

Abstract
Energy intake is strongly influenced by vagal afferent signals from the stomach, and is also modulated by leptin. Leptin may be secreted from gastric epithelial cells, so we aimed to determine the direct effect of leptin on gastric vagal afferents under different feeding conditions. Female C57BL/6 mice were fed standard laboratory diet, high-fat diet or were food restricted. The expression of leptin receptor (Lep-R) and its signal transduction molecules in vagal afferents was determined by retrograde tracing and reverse-transcription polymerase chain reaction, and the relationship between leptin-immunopositive cells and gastric vagal afferent endings determined by anterograde tracing and leptin immunohistochemistry. An in vitro preparation was used to determine the functional effects of leptin on gastric vagal afferents and the second messenger pathways involved. Leptin potentiated vagal mucosal afferent responses to tactile stimuli, and epithelial cells expressing leptin were found close to vagal mucosal endings. After fasting or diet-induced obesity, potentiation of mucosal afferents by leptin was lost and Lep-R expression reduced in the cell bodies of gastric mucosal afferents. These effects in diet-induced obese mice were accompanied by a reduction in anatomical vagal innervation of the gastric mucosa. In striking contrast, after fasting or diet-induced obesity, leptin actually inhibited responses to distension in tension receptors. The inhibitory effect on gastric tension receptors was mediated through phosphatidylinositol 3-kinase-dependent activation of large-conductance calcium-activated potassium channels. The excitatory effect of leptin on gastric mucosal vagal afferents was mediated by phospholipase C-dependent activation of canonical transient receptor potential channels. These data suggest the effect of leptin on gastric vagal afferent excitability is dynamic and related to the feeding state. Paradoxically, in obesity, leptin may reduce responses to gastric distension following food intake.
Key points
Obesity occurs when energy intake exceeds expenditure, and the excess energy is stored as fat.
We show that, after a 14 h food deprivation or 12 weeks consumption of a high-fat diet, gastric vagal afferent responses to mechanical stimulation in the presence of the satiety peptide leptin are altered.
Leptin has an excitatory effect on gastric mucosal vagal afferents, which is abolished after food restriction or prolonged excess.
In contrast, leptin has an inhibitory effect on gastric tension-sensitive afferents, but only after food restriction or energy excess conditions.
These changes in the response to leptin in the stomach, after food restriction or prolonged high-fat feeding, occur in such a manner as to facilitate an increase in food intake in both conditions.
Introduction
The satiety hormone, leptin, which is secreted from adipose tissue, provides a signal to the hypothalamus reflecting the amount of fat stored (Considine et al. 1996; Yannakoulia et al. 2003) and serves as a long-term regulator of nutrient intake, adiposity and body weight (Owyang & Heldsinger, 2011). Levels of circulating leptin reflect the degree of adiposity (Considine et al. 1996). However, despite elevated leptin levels obese individuals do not show diminished appetite, suggesting a relative resistance to leptin (Frederich et al. 1995). An alternative explanation may be that leptin permits fat stores to be maintained at or around a set point, that is genetically determined but susceptible to environmental modification (Speakman et al. 2011).
Leptin is also expressed in, and secreted by, chief cells (Bado et al. 1998) and parietal (P) cells (Mix et al. 2000) in the gastric mucosa. Leptin released from this site could act on adjacent gastric vagal afferent endings to modulate peripheral signals in response to food intake. Indeed, leptin has been shown to directly activate cultured gastric and duodenal vagal afferents from rats (Peters et al. 2004, 2006), and the leptin receptor (Lep-R) is expressed in rat vagal afferent neurons (Buyse et al. 2001; Burdyga et al. 2002), many of which terminate in the stomach. A vagal action of gastric leptin may serve to augment the acute appetite-suppressant effects of circulating leptin (Peters et al. 2005). However, recent evidence shows that the effects of leptin on vagal afferents are lost with chronic high-fat diet (HFD) feeding (de Lartigue et al. 2011a, 2012).
Within the stomach wall there are two classes of mechanically sensitive gastric vagal afferents. In the muscular layer, tension receptors detect fullness by responding to distension and contraction of the stomach wall (Blackshaw et al. 1987; Wang et al. 2008a). In contrast, mucosal receptors are excited by mechanical contact of larger food particles with the epithelium, and may contribute to the discrimination of particle size. The net effect of activating mucosal afferents is to trigger vagal reflexes that slow gastric emptying and facilitate mechanical digestion in the stomach (Becker & Kelly, 1983). It is currently unknown whether leptin has differential effects on gastric vagal afferent subclasses or whether any such effects can be modified by caloric restriction or excess. Furthermore, the mechanism by which Lep-Rs trigger changes in the electrical activity of vagal afferent endings has not been established.
We therefore determined the effect of leptin on the mechanosensitivity of gastric vagal afferents under different feeding conditions, including a short-term restriction in food intake and long-term consumption of a HFD, and examined the second messenger signalling pathways activated by leptin. In addition, we established the relationship between leptin-containing cells in the gastric mucosa and vagal afferent endings.
Methods
Ethical approval
All studies were approved and performed in accordance with the guidelines of the Animal Ethics Committees of the University of Adelaide and SA Pathology, Adelaide, Australia.
Mice
All mice used for this study were housed with littermates in groups of four, unless otherwise stated. For the fed and fasted treatment groups, 8-week-old female C57BL/6 mice, housed individually, were fed ad libitum or fasted for 14 h prior to experimentation. In diet studies, 7-week-old female C57BL/6 mice were allowed to acclimatize for a period of 1 week, after which control mice were continued on a standard laboratory mouse diet (SLD; 7% energy from fat, n= 52), whilst the HFD mice (n= 51) were placed on a diet high in fat (60% of energy from fat; Specialty Feeds, Glen Forrest, WA, Australia) for a further 12 weeks. During this period the mice were weighed weekly. We have previously determined that female C57BL/6 mice fed a HFD have increased total body weight and fat mass over a 12 week feeding period compared with SLD mice (Kentish et al. 2012). Reduced mechanosensitivity of gastric afferents observed in female HFD mice (Kentish et al. 2012) has also been observed in intestinal afferents of male mice (Daly et al. 2011), suggesting that diet-induced modulation of vagal afferent satiety signals is independent of gender.
In vitro mouse gastro-oesophageal afferent preparation
This preparation has been described in detail previously (Page et al. 2002). In short, C57BL/6 mice were killed via CO2 inhalation and the thorax opened by a midline incision. The stomach and oesophagus, with intact vagal nerves, were removed. The stomach and oesophagus were opened out longitudinally midway between the two main vagal branches, either side of the oesophagus, and along the greater curvature of the stomach. The tissue was then pinned down mucosa side up in an organ bath containing a modified Krebs solution composed of (in mm): NaCl, 118.1; KCl, 4.7; NaHCO3, 25.1; NaH2PO4, 1.3; MgSO4·7H2O, 1.2; CaCl2, 1.5; citric acid, 1.0; glucose, 11.1; nifidipine, 0.001; bubbled with 95% O2–5% CO2. The dissection process was carried out at 4°C.
Characterization of gastric vagal afferent properties
Receptive fields of mechanically sensitive vagal afferents, described earlier, were initially located using mechanical stimulation by brushing gently along the mouse gastro-oesophageal preparation. Once located, specific stimuli were then applied. Mucosal stroking was performed using calibrated von Frey hairs (10–1000 mg), which were stroked across the mucosa at a rate of 5 mm s−1. Each receptive field was stroked 10 times, and mechanical responses from the middle eight strokes were taken for analysis. Circular tension was applied using a threaded hook attached to an underpinned point adjacent to the receptive field. The threaded hook was attached to a cantilever via a pulley close to the preparation. Standard weights (0.5–5 g) were then placed on the opposite end of the cantilever. Each weight was applied for 1 min with a break of 1 min between removing one weight and applying the next. After analysing the two stimulus–response curves, we classified a receptive field as either a mucosal or tension receptor.
Single-unit vagal afferent recordings
Single units were discriminated on the basis of action potential shape, duration and amplitude by use of Spike 2 software (Cambridge Electronic Design, Cambridge, UK). After mechanosensitivity of a receptive field was established, the effect of leptin on this mechanosensitivity was assessed by adding leptin (0.1 nm) to the Krebs solution and allowing it to equilibrate for 20 min. This equilibration period was observed so as to ensure penetration of the drug into all layers of the tissue. After this time the tension–response and stroke–response curves were redetermined. This procedure was repeated for leptin at increasingly higher doses (1–10 nm). Time-controlled experiments were performed in which there was no significant change in the mechanical responses over a comparable duration. The second messenger system used by leptin to elicit its effects on mechanosensitivity was established using inhibitors of Janus kinase 2 (JAK2)–AG490 (5 μm; Liu et al. 2011), phosphatidylinositol 3-kinase (PI3K)–wortmannin (5 nm; Shyu et al. 2010), phosphodiesterase 3 (PDE3)–cilostamide (5 μm; Williams & Malik, 1990), phospholipase C (PLC)–U73122 (10 μm; Rogers & Hermann, 2008), large-conductance calcium-activated potassium channels (BKCa)–iberiotoxin (100 nm; Womack & Khodakhah, 2002) and canonical transient receptor potential cation (TRPC) channels–2-APB (30 μm; Lievremont et al. 2005), at concentrations previously reported in the literature before and after the addition of leptin (10 nm) in a separate series of experiments to the leptin dose–response experiments.
Drugs
Stock solutions of all drugs were kept frozen at −80°C and diluted to their final concentration in Krebs solution immediately prior to their use in the experiment. Leptin was obtained from Sigma-Aldrich (Sydney, Australia). Iberiotoxin, wortmannin, cilostamide, U73122, 2-APB and AG490 were obtained from Tocris Bioscience (Bristol, UK).
Nodose ganglia (NDG) quantitative reverse-transcription polymerase chain reaction (RT-PCR)
NDG were removed bilaterally from mice from all four experimental groups. Total RNA was extracted using an RNeasy Micro Kit (Qiagen, Doncaster, Australia) according to the manufacturer's instructions. RNA was quantified by measuring the absorbance at 260 nm (A260) using a NanoDrop™ ND 1000 spectrophotometer (Thermo Scientific), and RNA purity was estimated via the 260/280 absorbance ratio. Quantitative RT-PCR reactions were performed as described in detail previously (Hughes et al. 2007). Primers targeting Lep-R, TRPC1, TRPC5 and β-tubulin were pre-designed Quantitect Primer assays (Qiagen). The primer pairs for KCa1.1 were designed using Primer 3.0 software (Applied Biosystems, Foster City, USA). Forward primer (TCTACTTTGGCTTGCGGTTT) and reverse primer (CAAGCCAAGCCAACTTCTGT) for KCa1.1 were supplied by Geneworks (Adelaide, Australia). RT-PCR reactions were carried out using 10 ng of RNA under the following conditions: reverse transcription, 50°C for 30 min; initial PCR activation, 95°C for 15 min: PCR cycles 94°C for 15 s, 55°C for 30 s and 72°C for 30 s repeated for 50 cycles. A melt curve was obtained to confirm the specificity of the products produced, and products were visualised using 3% agarose gel electrophoresis with ethidium bromide staining. Relative RNA levels were calculated using the comparative computed tomography method as described previously (Pfaffl et al. 2002). Quantitative data are expressed as mean ± SEM.
Anterograde tracing
This procedure has been previously documented (Page et al. 2009). In brief, mice from the SLD (n= 7) and HFD (n= 7) groups were anaesthetised with isoflurane (1–1.5% in oxygen). The left NDG was exposed, and 0.5 μl of a horseradish peroxidase conjugate of wheat-germ agglutinin (WGA-HRP; 4 mg ml−1; Vector Laboratories) was pressure injected into the NDG via a glass micropipette (i.d. 25 μm). The injection site was dried and skin incision closed. Antibiotic (terramycin; 10 mg kg−1) and analgesic (butorphanol; 5 mg kg−1) were administered subcutaneously. Two days following injection of tracer (WGA-HRP), into the left nodose, the stomach was removed and dissected into ventral and dorsal flat sheets along the greater and lesser curvature. Frozen serial transverse sections of ventral flat sheet (10 μm) were then cut for immunohistochemistry. The dorsal flat sheet had submucosa and mucosa gently removed leaving the smooth muscle layers and myenteric plexus intact.
Permanent visualization of WGA-HRP was carried out using tyramide signal amplification (Perkin Elmer, USA; Page et al. 2009). Briefly, dorsal sheet whole-mounts and on-slide sections were rinsed in TNT buffer (0.05% Tween 20, 0.15 m NaCl and 0.1 m Tris–HCl, pH 7.5), blocked for 30 min in TNB (supplied with Perkin-Elmer TSA reagent kit) and then reacted for 20 min with tyramide-biotin, diluted 1:50 in amplification diluent. The tissue was then rinsed in TNT buffer and reacted with streptavidin conjugated to Alexa Fluor® 647 (1:200; Invitrogen) for 1–2 h at room temperature. Frozen sections were subsequently processed for leptin immunohistochemistry, then both sections and whole-mount specimens were mounted on slides and coverslipped using ProLong antifade (Invitrogen).
Retrograde tracing
Cell bodies of gastric vagal afferents innervating specific stomach layers were identified using differential tracing from the stomach as previously documented (Page et al. 2009).
Gastric muscle
SLD (n= 5) and HFD (n= 5) mice were anaesthetised with isoflurane (1–1.5% in oxygen), a laparotomy performed, and an Alexa Fluor® 555 conjugate of cholera toxin β-subunit (CTB-AF555 (0.5%); Invitrogen) injected subserosally into the muscularis externa of the proximal stomach using a 30-gauge Hamilton syringe. Multiple equally spaced injections of 2 μl were made parallel to and 1–2 mm from the lesser curvature on both dorsal and ventral surfaces (total volume 10 μl). The injection sites were dried, the laparotomy incision was closed, and antibiotic and analgesic were administered as above.
Gastric mucosa
SLD (n= 5) and HFD (n= 5) mice were anaesthetised with isoflurane (1–1.5% in oxygen), a laparotomy performed and a mucolytic (10%N-acetylcysteine; 200 μl) was injected into the stomach lumen, then removed via syringe after 5 min, followed by two saline rinses (200 μl each). Subsequently, 10 μl of 0.5% CTB-AF555 was injected into the proximal gastric lumen via a 30-gauge Hamilton syringe, and the proximal stomach walls gently opposed to expose the dorsal and ventral surfaces to tracer. The laparotomy incision was then closed, and antibiotic and analgesic administered as above. Food and water were withheld for 2 h postoperatively to maximize exposure of tracer.
After 2 days, traced mice were anaesthetised and perfused as indicated above. The left and right NDG were then removed and fixed in 4% paraformaldehyde/phosphate buffer at room temperature for 4 h and subsequently cryoprotected in 30% sucrose at 4°C for 24 h. Frozen serial transverse sections (10 μm) were then cut for immunohistochemistry.
Due to the ethical consideration of restricting food intake in mice a few days after surgery, these studies were only performed on SLD and HFD mice. We use the tracer WGA-HRP because WGA is widely accepted as one of the optimal anterograde tracers for filling of fine varicosity endings in vagal afferent pathways, while HRP allows for tyramide signal amplification to resolve fine anatomical structures (Kressel, 1998).
Laser capture microdissection
Retrogradely traced NDG were removed and dissociated before being cultured on a duplex dish for 2 h at 37°C in 5% CO2. Cells were then subject to laser-capture microdissection, performed on a P.A.L.M.® microbeam microdissection system (Carl Zeiss, Jena, Germany). Fluorescent-labelled nodose neurons were microdissected and catapulted directly into a lysis and stabilisation buffer (Buffer RLT, RNeasy Micro RNA extraction Kit, Qiagen) containing 0.14 mβ-mercaptoethanol (Sigma-Aldrich, Australia). The number of neurons captured varied with diet so that the maximum neurons possible were captured in order to maximise the RNA yield. RNA was extracted from these cells using the same protocol as for whole NDG (Hughes et al. 2007). There was no significant difference in RNA yield between the diet groups.
Immunohistochemistry
Immunohistochemistry was carried out in anterograde-traced stomach sections as described previously (Page et al. 2009). The primary anti-leptin antibody (rabbit polyclonal, Santa Cruz) was diluted 1:200 and a chicken anti-rabbit Alexa Fluor® 488 secondary antibody (1:200; Invitrogen) was used. Slide sections and whole-mounts were visualized using an epifluorescence microscope (BX-51, Olympus, Australia) equipped with filters for Alexa Fluor® 488 and 647, with images acquired by a CoolSnapfx monochrome digital camera (Roper Scientific, Tuscon, AZ, USA). Pseudocoloured fluorescence images were overlaid and fluorescence imaging software (Analysis LifeScience, Olympus) used to calculate the percentage of complete traced NDG neurons and density of traced vagal afferent endings in the stomach. Luminance of images was not adjusted. Counts were performed on a minimum of 10 sections per animal.
Statistical analysis
All data in graphs are expressed as mean ± SEM with n= the number of individual animals used. Vagal afferent stimulus–response curves and weight change were analysed using two-way ANOVA and Bonferroni post hoc tests. We compared the effects of leptin and mechanical stimuli on the response of tension and mucosal receptors using a two-way ANOVA, to establish if leptin affected the response to mechanical stimulation. The effect of diet on the modulatory action of leptin on responses to mechanical stimulation was determined by assessing the response to either stroking (50 mg von Frey hair; mucosal receptors) or tension (3 g; tension receptors) at different concentrations of leptin (0.1, 1 and 10 nm). Significant differences between diets and leptin concentration were assessed using two-way ANOVA to determine if diet caused a change in leptin effect. RNA levels, fat mass and vagal afferent profiles were analysed using unpaired t tests. Significance was defined at P < 0.05.
Results
Effect of leptin on gastric vagal afferent mechanosensitivity in fed and fasted mice
As previously reported (Kentish et al. 2012), short-term restriction of food intake consistently decreased the mechanosensitivity of tension receptors to circumferential stretch (P < 0.05; two-way ANOVA: data not shown), but not mucosal afferent responses to stroking.
In normal fed mice, leptin (0.1–10 nm) potentiated the mechanosensitivity of mucosal receptors (P < 0.01; leptin effect: two-way ANOVA; Fig. 1Aa and C). This effect was completely lost in fasted mice (Fig. 1Ab). When the response to mucosal stroking (50 mg) at varying concentrations of leptin was plotted (Fig. 1Ac), it was evident that fasting reduces mucosal receptor sensitivity to leptin (P < 0.001; diet effect; two-way ANOVA). Leptin had no effect on tension receptor mechanosensitivity in fed mice (Fig. 1Ba); however, in fasted mice the mechanosensitivity of these receptors was reduced by leptin (P < 0.05; leptin effect: two-way ANOVA; Fig. 1Bb and D). When the percentage change in response to a 3 g load was plotted against varying concentrations of leptin (Fig. 1Bc), it was evident that a restricted diet increased sensitivity of tension receptors to inhibition by leptin (P < 0.01; diet effect; two-way ANOVA). Recordings were also made from oesophageal receptors that showed effects of leptin similar to those in gastric afferents (Supplementary Fig. 1A and B).
Figure 1. Fasting changes the effects of leptin on gastric vagal afferent mechanosensitivity.
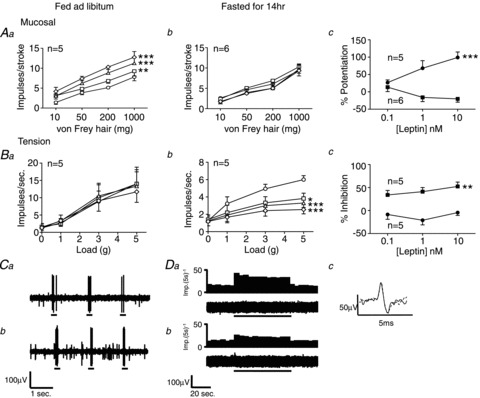
Single fibre recording stimulus–response curves of mucosal (Aa, n= 5; Ab, n= 6) and tension (Ba, n= 5; Bb, n= 5) receptors to mucosal stroking and circular tension, respectively, in fed (Aa and Ba) and fasted mice (Ab and Bb) before ○ and after exposure to leptin 0.1 nm□, 1 nmΔ and 10 nm◊. Ac and Bc, percentage change in response to a 50 mg von Frey hair and 3 g tension, respectively, compared with control at varying concentrations of leptin in fed (•) and fasted (▪) mice. *P < 0.05, **P < 0.01, ***P < 0.001 vs. control (two-way ANOVA). Data are expressed as mean ± SEM. C, original recording, from a fed mouse, of a mucosal receptor response to mucosal stroking with a 50 mg von Frey hair (a) prior to leptin and (b) after addition of leptin (10 nm). D, original recording, from a fasted mouse, of a tension receptor response to 3 g circular tension (a) prior to leptin, (b) after addition of leptin (10 nm), and (c) the average spike shape of the tension receptor prior to leptin (continuous line) and after addition of leptin (dashed line) illustrating that both responses were obtained from the same unit.
Effect of leptin in diet-induced obese mice
Mice fed the HFD gained a greater amount of body weight (P < 0.001; two-way ANOVA) and fat mass (P < 0.01; unpaired t test) than age-matched controls (Supplementary Fig. 2).
As previously reported (Kentish et al. 2012), the mechanosensitivity of tension receptors was reduced in HFD compared with SLD mice (P < 0.05, two-way ANOVA), and there was no difference in the mechanosensitivity of gastric mucosal receptors between HFD and SLD mice (data not shown).
In SLD mice, leptin (0.1–10 nm) increased the mechanosensitivity of mucosal receptors (P < 0.001; leptin effect: two-way ANOVA; Fig. 2Aa and C), a result similar to that reported in 8-week-old fed mice (above), indicating no change in leptin effect with age. Leptin failed to increase the mechanosensitivity of mucosal receptors in HFD mice (Fig. 2Ab). When the response to mucosal stroking (50 mg) was plotted against leptin concentration (Fig. 2Ac), it was evident that a HFD diet reduced the sensitivity of gastric mucosal receptors to leptin (P < 0.05; diet effect; two-way ANOVA). Correspondingly, the effect of leptin on tension receptor mechanosensitivity was switched from no effect in SLD (Fig. 2Ba) to inhibition in HFD mice (P < 0.001; leptin effect: two-way ANOVA; Fig. 2Bb and D). When the percentage change in response to 3 g load was plotted against concentration of leptin (Fig. 2Bc), it was evident that a HFD diet increased the sensitivity of tension receptors to leptin (P < 0.01; diet effect; two-way ANOVA). Oesophageal vagal afferents were also recorded, and their modulation by leptin closely mirrored that reported for gastric afferents (Supplementary Fig. 1C and D).
Figure 2. The effect of leptin on gastric vagal afferent mechanosensitivity is modified by high fat diet (HFD)-induced obesity.
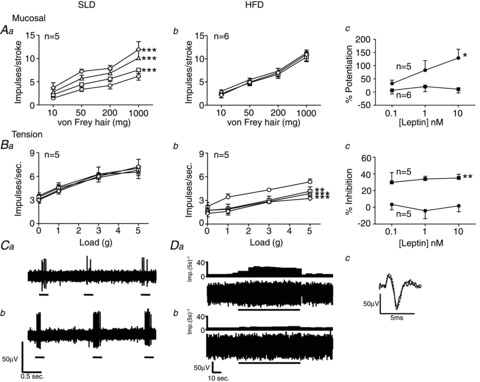
Single fibre recording stimulus–response curves of mucosal (Aa, n= 5; Ab, n= 6) and tension (Ba, n= 5; Bb, n= 5) receptors to mucosal stroking and circular tension, respectively, in standard laboratory diet (SLD; Aa and Ba) and high-fat diet (HFD)-fed mice (Ab and Bb) before ○ and after exposure to leptin 0.1 nm□, 1 nM Δ and 10 nm◊. Ac and Bc, percentage change in response to 50 mg von Frey hair and 3 g tension, respectively, compared with control at varying concentrations of leptin in SLD (•) and HFD (▪) mice. *P < 0.05, **P < 0.01, ***P < 0.001 vs. control (two-way ANOVA). Data are expressed as mean ± SEM. C, original recording, from a SLD mouse, of a mucosal receptor response to mucosal stroking with a 50 mg von Frey hair (a) prior to leptin and (b) after addition of leptin (10 nm). D, original recording, from a HFD mouse, of a tension receptor response to 3 g circular tension (a) prior to leptin, (b) after addition of leptin (10 nm), and (c) the average spike shape of the tension receptor prior to leptin (continuous line) and after addition of leptin (dashed line), illustrating that both responses were obtained from the same unit.
Lep-R expression
To determine if the differential effects of leptin were due to changes in Lep-R expression, we first compared mRNA levels of Lep-R in whole NDG. We found there was no difference in Lep-R expression in the fed and fasted mice (Fig. 3Aa), or SLD and HFD mice (Fig. 3Ab). However, following differential retrograde tracing to identify muscular or mucosal gastric afferents (Page et al. 2009), we observed a 94% reduction in Lep-R expression in mucosal afferents from HFD compared with SLD mice (P < 0.001; unpaired t test; Fig. 3Ba). In contrast, Lep-R expression in muscular afferents was similar in mice fed a HFD or SLD (P > 0.05; Fig. 3Bb). It appears, therefore, that Lep-R expression is regulated by feeding state only in mucosal afferents. Quantitative (Q)RT-PCR products were confirmed to be of appropriate size by agarose gel electrophoresis (Fig. 3C).
Figure 3. Changes in leptin receptor (Lep-R) expression and neuronal innervation of the mucosa induced by fasting and diet-induced obesity.
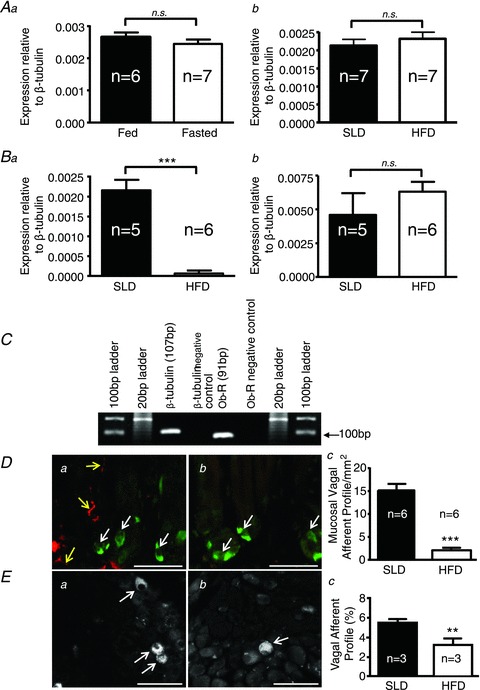
Aa, expression of Lep-R mRNA in whole nodose ganglia (NDG) of fed (n= 6) or fasted (n= 7) 8-week-old female C57BL/6 mice. Ab, expression of Lep-R mRNA in standard laboratory diet (SLD; n= 7) or high-fat diet (HFD; n= 7)-fed mice. Ba and b, expression of Lep-R mRNA in gastric mucosal and tension afferent neurons, respectively, in SLD (n= 5) or HFD (n= 6) mice. ***P < 0.001 vs. SLD (unpaired t test). C, agarose gel electrophoresis confirming quantitative reverse-transcription polymerase chain reaction (QRT-PCR) product size from the mucosal traced cells. D, combined leptin immunohistochemistry and anterogradely traced vagal afferent fibres in the mouse stomach (a: SLD; b: HFD), showing the relationship between leptin-containing epithelial cells (white arrows) and anterograde labelled vagal afferent fibres (yellow arrows). The stomach sections show a cross-section through glands at the base of villi. Dc, proportion of vagal afferent profiles in gastric mucosa of SLD (n= 6) or HFD (n= 6) mice following anterograde tracing (***P < 0.001; unpaired t test). E, vagal afferent cell bodies (white arrows) traced from the gastric mucosa in the NDG of SLD (a) or HFD (b) mice. Ec, proportion of vagal afferent cell bodies labelled in SLD (n= 3) or HFD (n= 3) mice (**P < 0.01; unpaired t test). Scale: 50 μm in all instances. Graphed data are expressed as mean ± SEM.
Leptin localization
To determine if the effects of leptin on mechanosensitivity of mucosal afferents can occur locally, we examined the relationship between leptin-immunopositive cells in the gastric mucosa and vagal afferent endings. Anterogradely traced mucosal vagal afferents were abundant in SLD mice and were closely associated with leptin-immunopositive cells (Fig. 3Da). In contrast, the number of mucosal afferents traced in HFD mice was reduced by 80% (P < 0.001; unpaired t test; Fig. 3Dc) and they were located at an increased distance from leptin-immunopositive cells (Fig. 3Db). In retrograde labelling experiments, the number of mucosal afferents that could be traced to the NDG was 50% lower in HFD compared with SLD mice (P < 0.01; unpaired t test; Fig. 3Ea–c), presumably due to the increased distance of the afferent endings from the lumen of the stomach where the tracer is added. In contrast, there was no change in the location or number of muscular afferents in either the stomach or NDG (Supplementary Fig. 3).
The second messaging systems utilized by leptin
The potentiating effect of leptin on mucosal afferents (fed (8 weeks old), SLD (20 weeks old) mice) and the inhibitory effect of leptin on tension-sensitive afferents (fasted (8 weeks old), HFD (20 weeks old) mice) suggests that the Lep-R is coupled to different second messenger systems. Evidence from previous work indicates that JAK2, PI3K, PDE3, PLC and TRPC channels may be involved in the potentiating effects of leptin on hypothalamic neuronal function (Zhoa et al. 2002; Hill et al. 2008; Jiang et al. 2008; Qiu et al. 2010). Using a range of blockers for these molecules, we show that the potentiating effect of leptin on afferent mechanosensitivity is abolished when any of these targets were inhibited individually (fed: Fig. 4Aa–e; SLD: Fig. 5Aa–e; indicating no age effect). However, the potentiating effect of leptin was not affected by the BKCa blocker iberiotoxin (fed: Fig. 4Af; SLD: Fig. 5Af). This suggests that the potentiating effect of leptin may involve in-series activation of signal transduction molecules illustrated in Fig. 5B. The presence of TRPC channels in vagal afferents was corroborated by QRT-PCR on whole NDG. We assessed levels of TRPC1 and TRPC5 previously shown to be expressed in NDG (Zhoa et al. 2009) and suggested to be activated by leptin in hypothalamic proopiomelanocortin (POMC) neurons (Qiu et al. 2010). There was no difference in mRNA transcript expression of TRPC5 in whole NDG of fed and fasted mice (P > 0.05; Fig. 4B). However, transcript expression of TRPC1 was higher in fed compared with fasted mice (P < 0.05; unpaired t test; Fig. 4C) and SLD compared with HFD mice (P < 0.01; unpaired t test: data not shown), suggesting this may be the channel subtype activated by leptin to potentiate function of mucosal afferents.
Figure 4. The excitatory effect of leptin on mucosal afferents from fed mice is conveyed through phospholipase C (PLC)-mediated activation of a canonical transient receptor potential cation (TRPC) channel.
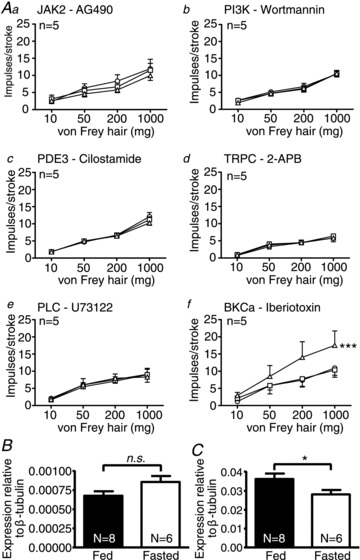
Single fibre recording stimulus–response curves of mucosal receptors to mucosal stroking in fed mice (Aa–f, n= 5) before ○ and after exposure to second messenger inhibitors (a: AG490 – 5 μm; b: wortmannin – 5 nm; c: cilostamide – 5 μm; d: 2-APB – 30 μm; e: U73122 – 10 μm; f: iberiotoxin – 100 nm) □, and in the presence of the respective second messenger inhibitor and leptin 10 nmΔ. ***P < 0.001 vs. control (leptin effect: two-way ANOVA). B and C, expression of TRPC5 and C1 mRNA, respectively, in whole nodose ganglia (NDG) of fed and fasted mice (*P < 0.05; unpaired t test). Data are expressed as mean ± SEM.
Figure 5. The excitatory effect of leptin on mucosal afferents from 20-week-old standard laboratory diet (SLD) mice is conveyed through phopholipase C (PLC)-mediated activation of a canonical transient receptor potential cation (TRPC) channel and therefore not affected by age.
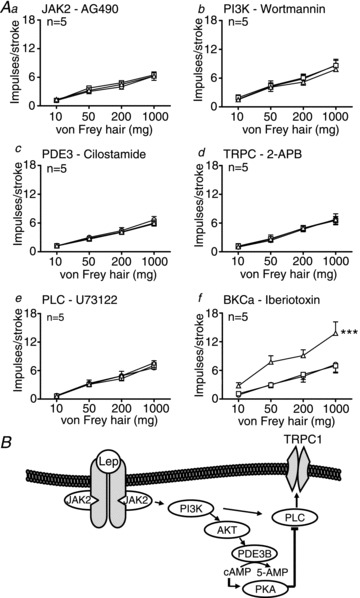
A Single fibre recording stimulus–response curves of mucosal receptors to mucosal stroking in SLD (Aa–f, n= 5) before ○ and after exposure to second messenger inhibitors (a: AG490 – 5 μm; b: wortmannin – 5 nm; c: cilostamide – 5 μm; d: 2-APB – 30 μm; e: U73122 – 10 μm; f: iberiotoxin – 100 nm) □, and in the presence of the respective second messenger inhibitor and leptin 10 nmΔ. ***P < 0.001 vs. control (leptin effect: two-way ANOVA). Data are expressed as mean ± SEM. B, schematic showing proposed signal transduction used by leptin to exert potentiation.
The inhibitory effect of leptin on mechanosensitivity of tension receptors (fasted, HFD mice) was eliminated by inhibitors targeting JAK2, PI3K and BKCa (fasted: Fig. 6Aa–c; HFD: Fig. 7Aa–c), but was unaffected by inhibitors targeting PDE3, PLC and TRPC (fasted: Fig. 6Ad–f; HFD: Fig. 7Ad–f). These data demonstrate a divergence in the signalling pathway prior to the effector channel, as illustrated in Fig. 7B. Performing QRT-PCR targeting the KCa1.1 subunit of BKCa, we found that transcript levels were higher in the whole NDG of fasted compared with fed mice (P < 0.05; unpaired t test; Fig. 6B) and HFD compared with SLD mice (P < 0.05; unpaired t test; data not shown).
Figure 6. Leptin inhibits gastric tension receptors through phosphatidylinositol 3-kinase (PI3K) activation of large-conductance calcium-activated potassium channel (BKCa) in fasted mice.
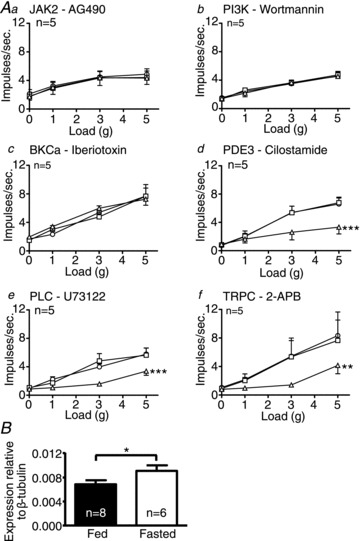
Single fibre recording stimulus–response curves of tension receptors to circular tension in fasted mice (Aa–f, n= 5) before ○ and after exposure to the second messenger inhibitors (a: AG490 – 5 μm; b: wortmannin – 5 nm; c: iberiotoxin – 100 nm; d: cilostamide – 5 μm; e: U73122 – 10 μm; f: 2-APB – 30 μm) □, and in the presence of the respective second messenger inhibitor and leptin 10 nmΔ. **P < 0.01, ***P < 0.001 vs. control (leptin effect: two-way ANOVA). B, expression of KCa1.1 mRNA in whole nodose ganglia (NDG) of fed and fasted mice (*P < 0.05; unpaired t test). Data are expressed as mean ± SEM.
Figure 7. Leptin-induced inhibition of tension receptor mechanosensitivity in mice fed a high-fat diet (HFD) is mediated by activation of large-conductance calcium-activated potassium channels (BKCa).
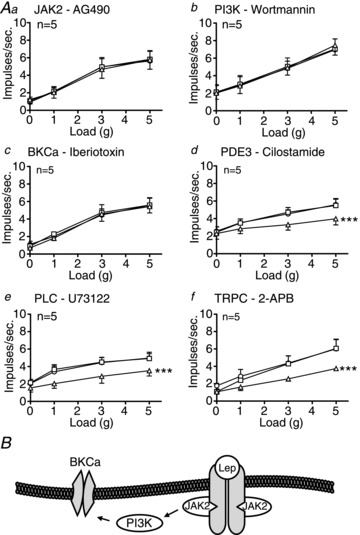
A, single fibre recording stimulus–response curves of tension receptors to circular tension in HFD mice (Aa–f, n= 5) before ○ and after exposure to second messenger inhibitors (a: AG490 – 5 μm; b: wortmannin – 5 nm; c: iberiotoxin – 100 nm; d: cilostamide – 5 μm; e: U73122 – 10 μm; f: 2-APB – 30 μm) □, and in the presence of the respective second messenger inhibitor and leptin 10 nmΔ. ***P < 0.001 vs. control (leptin effect: two-way ANOVA). Data are expressed as mean ± SEM. B, schematic showing proposed signal transduction used by leptin to exert inhibition.
Discussion
We have shown that the appetite-suppressant peptide leptin has a potent potentiating effect on mucosal vagal afferents during feeding, which is lost in response to fasting or HFD-induced obesity. In contrast, the mechanosensitivity of muscular vagal afferents was not affected by leptin during feeding, but was reduced by leptin after fasting or HFD-induced obesity. The varying effect of leptin on specific vagal afferent subtypes is due to coupling of the Lep-R to alternative second messenger systems. The changes in effect of leptin with feeding status are due to alterations in the coupling of Lep-R to these alternate second messenger systems and partly through reduced Lep-R expression. The data strongly suggest that leptin has important but opposite effects on vagal afferent signalling in normal and energy-imbalanced states.
The observation that leptin increases mechanosensitivity of mucosal receptors in fed mice, but not in fasted or HFD-fed mice, indicates that at least in part, the anorexigenic effect of leptin may be peripherally mediated. Consistent with this notion is the observation that leptin acts synergistically with cholecystokinin (CCK) on peripheral intestinal vagal afferent neurons to potentiate the effect of CCK on satiety (Barrachina et al. 1997; de Lartigue et al. 2010). In our preparation, leptin potentiates the mechanosensitivity of gastric vagal mucosal afferents. This mechanism of action makes sense, as it is well described that the tactile sensitivity of mucosal receptors facilitates detection of food particle size in the gastric lumen, and can initiate sensory feedback to delay gastric emptying and food intake via a vagal pathway (Becker & Kelly, 1983; Raybould & Tache, 1988). It has been reported that only 15% of vagal afferent cell bodies projecting to the rat fundus are Lep-R immunoreactive (Peiser et al. 2002), whereas in the current study all mucosal afferents from fed and SLD mice were potentiated by leptin. We speculate that, other than the possibility of species differences, Lep-R protein is rapidly trafficked to vagal peripheral endings, upon which leptin released from the gastric mucosa elicits modulation, and that the residual low levels of Lep-R in vagal cell bodies are difficult to detect with current immunohistochemical techniques.
The loss of effect of leptin on mucosal receptors in HFD mice is consistent with existing evidence of leptin resistance in obesity (Frederich et al. 1995; de Lartigue et al. 2011a, 2012), which we extend here to show is due to a specific reduction in Lep-R expression in mucosal vagal afferents, which may be paired with reduced expression of the putative effector channel, TRPC1. Furthermore, we identified a marked reduction in the close apposition of mucosal afferents to leptin-secreting cells in HFD mice. Such reorganisation of peripheral afferent endings would reduce the likelihood of gastric leptin–vagal signalling per se, further contributing to the net loss of signal from vagal afferents due to reduced leptin potentiation of individual mucosal afferents. We observed that Lep-R transcript expression in mouse whole NDG was unaffected by a short-term restriction in food intake or a HFD. In contrast, it has previously been demonstrated that Lep-R protein expression in the rat whole NDG is increased after fasting (Buyse et al. 2001). These differences may be species specific, or differences between transcript and protein regulation. In our traced vagal afferents we observed a quantitative reduction in Lep-R expression in mucosal afferents from HFD mice, which is in accord with the lack of effect of leptin on these afferents.
Evidence here of an inhibitory effect of leptin on tension receptors in fasting and HFD mice contrasts significantly with the extensive literature on leptin resistance in obesity (Frederich et al. 1995; de Lartigue et al. 2011b, 2012). There is evidence that leptin can inhibit neurons, for example, leptin has been shown to inhibit hypothalamic appetite-stimulatory neuropeptide Y neurons in rats, whilst also activating appetite-suppressing POMC neurons (Cowley et al. 2001; Morrison et al. 2005; Qiu et al. 2010). The novel observation in the current study is that the peripheral inhibitory effect of leptin on muscular afferents appears to occur in mutual exclusion to its excitatory effect on mucosal afferents, dependent on feeding status. However, due to the limited time points studied it is not possible to determine whether there is a point where both the potentiating and inhibiting effects of leptin are present. We have previously shown that mechanosensitivity of tension receptors is reduced after fasting or HFD (Kentish et al. 2012). Our current findings reveal that leptin causes a further decrease in the mechanosensitivity of tension receptors in both fasted and HFD mice. Tension receptors in the stomach detect distension (Powley & Phillips, 2004) following the ingestion of food. Gastric distension has been shown to activate satiety circuitry in the human brain and increase feelings of fullness (Wang et al. 2008b). Therefore, reduced mechanosensitivity of these afferents by leptin may permit ingestion of more food before satiation is reached, and although we have yet to undertake the behavioural studies to confirm this, rodents with diet-induced obesity have been shown to consume larger meals (Farley et al. 2003). This may be an adaptive function to allow an increase in energy intake to occur when food is readily available and perceived as ‘abundant’. The exact triggers for this ‘switch’ in the effect of leptin require further investigation, but lead candidates are specific nutrients and appetite-regulating hormones/peptides.
It is well established that the Lep-R is linked to JAK2 (Jiang et al. 2008) signalling through a PI3K pathway (Hill et al. 2008). Our data indicate that the modulatory effect of leptin on gastric afferent mechanosensitivity is likewise mediated through JAK2 and PI3K. There appears, however, to be a divergence in the second messenger pathways utilised by tension and mucosal afferents after this point. Importantly, our study suggests that the excitatory effect of leptin on gastric mucosal afferents may be mediated through PLC activation of a TRP channel, possibly TRPC1, which has been shown to be preferentially expressed in capsaicin-sensitive vagal afferents (Zhoa et al. 2009). This view is further supported by studies showing that the short-term appetite-suppressive effect of leptin is abolished upon capsaicin pre-treatment, and by reports that TRPC1 is highly expressed in POMC neurons that are leptin activated (Qiu et al. 2010). TRPC1 knockout mice are also known to gain more weight than their wild-type counterparts, indicating a role for TRPC1 in appetite regulation (Dietrich et al. 2007). The potentiating effect of leptin on gastric mucosal afferents also appears to rely on activity of PDE3. This could be due to an indirect activation of TRPC1, as cAMP and protein kinase A can inhibit PLC (Yada et al. 1989; Campbell et al. 1990), and would therefore prevent PLC activation of TRP channels. Accordingly, PDE3 may assist in mediating excitation by inhibiting the activity of molecules that oppose excitatory responses to leptin.
Leptin can activate ATP-activated potassium channels, via a PDE3-mediated pathway (Harvey et al. 1997; Williams et al. 2007), which represents a candidate mechanism for the inhibitory action of leptin on tension receptors in HFD and fasted mice. However, in the present study, PDE3 inhibition did not alter the inhibitory effect of leptin and therefore this pathway is less likely to be involved. Another report demonstrated that leptin can cause neuronal inhibition via BKCa (Shanley et al. 2002). Consistent with this we found that the inhibitory effect of leptin on gastric tension-sensitive afferents was abolished by blockade of BKCas. This was further supported by an increase in KCa1.1 expression seen in the whole NDG of fasted and HFD mice, suggesting that the channel responsible for signalling the inhibitory effect of leptin is upregulated by energy restriction or excess.
The in vitro effects of leptin on gastric vagal afferents were also observed in oesophageal afferents indicating that changes in afferent sensitivity following a HFD extend more generally to gastrointestinal afferent populations. However, the close apposition of vagal afferent endings and leptin-containing P-cells in the stomach mean that only the gastric afferent populations would be likely to undergo in vivo signalling changes based on leptin sensitivity. This selectivity, however, may be reduced in obesity where the plasma levels of leptin are significantly increased (Frederich et al. 1995), and needs to be considered in any potential targeted pharmacotherapy for obesity.
In conclusion, we have established that leptin has an excitatory effect on gastric mucosal vagal afferents that is abolished after both food restriction and an excess of energy intake. In contrast, leptin has an inhibitory effect on gastric tension-sensitive vagal afferents, evident only after food restriction or energy excess. These data highlight the plasticity in the mechanism of action of leptin under different feeding conditions and provide potential new targets for novel peripherally acting pharmacotherapies to modify food intake in obesity.
Translational perspective
Obesity is resistant to behavioural intervention but, to date, pharmacological approaches mainly aimed at appetite control in the CNS have had limited efficacy or unacceptable adverse effects. Targeting the initiation of the satiety signal in the periphery is an attractive therapeutic approach, and in the gut this involves integration of gastric and intestinal feedback signalling. Discovery of the satiety hormone leptin promised a new approach in the pharmacotherapy of obesity, but after more than 30 trials it was not possible to demonstrate that leptin was effective to ameliorate obesity. We hypothesised that the effect of leptin on gastric vagal afferent satiety signals is dependent on feeding status. This article reveals plasticity of the second messenger pathways activated by leptin, whereby HFD-induced obesity is associated with a change in the signalling pathway so as to limit satiety. In addition, we have shown that the response of gastric afferents to meal-related stimuli is dramatically altered by both calorie restriction and calorie excess, and thus undergoes extensive plasticity. It is important that this plasticity in the satiety signal is taken into account in any treatment regimes. It remains to be determined whether any ‘switch back’ occurs with diet-induced weight loss, or whether there is a time point beyond which any changes persist. However, the second messenger pathways specifically activated by leptin in HFD conditions in the stomach are potential targets for the treatment of obesity.
Acknowledgments
This work was supported by a National Health and Medical Research Council Project Grant (no. 565186). S.M.B. is supported by an NHMRC Australian Biomedical Research Fellowship. All authors declare that there is no conflict of interest with regard to this manuscript.
Glossary
- BKCa
large-conductance calcium-activated potassium channel
- CCK
cholecystokinin
- CTB-AF555
cholera toxin β-subunit conjugated with Alexa Fluor 555
- HFD
high-fat diet
- JAK2
janus kinase 2
- Lep-R
leptin receptor
- NDG
nodose ganglia
- P
parietal
- PDE3
phosphodiesterase 3
- PI3K
phosphatidylinositol 3-kinase
- PLC
phospholipase C
- POMC
proopiomelanocortin
- QRT-PCR
quantitative reverse-transcription polymerase chain reaction
- RT-PCR
reverse-transcription polymerase chain reaction
- SLD
standard laboratory diet
- TRPC
canonical transient receptor potential cation
- WGA-HRP
horseradish peroxidase conjugate of wheat-germ agglutinin
Author contributions
S.K. generated the obese mouse model, performed experiments, collected data, analysed data and wrote the manuscript; T.O. performed experiments and analysed data; N.I. performed immunohistochemistry experiments; R.Y. performed anterograde tracing; H.L. generated the obese mouse model; A.H. and S.B. developed laser capture microdissection for isolation of gastric neurones and QRT-PCR protocols; G.W. commented and helped construct the manuscript; A.B. aided in experimental design and manuscript construction; A.P. designed and performed experiments, obtained funding, analysed data and aided in the construction of manuscript. All authors approved the final version of the manuscript for publication.
Supplementary material
Supplementary Fig. 1
Supplementary Fig. 2
Supplementary Fig. 3
References
- Bado A, Levasseur S, Attoub S, Kermorgant S, Laigneau J, Bortoluzzi MI, Moizo L, Lehy T, Guerre-Millo M, Le Marchand-Brustel Y, Lewin MJM. The stomach is a source of leptin. Nature. 1998;394:790–793. doi: 10.1038/29547. [DOI] [PubMed] [Google Scholar]
- Barrachina MD, Martinez V, Wang L, Wei JY, Tache Y. Synergistic interaction between leptin and cholecystokinin to reduce short-term food intake in lean mice. Proc Natl Acad Sci U S A. 1997;94:10455–10460. doi: 10.1073/pnas.94.19.10455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker JM, Kelly KA. Antral control of canine gastric emptying of solids. Am J Physiol Gastrointest Liver Physiol. 1983;8:334–338. doi: 10.1152/ajpgi.1983.245.3.G334. [DOI] [PubMed] [Google Scholar]
- Blackshaw LA, Grundy D, Scratcherd T. Vagal afferent discharge from gastric mechanoreceptors during contraction and relaxation of the ferret corpus. J Auton Nerv Syst. 1987;18:19–24. doi: 10.1016/0165-1838(87)90130-5. [DOI] [PubMed] [Google Scholar]
- Burdyga G, Spiller D, Morris R, Lal S, Thompson DG, Saeed S, Dimaline R, Varro A, Dockray GJ. Expression of the leptin receptor in rat and human nodose ganglion neurones. Neuroscience. 2002;109:339–357. doi: 10.1016/s0306-4522(01)00474-2. [DOI] [PubMed] [Google Scholar]
- Buyse M, Ovesjö M-L, Goïot H, Guilmeau S, Péranzi G, Moizo L, Walker F, Lewin MJM, Meister B, Bado A. Expression and regulation of leptin receptor proteins in afferent and efferent neurons of the vagus nerve. Eur J Neurosci. 2001;14:64–72. doi: 10.1046/j.0953-816x.2001.01628.x. [DOI] [PubMed] [Google Scholar]
- Campbell MD, Subramaniam S, Kotlikoff MI, Williamson JR, Fluharty SJ. Cyclic AMP inhibits inositol polyphosphate production and calcium mobilization in neuroblastoma x glioma NG1 08–15 cells. Mol Pharmacol. 1990;38:282–288. [PubMed] [Google Scholar]
- Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, Ohannesian JP, Marco CC, McKee LJ, Bauer TL, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med. 1996;334:292–295. doi: 10.1056/NEJM199602013340503. [DOI] [PubMed] [Google Scholar]
- Cowley MA, Smart JL, Rubinstein M, Cerdán MG, Diano S, Horvath TL, Cone RD, Low MJ. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411:480–484. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- Daly DM, Park SJ, Valinsky WC, Beyak MJ. Impaired intestinal afferent nerve satiety signalling and vagal afferent excitability in diet induced obesity in the mouse. J Physiol. 2011;589:2857–2870. doi: 10.1113/jphysiol.2010.204594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lartigue G, Barbier de la Serre C, Espero E, Lee J, Raybould HE. Diet-induced obesity leads to the development of leptin resistance in vagal afferent neurons. Am J Physiol Endocrinol Metab. 2011a;301:E187–E195. doi: 10.1152/ajpendo.00056.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lartigue G, Barbier de la Serre C, Espero E, Lee J, Raybould HE. Leptin resistance in vagal afferent neurons inhibits cholecystokinin signaling and satiation in diet induced obese rats. PLoS One. 2012;7:e32967. doi: 10.1371/journal.pone.0032967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lartigue G, de La Serre CB, Raybould HE. Vagal afferent neurons in high fat diet-induced obesity; intestinal microflora, gut inflammation and cholecystokinin. Phyiol Behav. 2011b;105:100–105. doi: 10.1016/j.physbeh.2011.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lartigue G, Lur G, Dimaline R, Varro A, Raybould H, Dockray GJ. EGR1 is a target for cooperative interactions between cholecystokinin and leptin, and inhibition by ghrelin, in vagal afferent neurons. Endocrinology. 2010;151:3589–3599. doi: 10.1210/en.2010-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich A, Kalwa H, Storch U, Schnitzler MM, Salanova B, Pinkenburg O, Dubrovska G, Essin K, Gollasch M, Birnbaumer L, Gudermann T. Pressure-induced and store-operated cation influx in vascular smooth muscle cells is independent of TRPC1. Pflugers Arch. 2007;455:465–477. doi: 10.1007/s00424-007-0314-3. [DOI] [PubMed] [Google Scholar]
- Farley C, Cook JA, Spar BD, Austin TM, Kowalski TJ. Meal pattern analysis of diet-induced obesity in susceptible and resistant rats. Obesity. 2003;11:845–851. doi: 10.1038/oby.2003.116. [DOI] [PubMed] [Google Scholar]
- Frederich RC, Hamann A, Anderson S, Löllmann B, Lowell BB, Flier JS. Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nat Med. 1995;1:1311–1314. doi: 10.1038/nm1295-1311. [DOI] [PubMed] [Google Scholar]
- Harvey J, McKenna F, Herson PS, Spanswick D, Ashford ML. Leptin activates ATP-sensitive potassium channels in the rat insulin-secreting cell line, CRI-G1. J Physiol. 1997;504:527–535. doi: 10.1111/j.1469-7793.1997.527bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JW, Williams KW, Ye C, Luo J, Balthasar N, Coppari R, Cowley MA, Cantley LC, Lowell BB, Elmquist JK. Acute effects of leptin require PI3K signaling in hypothalamic proopiomelanocortin neurons in mice. J Clin Invest. 2008;118:1796–1805. doi: 10.1172/JCI32964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes PA, Brierley SM, Young RL, Blackshaw LA. Localization and comparative analysis of acid-sensing ion channel (ASIC1, 2, and 3) mRNA expression in mouse colonic sensory neurons within thoracolumbar dorsal root ganglia. J Comp Neurol. 2007;500:863–875. doi: 10.1002/cne.21204. [DOI] [PubMed] [Google Scholar]
- Jiang L, Li Z, Rui L. Leptin stimulates both JAK2-dependent and JAK2-independent signaling pathways. J Biol Chem. 2008;283:28066–28073. doi: 10.1074/jbc.M805545200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kentish S, Li H, Philp LK, O’Donnell TA, Isaacs NJ, Young RL, Wittert GA, Blackshaw LA, Page AJ. Diet-induced adaptation of vagal afferent function. J Physiol. 2012;590:209–221. doi: 10.1113/jphysiol.2011.222158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kressel M. Tyramide amplification allows anterograde tracing by horseradish peroxidase-conjugated lectins in conjunction with simultaneous immunohistochemistry. J Histochem Cytochem. 1998;46:527–533. doi: 10.1177/002215549804600413. [DOI] [PubMed] [Google Scholar]
- Lievremont JP, Bird GS, Putney JW., Jr Mechanism of inhibition of TRPC cation channels by 2- aminoethoxydiphenylborane. Molec Pharmacol. 2005;68:758–762. doi: 10.1124/mol.105.012856. [DOI] [PubMed] [Google Scholar]
- Liu Z, Qiu YH, Li B, Ma SH, Peng YP. Neuroprotection of interleukin-6 against NMDA-induced apoptosis and its signal-transduction mechanisms. Neurotox Res. 2011;19:484–495. doi: 10.1007/s12640-010-9215-x. [DOI] [PubMed] [Google Scholar]
- Mix H, Widjaja A, Jandl O, Cornberg M, Kaul A, Göke M, Beil W, Kuske M, Brabant G, Manns MP, Wagner S. Expression of leptin and leptin receptor isoforms in the human stomach. Gut. 2000;47:481–486. doi: 10.1136/gut.47.4.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison CD, Morton GJ, Niswender KD, Gelling RW, Schwartz MW. Leptin inhibits hypothalamic Npy and Agrp gene expression via a mechanism that requires phosphatidylinositol 3-OH-kinase signaling. Am J Physiol Endocrinol Metab. 2005;289:1051–1057. doi: 10.1152/ajpendo.00094.2005. [DOI] [PubMed] [Google Scholar]
- Owyang C, Heldsinger A. Vagal control of satiety and hormonal regulation of appetite. J Neurogastroenterol Motil. 2011;17:338–348. doi: 10.5056/jnm.2011.17.4.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page AJ, Martin CM, Blackshaw LA. Vagal mechanoreceptors and chemoreceptors in mouse stomach and esophagus. J Neurophysiol. 2002;87:2095–2103. doi: 10.1152/jn.00785.2001. [DOI] [PubMed] [Google Scholar]
- Page AJ, O’Donnell T, Cooper NJ, Young RL, Blackshaw LA. Nitric oxide as an endogenous peripheral modulator of visceral sensory neuronal function. J Neurosci. 2009;29:7246–7255. doi: 10.1523/JNEUROSCI.6099-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiser C, Springer J, Groneberg DA, McGregor GP, Fischer A, Lang RE. Leptin receptor expression in nodose ganglion cells projecting to the rat gastric fundus. Neurosci Lett. 2002;320:41–44. doi: 10.1016/s0304-3940(02)00023-x. [DOI] [PubMed] [Google Scholar]
- Peters JH, Karpiel AB, Ritter RC, Simasko SM. Cooperative activation of cultured vagal afferent neurons by leptin and cholecystokinin. Endocrinology. 2004;145:3652–3657. doi: 10.1210/en.2004-0221. [DOI] [PubMed] [Google Scholar]
- Peters JH, McKay BM, Simasko SM, Ritter RC. Leptin- induced satiation mediated by abdominal vagal afferents. Am J Physiol Regul Integr Comp Physiol. 2005;288:879–884. doi: 10.1152/ajpregu.00716.2004. [DOI] [PubMed] [Google Scholar]
- Peters JH, Ritter RC, Simasko SM. Leptin and CCK selectively activate vagal afferent neurons innervating the stomach and duodenum. Am J Physiol Regul Integr Comp Physiol. 2006;290:1544–1549. doi: 10.1152/ajpregu.00811.2005. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW, Horgan GW, Dempfle L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002;30:e36. doi: 10.1093/nar/30.9.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powley TL, Phillips RJ. Gastric satiation is volumetric, intestinal satiation is nutritive. Physiol Behav. 2004;82:69–74. doi: 10.1016/j.physbeh.2004.04.037. [DOI] [PubMed] [Google Scholar]
- Qiu J, Fang Y, Rønnekleiv OK, Kelly MJ. Leptin excites proopiomelanocortin neurons via activation of TRPC channels. J Neurosci. 2010;30:1560–1565. doi: 10.1523/JNEUROSCI.4816-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raybould HE, Tache Y. Cholecystokinin inhibits gastric motility and emptying via a capsaicin-sensitive vagal pathway in rats. Am J Physiol Gastrointest Liver Physiol. 1988;255:G242–G246. doi: 10.1152/ajpgi.1988.255.2.G242. [DOI] [PubMed] [Google Scholar]
- Rogers RC, Hermann GE. Mechanisms of action of CCK to activate central vagal afferent terminals. Peptides. 2008;29:1716–1725. doi: 10.1016/j.peptides.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanley LJ, Irving AJ, Rae MG, Ashford MLJ, Harvey J. Leptin inhibits rat hippocampal neurons via activation of large conductance calcium activated K+ channels. Nat Neurosci. 2002;5:299–300. doi: 10.1038/nn824. [DOI] [PubMed] [Google Scholar]
- Shyu KG, Wang BW, Chen WJ, Kuan P, Hung CR. Mechanism of the inhibitory effect of atorvastatin on endoglin expression induced by transforming growth factor-beta1 in cultured cardiac fibroblasts. Eur J Heart Failure. 2010;12:219–226. doi: 10.1093/eurjhf/hfq011. [DOI] [PubMed] [Google Scholar]
- Speakman JR, Levitsky DA, Allison DB, Bray MS, de Castro JM, Clegg DJ, Clapham JC, Dulloo AG, Gruer L, Haw S, Hebebrand J, Hetherington MM, Higgs S, Jebb SA, Loos RJ, Luckman S, Luke A, Mohammed-Ali V, O’Rahilly S, Pereira M, Perusse L, Robinson TN, Rolls B, Symonds ME, Westerterp-Plantenga MS. Set points, settling points and some alternative models: theoretical options to understand how genes and environments combine to regulate body adiposity. Dis Mod Mech. 2011;4:733–745. doi: 10.1242/dmm.008698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Tomasi D, Backus W, Wang R, Telang F, Geliebter A, Korner J, Bauman A, Fowler JS, Thanos PK, Volkow ND. Gastric distention activates satiety circuitry in the human brain. Neuroimage. 2008a;39:1824–1831. doi: 10.1016/j.neuroimage.2007.11.008. [DOI] [PubMed] [Google Scholar]
- Wang GJ, Tomasi D, Backus W, Wang R, Telang F, Geliebter A, Korner J, Bauman A, Fowler JS, Thanos PK, Volkow ND. Gastric distention activates satiety circuitry in the human brain. NeuroImage. 2008b;39:1824–1831. doi: 10.1016/j.neuroimage.2007.11.008. [DOI] [PubMed] [Google Scholar]
- Williams JL, Jr, Malik KU. Forskolin stimulates prostaglandin synthesis in rabbit heart by a mechanism that requires calcium and is independent of cyclic AMP. Circ Res. 1990;67:1247–1256. doi: 10.1161/01.res.67.5.1247. [DOI] [PubMed] [Google Scholar]
- Williams KW, Zsombok A, Smith BN. Rapid inhibition of neurons in the dorsal motor nucleus of the vagus by leptin. Endocrinology. 2007;148:1868–1881. doi: 10.1210/en.2006-1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womack MD, Khodakhah K. Characterization of large conductance Ca2+-activated K+ channels in cerebellar Purkinje neurons. Eur J Neurosci. 2002;16:1214–1222. doi: 10.1046/j.1460-9568.2002.02171.x. [DOI] [PubMed] [Google Scholar]
- Yada Y, Nagao S, Okano Y, Nozawa Y. Inhibition by cyclic AMP of guanine nucleotide-induced activation of phosphoinositide-specific phospholipase C in human platelets. FEBS Lett. 1989;242:368–372. doi: 10.1016/0014-5793(89)80503-4. [DOI] [PubMed] [Google Scholar]
- Yannakoulia M, Yiannakouris N, Bluher S, Matalas AL, Klimis-Zacas D, Mantzoros CS. Body fat mass and macronutrient intake in relation to circulating soluble leptin receptor, free leptin index, adiponectin, and resistin concentrations in healthy humans. J Clin Endocrinol Metab. 2003;88:1730–1736. doi: 10.1210/jc.2002-021604. [DOI] [PubMed] [Google Scholar]
- Zhoa AZ, Huan J, Gupta S, Pal R, Sahu A. A phosphatidylinositol 3-kinase-phosphodiesterase 3B-cyclic AMP pathway in hypothalamic action of leptin on feeding. Nat Neurosci. 2002;5:727–728. doi: 10.1038/nn885. [DOI] [PubMed] [Google Scholar]
- Zhoa H, Sprunger LK, Simasko SM. Expression of transient receptor potential channels and two-pore potassium channels in subtypes of vagal afferent neurons in rat. Am J Physiol Gastrointest Liver Physiol. 2009;298:G212–G221. doi: 10.1152/ajpgi.00396.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.