

Abstract
Hypocretin (orexin), a neuropeptide synthesized exclusively in the perifornical/lateral hypothalamus, is critical for drug seeking and relapse, but it is not clear how the circuitry centred on hypocretin-producing neurons (hypocretin neurons) is modified by drugs of abuse and how changes in this circuit might alter behaviours related to drug addiction. In this study, we show that repeated, but not single, in vivo cocaine administration leads to a long-lasting, experience-dependent potentiation of glutamatergic synapses on hypocretin neurons in mice following a cocaine-conditioned place preference (CPP) protocol. The synaptic potentiation occurs postsynaptically and probably involves up-regulation of AMPA-type glutamate receptors on hypocretin neurons. Phosphorylation of cAMP response element-binding protein (CREB) is also significantly increased in hypocretin neurons in cocaine-treated animals, suggesting that CREB-mediated pathways may contribute to synaptic potentiation in these cells. Furthermore, the potentiation of synaptic efficacy in hypocretin neurons persists during cocaine withdrawal, but reverses to baseline levels after prolonged abstinence. Finally, the induction of long-term potentiation (LTP) triggered by a high-frequency stimulation is facilitated in hypocretin neurons in cocaine-treated mice, suggesting that long-lasting changes in synapses onto hypocretin neurons would probably be further potentiated by other stimuli (such as concurrent environmental cues) paired with the drug. In summary, we show here that hypocretin neurons undergo experience-dependent synaptic potentiation that is distinct from that reported in other reward systems, such as the ventral tegmental area, following exposure to cocaine. These findings support the idea that the hypocretin system is important for behavioural changes associated with cocaine administration in animals and humans.
Key points
Repeated, but not single, in vivo cocaine exposure leads to an experience-dependent potentiation of glutamatergic synapses on hypocretin-producing neurons (hypocretin neurons) in mice.
The locus of synaptic potentiation is at the postsynaptic site of glutamatergic synapses on hypocretin neurons and the up-regulation of AMPA-type glutamate receptors may be involved.
Cocaine-induced synaptic potentiation is long-lasting and exists during the abstinence of cocaine.
The expression of tetanus-induced long-term potentiation is facilitated in hypocretin neurons in cocaine-treated mice.
These results may help us better understand the role of the hypocretin system in behavioural changes related to cocaine addiction in animals and humans.
Introduction
The lateral hypothalamus (LH) is known to be critical for reward-seeking (Gallistel et al. 1981). Rats will work for electrical stimulation of the LH (Olds & Milner, 1954; Olds, 1958; Adams et al. 1972; Goodall & Carey, 1975). In addition, rats and mice will self-administer drugs of abuse infused directly into the LH (Olds & Williams, 1980; Cazala et al. 1987). The neuropeptide hypocretin is synthesized exclusively in the perifornical/LH area (de Lecea et al. 1998; Peyron et al. 1998; Sakurai et al. 1998) and hypocretin neurons project to reward-associated regions such as the ventral tegmental area (VTA) and nucleus accumbens (NAc) (Fadel & Deutch, 2002; Balcita-Pedicino & Sesack, 2007). These neurons are activated, and transcription of the gene encoding hypocretin is induced in hypocretin neurons by rewarding stimuli such as food and drugs of abuse (Georgescu et al. 2003; Boutrel et al. 2005; Harris et al. 2005; Zhou et al. 2006; Zheng et al. 2007). Both activation of hypocretin neurons and administration of hypocretin directly into the VTA can reinstate morphine or cocaine seeking after extinction of self-administration of morphine or cocaine (Boutrel et al. 2005; Harris et al. 2005). The development of morphine dependence is attenuated in knockout mice lacking hypocretin and systemic blockade of hypocretin receptor 1 (HcrtR1) inhibits drug seeking induced by environmental cues and stress in rats (Georgescu et al. 2003; Boutrel et al. 2005; Harris et al. 2005). Despite an increasing body of evidence supporting the role of hypocretin neurons in addiction-related behaviours, it is not yet known how hypocretin neurons alter their activity following exposure to drugs of abuse.
It is widely accepted that experience-dependent synaptic plasticity is important for learning and memory and occurs during the development of neuronal circuitry (Malenka & Bear, 2004). Synaptic plasticity is also critical for the development of behaviours related to drug addiction (reviewed by Badiani & Robinson, 2004; Kauer, 2004; Kelley, 2004; Jones & Bonci, 2005; Kalivas & Volkow, 2005; Hyman et al. 2006; Kalivas, 2007; Kauer & Malenka, 2007). Long-term potentiation (LTP) and long-term depression (LTD), two major forms of experience-dependent synaptic plasticity, occur in several brain structures in response to drugs of abuse in animal models. Both acute and repeated administration of cocaine induces an LTP-like potentiation of glutamatergic synapses on dopaminergic (DA) neurons in the VTA of rodents (Ungless et al. 2001; Borgland et al. 2004; Liu et al. 2005). Other drugs of abuse, including nicotine, morphine and amphetamine, all induce LTP in DA neurons in the VTA (Mansvelder & McGehee, 2000; Saal et al. 2003; Faleiro et al. 2004). In addition, experience-dependent neural plasticity induced by drugs of abuse has been reported in other brain areas important for drug reward, including the NAc, amygdala and medial prefrontal cortex (Goussakov et al. 2006; Fu et al. 2007; Huang et al. 2007; Kourrich et al. 2007; Lu et al. 2009).
Hypocretin plays a pivotal role in drug-seeking behaviours (reviewed by Boutrel & de Lecea, 2008; Bonci & Borgland, 2009; Cason et al. 2010; Thompson & Borgland, 2011); however, it is not yet clear what synaptic changes take place on hypocretin neurons following exposure to drugs of abuse. In this study, we examined the hypothesis that cocaine induces experience-dependent synaptic plasticity in hypocretin neurons, which may contribute to the role of the hypocretin system in promoting behavioural changes associated with cocaine administration in animals and humans.
Methods
Animals
C57BL/6 mice (6–8 weeks old) expressing green fluorescent protein (GFP) selectively in hypocretin neurons under the control of a specific hypocretin promoter were used in this study. Mice were group housed and maintained on a 12 h/12 h light/dark cycle (lights on at 6 am) with free access to food and water under the care of Yale Animal Resources personnel. All studies were approved by the Yale University Animal Care and Use Committee and followed the NIH Guide for the Care and Use of Laboratory Animals.
Conditioned place preference (CPP)
CPP was performed in a 3-chamber Med Associates apparatus (Med Associates, Inc., St. Albans, VT, USA) using a non-biased protocol as reported previously (Hawes et al. 2008). Briefly, mice were transported in their home cages to the testing room about one hour before the first session and returned to the colony room at the end of the day. Baseline preference was measured on day 1 (at 12.30 pm) by allowing the mice to explore the CPP chambers freely for a total of 15 min. No baseline preference for either chamber was observed, and animals were then counter-balanced so that drug administration was randomly paired with one of the two training chambers. During the training phase (days 2, 3 and 4), mice were injected (i.p.) with vehicle (saline) at 10 am and confined for 30 min in one chamber. In the afternoon (3 pm), animals were isolated in the conditioning chamber following an injection of cocaine (10 mg kg−1, i.p.) or saline in control subjects. Pre-administration of a selective HcrtR1 antagonist SB334867 (30 mg kg−1, i.p.) was performed in both morning and afternoon sessions 30 min before vehicle or cocaine injections to evaluate the contribution of HcrtR1 to cocaine CPP. On day 5 (testing at 12.30 pm, chosen to be intermediate between saline and drug training times), animals were placed in the neutral compartment of the place-conditioning apparatus, and were allowed free access to both conditioning chambers for 15 min. Total time spent in each chamber was recorded and a preference score was derived by calculating the difference between the time spent in the drug-paired chamber on the testing day (day 5) and the time spent in this same chamber at baseline (day 1). A decrease from baseline preference for the drug-paired chamber was considered as conditioned place aversion while increased time in the chamber was interpreted as conditioned place preference (CPP). Preference scores were analysed by ANOVA with three levels of treatment (controls, cocaine and cocaine + SB334867) as between-subject factors. Post hoc analyses were performed by one-way ANOVA. The design for the CPP experiment including both morning and afternoon sessions has been successfully used in many published studies using cocaine and other drugs by the current authors and others (Carlezon et al. 1997, 1998; Brunzell et al. 2009; Russo et al. 2009; Mineur et al. 2009; Maze et al. 2010). This design has not resulted in any changes in behaviour compared to alternate day protocols.
Double immunolabelling
One day after the 3-day regimen of cocaine (10 mg kg−1) or saline, mice were deeply anaesthetized with isoflurane through inhalation and perfused transcardially with 4% paraformaldehyde in 0.1 m sodium phosphate buffer (PB, pH 7.4). Sections (50 μm thick) were cut on a vibratome and were washed in a buffer containing 0.1% lysine, 1% bovine serum albumin, 0.1% Tris and 0.4% Triton X-100, after which they were blocked with 2% normal horse serum and were incubated overnight in primary antibodies for orexin-B (Santa Cruz Biotechnology; sc-8071, goat, 1:2500) and phospho-CREB (Ser133) (Cell Signalling Technology; no. 9191, rabbit, 1:100). After several washes with PB, sections were incubated in secondary antibodies (1:250) conjugated to Alexa Fluor 488 and 594 (Invitrogen) for 2–3 h. Specimens were examined with an FV 300 confocal laser scanning microscope (Olympus America, Melville, NY, USA). For cell counting, 3–4 sections from each animal were taken at the level of the lateral hypothalamus, and the number of CREB/hypocretin-positive neurons were counted and compared to the total number of hypocretin neurons.
Electrophysiology
Coronal hypothalamic slices, 300 μm thick, were cut from mice expressing GFP exclusively in hypocretin neurons (Li et al. 2002; Yamanaka et al. 2003). Mice were anaesthetized with isoflurane and then decapitated. The brains were rapidly removed and immersed in an oxygenated bath solution at 4°C containing (in mm): sucrose 220, KCl 2.5, CaCl2 1, MgCl2 6, NaH2PO4 1.25, NaHCO3 26, and glucose 10, pH 7.3 with NaOH. After preparation, slices were maintained in a holding chamber with artificial cerebrospinal fluid (bubbled with 5% CO2 and 95% O2) containing (in mm): NaCl 124, KCl 3, CaCl2 2, MgCl2 2, NaH2PO4 1.23, NaHCO3 26, glucose 10, pH 7.4 with NaOH, and were transferred to a recording chamber constantly perfused with bath solution (33°C) at 2 ml min−1 after at least a 1 h recovery.
Whole-cell voltage clamp (at −60 mV or at +40 mV) was performed to observe evoked and miniature postsynaptic currents with a Multiclamp 700A amplifier (Axon Instruments, Union City, CA, USA) as described previously (Rao et al. 2007, 2008). The patch pipettes with a tip resistance of 4–6 MΩ were made of borosilicate glass (World Precision Instruments) with a Sutter pipette puller (P-97) and filled with a pipette solution containing (in mm): potassium gluconate (or caesium gluconate) 135, MgCl2 2, Hepes 10, EGTA 1.1, Mg-ATP 2, Na2-phosphocreatine 10, and Na2-GTP 0.3, pH 7.3 with KOH. After a giga-ohm (GΩ) seal and whole-cell access were achieved, the series resistance (between 20 and 40 MΩ) was partially compensated by the amplifier. Miniature EPSCs (mEPSCs) mediated by AMPA receptors were recorded under voltage clamp at −60 mV in the presence of tetrodotoxin (TTX, 0.5 μm) and picrotoxin (50 μm). To monitor evoked EPSCs (eEPSCs), a bipolar stimulating electrode was placed at least 125 μm laterally from the recorded neurons (for AMPA receptor (AMPAR)/NMDAR ratio) or placed in the middle of the fibre bundle (for LTP experiments). eEPSCs recorded in hypocretin neurons under voltage-clamp at +40 mV in the presence of picrotoxin (50 μm) contained both AMPA- and NMDA-components and AMPAR-carried EPSC was obtained by applying d-aminophosphonovalerate (AP5; 50 μm) to the recorded neurons. The NMDAR-carried component of the EPSC was determined by subtracting the AMPA component from the whole EPSC recorded before the application of AP5. To determine the rectification of AMPAR-mediated EPSCs, eEPSCs were recorded at different steps of holding potential from −60 mV to +60 mV with an increment of 20 mV in the presence of AP5 (50 μm) and picrotoxin (50 μm). Spermine (0.1 mm) was included in the pipette solution in this set of experiments on rectification index as in Liu & Cull-Candy (2000). In some experiments, insufficient space clamp may lead to a slower decay of evoked EPSCs (particularly NMDAR-mediated EPSCs) in our experiments. In LTP experiments, neurons were switched to current clamp when four trains of high frequency stimuli (HFS, 100 Hz, 1 s duration, 10 s interval) were delivered to medial forebrain bundle. Both input resistance and series resistance were monitored throughout the experiments. Only recordings with stable series resistance were accepted (the change in series resistance was less than 20%).
All data were sampled at 10 kHz and filtered at 3 kHz with an Apple Macintosh computer using AxoGraph X (developed by Dr John Clements, AxoGraph, Inc.). Electrophysiological data were analysed with AxoGraph X and plotted with Igor Pro software (WaveMetrics, Lake Oswego, OR, USA). Specifically, mEPSC was analysed with an event-detection package provided in AxoGraph X as reported previously (Clements & Bekkers, 1997; Rao et al. 2007, 2008). User-defined templates with characteristics of miniature synaptic events were slid along the signal to test for matches and the templates were automatically offset to adjust for baseline drift. An event was detected when the mismatch between a template and the signal approached the level of the recording noise. By using this approach, there was no need to filter the data and the default threshold setting usually gave excellent performance. Glitches and artifacts in the data were rejected because they did not conform to the template time course. Due to the variation in the recording conditions among all cells, the parameters for templates for mEPSC were as follows: rise time, 0.3–1 ms; decay time, 1–5 ms; amplitude, 10–20 pA. The threshold for event detection was always 5 times the noise standard error.
Cocaine hydrochloride (NIDA, NIH, Bethesda, MD, USA) was diluted in saline and injected at 10 mg kg−1 (i.p.), and SB334867 (Tocris) was diluted in 2% (v/v) DMSO/saline and injected at 30 mg kg−1. Solutions were prepared fresh in the morning of each training session and the volume of injection was less than 0.2 ml per injection for i.p. application.
Results
Cocaine exposure induces experience-dependent synaptic plasticity in hypocretin neurons
We used mice expressing green fluorescent protein (GFP) exclusively in hypocretin neurons to investigate the physiological changes in these cells following cocaine CPP. Hypocretin-GFP mice show a similar cocaine (10 mg kg−1i.p.) place preference to wild-type C57BL/6J mice (Fig. 1 inset; Romieu et al. 2003; Kreibich & Blendy, 2004). There was an overall treatment effect for the time spent in the drug-paired compartment (F(2,29)= 4.36, P < 0.05). Mice in the cocaine group (n= 8, black bar) spent significantly more time in the cocaine-paired chamber following training than mice in the control group (n= 8, white bar). Post hoc analyses showed that cocaine increased the time spent in the cocaine-paired chamber compared to saline treatment (F(1,14)= 6.33, P < 0.05). To identify changes in hypocretin neurons after cocaine exposure, whole-cell patch clamp recording was performed in these mice one day after the completion of CPP. Our results in the subsequent experiments demonstrated that the same changes occurring in hypocretin neurons in CPP mice took place in hypocretin neurons in mice that do not express CPP due to the pre-treatment of the hypocretin-1 receptor antagonist (Fig. 3), indicating that the presence of CPP does not selectively affect plasticity induced by cocaine in hypocretin neurons. Therefore, a 3-day home cage exposure of cocaine at exactly the same time points of the day as in the CPP protocol was performed in hypocretin-GFP mice and pooled with those from CPP mice in the subsequent experiments in the study unless otherwise mentioned.
Figure 1. Potentiation of glutamatergic synapses on hypocretin neurons following the establishment of cocaine-conditioned place preference (CPP).
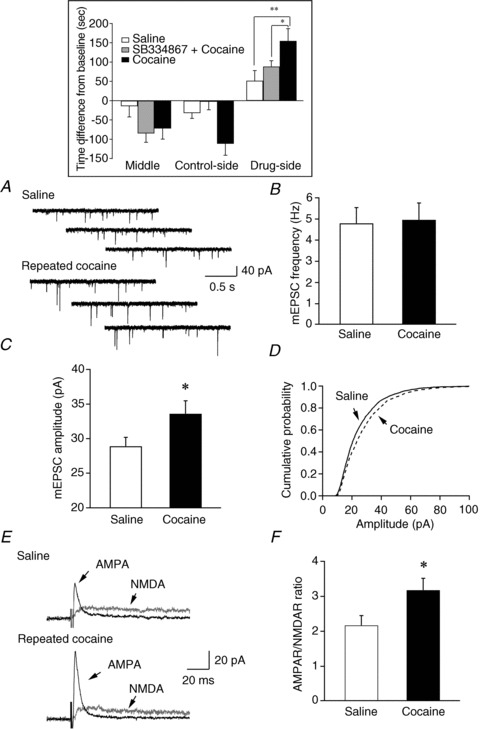
A, sample traces of mEPSCs recorded from control and cocaine-treated mice are presented. B, mean frequency of mEPSCs detected in saline- and cocaine-treated mice showed no significant difference between these two groups. C, a significant increase in the mean amplitude of mEPSC events detected in cocaine-treated mice was indicated as compared to the control group (*P < 0.05, t test). D, a significant right-shift in the cumulative probability of mEPSC amplitude was detected in cocaine-treated mice as compared to control mice (P < 0.001, Kolmogorov–Smirnov test). E, sample traces of evoked EPSCs carried by AMPA receptors (AMPAR) and NMDA receptors (NMDAR) in control and cocaine-treated mice. F, the ratio of evoked EPSCs carried by AMPAR and NMDAR was enhanced in mice treated with cocaine as compared to controls (*P < 0.05, t test). Inset, bar graph indicates that mice spent more time in the cocaine-paired side of the testing apparatus after a 3-day training session. Blockade of the hypocretin-1 receptor pathway blocks cocaine CPP in mice.
Figure 3. The synaptic efficacy of glutamatergic synapses on hypocretin neurons was not significantly different in cocaine-treated mice in the absence and presence of a selective hypocretin-1 receptor antagonist, SB334867 (SB).
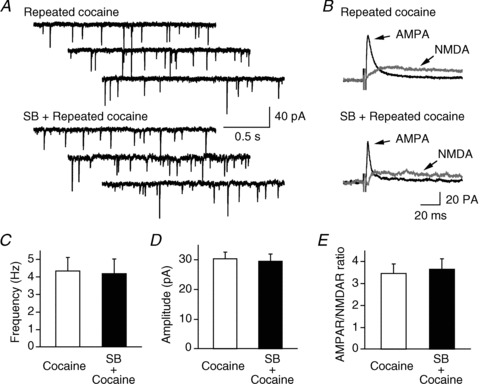
Sample traces of mEPSCs and evoked EPSCs are presented in A and B. The frequency, amplitude and AMPAR/NMDAR ratio in mice treated with cocaine alone and SB plus cocaine are presented in C, D and E.
One day after the CPP with cocaine (10 mg kg−1), we examined mEPSCs recorded in the presence of TTX (0.5 μm) and picrotoxin (50 μm) under voltage clamp (−60 mV) in hypocretin neurons in slices from saline- and cocaine-treated mice. The results from all recorded hypocretin neurons were included in the analysis no matter what their distribution was in the perifornical/LH area. The frequency of mEPSCs was comparable between saline- and cocaine-treated mice (saline, 4.8 ± 0.74 Hz, n= 24 from 8 mice; cocaine, 4.96 ± 0.79 Hz, n= 15 from 8 mice; P > 0.05; t test; Fig. 1A and B). Cocaine-treated mice showed a significant increase in the amplitude of mEPSCs (33.56 ± 1.86 pA, n= 14) compared to saline-treated controls (28.9 ± 1.28 pA, n= 24; P < 0.05, t test; Fig. 1A and C); the cumulative probability of the amplitude of mEPSCs in cocaine-treated mice was also shifted significantly to the right (control group: 2446 events; cocaine group: 1591 events; P < 0.001, Kolmogorov–Smirnov test; Fig. 1D), indicating that there was an up-regulation of postsynaptic glutamate receptors on hypocretin neurons after cocaine treatment. To investigate the change in AMPA receptor activity in hypocretin neurons following cocaine treatment further, we measured the relative contributions of AMPARs and NMDARs to evoked EPSCs in hypocretin neurons from saline- and cocaine-treated mice. Evoked EPSCs were recorded under voltage-clamp at +40 mV in the absence, and then presence, of the selective NMDA receptor antagonist dl-2-amino-5-phosphonovaleric acid (AP5, 50 μm) as described previously (Rao et al. 2007, 2008). Hypocretin neurons from mice treated with cocaine exhibited a significantly larger AMPAR/NMDAR ratio (3.18 ± 0.34, n= 14 from 4 mice) than mice injected with saline (2.17 ± 0.28, n= 9 from 4 mice; P < 0.05; one-tail t test; Fig. 1E and F), indicating a possible enhancement of the AMPAR-mediated component of EPSCs in these neurons in cocaine-treated mice.
It is important to note that AMPAR-mediated evoked EPSCs (AMPA-EPSCs) are inwardly rectifying in hypocretin neurons as compared to neighbouring melanin-concentrating hormone (MCH)-containing neurons (Fig. 2A–C). The recording of MCH neurons were made in C57/B6 mice exclusively expressing GFP in MCH cells under the control of a MCH promoter (this mouse strain was generated by Dr J. Friedman's group at Rockefeller Univ.). The rectification index (RI, defined as the ratio of the eEPSC amplitude at +40 mV to the predicted value at +40 mV from linear fitting of the eEPSC amplitudes at the negative holding potentials (Liu & Cull-Candy, 2000) was significantly lower in hypocretin neurons than in MCH neurons (hypocretin neurons: 0.43 ± 0.06, n= 8; MCH neurons: 1.02 ± 0.25, n= 9; P < 0.05, t test). Since GluR2-containing AMPA receptors are not calcium permeable and show less inward rectification, these data suggest that GluR2-lacking, Ca2+-permeable AMPARs are expressed in hypocretin neurons. Repeated cocaine treatment did not alter the rectification of AMPAR-EPSCs in hypocretin neurons (RI; control: 0.43 ± 0.06, n= 8 from 3 mice; cocaine: 0.45 ± 0.07, n= 9 from 3 mice; P > 0.05, t test) (Fig. 2D). Taken together, these data suggest that cocaine treatment induces increased insertion of AMPA receptors (including Ca2+-permeable AMPARs) at glutamatergic synapses on hypocretin neurons.
Figure 2. Repeated cocaine exposure does not induce changes in the current–voltage (I–V) relationship of AMPAR-mediated EPSC in hypocretin neurons.
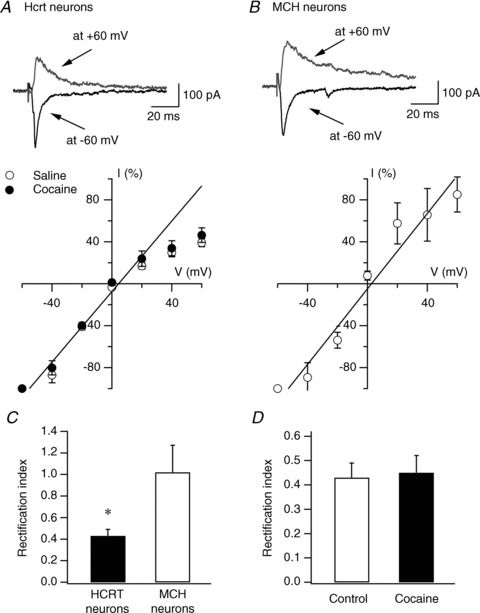
A, I–V relationship of evoked AMPAR-EPSCs recorded in hypocretin (Hcrt) neurons in naive (open circles) and cocaine-treated (filled circles) mice. Representative traces from naive mice are shown on the upper part of the panel. The straight line represents the linear regression of mean EPSC amplitude at negative potentials. B, representative traces (upper) and I–V relationship (lower) of evoked AMPAR-EPSCs recorded in melanin-concentrating hormone (MCH)-containing neurons in MCH-GFP mice. The straight line represents the linear regression of mean EPSC amplitude at negative potentials. C, mean RI in hypocretin and MCH neurons is presented. D, mean RI in hypocretin neurons from control and cocaine-treated mice is presented.
Consistent with a previous report (Harris et al. 2005), pretreatment with a selective hypocretin receptor 1 (HcrtR1) antagonist SB334867 (30 mg kg−1, i.p.) abolished conditioned place preference in the cocaine-treated group (Fig. 1, inset, grey bar). Mice treated with SB334867 plus cocaine spent less time in the cocaine-paired chamber than vehicle-plus-cocaine-treated mice (F(1,22)= 4.75, P < 0.05), and spent a comparable amount of time in the drug-paired chamber as compared to saline-treated mice (F(1,22)= 1.73, P > 0.05). In brain slices prepared from mice that received SB334876 followed by cocaine, or vehicle followed by cocaine as mentioned above, we found that neither the frequency (cocaine: 4.36 ± 0.74 Hz, n= 15 from 8 mice; SB334876 + cocaine: 4.21 ± 0.81 Hz, n= 15 from 8 mice; P > 0.05; t test; Fig. 3A) nor the amplitude (cocaine: 30.48 ± 2.06 pA, n= 15 from 8 mice; SB334876 + cocaine: 29.66 ± 2.28 pA, n= 15 from 8 mice; P > 0.05; t test; Fig. 3B) of mEPSCs were changed. There was also no significant difference in the AMPAR/NMDAR ratio between vehicle/cocaine- (3.48 ± 0.41, n= 9) and SB334876/cocaine-treated (3.67 ± 0.47, n= 11; P > 0.05; t test; Fig. 3C) animals. In a subset of experiments, we found that the application of SB334876 alone for 3 days did not induce any changes in synaptic efficacy of glutamatergic synapses on hypocretin neurons (data not shown). These data suggest that the HcrtR1 antagonist did not alter cocaine-induced synaptic potentiation in hypocretin neurons, even though it abolished cocaine-induced place preference when applied prior to cocaine exposure.
The transcription factor CREB is critical for many types of transcription and translation-dependent synaptic plasticity (Lamprecht, 1999; Wu et al. 2007). The phosphorylation of CREB is critical for mediating late-LTP and memory processes in the hippocampus, as well as regulating synaptic plasticity in the VTA in rats exposed to cocaine (Bourtchuladze et al. 1994; Yin et al. 1994; Impey et al. 1998; Hyman et al. 2006). Using a selective antibody against phosphorylated CREB (pCREB) at Ser133, we examined levels of pCREB in hypocretin neurons by dual immunostaining for pCREB and orexin-B in control and cocaine-treated mice. Levels of phosphorylated CREB increased significantly in hypocretin neurons after in vivo (home cage) administration of cocaine for 3 days in the CPP protocol (Fig. 4). In mice treated with saline (n= 3 mice), pCREB was only detected in a limited number of hypocretin neurons (5.59 ± 2.98%, n= 24 sections), in comparison to mice treated with 10 mg kg−1 cocaine (n= 3 mice), which demonstrated a robust increase in pCREB/hypocretin co-expressing neurons (62.04 ± 5.99%, n= 21 sections; P < 0.001, t test).
Figure 4. Enhanced phosphorylation of CREB (pCREB) induced by cocaine treatment in hypocretin neurons.
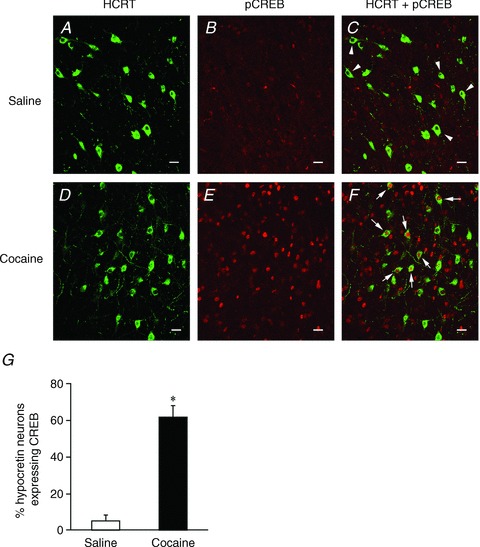
Dual immunostaining of pCREB and hypocretin (Hcrt) was performed in control (n= 3) and cocaine-treated mice (n= 3) after a 3-day treatment. A–C, images show that only hypocretin-immunopositive neurons (indicated by arrowheads) are present in the perifornical/LH area in saline-treated mice. D–F, images show that hypocretin and pCREB-immunopositive neurons (indicated by arrows) are present in the LH area in cocaine-treated mice. Scale bar: 15 μm. G, pooled data from all examined sections from saline- (n= 24 sections) and cocaine-treated (n= 21 sections) mice indicate a significant increase in pCREB expression in hypocretin neurons following a 3-day cocaine treatment. *P < 0.001, t test.
Experience-dependent synaptic plasticity in hypocretin neurons during cocaine withdrawal
It has been shown that cocaine-induced synaptic plasticity differs in its time course in brain areas involved in different aspects of cocaine self-administration and relapse (Thomas et al. 2008). We therefore explored the time course of cocaine-induced synaptic plasticity in hypocretin neurons. A single injection of cocaine is sufficient to induce long-term synaptic potentiation in dopaminergic cells in the VTA (Ungless et al. 2001). Therefore, we examined the frequency and amplitude of mEPSCs and the AMPAR/NMDAR ratio one day after a single treatment with cocaine (10 mg kg−1, i.p.) in the home cage. As shown in Fig. 5, the frequency (saline: 5.00 ± 0.87 Hz, n= 15 from 2 mice; cocaine: 4.44 ± 0.87 Hz, n= 16 from 2 mice; P > 0.05; t test; Fig. 5A) and amplitude (saline: 32.25 ± 2.93 pA, n= 15; cocaine: 33.23 ± 2.39 pA, n= 16; P > 0.05; t test; Fig. 5A) of mEPSCs in hypocretin neurons were comparable between control and cocaine-treated mice following a single cocaine injection. In addition, the AMPAR/NMDAR ratio did not change in mice that received a single injection of cocaine as compared to control animals (saline: 2.85 ± 0.38, n= 8 from 3 mice; cocaine: 2.91 ± 0.29, n= 9 from 3 mice; P > 0.05; t test; Fig. 5A). Thus, a single administration of cocaine is not sufficient to result in significant potentiation of glutamatergic synapses onto hypocretin neurons one day after treatment in mice.
Figure 5. Duration of experience-dependent synaptic plasticity at glutamatergic synapses on hypocretin neurons following cocaine exposure.
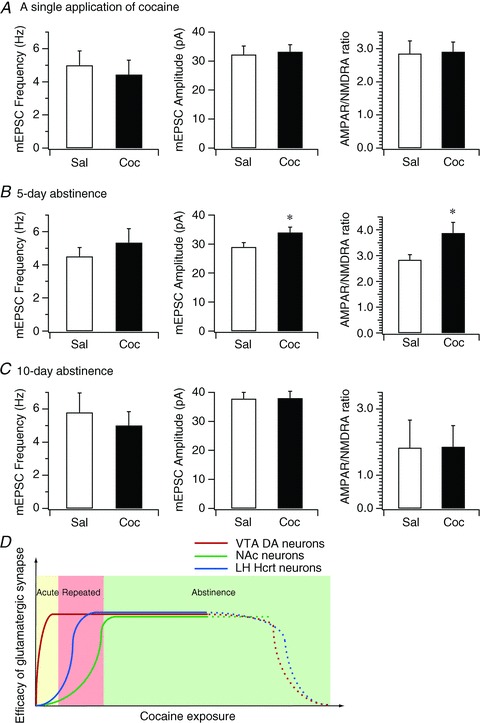
A, bar graph shows the mean frequency and amplitude of mEPSCs and mean AMPAR/NMDAR ratio in hypocretin neurons in control and cocaine-treated mice 1 day after a single exposure to cocaine. B, bar graph shows the mean frequency and amplitude of mEPSCs and mean AMPAR/NMDAR ratio in hypocretin neurons in control and cocaine-treated mice on the 5th day of withdrawal following the 3-day cocaine regimen. *P < 0.05, paired t test. C, bar graph shows the mean frequency and amplitude of mEPSCs and mean AMPAR/NMDAR ratio in hypocretin neurons in control and cocaine-treated mice on the 10th day of withdrawal after the 3-day cocaine regimen. D, the diagram summarizes the time courses of synaptic potentiation in the VTA, NAc and hypocretin neurons induced by exposure to cocaine. Our data suggest that the onset of expression of experience-dependent synaptic potentiation in hypocretin neurons (blue line) is later than synaptic potentiation induced in the VTA (red line) (Ungless et al. 2001) and earlier than in the NAc (green line) (Kourrich et al. 2007), which implicates the distinctive roles of synaptic plasticity in these areas in the development of animal behaviours relevant to cocaine addiction.
Since 3 days of cocaine treatments (CPP and home cage administration) result in potentiation of glutamatergic inputs to hypocretin neurons, we next determined whether this plasticity was long-lasting. We therefore examined synaptic parameters in these neurons from mice treated with the 3-day cocaine regimen (home cage administration) following 5 and 10 days of withdrawal in two separate cohorts of animals. After a 5-day withdrawal period, the amplitude of mEPSCs (saline: 28.99 ± 1.47 pA, n= 24 from 3 mice; cocaine 34.0 ± 1.80 pA, n= 23 from 3 mice; P < 0.05, t test; Fig. 5B) and the AMPAR/NMDAR ratio (saline: 2.84 ± 0.20, n= 8 from 3 mice; cocaine: 3.88 ± 0.41, n= 7 from 3 mice; P < 0.05, t test; Fig. 5B) onto hypocretin neurons still showed significant potentiation. After 10 days of withdrawal, the mEPSC frequency (saline: 5.79 ± 1.17 Hz, n= 16 from 3 mice; cocaine: 5.01 ± 0.83 Hz, n= 16 from 3 mice; P > 0.05, t test; Fig. 5C), amplitude (saline: 37.84 ± 2.15 pA, n= 16 from 3 mice; cocaine: 38.08 ± 2.37, n= 16 from 3 mice; P > 0.05, t test; Fig. 5C) and AMPAR/NMDAR ratio (saline: 1.83 ± 0.83, n= 8 from 3 mice; cocaine: 1.86 ± 0.64, n= 8 from 3 mice; P > 0.05, t test; Fig. 5C) were no longer significantly different from controls. Taken together, these data suggest that the synaptic potentiation of glutamatergic synapses at hypocretin neurons requires multiple exposures to cocaine and is long-lasting.
Facilitation of LTP induction in hypocretin neurons in cocaine-exposed mice
The occlusion of high frequency stimulation (HFS)-induced LTP in VTA DA neurons following a single administration of cocaine demonstrates that synaptic potentiation induced by cocaine in VTA DA neurons shares the same signalling pathways with HFS-LTP in these neurons (Ungless et al. 2001). We have tested the effects of repeated cocaine exposure on the induction of HFS-LTP in hypocretin neurons here. Evoked EPSCs were recorded in hypocretin neurons by stimulating the medial forebrain bundle (MFB) in the presence of picrotoxin (50 μm) as described previously (Rao et al. 2007). HFS-LTP has been induced in hypocretin neurons in a previous report by Xia et al. (2009). With a slightly different protocol (HFS, 100 Hz, 1 s duration, 10 trains, 10 s interval), HFS delivered to the MFB after stable recording of evoked EPSCs induced LTP in hypocretin neurons in this study (see inset in Fig. 6), while sub-threshold HFS (100 Hz, 1 s duration, 4 trains, 10 s interval) delivered to the MFB did not. As shown in Fig. 6A, only short-term potentiation of the amplitude of EPSCs was induced in slices from saline controls (Fig. 6A; 292.43 ± 55.02%, n= 9 from 5 mice immediately after HFS and 101.47 ± 13.2% (n= 9) 30 min after HFS). This was not significantly different from the amplitude of EPSCs before HFS (99.99 ± 2.98%; P > 0.05, paired t test). In contrast, mice treated with cocaine for 3 days in the home cage had an increase in EPSC amplitude (329.95 ± 54.72%, n= 9 from 5 mice, immediately after HFS and 143.3 ± 17.1%, n= 9, 30 min after HFS) that was significantly different from the baseline value (101.62 ± 2.11%) and lasted for the duration of the experiment (P < 0.05, paired t test; Fig. 6C). As shown above, a single administration of cocaine did not induce synaptic potentiation in hypocretin neurons and the induction of LTP was not facilitated in these neurons. Like saline-treated mice, following a single cocaine administration the amplitude of EPSCs was 238.87 ± 44.15% (n= 7 from 4 mice) immediately after HFS, but this decreased to 121.57 ± 13.53% (n= 7) 30 min after HFS, which was not significantly different from baseline (103.86 ± 2.65%, n= 7, P > 0.05; Fig. 6B). These data suggest that LTP induction in hypocretin neurons is only facilitated in mice treated with cocaine repeatedly.
Figure 6. Cocaine treatment facilitates the induction of LTP by HFS in hypocretin neurons.
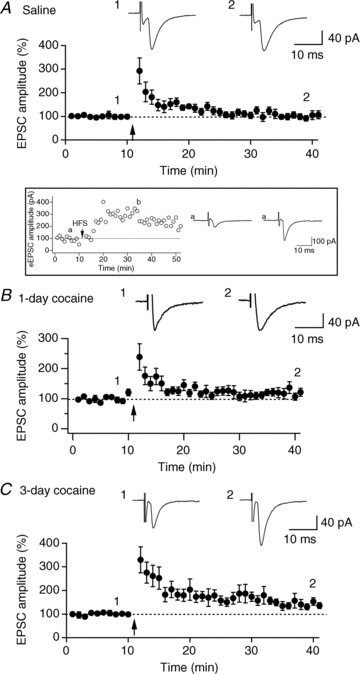
A, time course of evoked EPSCs recorded in hypocretin neurons from control mice treated with saline for 3 days. 1 and 2 are averaged traces of evoked EPSCs recorded before and 30 min after the HFS. The arrow indicates application of 4 trains of HFS (100 Hz, 1 s duration, 10 s interval) to the MFB when the recorded neurons were under current clamp. B, time course of evoked EPSCs in hypocretin neurons before and after HFS in mice given a single application of cocaine. Arrow, application of HFS. 1 and 2 are averaged traces (from 5 consecutive traces) of evoked EPSCs recorded before and 30 min after the application of HFS. C, time course of evoked EPSCs recorded in hypocretin neurons from mice treated with cocaine for 3 days. Arrow, application of HFS. 1 and 2 are averaged traces of evoked EPSCs recorded before and 30 min after the application of HFS. Inset, the time course of evoked EPSCs recorded in a control hypocretin neuron showing the expression of LTP induced by 10 trains of HFS. The arrow indicates application of 10 trains of high frequency stimulation (100 Hz, 1 s duration, 10 s interval) to the MFB when the recorded neurons were under current clamp. Symbols ‘a’ and ‘b’ indicate raw traces of evoked EPSCs recorded before and after high frequency stimulation.
Discussion
An accumulating body of evidence indicates the involvement of the hypocretin system in brain reward function (Cason et al. 2010; Thompson & Borgland, 2011). The increased expression of hypocretin mRNA in rats after morphine withdrawal and cocaine CPP suggests that there are adaptive changes in these neurons in response to drugs of abuse (Georgescu et al. 2003; Harris et al. 2005; Zhou et al. 2006, 2008). It was recently shown that the activation of μ-opioid receptors leads to inhibition of hypocretin neurons, providing a clue for how morphine can affect the function of these neurons (Li & van den Pol, 2008). In this study, we show that repeated rather than a single cocaine exposure leads to experience-dependent synaptic potentiation in hypocretin neurons. The synaptic potentiation occurs at the postsynaptic site and correlates with the phosphorylation of CREB in hypocretin neurons. The potentiation of synaptic efficacy after 3 days exposure to cocaine lasts at least 5 days following abstinence from cocaine and facilitates further induction of LTP in these neurons. These results are the first report of synaptic plasticity induced by drugs of abuse in hypocretin neurons, a newly established component of the brain rewarding circuitry.
Cocaine-triggered long-term synaptic potentiation in hypocretin neurons
Experience-dependent synaptic plasticity plays a prominent role in the adaptation of the CNS to the external environment and synaptic plasticity occurring in several brain areas (such as the VTA, amygdala and NAc) involved in drug abuse may underlie drug-seeking behaviours (Malenka & Bear, 2004; Hyman et al. 2006). The synaptic potentiation that we observe here in hypocretin neurons is therefore consistent with the idea that cocaine exposure results in plasticity in the brain reward circuitry. We found that synaptic plasticity of glutamatergic synapses on hypocretin neurons induced by cocaine exposure is expressed postsynaptically as indicated by the up-regulation of AMPA-mEPSC amplitude and the increase in AMPAR/NMDAR ratio in cocaine-treated mice, since the frequency of mEPSCs does not change after cocaine treatment. It is in contrast to the synaptic potentiation in VTA DA neurons induced by a single exposure to cocaine, in which both pre- and postsynaptic components of glutamatergic synapses are enhanced (Ungless et al. 2001). Moreover, the increase in AMPAR signalling after repeated cocaine treatment does not involve changes in the subunit composition of these receptors (including Ca2+-permeable AMPA receptors), as the rectification index of AMPAR-mediated synaptic currents does not change following cocaine exposure. This result is in contrast to recent reports showing enhancement of Ca2+-permeable AMPARs in VTA neurons after a single injection of cocaine and NAc neurons following prolonged withdrawal from cocaine self-administration (Bellone & Lücher, 2006; McCutcheon et al. 2011). In the current study we used a short-term cocaine exposure model (CPP and home cage administration), therefore our results do not exclude the possibility that long-term cocaine exposure may also lead to enhancement of Ca2+-permeable AMPARs in hypocretin neurons. Since AMPAR-mediated synaptic current significantly contributes more to glutamatergic transmission onto hypocretin neurons than the NMDAR-mediated component, the induction of LTP may be less NMDA receptor dependent in hypocretin neurons but probably depends on Ca2+-permeable AMPARs (Liu & Zukin, 2007). In a recent report, exposure to cocaine for a longer period of time (7 days) than in this study leads to an enhanced excitatory input onto perifornical/LH neurons including hypocretin neurons in rats (Yeoh et al. 2012). The difference between the studies of Yeoh et al. (2012) and our studies may be due to the different species (rats vs. mice) used in the experiments. The different behavioural protocol (cocaine self-administration vs. CPP/home cage injections) may also contribute to the distinctive response of hypocretin neurons to cocaine exposure. Nevertheless, our results reported here provide novel insights into the responses of hypocretin neurons to cocaine exposure. Firstly, we focused on the effects of cocaine on hypocretin neurons exclusively in transgenic mice. Secondly, we examined the time course of cocaine-induced effects on hypocretin neurons. Thirdly, we tested the effects of metaplasticity induced by cocaine on the function of hypocretin neurons.
The finding that phosphorylation of CREB is significantly increased in hypocretin neurons following repeated exposure to cocaine suggests that induction of transcriptional and translational processes may be responsible for neuronal plasticity occurring in hypocretin neurons following cocaine exposure. We should note that the current study does not provide a causal link between the changes in pCREB levels and effects on synaptic plasticity. Future studies will be necessary to connect the biochemical findings to the electrophysiological results. It has been proposed that drug exposure leads to initial responses represented by phosphorylation of transcription factors such as CREB, then activation of long-life transcription factors such as delta FosB, and lastly the remodelling of chromatin (modification of histones) (Hyman et al. 2006). This chain of reactions is believed to underlie the enduring changes (including experience-dependent synaptic plasticity) in the brain of animals exhibiting behaviours related to drug addiction. Therefore, it is essential to establish a temporal relationship between the development of drug-seeking behaviours and changes at the epigenetic, cellular and circuit levels in hypocretin neurons in the future. These studies should be critical in establishing a comprehensive understanding of the role of the hypocretin system in drug abuse. It is noteworthy that many non-hypocretin neurons in the LH area also express pCREB (Fig. 4) after the repeated cocaine exposure, suggesting the activation of non-hypocretin neurons by cocaine in this cocaine-responsive brain structure. It is both intriguing and essential to discover the ‘identities’ of these non-hypocretin neurons in future studies. It is also intriguing to postulate that the expression of pCREB in non-hypocretin neurons may be responsible for the potentiation of the presynaptic component on non-hypocretin LH neurons reported by Yeoh et al. (2012).
In this study we have found that the development of synaptic potentiation in hypocretin neurons has a distinctive time course as compared with synaptic plasticity in the VTA and NAc. In contrast to the report that a single injection of cocaine induces LTP-like potentiation in DA neurons in the VTA (Ungless et al. 2001), we found that repeated, but not acute, cocaine exposure leads to LTP-like potentiation in hypocretin neurons, which is similar to what is observed in the NAc (Kourrich et al. 2007). However, in NAc neurons synaptic potentiation develops neither immediately after repeated cocaine injection nor during early withdrawal, but rather, only after prolonged withdrawal (Kourrich et al. 2007). The expression of synaptic potentiation in hypocretin neurons is consistent with the time course of acquisition of CPP and lasts for 5 days of cocaine abstinence in our study. Since the VTA, NAc and hypocretin system may form a closely associated micro-circuit underlying behaviours related to cocaine addiction (Fadel & Deutch, 2002; Balcita-Pedicino & Sesack, 2007), the distinctive time course for the expression of synaptic plasticity in each of these brain structures may suggest specific roles for each brain area during the development of cocaine-seeking behaviours (Fig. 5D). The exact molecular and cellular mechanisms underlying the temporal changes in synaptic efficacy in hypocretin neurons following repeated cocaine exposure are not yet clear, but our results clearly provide a first clue to advance studies along this direction.
It is intriguing that synaptic potentiation of glutamatergic synapses on hypocretin neurons was not altered in mice treated with an HcrtR1 antagonist along with cocaine, even though cocaine-induced CPP was abolished in these mice. This result suggests that the expression of synaptic potentiation in hypocretin neurons does not depend on HcrtR1-mediated signalling pathways, consistent with a report of the predominance of HcrtR2 in the LH area (Sakurai, 2003). In the presence of HcrtR1 antagonists the functional connection between hypocretin neurons and its downstream targets is compromised, since the selective HcrtR1 antagonist applied in vivo blocks cocaine sensitization and experience-dependent potentiation of glutamatergic transmission onto DA neurons in rats (Borgland et al. 2006). Our results may further implicate that the blockade of adaptive changes in other cocaine-targeted brain areas such as the VTA does not have substantial effects on synaptic potentiation in hypocretin neurons. It is the existence of cocaine in the brain that may be required for the independent expression of synaptic plasticity in hypocretin neurons. It is intriguing to know in future whether the local exposure to cocaine is sufficient to induce plasticity in hypocretin neurons independent of other reward-seeking brain areas. It has been shown that HcrtR1 antagonism blocks or attenuates morphine CPP (Harris et al. 2005), stress-induced reinstatement of cocaine seeking (Boutrel et al. 2005), alcohol self-administration and cue-induced reinstatement of alcohol seeking (Lawrence et al. 2006), and stimulus-elicited (but not cocaine-induced) reinstatement of cocaine seeking (Aston-Jones et al. 2009). Therefore, our results may suggest that the expression of synaptic plasticity in hypocretin does not necessarily depend on the establishment of cocaine-seeking behaviours. It is therefore critical to address in the future whether the expression of synaptic plasticity in hypocretin neurons is required in the establishment of cocaine-seeking behaviours and whether the blockade of cocaine-induced plasticity in hypocretin neurons prevents these behaviours in animals.
Cocaine exposure and metaplasticity in hypocretin neurons
Synaptic plasticity induced by cocaine exposure may lead to different outcomes at excitatory synapses in different brain areas. In the VTA, synaptic potentiation induced by a single administration of cocaine occludes the further induction of LTP in DA neurons (Ungless et al. 2001). It has also been reported that repeated cocaine exposure facilitates the induction of LTP by reducing GABAergic tone on these neurons (Liu et al. 2005). In the NAc, synaptic potentiation induced by extended withdrawal from repeated cocaine can be reversed by a single administration of cocaine, but not by saline (Kourrich et al. 2007). In the current study, the induction of LTP triggered by HFS is facilitated in hypocretin neurons after repeated, but not a single, administration of cocaine. Since HFS-induced LTP expresses at both the pre- and postsynaptic sites of synapses while synaptic potentiation induced by cocaine exposure takes place at the postsynaptic site of synapses on hypocretin neurons, the facilitation of HFS-LTP in hypocretin neurons after cocaine exposure is not surprising. The same presynaptic release of glutamate is expected to induce a larger synaptic response in hypocretin neurons after cocaine exposure due to the enhanced postsynaptic response. Therefore, it is possible to lead to the induction of HFS-LTP by a stimulation protocol with a lesser intensity than that shown in this study. Furthermore, these results may also lead to the possibility that distinctive mechanisms may underlie the cocaine-induced synaptic potentiation and HFS-LTP in hypocretin neurons. The expression of cocaine-induced LTP is at the postsynaptic site, while HFS-LTP usually expresses at both pre- and postsynaptic sites. In summary, our results in this study suggest that metaplasticity occurring in hypocretin neurons during cocaine exposure may have important functional consequences.
Functional implications
Here we report prolonged changes (including experience-dependent synaptic plasticity) in hypocretin neurons after the acquisition of cocaine CPP or repeated cocaine exposure, providing a mechanism for how hypocretin neurons integrate with other brain areas to reshape the brain reward circuitry leading to behaviours related to addiction and addiction-associated mental disorders. The fact that synaptic plasticity in hypocretin neurons persists for more than 5 days after withdrawal from repeated cocaine exposure suggests that this may serve as a ‘memory trace’ of cocaine experience in the LH area, persisting beyond the withdrawal from cocaine use and contribute to the relapse to drug seeking. The formation of the ‘trace’ may be due to the direct effects of cocaine on DA-containing presynaptic terminals in the LH, since this process cannot be eliminated by the disruption of the communication between hypocretin neurons and the VTA with a selective HcrtR1 antagonist (Aston-Jones et al. 2009). It has been proposed that during the development of drug-seeking behaviour, hypocretin neurons are activated by cues associated with drug acquisition/exposure and are critical in stimulus-induced drug relapse (Aston-Jones et al. 2009). Our data showing that synaptic plasticity induced by cocaine experience facilitates the further induction of LTP are consistent with this idea. From a computational point of view, this feature may allow hypocretin neurons to integrate many other inputs or environmental cues activating hypocretin neurons (such as stress) to form a strong association with the cocaine experience. This may explain why hypocretin neurons are involved in stress-induced, but not context-elicited, drug-seeking behaviour (Boutrel et al. 2005; Aston-Jones et al. 2009).
In addition to their role in cocaine addiction, hypocretin neurons are a part of the wakefulness-promoting circuitry in the brain (Boutrel et al. 2010; Sakurai et al. 2010). The activity of hypocretin neurons, particularly patterns of action potentials, is closely associated with the behavioural state of the animal (Estabrooke et al. 2001; Zeitzer et al. 2003; Lee et al. 2005; Mileykovskiy et al. 2005; Adamantidis et al. 2007). The up-regulation of glutamatergic transmission onto hypocretin neurons through potentiated synaptic efficacy facilitates action potential generation and probably contributes to a lowered wake-promoting threshold in mice (Rao et al. 2008). Synaptic plasticity in hypocretin neurons in the reward-relevant area may lead to a lowered threshold of activation for the overall network of hypocretin neurons via inter-hypocretin neuronal connections (Guan et al. 2002). Although promoting and maintaining wakefulness requires a wide array of brain areas, our data imply that synaptic potentiation in hypocretin neurons induced by chronic cocaine could contribute to a lowered threshold for arousal, which may partly explain the physiological arousal and disturbed sleep in cocaine addicts (Morgan & Malison, 2007; Valladares & Irwin, 2007).
In summary, long-lasting experience-dependent synaptic plasticity of hypocretin neurons occurs following repeated cocaine exposure. The increase in activity and plasticity in the hypocretin system may, in turn, contribute to cocaine seeking following drug withdrawal, as well as sleep disturbance associated with cocaine addiction.
Acknowledgments
This work is supported by NIH grant DK 070723 and DA033595 (X.-B.G.). M.R.P. was supported by DA14241 and DA15425.
Glossary
- AMPAR
AMPA receptor
- CPP
conditioned place preference
- CREB
cAMP response element-binding protein
- DA neuron
dopaminergic neuron
- eEPSCs
evoked EPSCs
- GFP
green fluorescent protein
- HcrtR1
hypocretin receptor 1
- HFS
high frequency stimuli
- LH
lateral hypothalamus
- LTD
long-term depression
- LTP
long-term potentiation
- MCH
melanin-concentrating hormone
- mEPSCs
miniature EPSCs
- MFB
medial forebrain bundle
- NAc
nucleus accumbens
- NMDAR
NMDA receptor
- pCREB
phosphorylated CREB
- RI
rectification index
- TTX
tetrodotoxin
- VTA
ventral tegmental area
Author contributions
X.-B.G., M.R.P., T.L.H. and L.deL. conceived the project. Y.R. and X.-B.G. designed the electrophysiological experiments. Y.R., G.G., Z.-W.L. and S.S. performed electrophysiological experiments and analysed the data. Y.S.M. and M.R.P. designed the CPP experiment and Y.S.M. performed and analysed the CPP experiment. Y.R., A.H.W. and X.W. performed the immunocytochemical experiment. Y.R. and X.-B.G. drafted the article. L.deL., T.L.H. and M.R.P. contributed to the drafting of the manuscript. All authors approved the final version.
Author's present address
Y. Rao: Department of Biological Sciences, University of Illinois at Chicago, 840 West Taylor Street, Chicago, IL 60607, USA. Email: yanrao@uic.edu
References
- Adamantidis AR, Zhang F, Aravanis AM, Deisseroth K, de Lecea L. Neural substrates of awakening probed with optogenetic control of hypocretin neurons. Nature. 2007;450:420–424. doi: 10.1038/nature06310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams WJ, Lorens SA, Mitchell CL. Morphine enhances lateral hypothalamic self-stimulation in the rat. Proc Soc Exp Biol Med. 1972;140:770–771. doi: 10.3181/00379727-140-36549. [DOI] [PubMed] [Google Scholar]
- Aston-Jones G, Smith RJ, Moorman DE, Richardson KA. Role of lateral hypothalamic orexin neurons in reward processing and addiction. Neuropharmacol. 2009;56(Suppl. 1):112–121. doi: 10.1016/j.neuropharm.2008.06.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badiani A, Robinson TE. Drug-induced neurobehavioral plasticity: the role of environmental context. Behav Pharmacol. 2004;15:327–339. doi: 10.1097/00008877-200409000-00004. [DOI] [PubMed] [Google Scholar]
- Balcita-Pedicino JJ, Sesack SR. Orexin axons in the rat ventral tegmental area synapse infrequently onto dopamine and γ-aminobutyric acid neurons. J Comp Neurol. 2007;503:668–684. doi: 10.1002/cne.21420. [DOI] [PubMed] [Google Scholar]
- Bellone C, Lüscher C. Cocaine triggered AMPA receptor redistribution is reversed in vivo by mGluR-dependent long-term depression. Nat Neurosci. 2006;9:636–641. doi: 10.1038/nn1682. [DOI] [PubMed] [Google Scholar]
- Borgland SL, Malenka RC, Bonci A. Acute and chronic cocaine-induced potentiation of synaptic strength in the ventral tegmental area: electrophysiological and behavioral correlates in individual rats. J Neurosci. 2004;24:7482–7490. doi: 10.1523/JNEUROSCI.1312-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgland SL, Taha SA, Sarti F, Fields HL, Bonci A. Orexin A in the VTA is critical for the induction of synaptic plasticity and behavioral sensitization to cocaine. Neuron. 2006;49:589–601. doi: 10.1016/j.neuron.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Bonci A, Borgland S. Role of orexin/hypocretin and CRF in the formation of drug-dependent synaptic plasticity in the mesolimbic system. Neuropharmacology. 2009;56(Suppl. 1):107–111. doi: 10.1016/j.neuropharm.2008.07.024. [DOI] [PubMed] [Google Scholar]
- Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ. Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell. 1994;79:59–68. doi: 10.1016/0092-8674(94)90400-6. [DOI] [PubMed] [Google Scholar]
- Boutrel B, Cannella N, de Lecea L. The role of hypocretin in driving arousal and goal-oriented behaviors. Brain Res. 2010;1314:103–111. doi: 10.1016/j.brainres.2009.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutrel B, de Lecea L. Addiction and arousal: the hypocretin connection. Physiol Behav. 2008;93:947–951. doi: 10.1016/j.physbeh.2007.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutrel B, Kenny PJ, Specio SE, Martin-Fardon R, Markou A, Koob GF, de Lecea L. Role for hypocretin in mediating stress-induced reinstatement of cocaine-seeking behavior. Proc Natl Acad Sci U S A. 2005;102:19168–19173. doi: 10.1073/pnas.0507480102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunzell D, Mineur Y, Neve R, Picciotto M. Nucleus accumbens CREB activity is necessary for nicotine conditioned place preference. Neuropsychopharmacology. 2009;34:1993–2001. doi: 10.1038/npp.2009.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlezon WA, Jr, Boundy VA, Haile CN, Lane SB, Kalb RG, Neve RL, Nestler EJ. Sensitization to morphine induced by viral-mediated gene transfer. Science. 1997;277:812–814. doi: 10.1126/science.277.5327.812. [DOI] [PubMed] [Google Scholar]
- Carlezon WA, Thome J, Olson VG, Lane-Ladd SB, Brodkin ES, Hiroi N, Duman RS, Neve RL, Nestler EJ. Regulation of cocaine reward by CREB. Science. 1998;282:2272–2275. doi: 10.1126/science.282.5397.2272. [DOI] [PubMed] [Google Scholar]
- Cason AM, Smith RJ, Tahsili-Fahadan P, Moorman DE, Sartor GC, Aston-Jones G. Role of orexin/hypocretin in reward-seeking and addiction: implications for obesity. Physiol Behav. 2010;100:419–428. doi: 10.1016/j.physbeh.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazala P, Darrac C, Saint-Marc M. Self-administration of morphine into the lateral hypothalamus in the mouse. Brain Res. 1987;416:283–288. doi: 10.1016/0006-8993(87)90908-5. [DOI] [PubMed] [Google Scholar]
- Clements JD, Bekkers JM. Detection of spontaneous synaptic events with an optimally scaled template. Biophys J. 1997;73:220–229. doi: 10.1016/S0006-3495(97)78062-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lecea L, Kilduff TS, Peyron C, Gao X, Foye PE, Danielson PE, Fukuhara C, Battenberg EL, Gautvik VT, Bartlett FS, 2nd, Frankel WN, van den Pol AN, Bloom FE, Gautvik KM, Sutcliffe JG. The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci U S A. 1998;95:322–327. doi: 10.1073/pnas.95.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estabrooke IV, McCarthy MT, Ko E, Chou TC, Chemelli RM, Yanagisawa M, Saper CB, Scammell TE. Fos expression in orexin neurons varies with behavioral state. J Neurosci. 2001;21:1656–1662. doi: 10.1523/JNEUROSCI.21-05-01656.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadel J, Deutch AY. Anatomical substrates of orexin-dopamine interactions: lateral hypothalamic projections to the ventral tegmental area. Neuroscience. 2002;111:379–387. doi: 10.1016/s0306-4522(02)00017-9. [DOI] [PubMed] [Google Scholar]
- Faleiro LJ, Jones S, Kauer JA. Rapid synaptic plasticity of glutamatergic synapses on dopamine neurons in the ventral tegmental area in response to acute amphetamine injection. Neuropsychopharmacology. 2004;29:2115–2125. doi: 10.1038/sj.npp.1300495. [DOI] [PubMed] [Google Scholar]
- Fu Y, Pollandt S, Liu J, Krishnan B, Genzer K, Orozco-Cabal L, Gallagher JP, Shinnick-Gallagher P. Long-term potentiation (LTP) in the central amygdala (CeA) is enhanced after prolonged withdrawal from chronic cocaine and requires CRF1 receptors. J Neurophysiol. 2007;97:937–941. doi: 10.1152/jn.00349.2006. [DOI] [PubMed] [Google Scholar]
- Gallistel CR, Shizgal P, Yeomans JS. A portrait of the substrate for self-stimulation. Psychol Rev. 1981;88:228–273. [PubMed] [Google Scholar]
- Georgescu D, Zachariou V, Barrot M, Mieda M, Willie JT, Eisch AJ, Yanagisawa M, Nestler EJ, DiLeone RJ. Involvement of the lateral hypothalamic peptide orexin in morphine dependence and withdrawal. J Neurosci. 2003;23:3106–3111. doi: 10.1523/JNEUROSCI.23-08-03106.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodall EB, Carey RJ. Effects of D- versus L-amphetamine, food deprivation, and current intensity on self-stimulation of the lateral hypothalamus, substantia nigra, and medial frontal cortex of the rat. J Comp Physiol Psychol. 1975;89:1029–1045. doi: 10.1037/h0077187. [DOI] [PubMed] [Google Scholar]
- Goussakov I, Chartoff EH, Tsvetkov E, Gerety LP, Meloni EG, Carlezon WA, Jr, Bolshakov VY. LTP in the lateral amygdala during cocaine withdrawal. Eur J Neurosci. 2006;23:239–250. doi: 10.1111/j.1460-9568.2005.04538.x. [DOI] [PubMed] [Google Scholar]
- Guan JL, Uehara K, Lu S, Wang QP, Funahashi H, Sakurai T, Yanagizawa M, Shioda S. Reciprocal synaptic relationships between orexin- and melanin-concentrating hormone-containing neurons in the rat lateral hypothalamus: a novel circuit implicated in feeding regulation. Int J Obes Relat Metab Disord. 2002;26:1523–1532. doi: 10.1038/sj.ijo.0802155. [DOI] [PubMed] [Google Scholar]
- Harris GC, Wimmer M, Aston-Jones G. A role for lateral hypothalamic orexin neurons in reward seeking. Nature. 2005;437:556–559. doi: 10.1038/nature04071. [DOI] [PubMed] [Google Scholar]
- Hawes JJ, Brunzell DH, Narasimhaiah R, Langel U, Wynick D, Picciotto MR. Galanin protects against behavioral and neurochemical correlates of opiate reward. Neuropsychopharmacol. 2008;33:1864–1873. doi: 10.1038/sj.npp.1301579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Lin HJ, Hsu KS. Repeated cocaine administration promotes long-term potentiation induction in rat medial prefrontal cortex. Cereb Cortex. 2007;17:1877–1888. doi: 10.1093/cercor/bhl096. [DOI] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward–related learning and memory. Annu Rev Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- Impey S, Smith DM, Obrietan K, Donahue R, Wade C, Storm DR. Stimulation of cAMP response element (CRE)-mediated transcription during contextual learning. Nat Neurosci. 1998;1:595–601. doi: 10.1038/2830. [DOI] [PubMed] [Google Scholar]
- Jones S, Bonci A. Synaptic plasticity and drug addiction. Curr Opin Pharmacol. 2005;5:20–25. doi: 10.1016/j.coph.2004.08.011. [DOI] [PubMed] [Google Scholar]
- Kalivas PW. Neurobiology of cocaine addiction: implications for new pharmacotherapy. Am J Addict. 2007;16:71–78. doi: 10.1080/10550490601184142. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Volkow ND. The neural basis of addiction: a pathology of motivation and choice. Am J Psychiatry. 2005;162:1403–1413. doi: 10.1176/appi.ajp.162.8.1403. [DOI] [PubMed] [Google Scholar]
- Kauer JA. Learning mechanisms in addiction: synaptic plasticity in the ventral tegmental area as a result of exposure to drugs of abuse. Annu Rev Physiol. 2004;66:447–475. doi: 10.1146/annurev.physiol.66.032102.112534. [DOI] [PubMed] [Google Scholar]
- Kauer JA, Malenka RC. Synaptic plasticity and addiction. Nat Rev Neurosci. 2007;8:844–858. doi: 10.1038/nrn2234. [DOI] [PubMed] [Google Scholar]
- Kelley AE. Memory and addiction: shared neural circuitry and molecular mechanisms. Neuron. 2004;44:161–179. doi: 10.1016/j.neuron.2004.09.016. [DOI] [PubMed] [Google Scholar]
- Kourrich S, Rothwell PE, Klug JR, Thomas MJ. Cocaine experience controls bidirectional synaptic plasticity in the nucleus accumbens. J Neurosci. 2007;27:7921–7928. doi: 10.1523/JNEUROSCI.1859-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreibich AS, Blendy JA. cAMP response element-binding protein is required for stress but not cocaine-induced reinstatement. J Neurosci. 2004;24:6686–6692. doi: 10.1523/JNEUROSCI.1706-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamprecht R. CREB: a message to remember. Cell Mol Life Sci. 1999;55:554–563. doi: 10.1007/s000180050314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence AJ, Cowen MS, Yang HJ, Chen F, Oldfield B. The orexin system regulates alcohol-seeking in rats. Br J Pharmacol. 2006;148:752–759. doi: 10.1038/sj.bjp.0706789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MG, Hassani OK, Jones BE. Discharge of identified orexin/hypocretin neurons across the sleep-waking cycle. J Neurosci. 2005;25:6716–6720. doi: 10.1523/JNEUROSCI.1887-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Gao X-B, Sakurai T, van den Pol AN. Hypocretin/orexin excites hypocretin neurons via a local glutamate neuron–A potential mechanism for orchestrating the hypothalamic arousal system. Neuron. 2002;36:1169–1181. doi: 10.1016/s0896-6273(02)01132-7. [DOI] [PubMed] [Google Scholar]
- Li Y, van den Pol AN. μ-Opioid receptor-mediated depression of the hypothalamic hypocretin/orexin arousal system. J Neurosci. 2008;28:2814–2829. doi: 10.1523/JNEUROSCI.5447-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S-Q, Cull-Candy SG. Synaptic activity at calcium-permeable AMPA receptors induces a switch in receptor subtype. Nature. 2000;405:454–458. doi: 10.1038/35013064. [DOI] [PubMed] [Google Scholar]
- Liu QS, Pu L, Poo MM. Repeated cocaine exposure in vivo facilitates LTP induction in midbrain dopamine neurons. Nature. 2005;437:1027–1031. doi: 10.1038/nature04050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SJ, Zukin RS. Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci. 2007;30:126–134. doi: 10.1016/j.tins.2007.01.006. [DOI] [PubMed] [Google Scholar]
- Lu H, Lim B, Poo MM. Cocaine exposure in utero alters synaptic plasticity in the medial prefrontal cortex of postnatal rats. J Neurosci. 2009;29:12664–12674. doi: 10.1523/JNEUROSCI.1984-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCutcheon JE, Wang X, Tseng KY, Wolf ME, Marinelli M. Calcium-permeable AMPA receptors are present in nucleus accumbens synapses after prolonged withdrawal from cocaine self-administration but not experimenter-administered cocaine. J Neurosci. 2011;31:5737–5743. doi: 10.1523/JNEUROSCI.0350-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, McGehee DS. Long-term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron. 2000;27:349–357. doi: 10.1016/s0896-6273(00)00042-8. [DOI] [PubMed] [Google Scholar]
- Maze I, Covington HE, 3rd, Dietz DM, LaPlant Q, Renthal W, Russo SJ, Mechanic M, Mouzon E, Neve RL, Haggarty SJ, Ren Y, Sampath SC, Hurd YL, Greengard P, Tarakhovsky A, Schaefer A, Nestler EJ. Essential role of the histone methyltransferase G9a in cocaine-induced plasticity. Science. 2010;327:213–216. doi: 10.1126/science.1179438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mileykovskiy BY, Kiyashchenko LI, Siegel JM. Behavioral correlates of activity in identified hypocretin/orexin neurons. Neuron. 2005;46:787–798. doi: 10.1016/j.neuron.2005.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mineur Y, Brunzell D, Grady S, Lindstrom J, McIntosh J, Marks M, King S, Picciotto M. Localized low level re-expression of high affinity mesolimbic nicotinic acetylcholine receptors restores nicotine-induced locomotion but not place conditioning. Genes Brain Behav. 2009;8:257–266. doi: 10.1111/j.1601-183X.2008.00468.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan PT, Malison RT. Cocaine and sleep: early abstinence. ScientificWorldJournal. 2007;7:223–230. doi: 10.1100/tsw.2007.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olds J. Self-stimulation of the brain. Science. 1958;127:315–324. doi: 10.1126/science.127.3294.315. [DOI] [PubMed] [Google Scholar]
- Olds J, Milner P. Positive reinforcement produced by electrical stimulation of septal area and other regions of rat brain. J Comp Physiol Psychol. 1954;47:419–427. doi: 10.1037/h0058775. [DOI] [PubMed] [Google Scholar]
- Olds ME, Williams KN. Self-administration of D-Ala2-Met-enkephalinamide at hypothalamic self-stimulation sites. Brain Res. 1980;194:155–170. doi: 10.1016/0006-8993(80)91325-6. [DOI] [PubMed] [Google Scholar]
- Peyron C, Tighe DK, van den Pol AN, de Lecea L, Heller HC, Sutcliffe JG, Kilduff TS. Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci. 1998;18:9996–10015. doi: 10.1523/JNEUROSCI.18-23-09996.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao Y, Liu Z-W, Borok E, Rabenstein RL, Shanabrough M, Lu M, Picciotto MR, Horvath TL, Gao X-B. Prolonged wakefulness induces experience-dependent synaptic plasticity in hypocretin/orexin neurons. J Clin Invest. 2007;117:4022–4033. doi: 10.1172/JCI32829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao Y, Lu M, Ge F, Marsh DJ, Qian S, Wang AH, Picciotto MR, Gao X-B. Regulation of synaptic efficacy in hypocretin/orexin containing neurons by melanin concentrating hormone in the lateral hypothalamus. J Neurosci. 2008;28:9101–9110. doi: 10.1523/JNEUROSCI.1766-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romieu P, Martin-Fardon R, Bowen WD, Maurice T. Sigma 1 receptor-related neuroactive steroids modulate cocaine-induced reward. J Neurosci. 2003;23:3572–3576. doi: 10.1523/JNEUROSCI.23-09-03572.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo SJ, Wilkinson MB, Mazei-Robison MS, Dietz DM, Maze I, Krishnan V, Renthal W, Graham A, Birnbaum SG, Green TA, Robison B, Lesselyong A, Perrotti LI, Bolaños CA, Kumar A, Clark MS, Neumaier JF, Neve RL, Bhakar AL, Barker PA, Nestler EJ. Nuclear factor κB signaling regulates neuronal morphology and cocaine reward. J Neurosci. 2009;29:3529–3537. doi: 10.1523/JNEUROSCI.6173-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saal D, Dong Y, Bonci A, Malenka RC. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron. 2003;37:577–582. doi: 10.1016/s0896-6273(03)00021-7. [DOI] [PubMed] [Google Scholar]
- Sakurai T. Orexin: a link between energy homeostasis and adaptive behaviour. Curr Opin Clin Nutr Metab Care. 2003;6:353–360. doi: 10.1097/01.mco.0000078995.96795.91. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, Williams SC, Richardson JA, Kozlowski GP, Wilson S, Arch JR, Buckingham RE, Haynes AC, Carr SA, Annan RS, McNulty DE, Liu WS, Terrett JA, Elshourbagy NA, Bergsma DJ, Yanagisawa M. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92:573–585. doi: 10.1016/s0092-8674(00)80949-6. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Mieda M, Tsujino N. The orexin system: roles in sleep/wake regulation. Ann N Y Acad Sci. 2010;1200:149–161. doi: 10.1111/j.1749-6632.2010.05513.x. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Kalivas PW, Shaham Y. Neuroplasticity in the mesolimbic dopamine system and cocaine addiction. Br J Pharmacol. 2008;154:327–342. doi: 10.1038/bjp.2008.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JL, Borgland SL. A role for hypocretin/orexin in motivation. Behav Brain Res. 2011;217:446–453. doi: 10.1016/j.bbr.2010.09.028. [DOI] [PubMed] [Google Scholar]
- Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–587. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- Valladares EM, Irwin MR. Polysomnographic sleep dysregulation in cocaine dependence. ScientificWorldJournal. 2007;7:213–216. doi: 10.1100/tsw.2007.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Zhou Y, Xiong ZQ. Transducer of regulated CREB and late phase long-term synaptic potentiation. FEBS J. 2007;274:3218–3223. doi: 10.1111/j.1742-4658.2007.05891.x. [DOI] [PubMed] [Google Scholar]
- Xia J, Chen F, Ye J, Yan J, Wang H, Duan S, Hu Z. Activity-dependent release of adenosine inhibits the glutamatergic synaptic transmission and plasticity in the hypothalamic hypocretin/orexin neurons. Neuroscience. 2009;162:980–988. doi: 10.1016/j.neuroscience.2009.05.033. [DOI] [PubMed] [Google Scholar]
- Yamanaka A, Beuckmann CT, Willie JT, Hara J, Tsujino N, Mieda M, Tominaga M, Yagami K, Sugiyama F, Goto K, Yanagisawa M, Sakurai T. Hypothalamic orexin neurons regulate arousal according to energy balance in mice. Neuron. 2003;38:701–713. doi: 10.1016/s0896-6273(03)00331-3. [DOI] [PubMed] [Google Scholar]
- Yeoh JW, James MH, Jobling P, Bains JS, Graham BA, Dayas CV. Cocaine potentiates excitatory drive in the perifornical/lateral hypothalamus. J Physiol. 2012;590:3677–3689. doi: 10.1113/jphysiol.2012.230268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin JC, Wallach JS, Del Vecchio M, Wilder EL, Zhou H, Quinn WG, Tully T. Induction of a dominant negative CREB transgene specifically blocks long-term memory in Drosophila. Cell. 1994;79:49–58. doi: 10.1016/0092-8674(94)90399-9. [DOI] [PubMed] [Google Scholar]
- Zeitzer JM, Buckmaster CL, Parker KJ, Hauck CM, Lyons DM, Mignot E. Circadian and homeostatic regulation of hypocretin in a primate model: Implications for the consolidation of wakefulness. J Neurosci. 2003;23:3555–3560. doi: 10.1523/JNEUROSCI.23-08-03555.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Patterson LM, Berthoud HR. Orexin signaling in the ventral tegmental area is required for high-fat appetite induced by opioid stimulation of the nucleus accumbens. J Neurosci. 2007;27:11075–11082. doi: 10.1523/JNEUROSCI.3542-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Bendor J, Hofmann L, Randesi M, Ho A, Kreek MJ. Mu opioid receptor and orexin/hypocretin mRNA levels in the lateral hypothalamus and striatum are enhanced by morphine withdrawal. J Endocrinol. 2006;191:137–145. doi: 10.1677/joe.1.06960. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Cui CL, Schlussman SD, Choi JC, Ho A, Han JS, Kreek MJ. Effects of cocaine place conditioning, chronic escalating-dose “binge” pattern cocaine administration and acute withdrawal on orexin/hypocretin and preprodynorphin gene expressions in lateral hypothalamus of Fischer and Sprague–Dawley rats. Neuroscience. 2008;153:1225–1234. doi: 10.1016/j.neuroscience.2008.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]