

Abstract
Summary: Limitations of genome sequencing techniques have led to dozens of assembly algorithms, none of which is perfect. A number of methods for comparing assemblers have been developed, but none is yet a recognized benchmark. Further, most existing methods for comparing assemblies are only applicable to new assemblies of finished genomes; the problem of evaluating assemblies of previously unsequenced species has not been adequately considered. Here, we present QUAST—a quality assessment tool for evaluating and comparing genome assemblies. This tool improves on leading assembly comparison software with new ideas and quality metrics. QUAST can evaluate assemblies both with a reference genome, as well as without a reference. QUAST produces many reports, summary tables and plots to help scientists in their research and in their publications. In this study, we used QUAST to compare several genome assemblers on three datasets. QUAST tables and plots for all of them are available in the Supplementary Material, and interactive versions of these reports are on the QUAST website.
Availability: http://bioinf.spbau.ru/quast
Contact: gurevich@bioinf.spbau.ru
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Modern DNA sequencing technologies cannot produce the complete sequence of a chromosome. Instead, they generate large numbers of reads, ranging from dozens to thousands of consecutive bases, sampled from different parts of the genome. Genome assembly software combines the reads into larger regions called contigs. However, current sequencing technologies and software face many complications that impede reconstruction of full chromosomes, including errors in reads and large repeats in the genome.
Different assembly programs use different heuristic approaches to tackle these challenges, resulting in many differences in the contigs they output. This leads to the questions of how to assess the quality of an assembly and how to compare different assemblies.
Recently, there has been a lot of work on developing comprehensive ways to compare different assemblers.
Plantagora (Barthelson et al., 2011) is a web-based platform aimed at helping scientists view characteristics of the most popular sequencing strategies (including sequencing platforms and assembly software) for plant genomes. Plantagora has a well-designed interface to browse their database of evaluation results. Researchers may run the Plantagora assessment tool on their own assembly, but the results cannot be viewed through the friendly user-interface; instead, the user has to parse a large log file.
The Assemblathon competition (Earl et al., 2011) compared 41 de novo assemblies on >100 evaluation metrics. The Assemblathon assessment scripts are freely available, but they are highly focused on the genomes used in the competition, and normal users cannot easily apply them to other genomes.
Another freely available genome assembly assessment tool is GAGE (Salzberg et al., 2011). In Salzberg et al. (2011), it was used to evaluate several leading genome assemblers on four datasets. GAGE evaluates a set of metrics, including different types of misassembly errors (inversions, relocations and translocations).
Plantagora and GAGE can only be used to evaluate assemblies of datasets with a known reference genome; thus, they are not suitable for evaluating assemblies of previously unsequenced genomes. Additionally, GAGE can only be run on one dataset at a time; therefore, to compare multiple assemblers on the same dataset, one has to manually combine output from separate GAGE reports into a table.
We introduce QUAST, a new assembly quality assessment tool. QUAST evaluates a full range of metrics needed by various users. However, the number of metrics is not so large that it would become difficult to interpret all of them. The interface and visualizations are easy to use, representative and informative. QUAST can evaluate assembly quality even without a reference genome, so that researchers can assess the quality of assemblies of new species that do not yet have a finished reference genome. In addition, QUAST is rather fast, and its most time-consuming steps are parallelized; therefore, it can be effectively run on multi-core processors. See Supplementary Table S1 for QUAST’s performance on different genomes.
2 METHODS
2.1 Metrics
QUAST aggregates methods and quality metrics from existing software, such as Plantagora, GAGE, GeneMark.hmm (Lukashin and Borodovsky 1998) and GlimmerHMM (Majoros et al., 2004), and it extends these with new metrics. For example, the well-known N50 statistic can be artificially increased by concatenating contigs, at the expense of increasing the number of misassemblies; QUAST introduces a new statistic, NA50, to counter this.
QUAST uses the Nucmer aligner from MUMmer v3.23 (Kurtz et al., 2004) to align assemblies to a reference genome and evaluate metrics depending on alignments. QUAST also computes metrics that are useful for assessing assemblies of previously unsequenced species, whereas most other assembly assessment software require a reference genome.
We will split the metrics evaluated by QUAST into several groups. Most have been used in previous studies, but some are new to QUAST.
2.1.1 Contig sizes
The following metrics (except for NGx) can be evaluated with or without a reference genome. We also provide filtered versions of them, restricted to contigs of length above a specified minimum size, to exclude short contigs that may not be of much use.
No. of contigs: The total number of contigs in the assembly.
Largest contig: The length of the largest contig in the assembly.
Total length: The total number of bases in the assembly.
Nx (where
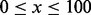 ): The largest contig length, L, such that using contigs of length
): The largest contig length, L, such that using contigs of length 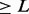 accounts for at least x% of the bases of the assembly.
accounts for at least x% of the bases of the assembly.NGx, Genome Nx: The contig length such that using equal or longer length contigs produces x% of the length of the reference genome, rather than x% of the assembly length.
2.1.2 Misassemblies and structural variations
The following metrics describe structural errors in the contigs. QUAST can evaluate them only with respect to a known reference genome. If the reference genome exactly matches the dataset being assembled, differences may be attributed to misassemblies by the software or to sequencing errors, such as chimeric reads. Sometimes one uses a reference genome that is related to but different than the dataset being sequenced. In this case, the differences may still be misassemblies, but they may also be true structural variations, such as rearrangements, large indels, different repeat copy numbers and so forth.
No. of misassemblies: The number of misassemblies, using Plantagora’s definition. Plantagora defines a misassembly breakpoint as a position in the assembled contigs where the left flanking sequence aligns over 1 kb away from the right flanking sequence on the reference, or they overlap by >1 kb, or the flanking sequences align on opposite strands or different chromosomes. QUAST also generates a report with the number of misassemblies because of each of these reasons. See the Supplementary Methods for details.
No. of misassembled contigs: The number of contigs that contain misassembly breakpoints.
Misassembled contigs length: The total number of bases contained in all contigs that have one or more misassemblies.
No. of unaligned contigs: The number of contigs that have no alignment to the reference sequence.
No. of ambiguously mapped contigs: The number of contigs that have high-scoring reference alignments of equal quality in multiple locations on the reference genome.
In addition to these summary statistics, QUAST also generates reports with detailed information about each contig, including whether the contig is unaligned, ambiguously mapped, misassembled or correct.
2.1.3 Genome representation and its functional elements
This section lists metrics evaluating genome representation in contigs and the number of assembled functional elements, such as genes and operons. Most of these require a reference genome.
Genome fraction (%): The total number of aligned bases in the reference, divided by the genome size. A base in the reference genome is counted as aligned if at least one contig has at least one alignment to this base. Contigs from repeat regions may map to multiple places, and thus may be counted multiple times in this quantity.
Duplication ratio: The total number of aligned bases in the assembly (i.e. total length minus unaligned contigs length), divided by the total number of aligned bases in the reference [see the genome fraction (%) metric]. If the assembly contains many contigs that cover the same regions of the reference, its duplication ratio may be much >1. This may occur due to overestimating repeat multiplicities and due to small overlaps between contigs, among other reasons.
GC (%): The total number of G and C nucleotides in the assembly, divided by the total length of the assembly. This metric can be computed without a reference genome.
No. of mismatches per 100 kb: The average number of mismatches per 100 000 aligned bases. QUAST also generates a more detailed report with the coordinates of mismatches. This metric does not distinguish between single-nucleotide polymorphisms, which are true differences in the assembled genome versus the reference genome, and single-nucleotide errors, which are due to errors in reads or errors in the assembly algorithm.
No. of indels per 100 kb: The average number of single nucleotide insertions or deletions per 100 000 aligned bases. A detailed report with coordinates of indels for all assemblies is also available.
No. of genes: The number of genes in the assembly (complete and partial), based on a user-provided annotated list of gene positions in the reference genome. A gene is partially covered if the assembly contains at least 100 bp of the gene but not the whole gene. QUAST also reports a list of the completely covered genes for each assembly. If an annotated list of gene positions is not available, this metric cannot be computed, but the number of predicted genes (see later in the text) can be used instead.
No. of operons: Complete and partial operons are counted in a similar fashion to genes, using a user-provided annotated list of operon positions in the reference genome.
No. of predicted genes: The number of genes in the assembly predicted by QUAST’s gene-finding module, which is based on GeneMark.hmm (Lukashin and Borodovsky 1998) for prokaryotes and GlimmerHMM (Majoros et al., 2004) for eukaryotes. The GeneMark.hmm authors have kindly allowed use of their software inside QUAST, and GlimmerHMM is an open-source tool. If the user provides a reference genome with an annotated list of genes, we use the number of genes statistic instead. Otherwise, QUAST counts the number of genes annotated by GeneMark.hmm or GlimmerHMM and then filters them to count only those with lengths above one or more specified minimum thresholds.
2.1.4 Variations of N50 based on aligned blocks
The following metrics in QUAST are new, but they have similarities with GAGE’s ‘corrected Nx’ (Salzberg et al., 2011), Assemblathon’s ‘contig path Nx over alignment graph’ (Earl et al., 2011) and the ‘normalized N50’ (Makinen et al., 2012) metric. Here, we give short descriptions for these metrics. See the Supplementary Methods for more detailed information.
NAx (A stands for aligned; x ranges from 0–100): This is a combination of the well-known Nx metric and Plantagora’s number of misassemblies metric. It is computed in two steps. First, we break the contigs into aligned blocks. If a contig has misassembly breakpoints (per the previous definition from Plantagora), it is broken into multiple blocks at these breakpoints. Additionally, if there are unaligned regions within a contig, these regions are removed, and the contig is split into blocks. Next, we compute the ordinary Nx statistic on these blocks instead of on the original contigs.
NGAx: We break contigs into aligned blocks as described for NAx, and then we compute the NGx statistic (instead of Nx) on these blocks.
Both the NAx and NGAx metrics require a reference genome. If the reference genome is different than the sample being assembled, some breakpoints and indels may represent true structural differences.
2.2 Visualization
QUAST presents a number of statistics in graphical form and supports SVG, PNG and PDF formats. Sample plots are presented in the Supplementary Material. These plots are divided into several groups:
Nx-like plots: These show the trends of Nx, NGx, NAx or NGAx metrics as x varies. This is more informative than just using N50.
Cumulative plots: Contigs are ordered from largest to smallest (in number of bases) for all the types of cumulative plots considered. The cumulative length plot shows the number of bases in the first x contigs, as x varies from zero to the number of contigs. The cumulative number of complete genes and cumulative number of complete operons plots are computed similarly.
GC content plots: These show the distribution of GC content in the contigs. The x value shows the per cent of GC (from 0 to 100). The y value shows the number of non-overlapping 100 bp windows whose GC content is x. This distribution is often Gaussian (Bohlin et al., 2010); however, if there are contaminants with a different GC content, there will often be a superposition of multiple Gaussians.
Contig alignment plots (Fig. 1): These show alignment of contigs to the reference genome and the positions of misassemblies in these contigs. Colour coding indicates when block boundaries are shared by multiple assemblies, and to show misassembled blocks. An optional track shows the read coverage along the reference genome.
QUAST also makes comparative histograms of several metrics: the number of complete genes, the number of complete operons and the genome fraction (%). Histograms of other metrics can be added as well.
Fig. 1.
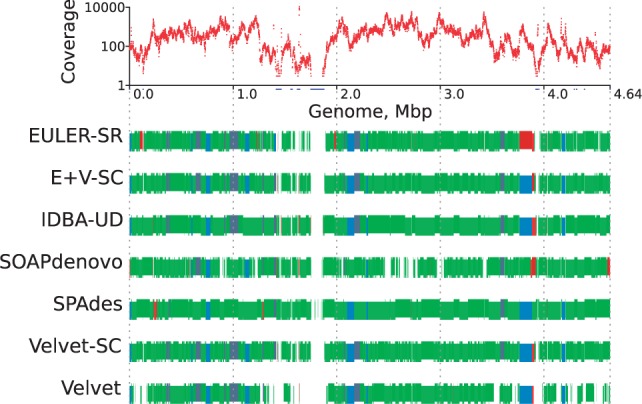
Alignment of single-cell E.coli assemblies to the reference genome. On all tracks, the x-axis is genome position. Top track: Read coverage on a logarithmic scale. The red curve shows coverage binned in 1000 bp windows. Blue positions on the x-axis have zero coverage, even if their bin has some coverage. Coverage is highly non-uniform, ranging from 0 to near 10 000. All other tracks: Comparison of positions of aligned contigs. Contigs that align correctly are coloured blue if the boundaries agree (within 2000 bp on each side) in at least half of the assemblies, and green otherwise. Contigs with misassemblies are broken into blocks and coloured orange if the boundaries agree in at least half of the assemblies, and red otherwise. Contigs are staggered vertically and are shown in different shades of their colour to distinguish the separate contigs, including small ones
2.3 Comparing assemblers
In this study, we evaluated several of the leading genome assemblers on three datasets: Escherichia coli (a single-cell sample), Homo sapiens chromosome 14 and Bombus impatiens (the bumble bee, which at publication time does not have a finished assembly). The E.coli dataset and some of its assemblies are taken from Chitsaz et al. (2011). The SPAdes and IDBA-UD assemblies are new. All assemblies of H.sapiens and B.impatiens and both datasets are taken from Salzberg et al. (2011). In this article, we present some of QUAST’s comparison statistics and a sample plot comparing E.coli assemblies. See Supplementary Figures S3–S29 and Supplementary Tables S2–S8 for more plots and extended tables for E.coli and for comparisons of assemblers on the other two datasets.
2.3.1 Comparison of E.coli assemblies
The reference genome is E.coli str. K-12 substr. MG1655 (Blattner et al., 1997), available at the NCBI website. Gene annotations were taken from http://www.ecogene.org/.
We include several well-known assemblers designed for cultured bacterial datasets: EULER-SR (Pevzner et al., 2001), Velvet (Zerbino and Birney, 2008), and SOAPdenovo (Li et al., 2010). We also include several recently introduced assemblers that have been adapted or designed from scratch to handle single-cell datasets: Velvet-SC and EULER + Velvet-SC (Chitsaz et al., 2011), our assembler, SPAdes (Bankevich et al., 2012) and IDBA-UD (Peng et al., 2012).
Table 1 shows that SPAdes and IDBA-UD have the best results in almost all metrics. IDBA-UD assembled the largest contig (224 018 bp) and has the smallest number of contigs (283), but SPAdes has a larger NGA50 than IDBA-UD (99 913 versus 90 607 bp) and assembled a higher percentage of the genome (96.99 versus 95.90%). SPAdes also assembled the highest number of complete genes (4071 of 4324), with IDBA-UD a close second (4030). However, both SPAdes and IDBA-UD have more misassemblies than the three Velvet-based assemblers.
Table 1.
Comparison of assemblies of a single-cell sample of E.coli (for contigs 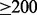 bp)
bp)
| Assembler | No. of contigs | NGA50 (bp) | Largest (bp) | Total (bp) | Genome fraction (%) | No. of misassemblies | No. of complete genes |
|---|---|---|---|---|---|---|---|
| EULER-SR | 610 | 26 580 | 140 518 | 4 306 898 | 86.54 | 19 | 3442 |
| E+V-SC | 396 | 32 051 | 132 865 | 4 555 721 | 93.58 | 2 | 3816 |
| IDBA-UD | 283 | 90 607 | 224 018 | 4 734 432 | 95.90 | 9 | 4030 |
| SOAPdenovo | 817 | 16 606 | 87 533 | 4 183 037 | 81.36 | 6 | 3060 |
| SPAdes | 532 | 99 913 | 211 020 | 4 975 641 | 96.99 | 11 | 4071 |
| Velvet | 310 | 22 648 | 132 865 | 3 517 182 | 75.53 | 2 | 3121 |
| Velvet-SC | 617 | 19 791 | 121 367 | 4 556 809 | 93.31 | 2 | 3662 |
The best value for each column is indicated in bold.
Figure 1 shows how the contigs align to the reference genome and reveals high similarity between some of the assemblies. E+V-SC, Velvet and Velvet-SC generated assemblies with dozens of similar contigs; this is natural because all of these assemblers are modifications of Velvet. The top track shows the read coverage along the genome. Velvet was not able to assemble low-coverage regions of the genome, whereas the assemblers designed for single-cell datasets (Velvet-SC, E+V-SC, SPAdes and IDBA-UD) did much better, although, of course, none of them can assemble the regions that literally have zero coverage.
3 CONCLUSION
Many assembly algorithms have been developed for the challenging problem of genome assembly from short reads. Our new open-access quality assessment tool QUAST will help scientists to assess different assembly software to choose the best pipeline for their research, and it will help developers of genome assemblers to improve their software and algorithms.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank the SPAdes team (Bankevich et al., 2012) for productive collaboration, helpful comments and feedback on using our software. The authors are especially grateful to Andrey Prjibelski for his help in developing the plots in QUAST and to Dmitry Antipov for his help in testing QUAST.
Funding: Government of the Russian Federation (11.G34.31.0018); NIH (3P41RR024851-02S1).
Conflict of Interest: none declared.
REFERENCES
- Bankevich A, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthelson R, et al. Plantagora: modeling whole genome sequencing and assembly of plant genomes. PLoS One. 2011;6:e28436. doi: 10.1371/journal.pone.0028436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blattner FR, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- Bohlin J, et al. Analysis of intra-genomic GC content homogeneity within prokaryotes. BMC Genomics. 2010;11:464. doi: 10.1186/1471-2164-11-464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitsaz H, et al. Efficient de novo assembly of single-cell bacterial genomes from short-read data sets. Nat. Biotechnol. 2011;29:915–921. doi: 10.1038/nbt.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earl D, et al. Assemblathon 1: a competitive assessment of de novo short read assembly methods. Genome Res. 2011;21:2224–2241. doi: 10.1101/gr.126599.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010;20:265–272. doi: 10.1101/gr.097261.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukashin AV, Borodovsky M. GeneMark.hmm: new solutions for gene finding. Nucleic Acids Res. 1998;26:1107–1115. doi: 10.1093/nar/26.4.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majoros WH, et al. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics. 2004;20:2878–2879. doi: 10.1093/bioinformatics/bth315. [DOI] [PubMed] [Google Scholar]
- Makinen V, et al. Normalized N50 assembly metric using gap-restricted co-linear chaining. BMC Bioinformatics. 2012;13:255. doi: 10.1186/1471-2105-13-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, et al. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics. 2012;28:1–8. doi: 10.1093/bioinformatics/bts174. [DOI] [PubMed] [Google Scholar]
- Pevzner PA, et al. An Eulerian path approach to DNA fragment assembly. Proc. Natl. Acad. Sci. USA. 2001;98:9748–9753. doi: 10.1073/pnas.171285098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzberg SL, et al. GAGE: a critical evaluation of genome assemblies and assembly algorithms. Genome Res. 2011;22:557–567. doi: 10.1101/gr.131383.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino D, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.