9 (s, 9H); 13C NMR (125 MHz, DMSO-d6): δ 180.4, 170.5, 163.1, 137.2, 136.0, 134.4, 131.7(2C), 131.6, 130.5, 129.2, 128.3(3C), 127.5(2C), 127.3(3C), 113.6, 109.8, 106.0, 102.2, 63.7, 15.8, 12.0, 10.3; LCMS m/z 481.17 (M + 1); HRMS m/z 481.1736 ([M + H]+; calcd for C27H24F3N2O3+ 481.1734).
Acknowledgments
This investigation was done for and received support from the UNICEF/UNDP/World Bank/WHO Special Programme for Research and Training in Tropical Diseases (TDR). The University of Dundee would also like to acknowledge the Wellcome Trust (Grant 083481) for support.
Glossary
Abbreviations
- CLint
intrinsic clearance
- CYP
cytochrome P450
- DMPK
drug metabolism and pharmacokinetics
- SAR
structure–activity relationship
- UDPGA
uridine-5′-diphospho-glucuronic acid
- WHO
World Health Organisation
- WHO-TDR
World Health Organisation Programme for Research and Training in Tropical Diseases
Supporting Information Available
Synthetic routes for the synthesis of intermediates, methods for physicochemical evaluation of the compounds and the data generated, methods for the drug metabolism, pharmacokinetics, in vitro and in vivo parasite testing; chemistry experimental for compounds not included in the main text. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
○ D.M. and A.M. contributed equally to the work.
The authors declare no competing financial interest.
Footnotes
Some of the compounds had a trace, inseparable amount of Z isomer. This could not be detected in the 13C NMR spectra and was only detected using UPLC.
Supplementary Material
References
- Badawey E.; Rida S. M.; Soliman F. S. G.; Kappe T. Benzimidazole condensed ring-systmes. 4. New approaches to the synthesis of substituted pyrimido 1,6-A-benzimidazole-1,3(2H,5H)-diones. J. Heterocyclic Chem. 1989, 26, 405–408. [Google Scholar]
- Haynes R. K.Preparation of antiparasitic artemisinin derivatives (sesquiterpene endoperoxides). WO2003076446A1, 2003.
- Haynes R. K.; Chan H.-W.; Lam W.-L.; Tsang H.-W.; Cheung M.-K.. Synthesis and antiparasitic activity of artemisinin derivatives (endoperoxides). WO2000004024A1, 2000.
- Haynes R. K.; Fugmann B.; Stetter J.; Rieckmann K.; Heilmann H. D.; Chan H. W.; Cheung M. K.; Lam W. L.; Wong H. N.; Croft S. L.; Vivas L.; Rattray L.; Stewart L.; Peters W.; Robinson B. L.; Edstein M. D.; Kotecka B.; Kyle D. E.; Beckermann B.; Gerisch M.; Radtke M.; Schmuck G.; Steinke W.; Wollborn U.; Schmeer K.; Romer A. Artemisone - A highly active antimalarial drug of the artemisinin class. Angew. Chem., Int. Ed. 2006, 45, 2082–2088. [DOI] [PubMed] [Google Scholar]
- Haynes R. K.; Ho W. Y.; Chan H. W.; Fugmann B.; Stetter J.; Croft S. L.; Vivas L.; Peters W.; Robinson B. L. Highly antimalaria-active artemisinin derivatives: Biological activity does not correlate with chemical reactivity. Angew. Chem., Int. Ed. 2004, 43, 1381–1385. [DOI] [PubMed] [Google Scholar]
- Veiga M. I.; Ferreira P. E.; Jornhagen L.; Malmberg M.; Kone A.; Schmidt B. A.; Petzold M.; Bjorkman A.; Nosten F.; Gil J. P. Novel Polymorphisms in Plasmodium falciparum ABC Transporter Genes Are Associated with Major ACT Antimalarial Drug Resistance. PLoS One 2011, 6, e20212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badawey E.; Rida S. M.; Soliman F. S. G.; Kappe T. Benzimidazole condensed ring-systems. 5. Studies on the synthesis of pyrimido 1,6-A benzimidazole-1,3(2H,5H)-diones. J. Heterocyclic Chem. 1989, 26, 1401–1404. [Google Scholar]
- Olliaro P.; Wells T. N. C. The Global Portfolio of New Antimalarial Medicines Under Development. Clin. Pharmacol. Ther. 2009, 85, 584–595. [DOI] [PubMed] [Google Scholar]
- Renslo A. R.; McKerrow J. H. Drug discovery and development for neglected parasitic diseases. Nat. Chem. Biol. 2006, 2, 701–710. [DOI] [PubMed] [Google Scholar]
- Braibante M. E. F.; Braibante H. T. S.; Costa C. C.; Martins D. B. Reactivity of chloroacetylated beta-enamino compounds. Synthesis of heterocycles. Tetrahedron Lett. 2002, 43, 8079–8081. [Google Scholar]
- Galdino S. L.; Pitta I. R.; Luu-Duc C.; Lucena B.; Oliveria C. L.; Rosalia M. 5-Benzylidene pyrrolones, furanones and thiophenones. I. Synthesis, structure, and pharmacological activities. Eur. J. Med. Chem. 1985, 20, 439–442. [Google Scholar]
- Higino J. S.; Linsgaldino S.; Darochapitta I.; Delima J. G.; Luu-Duc C. 5-Benzylidene Pyrrolones. 4. Synthesis and Antifungal Activity of Some 5-Benzylidene Derivatives of 1,2-Dimethyl-3-Carbethoxy-Pyrrol-4-One. Farmaco 1990, 45, 1283–1287. [PubMed] [Google Scholar]
- Ragan J. A.; Jones B. P.; Castaldi M. J.; Hill P. D.; Makowaki T. W. Ullman methoxylation in the presence of a 2,5-dimethykpyrrole-blocked aniline; preparation of 2-fuloro-4-methoxyaniline. Org. Syn. 2004, 10, 418. [Google Scholar]
- Vorkapic-Furac J.; Mintas M.; Burgemeister T.; Mannschreck A. Sterically hindered N-aryl pyrroles: chromatographic separation of enantiomers and barriers to racemization. J. Chem. Soc., Perkin Trans. 2 1989, 713–717. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.