

Abstract
Voltage-dependent calcium channels (VDCCs) serve a wide range of physiological functions and their activity is modulated by different neurotransmitter systems. GABAergic inhibition of VDCCs in neurons has an important impact in controlling transmitter release, neuronal plasticity, gene expression and neuronal excitability. We investigated the molecular signalling mechanisms by which GABAB receptors inhibit calcium-mediated electrogenesis (Ca2+ spikes) in the distal apical dendrite of cortical layer 5 pyramidal neurons. Ca2+ spikes are the basis of coincidence detection and signal amplification of distal tuft synaptic inputs characteristic for the computational function of cortical pyramidal neurons. By combining dendritic whole-cell recordings with two-photon fluorescence Ca2+ imaging we found that all subtypes of VDCCs were present in the Ca2+ spike initiation zone, but that they contribute differently to the initiation and sustaining of dendritic Ca2+ spikes. Particularly, Cav1 VDCCs are the most abundant VDCC present in this dendritic compartment and they generated the sustained plateau potential characteristic for the Ca2+ spike. Activation of GABAB receptors specifically inhibited Cav1 channels. This inhibition of L-type Ca2+ currents was transiently relieved by strong depolarization but did not depend on protein kinase activity. Therefore, our findings suggest a novel membrane-delimited interaction of the Gi/o-βγ-subunit with Cav1 channels identifying this mechanism as the general pathway of GABAB receptor-mediated inhibition of VDCCs. Furthermore, the characterization of the contribution of the different VDCCs to the generation of the Ca2+ spike provides new insights into the molecular mechanism of dendritic computation.
Key points
Voltage-dependent Ca2+ channels mediate a large repertoire of physiological actions, including the generation of dendritic spikes in neocortical pyramidal neurons; however, the type of Ca2+ channels involved in their generation remains unknown.
We found that L-type Ca2+ currents generate the sustained plateau potential of the Ca2+ spike. GABAB receptors inhibit Ca2+ spikes by specifically blocking dendritic L-type currents.
This inhibition is mediated by a direct Gi/o-βγ-subunit interaction with the Cav1 channels.
Protein kinases (protein kinase C and A) have an important influence on the generation and sustaining of dendritic Ca2+ spikes; however, their activity is not involved in the GABAB-mediated inhibition of Ca2+ spikes.
Inhibitory modulation of dendritic activity is important to understand the transformation of synaptic inputs into neuronal output activity. Our results shed light on the molecular mechanisms by which GABA acting via its GABAB receptors can exert this inhibitory action.
Introduction
Voltage-dependent calcium channels (VDCCs) are a major source of calcium influx into neurons. They serve versatile roles in neuronal signaling, such as transmitter release, neuronal plasticity, gene expression and, very importantly, neuronal excitability (Tsien et al. 1988; Catterall, 2000; Catterall & Few, 2008). Different neurotransmitters regulate these fundamental physiological aspects by modulating the activity of VDCCs through their G-protein-coupled receptors (Catterall, 2000; Dolphin, 2003; Catterall & Few, 2008).
In particular, the neurotransmitter GABA acting through metabotropic GABAB receptors can inhibit VDCCs (Dolphin & Scott, 1987) with important implications for neuronal function. Upon activation, GABAB receptors inhibit presynaptic Cav 2.1 and 2.2 VDCCs (P/Q- and N-type Ca2+ currents, respectively), modulating synaptic release. This mechanism is exerted through a membrane-delimited pathway involving the βγ-subunit of a Gi/o protein (Gi/o-βγ; Kavalali et al. 1997; Bettler et al. 2004; Padgett & Slesinger, 2010). GABAB receptor activation can also inhibit Cav1 VDCCs (L-type Ca2+ currents) in native neurons (Maguire et al. 1989; Marchetti et al. 1991; Scholz & Miller, 1991; Amico et al. 1995; Chalifoux & Carter, 2011), resulting in a reduction in somatic and dendritic Ca2+ influx. However, the mechanism by which this inhibition takes place is still unresolved. The possibility that this action is mediated by the Gi/o-βγ-subunit in a membrane-delimited fashion might a priori be discarded, as it has been shown that Cav1 VDCCs expressed in heterologous systems (Bourinet et al. 1996; Toth et al. 1996; Zhang et al. 1996) or in peripheral neurons (Plummer et al. 1989; Cox & Dunlap, 1992) can not be downregulated by neuromodulators that exert their physiological actions through this pathway. The remaining possibility would be that the GABAB modulation of the Cav1 is exerted by the activation of cytosolic protein kinases (Catterall, 2000; Catterall & Few, 2008).
Here, we investigated this important functional link between GABAB receptor activation and the inhibition of Cav1 channels. In the distal apical dendrite of neocortical pyramidal neurons, activation of GABAB receptors leads to the inhibition of calcium-mediated electrogenesis, the so-called Ca2+ spike (Pérez-Garci et al. 2006; Breton & Stuart, 2012). These events are generated in a specific region of the apical dendrite and initial tuft dendrites about 600–900 μm from the pyramidal cell soma (Amitai et al. 1993; Yuste et al. 1994; Schiller et al. 1997; Larkum & Zhu, 2002; Larkum et al. 2009), suggesting a compartmentalization of the underlying VDCCs. Ca2+ spikes serve important functions in dendritic computation and can transiently switch action potential (AP) output from regular to burst firing mode (Larkum et al. 2001). They are suggested to act as coincidence detectors for correlating feedback inputs from higher cortical areas, preferentially innervating distal tuft inputs, with feedforward sensory information. Furthermore, Ca2+ spikes can amplify distal tuft synaptic inputs that are received in specific spatio-temporal sequences (Larkum et al. 1999b, 2009; Schaefer et al. 2003; Larkum & Nevian, 2008). Their actual influence on neuronal activity is tightly controlled by inhibitory inputs (Larkum et al. 1999b; Murayama et al. 2009; Gidon & Segev, 2012). GABAB-mediated inhibition of Ca2+ spikes can occur by synaptic release of GABA from local interneurons (Pérez-Garci et al. 2006). However, very little is known about the composition of VDCCs in the Ca2+ spike initiation zone that contributes to the initiation and sustaining of the dendritic plateau potentials, as well as how GABAB receptors interact with them to inhibit the Ca2+ spike. Given that Ca2+ spikes result in large elevations of intracellular Ca2+ concentration in this particular region and that they are strongly modulated by GABAB receptor activation renders the apical dendrite of cortical layer 5 pyramidal neurons an ideal model system to study the interaction of GABAB receptors with VDCCs in native central neurons.
Methods
Ethical approval
All experiments were approved by the veterinary office of the canton of Berne, Switzerland; and were carried out in compliance with The Journal of Physiology guidelines (Drummond, 2009).
Slice preparation
Acute cortical brain slices were obtained from Wistar rats (P28–P34). The animals were rapidly decapitated prior to brain extraction from the skull.
Parasagittal slices of the primary somatosensory cortex (300 μm thick) were cut with a vibrating microslicer on a block angled at 15 deg to the horizontal in ice-cold oxygenated artificial cerebral spinal fluid (ACSF) and then maintained at 37°C in ACSF for 15–120 min. Slices were perfused continuously with ACSF at 35 ± 2°C throughout the experiments. ACSF contained (in mm): NaCl, 125; NaHCO3, 25; KCl, 2.5; NaH2PO4, 1.25; MgCl2, 1; glucose, 25; CaCl2, 2; pH 7.4.
Electrophysiology
Whole-cell recordings from somata and/or from dendrites were obtained with the aid of oblique illumination on a Nikon Eclipse E600FN or by infrared gradient contrast video microscopy on a Leica DMLFS microscope equipped with a 40× objective (Leica, HCX Apo, W40×, UVI, NA 0.8).
Somatic (4–6 MΩ) and dendritic (10–20 MΩ) recording pipettes were filled with an intracellular solution containing (in mm): potassium gluconate, 135; KCl, 7; Hepes, 10; Na2-phosphocreatine, 10; Mg-ATP, 4; GTP, 0.3; 0.2% biocytin; pH 7.2 (with KOH); 291–293 mosmol l−1. For double patch-clamp recordings, 10–20 μm Alexa 594 was added to the intracellular solution. For Ca2+ imaging, 300 μm Fluo-5F and 30 μm Alexa 594 were added to the internal solution. Dendritic Ca2+ spikes were evoked by injecting an EPSP-shaped current wave form: (double exponential shape: f(t) = (1 − e−t/τ1)e−t/τ2; where τ1 = 4 ms and τ2 = 10 ms; time to peak: 5 ms) via the dendritic pipette.
Two-photon calcium imaging
Two-photon-excited fluorescence was generated with a titanium-sapphire laser (Tsunami, Spectra-Physics) with laser pulses of 100 fs at a wavelength of 800 nm. The laser was coupled to a confocal scanning unit (TCS-SP2, Leica Microsystems) attached to an upright microscope (DMLFS, Leica), equipped with a 40× objective and non-descanned detectors. Red and green fluorescence signals were separated using a dichroic mirror (560DCXR, AHF) and corresponding bandpass filters (HQ520/25 and HQ585/40, AHF; Nevian & Sakmann, 2006). Raw fluorescence G(t) and R(t) was derived from line scans recorded at 800 Hz by integrating the fluorescence in regions of interest enclosing the fluorescent structures. Relative fluorescence changes were calculated as ΔG/R(t) = (G(t) − G0)/Rave, where G0 is the mean resting green fluorescence during 50 ms before stimulation. The Ca2+-insensitive red fluorescence Rave was calculated as the mean over the entire time course of the line scan recording (Nevian & Helmchen, 2007).
Pharmacology
Drugs were freshly prepared the same day of the experiment and consisted of (in μm): RS-baclofen, 10; nimodipine, 10; Gö6850 bisindolylmaleimide I, 1; KT 5720, 0.5; wortmannin, 0.2; Rp-cAMPS triethylammonium salt, 1000 (from TOCRIS Bioscience, Switzerland); Sp-cAMPS triethylammonium salt, 30 (from SIGMA-ALDRICH); SNX-482, 0.23; ω-agatoxin TK, 0.4; ω-conotoxin GVIA, 1 (from Alomone Labs, Israel).
For isolated dendritic L-type Ca2+ currents, the internal solution consisted of (in mm): caesium- methanesulphonate, 108; Hepes, 9; Na2-phosphocreatine, 14; Mg-ATP, 4; Na-GTP, 0.3; 0.2% biocytin; pH 7.3 (with CsOH). The external solution included (in mm): TTX, 0.001; tetraethylammonium-chloride (TEA), 30; 4-aminopyridine (4-AP), 5; BaCl2, 0.100; NiCl2, 0.05 (Sigma-Aldrich); ω-conotoxin MVIIC, 0.0005 (from Alomone Labs).
When bath-applied, a minimum wait of 15 min was allowed to assess for the effect of the drug.
Local perfusion of baclofen (50 μm) onto the dendritic recording site was achieved by means of a puffing pipette (somatic pipette) located 50–100 μm lateral from the dendritic recording electrode. The range of area covered by the bolus expelled from the pipette was estimated to have a diameter of ∼180 μm, as measured by a test application of the fluorescent indicator Alexa 594 into a brain slice.
Data analysis
For each cell tested, the sweep showing the maximal effect of baclofen on the Ca2+ spike was used for analysis. The effectiveness of baclofen was quantified as the area underneath the control Ca2+ spike sensitive to the local application of baclofen to the apical tuft. This area was estimated by subtracting the baclofen-reduced Ca2+ spike from the control Ca2+ spike. For simplification, the area underneath the Ca2+ spike contributed by the back-propagating somatic AP was not considered in the analysis (although this may well have led to an underestimation of the physiological effect of activating GABAB receptors in the apical tuft). Wilcoxon signed-rank tests for repeated measurements and Mann–Whitney U tests for independent samples were performed for statistical comparisons.
Results
Subcellular compartmentalization of VDCCs in the Ca2+ spike initiation zone
We studied the contribution of the different VDCCs to the generation of dendritic Ca2+ spikes as a first step to study their inhibition by GABAB receptors. Dendritic whole-cell recordings were performed within the Ca2+ spike initiation zone of layer 5 neocortical pyramidal neurons around the main bifurcation of the apical dendrite (Fig. 1A). Dendrites were loaded with a Ca2+ indicator (Fluo-5F, 300 μm) and Alexa-594 (30 μm), and Ca2+ spikes were evoked by injecting an EPSP-shaped current with peak amplitudes of 800–1500 pA via the patch pipette into the dendrite. Ca2+ transients were measured in spines and adjacent dendritic shafts 50–150 μm distal from the recording pipette (Fig. 1B and C). Bath application of the Cav1 (L-type) VDCC blocker nimodipine (10 μm) resulted in an inhibition of the sustained depolarizing plateau that followed the initial depolarization of the Ca2+ spike and a large reduction of the dendritic Ca2+ transient by 55 ± 4% (n = 10; Fig. 1C and F). Ni2+ (50 μm), which blocks both Cav 3 (T-type) and Cav 2.3 (R-type) VDCCs prevented Ca2+ spike initiation in most cases, and abolished Ca2+ transients in both the spine and dendrite (Fig. 1D). Extra current (∼400 pA) injected into the dendrite recovered the Ca2+ spike, albeit with a significant decrease in the dendritic Ca2+ transient by 31 ± 8% (n = 7). SNX-482 (230 nm), a selective blocker of Cav 2.3 (R-type) VDCCs, had no effect on the initiation of the Ca2+ spike, but it resulted in a similar reduction of the dendritic Ca2+ transient (26 ± 1%; n = 3), indicating a large contribution of R-type conductances within the Ni2+-sensitive component. Cav 2.1 (P/Q-type) VDCCs, blocked by ω-agatoxin TK (400 nm), contributed 21 ± 1% (n = 4); and Cav 2.2 (N-type) VDCCs, blocked by ω-conotoxin GVIA (1 μm), contributed 20 ± 1% (n = 8) to the Ca2+ spike-evoked Ca2+ transients in the dendrite. The relative contribution of each VDCC subtype to the Ca2+ transients in spines was different (Fig. 1G). Cav1 VDCCs contributed 45 ± 4% to the Ca2+ transient in the spine, which was significantly less than in the parent dendrite (P < 0.05). Cav2.3 VDCCs contributed 34 ± 6% to the Ca2+ transient accounting for most of the combined R- and T-type contribution determined by application of Ni2+ (41 ± 7%). Cav2.1 VDCCs were found to contribute 21 ± 2% to the spine Ca2+ transient, whereas Cav2.2 VDCCs made no significant contribution (9 ± 5%).
Figure 1. Subcellular compartmentalized distribution of voltage-dependent calcium channels (VDCCs) and action of GABAB receptors.
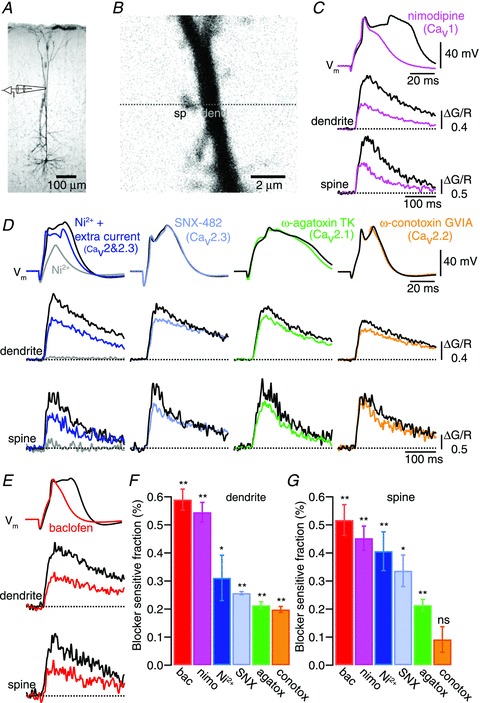
A, micrograph of a biocytin-filled L5 pyramidal neuron indicating the dendritic recording site. B, 2-photon fluorescence image (Alexa 594) of the recording site at the distal apical dendrite and a magnified view showing a spiny region in the apical tuft. Ca2+ imaging (Fluo-5) was measured by establishing a line scan (dashed line) covering a spine and its adjacent dendrite. C and D, dendritic Ca2+ spikes (top sweep) evoked by current injection via the recording pipette elicited Ca2+ transients in both the dendrite (middle panel) and in the spines (bottom panel; black sweeps). Coloured sweeps show representative examples of the action of different VDCCs blockers on the Ca2+ spikes and the resulting inhibition of Ca2+ fluorescence transients in both the dendrites and in the spines: nimodipine (10 μm, magenta traces), Ni2+ (50 μm, grey and blue sweeps, respectively, show recordings before and after injection of additional current via the recording pipette), SNX-482 (230 nm, light blue), ω-agatoxin TK (400 nm, green sweeps) and ω-conotoxin GVIA (1 μm, orange sweeps). E, baclofen added to the bath (10–30 μm) partially inhibited the Ca2+ spikes and their associated Ca2+ transients (red sweeps). F, summary bar plot showing the relative block of Ca2+ transients recorded in the dendrites for each drug tested. G, summary bar plot showing the relative block of Ca2+ transients recorded in the spines for each drug tested. Error bars indicate SEM. *P < 0.05, **P < 0.01.
GABAB receptor activation inhibits Cav1 VDCCs
Ca2+ spikes initiated in the distal main apical dendrite of layer 5 pyramidal neurons are modulated by the local activation of GABAB receptors inhibiting Ca2+ conductances (Pérez-Garci et al. 2006; Breton & Stuart, 2012). Therefore we assessed the effect of activating GABAB receptors on the Ca2+ transients evoked by Ca2+ spikes. Bath application of the GABAB agonist baclofen (10–30 μm) blocked the dendritic plateau potential that followed the upstroke of the Ca2+ spike and reduced the Ca2+ transients in dendrites by 59 ± 4% (P < 0.01; n = 9) and in spines by 52 ± 5% (P < 0.01; Fig. 1E–G). The effect of baclofen on the Ca2+ spike was similar to the effect of nimodipine in blocking the characteristic plateau potential and in the reduction of the Ca2+ transients associated to these events. These similarities suggested that GABAB receptors might exert their modulatory effect on the Ca2+ spike by inhibiting Cav1 VDCCs.
To test this hypothesis, double patch-clamp recordings were performed from the soma and the distal apical dendrite (>700 μm from soma; Fig. 2) and Ca2+ spikes were evoked by dendritic current injection. The GABAB agonist baclofen (50 μm) was applied by pressure ejection to the Ca2+ spike initiation zone using a third pipette located 50–100 μm lateral from the dendritic recording electrode to determine the effectiveness of GABAB receptor inhibition on the Ca2+ spike after blockade of the various VDCCs. Under control conditions, dendritic depolarization evoked Ca2+ spikes that propagated towards the soma, evoking somatic APs. Subsequent transient activation of GABAB receptors resulted in the inhibition of the depolarizing plateau that followed the initial upstroke of the Ca2+ spike and blocked somatic firing. The action of baclofen was completely reversed within 6 s after ceasing the pressure application. Block of Cav1 VDCCs either with nicardipine (5 μm; n = 6) or nimodipine (10 μm; n = 7) resulted in the inhibition of the plateau component of the Ca2+ spike leaving the fast initial depolarization intact. Under these conditions, baclofen reapplied locally to the apical dendrite did not cause further inhibition, indicating that blocking Cav1 VDCCs occluded the effect of baclofen (Fig. 2A). To test the possibility that the decrease in dendritic depolarization after blocking Cav1 channels affected the activation of other VDCCs that might be susceptible to modulation by GABAB receptors, extra current (additional ∼400 pA) was injected in some experiments. Under these conditions somatic APs were reestablished; however, the effect of baclofen was still occluded (n = 5).
Figure 2. Activation of dendritic GABAB receptors inhibits L-type Ca2+ conductances involved in the generation of Ca2+ spikes.
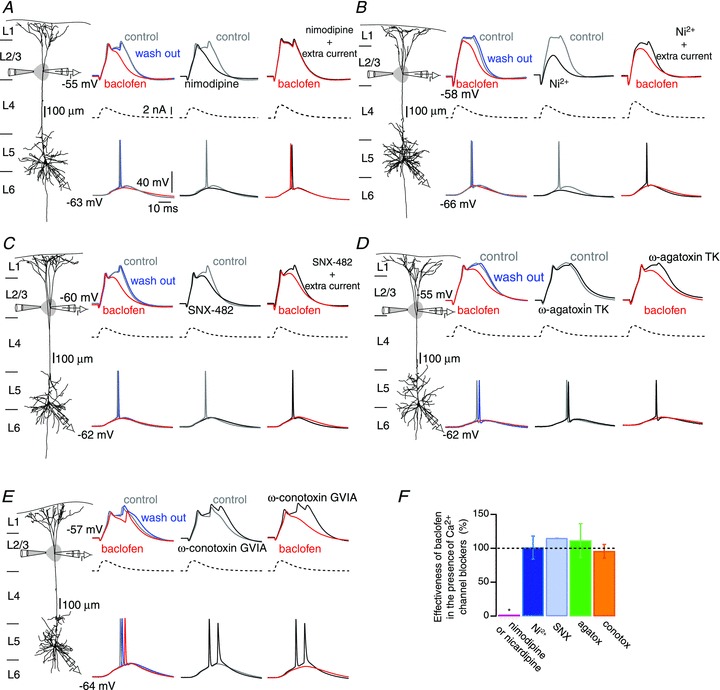
A–E, left columns: reconstruction of biocytin-filled L5 pyramidal neurons showing simultaneous somatic and distal (>700 μm) patch recordings, while a puff pipette expelled baclofen (50 μm) onto the apical tuft. Second columns: dendritic Ca2+ spikes (top, grey sweeps) were evoked by injecting an EPSP-shaped current waveform (middle, dashed sweeps) via the distal pipette. Dendritic spikes propagated towards the soma evoking APs (bottom, grey sweeps). Puffing baclofen on the dendrite shortened and reduced the amplitude of the Ca2+ spikes (red sweeps). The effect of baclofen was fully reversed after ceasing the local application of baclofen (blue sweeps). Third columns: control recordings and recordings obtained after bath application of different voltage-dependent calcium channels (VDCCs) blockers (black sweeps; A, nimodipne 10 μm; B, Ni2+ 50 μm; C, SNX-482, 230 nm; D, ω-agatoxin TK, 400 nm; E, ω-conotoxin GVIA, 1 μm). Fourth columns: reapplication of baclofen to the tuft in the presence of VDCCs blockers. In some cases larger current peaks were injected to the dendrite to reestablish the dendritic depolarization (see A–C). F, effectiveness of baclofen estimated as the area underneath the control Ca2+ spike sensitive to the local application of baclofen. Values obtained after blockade of VDCCs are normalized to those obtained before bath application of VDCCs blockers (see SI Materials and methods). Error bars indicate SEM. *P < 0.01.
As shown above, Ni2+ (50 μm) prevented the initiation of dendritic Ca2+ spikes near threshold current injection, but extra current (∼400 pA) injected into the dendrite reactivated the remaining VDCCs (Fig. 2B). Under these conditions, application of baclofen to the apical dendrite was still effective in inhibiting the Ca2+ spike (n = 5), indicating that the action of baclofen on the sustained component of the Ca2+ spike was not mediated by inhibition of Ni2+-sensitive (Cav3 and Cav2.3) VDCCs. Similarly, blocking Cav2.1, Cav2.2 and Cav2.3 VDCCs with ω-agatoxin TK (400 nm), ω-conotoxin GVIA (1 μm) and SNX-482 (230 nm), respectively, did not influence the effectiveness of baclofen to inhibit Ca2+ spikes (Fig. 2C–E). These results therefore showed that activation of GABAB receptors in the distal apical dendrite exerted its inhibitory effect on dendritic Ca2+ spikes by specifically inhibiting Cav1 VDCCs (Fig. 2F).
After having identified the predominant effect of GABAB-mediated inhibition of Ca2+ spikes to act on Cav1, we tested the action of baclofen on pharmacologically isolated L-type Ca2+ currents recorded from the distal apical dendrite directly (Fig. 3). Because voltage-clamp recordings in dendrites of neurons with an extensive dendritic arborization are limited by an incomplete space clamp of the dendritic membrane (Williams & Mitchell, 2008), we used this method only to phenomenologically assess the interaction of GABAB receptors with L-type Ca2+ currents by comparing the relative effect of baclofen with control conditions. Dendritic patch-clamp recordings were performed with pipettes containing an internal Cs+-methanesulphonate-based solution. We first confirmed the proximity of the recording site to the dendritic Ca2+ spike initiation zone using current steps in current-clamp mode to evoke Ca2+ spikes in normal ACSF (Fig. 3A). The extracellular solution was then supplemented with a ‘cocktail’ for isolating L-type Ca2+ currents by blocking all other VDCCs, K+ channels and Na+ channels. In addition, Cs+ applied intracellularly effectively blocks Ih currents (Harris et al. 1994) and GIRK channels (Sodickson & Bean, 1996). L-type Ca2+ currents were then evoked in voltage-clamp mode by changing the dendritic command potential from −80 mV to 20 mV (Fig. 3B). Under these conditions, baclofen applied locally to the dendrite reversibly inhibited the L-type Ca2+ currents by 82 ± 12% (P < 0.01; n = 4; Fig. 3C). L-type Ca2+ currents subsequently recovered fully within about 3 s. Because the application of baclofen was spatially restricted (∼180 μm), the remaining ∼20% of the Ca2+ currents may be explained by L-type Ca2+ influx beyond the area affected. This suggested that under our experimental conditions the voltage-clamp step command still depolarized a stretch of dendrite beyond 180 μm sufficiently to activate L-type Ca2+ currents at even more distal sites. During the puff-application of baclofen we did not observe any change in the holding current required to clamp the dendrite at −80 mV, suggesting that baclofen did not activate or inactivate any potential leak conductances that would alter the spatial extend of the voltage-clamp. Subsequent bath application of nimodipine (10 μm) blocked the Ca2+ currents by 98.2 ± 8% (P < 0.01; n = 4; Fig. 3D), demonstrating the L-type nature of the recorded current. After having blocked L-type currents with nimodipine (and in the presence of the cocktail containing unspecific blockers for the common Na+, K+ and Ca2+ currents), a final application of baclofen was ineffective to evoke any detectable current corroborating the result that in this set of voltage-clamp experiments GABAB activation did not modulate any other currents than L-type currents under our experimental conditions (data not shown).
Figure 3. Activation of dendritic GABAB receptors inhibits dendritic L-type Ca2+ currents.
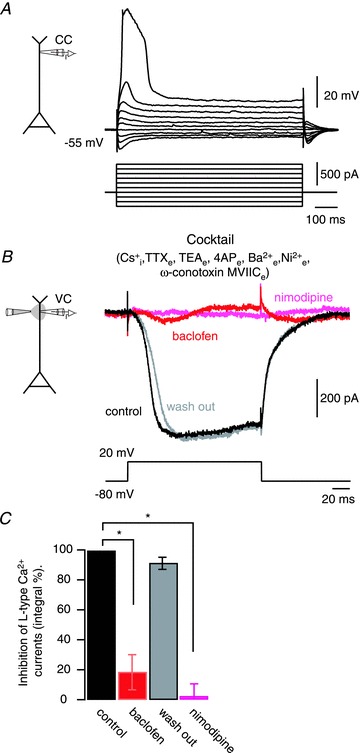
A, voltage recordings performed in current-clamp mode at a distal dendritic site (700 μm) immediately after seal rupture. The internal pipette solution included 108 mm Cs+. Dendritic Ca2+ spikes occurred with depolarizing current steps (lower panel). B, a depolarizing voltage command in voltage-clamp mode was applied to the dendrite in an external medium containing TTX (1 μm), tetraethylammounium-chloride (TEA; 30 mm), 4-aminopyridine (4-AP; 5 mm) and Ba2+ (100 μm). Voltage-dependent calcium channels (VDCCs) other than Cav1 were also blocked with Ni2+ (50 μm; T- and R-type) and ω-conotoxin MVIIC (500 nm; N- and P/Q-type). Under these conditions, inward L-type Ca2+ (after leak subtraction) currents were recorded (black sweep). Local application of the GABAB agonist baclofen (50 μm) to the apical tuft inhibited the L-type Ca2+ currents (red sweep). The action of baclofen was reversed after 5 s ceasing the pressure application of baclofen (grey sweep). Subsequent bath application of nimodipine (10 μm) effectively blocked the L-type Ca2+ current (pink sweep). C, summary of the inhibitory effect of baclofen and nimodipine. Error bars indicate SEM. *P < 0.01.
These results show unequivocally that activation of GABAB receptors in the distal apical dendrite directly inhibit Cav1 channels, which constitute the predominant conductance activated during the plateau potential of the dendritic Ca2+ spike.
Signalling mechanisms underlying the action of GABAB inhibition on dendritic Ca2+ spikes
How is the observed interaction between GABAB receptors and Cav1 channels mediated? GABAB receptors act via a pertussis toxin-sensitive Gi/o protein (Bettler et al. 2004). They are known to inhibit Cav2 VDCCs (Kavalali et al. 1997; Bettler et al. 2004; Padgett & Slesinger, 2010) by a membrane-delimited pathway involving the βγ-subunit of the Gi/o protein. However, this form of inhibition has not been found for Cav1 VDCCs when studied in heterologous expression systems (Bourinet et al. 1996; Toth et al. 1996; Zhang et al. 1996) or in peripheral neurons (Plummer et al. 1989; Cox & Dunlap, 1992) so far. Alternatively, GABAB receptors might inhibit Cav1 channels by cytosolic signalling pathways acting on protein kinases. Thus, we investigated the potential signalling mechanisms downstream of GABAB receptor activation that led to the inhibition of Cav1 channels.
We first sought to test a direct interaction of the βγ-subunit with Cav1 channels. To this end we exploited the fact that the βγ-subunit inhibitory interaction with VDCCs is voltage dependent and can be transiently relieved by a relatively strong depolarization, or prepulse facilitation voltage command sequence (Bourinet et al. 1996; Herlitze et al. 1996; Ikeda, 1996; Toth et al. 1996; Zhang et al. 1996; Zamponi et al. 1997; De Waard et al. 2005). Therefore, we tested whether a prepulse voltage command was able to reverse the inhibitory effect of baclofen on isolated dendritic L-type Ca2+ currents (Fig. 4). Bath application of baclofen (10–30 μm) abolished the L-type Ca2+ current evoked by a voltage command from −80 mV to 20 mV (current integral compared with control; 2.3 ± 1%; P < 0.01; n = 4; Fig. 4B and C). Thereafter, a sequence of prepulse voltage commands consisting of 50 ms depolarizations from −80 mV to 70–100 mV preceding a 200 ms-long voltage step from −80 mV to 20 mV by 60 ms repeated five times every 5 s was applied to disrupt the potential βγ-subunit interaction with the Cav1 VDCCs. Indeed, after sessation of this prepulse stimulation protocol, we observed a partial recovery of the L-type Ca2+ current (57.5 ± 18%; Fig. 4B and C). This recovery lasted 10–30 s in the absence of the prepulse voltage commands, before baclofen exerted its inhibitory effect again (3.4 ± 2%; Fig. 4B and C). Addition of nimodipine to the bath solution maintained the complete block of the measured Ca2+ current as expected (3.1 ± 3%; Fig. 4B and C). However, in the presence of nimodipine, prepulse voltage commands were ineffective in rescuing the L-type Ca2+ current from GABAB-mediated inhibition (5.8 ± 2.8%).
Figure 4. GABAB inhibition of dendritic L-type Ca2+ currents is mediated by a direct Gi/o-βγ-subunit interaction.
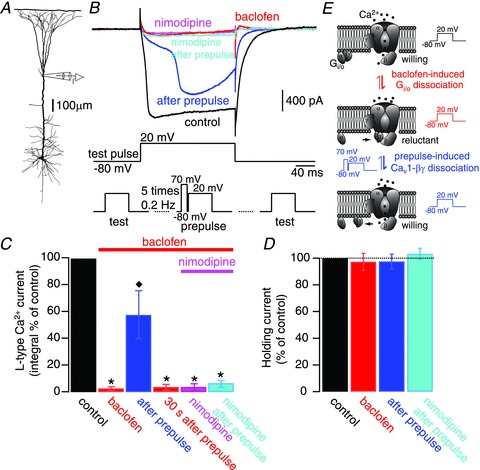
A, experimental configuration of dendritic voltage-clamp experiments. B, pharmacologically isolated dendritic L-type Ca2+ currents (black trace) were evoked by injecting a test voltage command from −80 to 20 mV (test pulse; middle trace). Bath application of baclofen (10–30 μm) inhibited the L-type Ca2+ currents (red trace). A series of 5 prepulse voltage commands from −80 mV to 70 mV (50 ms duration) preceding a depolarization from −80 mV to 20 mV (200 ms) by 60 ms (prepulse; bottom middle trace), partially and transiently relieved the L-type Ca2+ current from its inhibition (blue trace). After recovery of the GABAB-induced inhibition, nimodipine (10 μm) was supplemented to the bath (pink trace). In the presence of the Cav1 blocker, a new set of 5 prepulse voltage commands was ineffective to reverse the inhibition of L-type Ca2+ currents (cyan trace). The lower traces illustrate the sequence of test pulses and the prepulse protocol. C, summary of results. Error bars indicate SEM. *P < 0.01, relative to control; ♦P < 0.01, relative to baclofen inhibition (n = 4). D, bar graph showing the lack of variation in holding current values (at −80 mV) during the different experimental manipulations. E, sketch of the prepulse-induced relief of inhibition illustrating the interpretation of the current traces depicted in B.
During the course of the experiment, neither the tonic activation of GABAB receptors by bath application of baclofen nor the prepulse voltage commands modified the holding current for a holding potential of −80 mV (Fig. 4D), arguing against the activation or inactivation of leak conductances that would compromise the extend of our voltage-clamp recordings and that would change the membrane length constant and therefore the number of activated Cav1 VDCCs. Thus, we conclude that our voltage-clamp conditions were stable and faithfully reflected the behaviour of the L-type current.
Not only was the prepulse protocol effective in recovering the L-type current, but also a 200 ms-long voltage step from −80 mV to 50–80 mV was able to reestablish the L-type Ca2+ currents, consistent with the voltage-dependence of the βγ-subunit interaction with VDCCs (Fig. S1; n = 3). After bath application of nimodipine the voltage steps applied did not uncover any additional currents demonstrating the L-type nature of the recorded currents. Even long (200 ms) current steps to +80 mV failed to activate any further currents (Fig. S1), arguing against the possibility that the activation of L-type currents at potentials of +20 mV resulted in a regenerative Ca2+-mediated event that escaped the voltage clamp and depolarized the membrane beyond the set level of +20 mV and thereby activated additional currents that required a higher threshold for activation.
These results demonstrated that the recovery from inhibition due to strong depolarization was indeed specific to the relief of the Cav1 VDCC. The most parsimonious explanation for the voltage dependency of GABAB receptor-mediated inhibition of Cav1 channels is therefore a membrane-delimited pathway involving the Gi/o-βγ-subunit (Fig. 4E).
Next, we investigated if the GABAB receptor-mediated inhibition of Ca2+ spikes could additionally be mediated by cytosolic signalling cascades. These pathways involve different enzymes that have VDCCs as their main targets: protein kinase C (PKC), cAMP-dependent protein kinases I and II (PKA), phosphatidylinositol 3-kinase (PI3K; Bettler et al. 2004; Deng et al. 2009) and phospholipase C (PLC; Sickmann et al. 2008).
We performed six pharmacological manipulations in order to test the influence of the activity state of these enzymes on the properties of dendritic Ca2+ spikes directly: pre-incubation of the slices with the inhibitor of PKC, bisindolylmaleimide I (1 μm); pre-incubation of the slices with the inhibitor of PKA, KT 5720 (500 nm); inclusion in the intracellular solution of the inhibitor of PKA, Rp-cAMPS (1 mm); inclusion in the intracellular solution of the activator of PKA, Sp-cAMPS (60–100 μm); inclusion in the intracellular solution of the PI3K inhibitor wortmannin (400 nm); and inclusion in the intracellular solution of the PLC inhibitor U73122 (1 μm). Of all these manipulations, only inhibition of PKC and inhibition of PKA affected dendritic Ca2+ spikes (Figs S2 and S3).
We next examined if these enzymes are activated by GABAB receptors to inhibit dendritic Ca2+ spikes. Irreversible inhibition of PKC by bisindolylmaleimide I (1 μm) led to prolonged dendritic Ca2+ spikes (Fig. S2). This indicated that activation of PKC could be a signalling cascade to inhibit Ca2+ spikes. If GABAB receptor activation upregulated the activity of PKC to inhibit dendritic Ca2+ spikes, pretreatment with bisindolylmaleimide I would occlude the effect of GABAB activation with baclofen. However, we found that local application of baclofen to the apical tuft of neurons pretreated with bisindolylmaleimide I inhibited Ca2+ spikes to a similar extent as in untreated cells (untreated cells, 10.5 ± 1.6%; n = 10; bisindolylmaleimide I, 11.9 ± 0.2%; n = 3; P > 0.05; Fig. 5B and H).
Figure 5. GABAB receptors inhibit Ca2+ spikes independent of protein kinases.
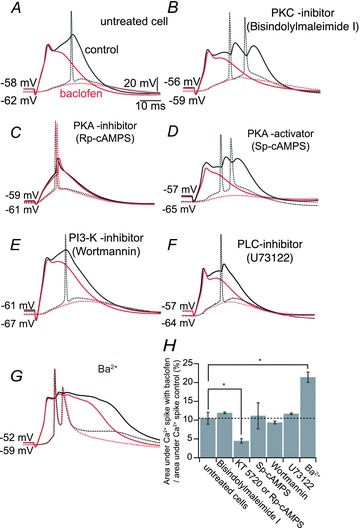
A, dendritic Ca2+ spike (black solid sweep) leading to somatic AP (black dashed sweep). Baclofen (50 μm; red sweeps) applied locally to the apical tuft induced inhibition of the Ca2+ spike. B–F, simultaneous double patch-clamp recording from the apical tuft and soma as in A, showing the baclofen-induced inhibition of Ca2+ spikes in cells pretreated with bisindolylmaleimide I (1 μm) to inhibit protein kinase C (PKC; B); Rp-cAMPS (1 mm) to inhibit cAMP-dependent protein kinases I and II (PKA; C); Sp-cAMPS (60–100 μm) to overactivate PKA (D); wortmannin (400 nm) to inhibit phosphatidylinositol 3-kinase (PI3K; E); and U73122 (1 μm) to inhibit phospholipase C (PLC; F). G, simultaneous double patch-clamp recording from the apical tuft and soma as in A, showing the baclofen-induced inhibition of Ca2+ spikes in the presence of Ba2+ (200 μm). H, summary of the inhibitory effect of baclofen (normalized to the area underneath the Ca2+ spike control) for each pharmacological condition. Error bars indicate SEM. *P < 0.05.
Irreversible inhibition of PKA with either KT 5720 or Rp-cAMPS decreased the duration or inhibited the generation of Ca2+ spikes (Figs S2 and S3). Under these conditions, the effect of baclofen could not be determined (Fig. 5C). However, the irreversible activation of PKA by adding Sp-cAMPS (60–100 μm) to the recording pipette did not occlude the effect of baclofen in inhibiting the Ca2+ spike (11.2 ± 3.4%, n = 4; P > 0.05; Fig. 5D and H). This shows that GABAB-induced downregulation of the activity of PKA cannot explain the inhibitory action of baclofen. The inhibition of PI3K, a protein kinase known to be activated by Gi/o proteins (Salgado et al. 2007), was also ineffective in modifying the inhibitory effect of baclofen (9.4 ± 0.4%; n = 3; P > 0.05; Fig. 5E and H). Inhibition of PLC by U73122 has been reported to reduce the amplitude of GABAB-mediated Kir3 responses in dissociated neocortical pyramidal neurons (Sickmann et al. 2008); however, intracellular application of U73122 did not modify the effect of baclofen on dendritic Ca2+ spikes, (11.8 ± 3.5%; n = 4; P > 0.05; Fig. 5F and H).
Finally, selective blockade of Kir3 K+ channels by Ba2+ (Newberry & Nicoll, 1984) did not prevent the action of baclofen (control 10.5 ± 1.6 vs. 21.4 ± 1.4% in 200 μm Ba2+; Fig. 5G and H), ruling out that the inhibition of this conductance by GABAB receptors was responsible for the inhibition of the Ca2+ spike. The increase in dendritic Ca2+ spikes under Ba2+ could be attributed to the increased permeability of VDCCs to Ba2+ versus Ca2+ ions and/or the increase in local input resistance due to blockade of K+ channels.
Taken together our results indicate that the inhibitory effect of GABAB receptors on Ca2+ spikes is mediated predominantly by the downregulation of Cav1 VDCCs rather than the activation of Kir3 K+ currents. Moreover, this modulation did not depend on cytosolic signalling cascades commonly associated with GABAB receptors, but is likely mediated by a direct interaction of the Cav1 VDCCs with the Gi/o-βγ-subunit.
Discussion
Inhibitory modulation of dendritic function is gaining much attention as a key mechanism to influence dendritic computation (Murayama et al. 2009; Lovett-Barron et al. 2012; Palmer et al. 2012). In particular, the influence of inhibition on non-linear dendritic integration by the modulation of voltage-dependent ion channels in dendrites is important in order to understand the transformation of synaptic inputs to AP output. This study revealed a direct interaction of the GABAB receptor-activated G-protein βγ-subunit with Cav1 channels in the distal apical dendrite of cortical layer 5 pyramidal neurons. Furthermore, we demonstrated that this modulation of Cav1 channels is the molecular mechanism for the inhibition of Ca2+ spikes, which are regenerative dendritic depolarizations important for signal integration in cortical pyramidal neurons.
Differential roles of VDCCs in the generation of Ca2+ spikes
We found that all VDCCs are present in the distal Ca2+ spike initiation zone of cortical pyramidal neurons but with different relative contributions to the evoked Ca2+ transients. Similarly, it has been found that all VDCCs are present in the proximal stretch of the apical dendrites of various pyramidal cells and are activated by somatically evoked back-propagating APs (Jaffe et al. 1992; Yuste et al. 1994; Christie et al. 1995; Markram et al. 1995; Sabatini & Svoboda, 2000) or synaptic activity (Markram & Sakmann, 1994; Yuste et al. 1994; Magee et al. 1995; Magee & Johnston, 1995b; Bloodgood & Sabatini, 2007). Comparison of our results to the previous findings indicates that the VDCC subtypes are differentially distributed along the apical dendrite of layer 5 pyramidal neurons. The contribution of Cav1 channels to evoked Ca2+ transients is much larger (>50% contribution) in the distal parts of the apical dendrite, in which Ca2+ spikes can be generated as compared with more proximal parts (20% contribution; Markram et al. 1995). This suggests that the Ca2+ spike initiation zone is a special compartment in terms of ion channel composition (Larkum et al. 2009).
The use of non-specific high-VDCC blockers such as cadmium and cobalt has been shown to block the sustained plateau potential of dendritic Ca2+ spikes (Kim & Connors, 1993; Schiller et al. 1997; Larkum et al. 1999a, 2001; Larkum & Zhu, 2002). TTX and Ni2+ affect the initial fast component of Ca2+ spikes, pointing to Na+ and low-voltage-activated T-type Ca2+ currents as being responsible for the initiation of Ca2+ spikes (Larkum et al. 2001; Schaefer et al. 2003); however, the concentration of Ni2+ used in previous reports (>50 μm) is also known to block Cav2.3 channels (R-type; Soong et al. 1993; Randall & Tsien, 1997). Furthermore, Cav3 (T-type) conductances on their own can not account for the generation and duration of the Ca2+ spike (up to 50 ms; Larkum et al. 2001) given the fast inactivation kinetics (25 ms at 0 mV) and low percentage of available Cav3 channels at the dendritic resting membrane potential (<20% at −55 mV; Magee & Johnston, 1995a; Magee, 2008). We found that Ni2+ (50 μm) inhibited the generation of Ca2+ spikes to a large extent. The effect of Ni2+ was to increase the threshold for Ca2+ spike initiation because extra current injected to the distal dendrite reactivated Ca2+ spikes and their associated Ca2+ transients. Blockade of one fraction of the Ni2+-sensitive conductance (Cav2.3 channels) revealed a significant contribution of R-type conductances to the Ca2+ spike not previously reported. Our results are consistent with the apparent lack of T-type Ca2+ currents observed in nucleated patch-clamp recordings from somatosensory layer 5 pyramidal neurons (Almog & Korngreen, 2009).
Here we show that Cav1 (L-type) channels contribute predominantly to the sustained (>30 ms) dendritic plateau potential. It is this slow and long-lasting part of the dendritic Ca2+ spike that determines the number and pattern of somatic APs (Larkum et al. 2001). In this respect, Ca2+ spikes and Cav1 VDCCs are important for the computational properties of the thick tufted pyramidal neurons, such as performing coincidence detection and non-linear integration of synaptic inputs (Larkum et al. 1999b, 2009; Schaefer et al. 2003). All other VDCCs contribute to the Ca2+ signal, but they do have only a minor effect on the electrogenesis.
GABAB modulation of VDCCs
Spines from apical dendrites are biochemically independent compartments, well isolated from their parent dendrites (Muller & Connor, 1991; Yuste & Denk, 1995; Svoboda et al. 1996; Sabatini & Svoboda, 2000) that contain VDCCs (Sabatini & Svoboda, 2000; Sabatini et al. 2001; Bloodgood & Sabatini, 2007). Evidence from hippocampal pyramidal neurons has shown a differential compartmentalization of VDCCs between shafts and spines where Cav2.3 channels form functional microdomains within the spine (Sabatini & Svoboda, 2000; Bloodgood & Sabatini, 2007). In particular, GABAB receptor activation has been shown to inhibit Ca2+ transients evoked by somatic back-propagating APs only in the spines of CA1 pyramidal neurons, by inhibiting Cav2.3 channels (Sabatini & Svoboda, 2000). Our findings show that activation of GABAB receptors or blockade of Cav1 VDCCs both strongly inhibited Ca2+ transients evoked by Ca2+ spikes in the distal apical dendritic shaft as well as the neighbouring spines. This result points to a fundamental difference between cortical and hippocampal pyramidal neurons in terms of Ca2+ signalling and its modulation by GABAB receptors. In agreement with our results, recent work suggests that GABAB receptor activation inhibits Ca2+ transients in the dendritic shafts of L2/3 cortical pyramidal neurons, evoked by back-propagating APs. This effect was only partially occluded by nimodipine, indicating a GABAB-mediated inhibition of Cav1 VDCCs in addition to other VDCCs in these cells (Chalifoux & Carter, 2011). In the distal dendrites of L5 pyramidal neurons, only the blockade of Cav1 VDCCs occluded the postsynaptic actions of GABAB receptors. Furthermore, local activation of GABAB receptors inhibited isolated L-type Ca2+ currents recorded directly at the site of generation of Ca2+ spikes, showing unequivocally that GABAB receptors exert their inhibitory action on dendritic Ca2+ spikes by inhibiting L-type Ca2+ conductances. Thus, the functional coupling of GABAB receptors to VDCCs depends on cell type and/or the subcellular compartment.
GABAB receptors are localized along the apical dendrite of pyramidal neurons. At the subcellular level they are present in both dendritic spines and their parent dendritic shafts (Kulik et al. 2003). At the spine level, GABAB receptors form clusters together with Kir3.2 inwardly rectifying K+ channels, whereas in the shaft both proteins are segregated (Kulik et al. 2006). This raises the possibility that GABAB receptors in the shaft might be co-localized to Cav1 channels, although this has to be determined at the electron-microscopic level. Such a degree of co-localization is consistent with a membrane-delimited coupling between these proteins via a Gi/o protein (Karschin, 1999; Bettler et al. 2004; Pinard et al. 2010). Although this form of regulation has been shown for GABAB–Kir3.2 and GABAB–Cav2 interactions, it has never been demonstrated for GABAB–Cav1. Previous evidence already suggested an inhibitory role of GABAB receptors on L-type Ca2+ conductances even though the mechanism for this interaction remained elusive (Maguire et al. 1989; Marchetti et al. 1991; Scholz & Miller, 1991; Amico et al. 1995; Chalifoux & Carter, 2011). Our results now show for the first time that GABAB receptors can exert this modulatory action on dendritic Cav1 channels (L-type currents) via a membrane-delimited pathway. Three lines of evidence support this view. Firstly, GABAB-mediated inhibition persisted after pharmacological manipulation of different cytosolic protein kinases known to be associated with the activation of Gi/o proteins, namely PKA, PKC and PIK3 (Bettler et al. 2004; Salgado et al. 2007; Deng et al. 2009). Secondly, prepulse facilitation relieved the GABAB-mediated inhibition of dendritic L-type Ca2+ currents and, thirdly, L-type Ca2+ currents could be recovered by strong depolarizing voltage commands. The prepulse facilitation protocol has been shown to be effective for reversing the Gi/o-βγ-mediated inhibition of Cav2, but so far not for Cav1 VDCCs (Bourinet et al. 1996; Herlitze et al. 1996; Ikeda, 1996; Toth et al. 1996; Zhang et al. 1996; Zamponi et al. 1997; De Waard et al. 2005). Strong depolarizing voltages have been assumed to promote the uncoupling of the Gi/o-βγ-dimer from the βγ-binding site located in the intracellular loop I–II of the pore-forming α-subunit of the channel (De Waard et al. 2005). Indeed, this loop has been shown to act as a voltage sensor (Sandoz et al. 2004), and its movement during membrane depolarization might promote the dissociation. In addition, the recovery of L-type currents at higher depolarizations is consistent with a model by which the Gi/o-βγ complex shifts the activation curve of Cav1 VDCCs to more depolarized values, indicating their transition to a ‘reluctant state’ (Fig. 4D; De Waard et al. 2005).
The Gi/o-βγ-mediated regulation of VDCCs has mainly been tested in heterologous expression systems. This raises the possibility that native central neurons possess preserved Cav1 VDCCs susceptible to regulation through this pathway (Bourinet et al. 1996). Alternatively, Gi/o-βγ-mediated modulation might require additional factors like accessory proteins that were present in the specialized region of the neuron we investigated, but which were absent in the heterologous expression systems previously used. Our findings expand the repertoire of interactions between G-protein-coupled receptors and VDCCs opening new vistas in neuromodulation. Furthermore, our result implies that the Gi/o-βγ-subunit interaction with VDCCs is a common mechanism of action by which GABAB receptors inhibit VDCCs.
Acknowledgments
The authors thank Drs Lucy Palmer, Rogier Min and Thomas Gener for their comments on the manuscript; Dr Humberto Salgado for discussion; Natalie Nevian for Neurolucida reconstructions; and The Institute of Physics University of Bern for providing material for the construction of an electrophysiological set-up. This work was supported by: E.P.-G., Spanish Ministry of Science and Innovation (Spain) (SAF2010-18884); Swiss National Science Foundation (T.N., grant 3100A0-118395 and M.E.L., 3100A0-118395); M.E.L., SystemsX.ch (Neurochoice); T.N., equipment grant from the Berne University Research Foundation; and E.P.-G. and T.N., Hans-Sigrist-Foundation.
Glossary
- 4-AP
4-aminopyridine
- ACSF
artificial cerebral spinal fluid
- AP
action potential
- Gi/o-βγ
βγ-subunit of a Gi/o protein
- PI3K
phosphatidylinositol 3-kinase
- PKA
cAMP-dependent protein kinases I and II
- PKC
protein kinase C
- PLC
phospholipase C
- TEA
tetraethylammonium-chloride
- VDCCs
voltage-dependent calcium channels
Author contributions
E.P.-G., M.E.L. and T.N.: designed the experiments; E.P.-G. and T.N.: performed and analysed the experiments; E.P.-G., M.E.L. and T.N. wrote the manuscript. All authors approved the final version for publication.
References
- Almog M, Korngreen A. Characterization of voltage-gated Ca2+ conductances in layer 5 neocortical pyramidal neurons from rats. PLoS One. 2009;4:e4841. doi: 10.1371/journal.pone.0004841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amico C, Marchetti C, Nobile M, Usai C. Pharmacological types of calcium channels and their modulation by baclofen in cerebellar granules. J Neurosci. 1995;15:2839–2848. doi: 10.1523/JNEUROSCI.15-04-02839.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amitai Y, Friedman A, Connors BW, Gutnick MJ. Regenerative activity in apical dendrites of pyramidal cells in neocortex. Cereb Cortex. 1993;3:26–38. doi: 10.1093/cercor/3.1.26. [DOI] [PubMed] [Google Scholar]
- Bettler B, Kaupmann K, Mosbacher J, Gassmann M. Molecular structure and physiological functions of GABAB receptors. Physiol Rev. 2004;84:835–867. doi: 10.1152/physrev.00036.2003. [DOI] [PubMed] [Google Scholar]
- Bloodgood BL, Sabatini BL. Nonlinear regulation of unitary synaptic signals by CaV(2.3) voltage-sensitive calcium channels located in dendritic spines. Neuron. 2007;53:249–260. doi: 10.1016/j.neuron.2006.12.017. [DOI] [PubMed] [Google Scholar]
- Bourinet E, Soong TW, Stea A, Snutch TP. Determinants of the G protein-dependent opioid modulation of neuronal calcium channels. Proc Natl Acad Sci U S A. 1996;93:1486–1491. doi: 10.1073/pnas.93.4.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breton JD, Stuart GJ. Somatic and dendritic GABAB receptors regulate neuronal excitability via different mechanisms. J Neurophysiol. 2012;108:2810–2818. doi: 10.1152/jn.00524.2012. [DOI] [PubMed] [Google Scholar]
- Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol. 2000;16:521–555. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Few AP. Calcium channel regulation and presynaptic plasticity. Neuron. 2008;59:882–901. doi: 10.1016/j.neuron.2008.09.005. [DOI] [PubMed] [Google Scholar]
- Chalifoux JR, Carter AG. GABAB receptor modulation of voltage-sensitive calcium channels in spines and dendrites. J Neurosci. 2011;31:4221–4232. doi: 10.1523/JNEUROSCI.4561-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie BR, Eliot LS, Ito K, Miyakawa H, Johnston D. Different Ca2+ channels in soma and dendrites of hippocampal pyramidal neurons mediate spike-induced Ca2+ influx. J Neurophysiol. 1995;73:2553–2557. doi: 10.1152/jn.1995.73.6.2553. [DOI] [PubMed] [Google Scholar]
- Cox DH, Dunlap K. Pharmacological discrimination of N-type from L-type calcium current and its selective modulation by transmitters. J Neurosci. 1992;12:906–914. doi: 10.1523/JNEUROSCI.12-03-00906.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Waard M, Hering J, Weiss N, Feltz A. How do G proteins directly control neuronal Ca2+ channel function. Trends Pharmacol Sci. 2005;26:427–436. doi: 10.1016/j.tips.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Deng PY, Xiao Z, Yang C, Rojanathammanee L, Grisanti L, Watt J, Geiger JD, Liu R, Porter JE, Lei S. GABAB receptor activation inhibits neuronal excitability and spatial learning in the entorhinal cortex by activating TREK-2 K+ channels. Neuron. 2009;63:230–243. doi: 10.1016/j.neuron.2009.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin AC. G protein modulation of voltage-gated calcium channels. Pharmacol Rev. 2003;55:607–627. doi: 10.1124/pr.55.4.3. [DOI] [PubMed] [Google Scholar]
- Dolphin AC, Scott RH. Calcium channel currents and their inhibition by (−)-baclofen in rat sensory neurones: modulation by guanine nucleotides. J Physiol. 1987;386:1–17. doi: 10.1113/jphysiol.1987.sp016518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in the Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gidon A, Segev I. Principles governing the operation of synaptic inhibition in dendrites. Neuron. 2012;75:330–341. doi: 10.1016/j.neuron.2012.05.015. [DOI] [PubMed] [Google Scholar]
- Harris NC, Libri V, Constanti A. Selective blockade of the hyperpolarization-activated cationic current (Ih) in guinea pig substantia nigra pars compacta neurones by a novel bradycardic agent, Zeneca ZM 227189. Neurosci Lett. 1994;176:221–225. doi: 10.1016/0304-3940(94)90087-6. [DOI] [PubMed] [Google Scholar]
- Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA. Modulation of Ca2+ channels by G-protein βγ subunits. Nature. 1996;380:258–262. doi: 10.1038/380258a0. [DOI] [PubMed] [Google Scholar]
- Ikeda SR. Voltage-dependent modulation of N-type calcium channels by G-protein βγ subunits. Nature. 1996;380:255–258. doi: 10.1038/380255a0. [DOI] [PubMed] [Google Scholar]
- Jaffe DB, Johnston D, Lasser-Ross N, Lisman JE, Miyakawa H, Ross WN. The spread of Na+ spikes determines the pattern of dendritic Ca2+ entry into hippocampal neurons. Nature. 1992;357:244–246. doi: 10.1038/357244a0. [DOI] [PubMed] [Google Scholar]
- Karschin A. G protein regulation of inwardly rectifying K+ channels. News Physiol Sci. 1999;14:215–220. doi: 10.1152/physiologyonline.1999.14.5.215. [DOI] [PubMed] [Google Scholar]
- Kavalali ET, Zhuo M, Bito H, Tsien RW. Dendritic Ca2+ channels characterized by recordings from isolated hippocampal dendritic segments. Neuron. 1997;18:651–663. doi: 10.1016/s0896-6273(00)80305-0. [DOI] [PubMed] [Google Scholar]
- Kim HG, Connors BW. Apical dendrites of the neocortex: correlation between sodium- and calcium-dependent spiking and pyramidal cell morphology. J Neurosci. 1993;13:5301–5311. doi: 10.1523/JNEUROSCI.13-12-05301.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulik A, Vida I, Fukazawa Y, Guetg N, Kasugai Y, Marker CL, Rigato F, Bettler B, Wickman K, Frotscher M, Shigemoto R. Compartment-dependent colocalization of Kir3.2-containing K+ channels and GABAB receptors in hippocampal pyramidal cells. J Neurosci. 2006;26:4289–4297. doi: 10.1523/JNEUROSCI.4178-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulik A, Vida I, Lujan R, Haas CA, Lopez-Bendito G, Shigemoto R, Frotscher M. Subcellular localization of metabotropic GABAB receptor subunits GABAB1a/b and GABAB2 in the rat hippocampus. J Neurosci. 2003;23:11026–11035. doi: 10.1523/JNEUROSCI.23-35-11026.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkum ME, Kaiser KM, Sakmann B. Calcium electrogenesis in distal apical dendrites of layer 5 pyramidal cells at a critical frequency of back-propagating action potentials. Proc Natl Acad Sci U S A. 1999a;96:14600–14604. doi: 10.1073/pnas.96.25.14600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkum ME, Nevian T. Synaptic clustering by dendritic signalling mechanisms. Curr Opin Neurobiol. 2008;18:321–331. doi: 10.1016/j.conb.2008.08.013. [DOI] [PubMed] [Google Scholar]
- Larkum ME, Nevian T, Sandler M, Polsky A, Schiller J. Synaptic integration in tuft dendrites of layer 5 pyramidal neurons: a new unifying principle. Science. 2009;325:756–760. doi: 10.1126/science.1171958. [DOI] [PubMed] [Google Scholar]
- Larkum ME, Zhu JJ. Signaling of layer 1 and whisker-evoked Ca2+ and Na+ action potentials in distal and terminal dendrites of rat neocortical pyramidal neurons in vitro and in vivo. J Neurosci. 2002;22:6991–7005. doi: 10.1523/JNEUROSCI.22-16-06991.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkum ME, Zhu JJ, Sakmann B. A new cellular mechanism for coupling inputs arriving at different cortical layers. Nature. 1999b;398:338–341. doi: 10.1038/18686. [DOI] [PubMed] [Google Scholar]
- Larkum ME, Zhu JJ, Sakmann B. Dendritic mechanisms underlying the coupling of the dendritic with the axonal action potential initiation zone of adult rat layer 5 pyramidal neurons. J Physiol. 2001;533:447–466. doi: 10.1111/j.1469-7793.2001.0447a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovett-Barron M, Turi GF, Kaifosh P, Lee PH, Bolze F, Sun XH, Nicoud JF, Zemelman BV, Sternson SM, Losonczy A. Regulation of neuronal input transformations by tunable dendritic inhibition. Nature Neurosci. 2012;15:423–430. doi: 10.1038/nn.3024. S421–S423. [DOI] [PubMed] [Google Scholar]
- Magee JC. Dendritic voltage-gated ion channels. In: Stuart G, Spruston N, Häusser M, editors. Dendrites. Oxford: Oxford University Press; 2008. pp. 225–250. [Google Scholar]
- Magee JC, Christofi G, Miyakawa H, Christie B, Lasser-Ross N, Johnston D. Subthreshold synaptic activation of voltage-gated Ca2+ channels mediates a localized Ca2+ influx into the dendrites of hippocampal pyramidal neurons. J Neurophysiol. 1995;74:1335–1342. doi: 10.1152/jn.1995.74.3.1335. [DOI] [PubMed] [Google Scholar]
- Magee JC, Johnston D. Characterization of single voltage-gated Na+ and Ca2+ channels in apical dendrites of rat CA1 pyramidal neurons. J Physiol. 1995a;487:67–90. doi: 10.1113/jphysiol.1995.sp020862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC, Johnston D. Synaptic activation of voltage-gated channels in the dendrites of hippocampal pyramidal neurons. Science. 1995b;268:301–304. doi: 10.1126/science.7716525. [DOI] [PubMed] [Google Scholar]
- Maguire G, Maple B, Lukasiewicz P, Werblin F. Gamma-aminobutyrate type B receptor modulation of L-type calcium channel current at bipolar cell terminals in the retina of the tiger salamander. Proc Natl Acad Sci U S A. 1989;86:10144–10147. doi: 10.1073/pnas.86.24.10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetti C, Carignani C, Robello M. Voltage-dependent calcium currents in dissociated granule cells from rat cerebellum. Neuroscience. 1991;43:121–133. doi: 10.1016/0306-4522(91)90422-k. [DOI] [PubMed] [Google Scholar]
- Markram H, Helm PJ, Sakmann B. Dendritic calcium transients evoked by single back-propagating action potentials in rat neocortical pyramidal neurons. J Physiol. 1995;485(Pt 1):1–20. doi: 10.1113/jphysiol.1995.sp020708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Sakmann B. Calcium transients in dendrites of neocortical neurons evoked by single subthreshold excitatory postsynaptic potentials via low-voltage-activated calcium channels. Proc Natl Acad Sci U S A. 1994;91:5207–5211. doi: 10.1073/pnas.91.11.5207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller W, Connor JA. Dendritic spines as individual neuronal compartments for synaptic Ca2+ responses. Nature. 1991;354:73–76. doi: 10.1038/354073a0. [DOI] [PubMed] [Google Scholar]
- Murayama M, Pérez-Garci E, Nevian T, Bock T, Senn W, Larkum ME. Dendritic encoding of sensory stimuli controlled by deep cortical interneurons. Nature. 2009;457:1137–1141. doi: 10.1038/nature07663. [DOI] [PubMed] [Google Scholar]
- Nevian T, Helmchen F. Calcium indicator loading of neurons using single-cell electroporation. Pflugers Arch. 2007;454:675–688. doi: 10.1007/s00424-007-0234-2. [DOI] [PubMed] [Google Scholar]
- Nevian T, Sakmann B. Spine Ca2+ signaling in spike- timing-dependent plasticity. J Neurosci. 2006;26:11001–11013. doi: 10.1523/JNEUROSCI.1749-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newberry NR, Nicoll RA. Direct hyperpolarizing action of baclofen on hippocampal pyramidal cells. Nature. 1984;308:450–452. doi: 10.1038/308450a0. [DOI] [PubMed] [Google Scholar]
- Padgett CL, Slesinger PA. GABAB receptor coupling to G-proteins and ion channels. Adv Pharmacol. 2010;58:123–147. doi: 10.1016/S1054-3589(10)58006-2. [DOI] [PubMed] [Google Scholar]
- Palmer LM, Schulz JM, Murphy SC, Ledergerber D, Murayama M, Larkum ME. The cellular basis of GABAB-mediated interhemispheric inhibition. Science. 2012;335:989–993. doi: 10.1126/science.1217276. [DOI] [PubMed] [Google Scholar]
- Pérez-Garci E, Gassmann M, Bettler B, Larkum ME. The GABAB1b isoform mediates long-lasting inhibition of dendritic Ca2+ spikes in layer 5 somatosensory pyramidal neurons. Neuron. 2006;50:603–616. doi: 10.1016/j.neuron.2006.04.019. [DOI] [PubMed] [Google Scholar]
- Pinard A, Seddik R, Bettler B(2010) GABAB receptors: physiological functions and mechanisms of diversity. Adv Pharmacol. 58:231–255. doi: 10.1016/S1054-3589(10)58010-4. [DOI] [PubMed] [Google Scholar]
- Plummer MR, Logothetis DE, Hess P. Elementary properties and pharmacological sensitivities of calcium channels in mammalian peripheral neurons. Neuron. 1989;2:1453–1463. doi: 10.1016/0896-6273(89)90191-8. [DOI] [PubMed] [Google Scholar]
- Randall AD, Tsien RW. Contrasting biophysical and pharmacological properties of T-type and R-type calcium channels. Neuropharmacology. 1997;36:879–893. doi: 10.1016/s0028-3908(97)00086-5. [DOI] [PubMed] [Google Scholar]
- Sabatini BL, Maravall M, Svoboda K. Ca2+ signaling in dendritic spines. Curr Opin Neurobiol. 2001;11:349–356. doi: 10.1016/s0959-4388(00)00218-x. [DOI] [PubMed] [Google Scholar]
- Sabatini BL, Svoboda K. Analysis of calcium channels in single spines using optical fluctuation analysis. Nature. 2000;408:589–593. doi: 10.1038/35046076. [DOI] [PubMed] [Google Scholar]
- Salgado H, Bellay T, Nichols JA, Bose M, Martinolich L, Perrotti L, Atzori M. Muscarinic M2 and M1 receptors reduce GABA release by Ca2+ channel modulation through activation of PI3K/Ca2+-independent and PLC/Ca2+-dependent PKC. J Neurophysiol. 2007;98:952–965. doi: 10.1152/jn.00060.2007. [DOI] [PubMed] [Google Scholar]
- Sandoz G, Lopez-Gonzalez I, Stamboulian S, Weiss N, Arnoult C, De Waard M. Repositioning of charged I-II loop amino acid residues within the electric field by β subunit as a novel working hypothesis for the control of fast P/Q calcium channel inactivation. Eur J Neurosci. 2004;19:1759–1772. doi: 10.1111/j.1460-9568.2004.03216.x. [DOI] [PubMed] [Google Scholar]
- Schaefer AT, Larkum ME, Sakmann B, Roth A. Coincidence detection in pyramidal neurons is tuned by their dendritic branching pattern. J Neurophysiol. 2003;89:3143–3154. doi: 10.1152/jn.00046.2003. [DOI] [PubMed] [Google Scholar]
- Schiller J, Schiller Y, Stuart G, Sakmann B. Calcium action potentials restricted to distal apical dendrites of rat neocortical pyramidal neurons. J Physiol. 1997;505:605–616. doi: 10.1111/j.1469-7793.1997.605ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz KP, Miller RJ. GABAB receptor-mediated inhibition of Ca2+ currents and synaptic transmission in cultured rat hippocampal neurones. J Physiol. 1991;444:669–686. doi: 10.1113/jphysiol.1991.sp018900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sickmann T, Klose A, Huth T, Alzheimer C. Unexpected suppression of neuronal G protein-activated, inwardly rectifying K+ current by common phospholipase C inhibitor. Neurosci Lett. 2008;436:102–106. doi: 10.1016/j.neulet.2008.02.067. [DOI] [PubMed] [Google Scholar]
- Sodickson DL, Bean BP. GABAB receptor-activated inwardly rectifying potassium current in dissociated hippocampal CA3 neurons. J Neurosci. 1996;16:6374–6385. doi: 10.1523/JNEUROSCI.16-20-06374.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soong TW, Stea A, Hodson CD, Dubel SJ, Vincent SR, Snutch TP. Structure and functional expression of a member of the low voltage-activated calcium channel family. Science. 1993;260:1133–1136. doi: 10.1126/science.8388125. [DOI] [PubMed] [Google Scholar]
- Svoboda K, Tank DW, Denk W. Direct measurement of coupling between dendritic spines and shafts. Science. 1996;272:716–719. doi: 10.1126/science.272.5262.716. [DOI] [PubMed] [Google Scholar]
- Toth PT, Shekter LR, Ma GH, Philipson LH, Miller RJ. Selective G-protein regulation of neuronal calcium channels. J Neurosci. 1996;16:4617–4624. doi: 10.1523/JNEUROSCI.16-15-04617.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien RW, Lipscombe D, Madison DV, Bley KR, Fox AP. Multiple types of neuronal calcium channels and their selective modulation. Trends Neurosci. 1988;11:431–438. doi: 10.1016/0166-2236(88)90194-4. [DOI] [PubMed] [Google Scholar]
- Williams SR, Mitchell SJ. Direct measurement of somatic voltage clamp errors in central neurons. Nat Neurosci. 2008;11:790–798. doi: 10.1038/nn.2137. [DOI] [PubMed] [Google Scholar]
- Yuste R, Denk W. Dendritic spines as basic functional units of neuronal integration. Nature. 1995;375:682–684. doi: 10.1038/375682a0. [DOI] [PubMed] [Google Scholar]
- Yuste R, Gutnick MJ, Saar D, Delaney KR, Tank DW. Ca2+ accumulations in dendrites of neocortical pyramidal neurons: an apical band and evidence for two functional compartments. Neuron. 1994;13:23–43. doi: 10.1016/0896-6273(94)90457-x. [DOI] [PubMed] [Google Scholar]
- Zamponi GW, Bourinet E, Nelson D, Nargeot J, Snutch TP. Crosstalk between G proteins and protein kinase C mediated by the calcium channel α1 subunit. Nature. 1997;385:442–446. doi: 10.1038/385442a0. [DOI] [PubMed] [Google Scholar]
- Zhang JF, Ellinor PT, Aldrich RW, Tsien RW. Multiple structural elements in voltage-dependent Ca2+ channels support their inhibition by G proteins. Neuron. 1996;17:991–1003. doi: 10.1016/s0896-6273(00)80229-9. [DOI] [PubMed] [Google Scholar]