

Abstract
Activation of N-methyl-d-aspartate (NMDA) receptors (NMDARs) is a crucial mechanism underlying the development and maintenance of pain. Traditionally, the role of NMDARs in the pathogenesis of pain is ascribed to their activation and signalling cascades in postsynaptic neurons. In this study, we determined if presynaptic NMDARs in the primary afferent central terminals play a role in synaptic plasticity of the spinal first sensory synapse in a rat model of neuropathic pain induced by spinal nerve ligation. Excitatory postsynaptic currents (EPSCs) were recorded from superficial dorsal horn neurons of spinal slices taken from young adult rats. We showed that increased glutamate release from the primary afferents contributed to the enhanced amplitudes of EPSCs evoked by input from the primary afferents in neuropathic rats. Endogenous activation of presynaptic NMDARs increased glutamate release from the primary afferents in neuropathic rats. Presynaptic NMDARs in neuropathic rats were mainly composed of NR2B receptors. The action of presynaptic NMDARs in neuropathic rats was enhanced by exogenous d-serine and/or NMDA and dependent on activation of protein kinase C. In contrast, glutamate release from the primary afferents in sham-operated rats was not regulated by presynaptic NMDARs. We demonstrated that the lack of NMDAR-mediated regulation of glutamate release in sham-operated rats was not attributable to low extracellular levels of the NMDAR agonist and/or coagonist (d-serine), but rather was due to the insufficient function and/or number of presynaptic NMDARs. This was supported by an increase of NR2B receptor protein expression in both the dorsal root ganglion and spinal dorsal horn ipsilateral to the injury site in neuropathic rats. Hence, suppression of the presynaptic NMDAR activity in the primary sensory afferents is an effective approach to attenuate the enhanced glutamatergic response in the spinal first sensory synapse induced by peripheral nerve injury, and presynaptic NMDARs might be a novel target for the development of analgesics.
Key points
Activation of N-methyl-d-aspartate (NMDA) receptors (NMDARs) is a crucial mechanism underlying the development and maintenance of pain.
Little is known about the role of presynaptic NMDARs in regulating glutamate release from the spinal primary afferent terminals in neuropathic pain conditions in adult rats.
In this study we use electrophysiological recording from superficial dorsal horn neurons to show that endogenous activation of presynaptic NMDARs in neuropathic rats increases glutamate release from the primary afferents, which contributes to the enhanced amplitudes of EPSCs evoked by input from the primary afferents. In contrast, glutamate release from the primary afferents in sham-operated rats was not regulated by presynaptic NMDARs. These findings are supported by an increase of NR2B receptor protein expression in both the dorsal root ganglion and spinal dorsal horn ipsilateral to the injury site in neuropathic rats.
Our data demonstrated that suppression of the presynaptic NMDAR activity in the primary sensory afferents is an effective approach to attenuate the enhanced glutamatergic response in the spinal first sensory synapse induced by peripheral nerve injury, and presynaptic NMDARs might be a novel target for the development of analgesics.
Introduction
Neuropathic pain, i.e. pain caused by injury or dysfunction in the nervous system, is a major treatment challenge for both patients and healthcare providers. Ineffective treatment of neuropathic pain results from our incomplete understanding of mechanisms underlying abnormal neuronal activity along the nociceptive pathways. Excessive activation of glutamate receptors in neurons is a crucial mechanism leading to the enhancement of excitatory synaptic transmission in nociceptive pathways in chronic pain (Ren & Dubner, 2007; Salter & Pitcher, 2012). Increased glutamate release from primary afferent terminals in the spinal dorsal horn (Chen et al. 2009; Yang et al. 2011), decreased clearance of glutamate due to impairment of glutamate transporters (Sung et al. 2003; Nie & Weng, 2010), and increases in the number and/or function of glutamate receptors (Doolen et al. 2012; Salter & Pitcher, 2012) have all been ascribed to the excessive activation of glutamate receptors in spinal dorsal horn neurons.
The critical role of NMDARs (one type of glutamate receptor) in spinal nociceptive sensory processing was demonstrated by early reports that blockade of spinal NMDARs with intrathecal injection of NMDA antagonists has little effect on the responses to acute nociceptive stimuli in normal animals but markedly reduces touch- and heat- hyperalgesia following peripheral inflammation or nerve injury (Chapman & Dickenson, 1992; Yamamoto & Yaksh, 1992; Ma & Woolf, 1995; Lufty et al. 1997). Traditionally, NMDARs are considered to be located in postsynaptic neurons. Numerous studies have shown that the activation of postsynaptic NMDARs not only participates in glutamatergic sensory synaptic transmission in normal conditions, but more importantly is also involved in synaptic plasticity in the spinal dorsal horn in pathological pain conditions induced by tissue inflammation or nerve injury (Wu & Zhuo, 2009; Salter & Pitcher, 2012). It is generally believed that excessive activation of NMDARs results in enhanced influx of Ca2+ into the neuron. Inside the cell, Ca2+ triggers calcium-sensitive signalling cascades and synaptic plasticity (Wu & Zhuo, 2009; Salter & Pitcher, 2012). Despite extensive studies on the role of postsynaptic NMDARs in the development and maintenance of pathological pain including neuropathic pain, the role of presynaptic NMDARs of primary afferents in the spinal nociceptive sensory processing remains elusive.
NMDARs are present in primary afferent terminals in the spinal dorsal horn and dorsal root ganglion neurons, as repeatedly confirmed both anatomically and physiologically (Liu et al. 1994; Bardoni et al. 2004; Li et al. 2006; Zeng et al. 2006). Activation of presynaptic NMDARs with intrathecal injection of the NMDAR agonist NMDA results in increased release of substance P from primary afferents in adult rats (Liu et al. 1997). Selective knockdown of NMDARs in primary afferent neurons decreases pain behaviours in phase 2 of the formalin test in adult rats (McRoberts et al. 2010). In the naive neonatal spinal dorsal horn, activation of presynaptic NMDARs on primary afferent terminals by bath application of NMDA inhibits glutamate release as measured by EPSPs in dorsal horn neurons (Bardoni et al. 2004). However, activation of presynaptic NMDARs in morphine-tolerant neonatal rats increases glutamate release from primary afferents in the spinal dorsal horn (Liu et al. 1994; Bardoni et al. 2004; Zeng et al. 2006). Little is known about the role of presynaptic NMDARs in regulating glutamate release from the spinal primary afferent terminals in neuropathic pain conditions in adult rats.
In this study, using the whole cell voltage clamp recording technique, we demonstrated that injury of L5 spinal nerve resulted in an increased glutamate release from the primary afferents in the spinal dorsal horn, which, in part, is attributable to the endogenous activation of presynaptic NMDARs in the primary afferents. In contrast, release of glutamate from the primary terminals in the spinal dorsal horn of normal (sham-operated) animals is not regulated by functional presynaptic NMDARs. Our study reveals a novel synaptic mechanism underlying the plasticity induced by nerve injury in the first spinal sensory synapse.
Methods
Animals
Young adult male Sprague–Dawley rats (weight range, 160–230 g) were used. All experiments were approved by the Institutional Animal Care and Use Committees at the University of Georgia and the University of Texas M.D. Anderson Cancer Center, and were fully compliant with the National Institutes of Health Guidelines for the Use and Care of Laboratory Animals.
Ligation of L5 spinal nerve and behavioural tests
The animals were randomly divided into spinal nerve ligation injury (SNL) group and sham-operated (control) group. SNL injury was performed as previously described (Kim & Chung, 1992). Briefly, under 2–3% isoflurane anaesthesia, a midline incision above the lumbar spine and deep dissection through the paraspinal muscles were made to expose the left L6 transverse process, and the process was then removed. The L5 spinal nerve was isolated and tightly ligated with a 4-0 silk suture distal to the dorsal root ganglia and proximal to the formation of the sciatic nerve. The incisions were then closed. Sham-operated rats underwent the same operation and handling as the SNL group, but without nerve ligation. No drugs were used after the surgery. Behavioural tests were performed to determine mechanical sensitivity 1 day before the operation, and on days 7–14 post-surgery, prior to electrophysiological and molecular experiments. Behavioural tests were used to determine the hind paw mechanical sensitivity after the surgery as described earlier (Nie & Weng, 2010; Weng et al. 2003). Briefly, the animals were placed on wire mesh, loosely restrained under a plexiglass cage (12 cm × 20 cm × 15 cm), and allowed to accommodate for at least 30 min. Von Frey monofilaments with bending forces ranging from 0.1 to 12.4 g were applied from below through the mesh onto the mid-plantar side of each hind paw to evoke paw withdrawal responses. Each hind paw was stimulated 10 times with each Von Frey monofilament, and the frequency (percentage) of paw withdrawal responses to 10 stimulations was recorded. The least bending force that evoked withdrawal in more than half the trials was assigned as the 50% withdrawal threshold.
In vitro whole cell recordings and data analysis
Spinal slice preparation
Rats were deeply anaesthetized by isoflurane inhalation and underwent laminectomy for removal of the lumbar spinal cord. The lumbar spinal cord section was placed in ice-cold sucrose artificial cerebrospinal fluid (aCSF) pre-saturated with 95% O2 and 5% CO2. The sucrose aCSF contained 234 mm sucrose, 3.6 mm KCl, 1.2 mm MgCl2, 2.5 mm CaCl2, 1.2 mm NaH2PO4, 12.0 mm glucose, and 25.0 mm NaHCO3. The pia-arachnoid membrane was removed from the section. The L4–5 spinal segment, identified by the lumbar enlargement and large dorsal roots, was attached with cyanoacrylate glue to a cutting support, which was then glued onto the stage of a vibratome (Series 1000, Technical Products International, St Louis, MO, USA). Transverse spinal cord slices (400 μm) were cut in the ice-cold sucrose aCSF and then preincubated in Krebs solution oxygenated with 95% O2 and 5% CO2 at 35°C for at least 2 h before they were transferred to the recording chamber. The Krebs solution contained 117.0 mm NaCl, 3.6 mm KCl, 1.2 mm MgCl2, 2.5 mm CaCl2, 1.2 mm NaH2PO4, 11.0 mm glucose, and 25.0 mm NaHCO3.
Whole cell voltage-clamp recordings
Following preincubation, a single slice was placed in the recording chamber (volume, 1.5 ml), perfused with Krebs solution at 35°C, and saturated with 95% O2 and 5% CO2. Borosilicate glass recording electrodes (resistance, 3–5 MΩ) were pulled and filled with an internal solution containing (in mm) potassium gluconate, 135; KCl, 5; MgCl2, 2.0; CaCl2, 0.5; Hepes, 5.0; EGTA, 5.0; ATP-Mg, 5.0; Na-GTP, 0.5; QX-314, 10; and MK-801, 1; guanosine 5′-O-(2-thiodiphosphate) (GDP-β-S), 2. MK-801 was used to block postsynaptic NMDARs (Berretta & Jones, 1996; Drdla et al. 2009; Nie & Weng, 2009) and GDP-β-S (2 mm) used to block signalling pathways activated by G protein-coupled receptors. Live dorsal horn neurons in the spinal lamina I and outer lamina II were visualized using a microscope system and approached using a three-dimensional motorized manipulator (Sutter Instrument Company), and whole-cell configurations were established by applying moderate negative pressure after electrode contact (Nakatsuka et al. 2003). A seal resistance of at least 2 GΩ and an access resistance of 20–35 MΩ were considered acceptable (Wu et al. 2005; Weng et al. 2007). The series resistance was optimally compensated by at least 70% and constantly monitored throughout the experiments. Experiments showing any evidence of loss of voltage control were discarded. Signals were amplified using an Axopatch 700B (Molecular Devices) and displayed and stored in a personal computer.
Excitatory postsynaptic currents (EPSCs) were evoked using constant-current electrical stimuli (0.2 ms duration repeated every 40 s) at a fixed stimulating intensity (0.8 mA) applied with a concentric bipolar stimulating electrode placed at the dorsal root entry zone (Yoshimura & Nishi, 1993; Weng et al. 2006). To specifically determine the function of presynaptic NMDARs in the primary afferent terminals, only neurons receiving monosynaptic input from primary afferent input were recorded. Monosynaptic input was based on a constant latency with graded intensity and high-frequency repetitive stimulation (20 Hz) (Yoshimura & Jessell, 1989; Kohno et al. 2003). Miniature EPSCs (mEPSCs) were recorded in the presence of tetrodotoxin (TTX, 1 μm). In a subset of experiments, currents in superficial dorsal horn neurons were evoked by exogenous l-glutamate (50 μm) injected onto the recorded neuron by puff-application (pressure: 3 p.s.i.; duration: 20 ms, repeated every 60 s) through a glass pipette with opening tip size of 8–12 μm. All recordings were made in the presence of bicuculline (10 μm) and strychnine (5 μm) in the external solution, to block GABAA and glycine receptors, respectively, at a membrane potential of –70 mV.
Data analysis
Data were recorded using Axopatch 700B amplifier, digitized at 10 kHz, and analysed off-line. The means of four EPSCs evoked by electrical stimulation at baseline, in the presence of tested drugs, and after washout of tested drugs were measured using the Clampfit software program (version 10.2; Molecular Devices, Sunnyvale, CA, USA). In some neurons, we also measured the inverse squared coefficient of variation (CV−2) of the peak amplitudes of 10 evoked EPSCs, where CV represents the ratio of the standard deviation to the mean. The frequency and amplitude of mEPSCs in the 3 min before and during perfusion of tested drugs were analysed and averaged using a peak detection program (MiniAnalysis; Synaptosoft Inc., Decatur, GA, USA).
The data were presented as the mean ± SEM. Student's t test was used to determine statistical differences between data obtained in the absence and presence of tested drugs (paired t test) or between groups (non-paired t test). A P value less than 0.05 was considered statistically significant.
Western blot experiments
Animals were deeply anaesthetized with urethane (1.3–1.5 g kg−1, i.p.) 7 days after ligation of the L5 spinal nerve. The L4 and L5 dorsal root ganglions ipsilateral to the injury site and the L4 and L5 spinal segments were exposed by surgery and removed from the rats. The dorsal quadrant of the spinal cord of each segment ipsilateral to the operated side was isolated. The dorsal root ganglions and the dorsal quadrants of the spinal cord were quickly frozen in liquid nitrogen and stored at –80°C for later use. Frozen tissues were homogenized with a hand-held pellet in lysis buffer (50 mm Tris, pH 7.5, 150 mm NaCl, 1 mm EDTA, 0.1% SDS, 1% deoxycholic acid, 2 mm orthovanadate, 100 mm NaF, 1% Triton X-100, 0.5 mm phenylmethylsulfonyl fluoride, 20 μm leupeptin, 100 IU ml−1 aprotinin) for 0.5 h at 37°C. The samples were then centrifuged for 20 min at 12,000 g at 4°C and the supernatants containing proteins were collected. The quantification of protein contents was made by the BCA method. Protein samples (40 μg) were electrophoresed in 8% SDS polyacrylamide gels and transferred to a polyvinylidene difluoride membrane (Millipore, Bedford, MA, USA). The membranes were blocked with 5% milk and incubated overnight at 4°C with a polyclonal goat anti-NR2B (1:200, Santa Cruz Biotechnology, CA, USA) or a monoclonal mouse anti-β-actin (1:2000, Sigma-Aldrich, St Louis, MO, USA) primary antibody as a loading control. Then the blots were incubated for 1 h at room temperature with a corresponding HRP-conjugated secondary antibody (1:5000; Santa Cruz Biotechnology, CA, USA), visualized in enhanced chemiluminescence (ECL) solution (SuperSignal West Pico Chemiluminescent Substrate, Pierce, Rockford, IL, USA) for 1 min, and exposed onto FluorChem HD2 System. The intensity of immunoreactive bands was quantified using ImageJ 1.46 software (NIH). Results were expressed as the ratio of NR2B to β-actin control. Student's t test (non-paired t test) was used to determine statistical differences between the neuropathic and sham-operated groups. A P value less than 0.05 was considered statistically significant.
Materials
Bicuculline, strychnine, N-methyl-d-aspartic acid (NMDA), GDP-β-S, 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA), GF109203X, MK-801, l-glutamate and TTX were obtained from Sigma (St Louis, MO, USA). d-Aminophosphonovaleric acid (d-AP5) and Ro 25-6981 were obtained from Tocris Bioscience (Minneapolis, MN, USA). The protein kinase C inhibitor peptide PKCI 19–31 was obtained from EMD Biosciences (San Diego, CA, USA). All pharmacological agents were applied via perfusion into the recording chamber unless indicated otherwise.
Results
All rats receiving ligation of L5 spinal nerve had mechanical allodynia prior to undergoing the electrophysiological experiments performed on days 7–14 post surgery. The mechanical threshold ipsilateral to the L5 ligation side decreased significantly from 7.25 ± 0.37 g at baseline to 1.29 ± 0.16 g (P < 0.001) prior to the electrophysiological recordings and molecular experiments in 48 SNL rats. The mechanical threshold in 25 sham-operated rats was not significantly altered (from 6.93 ± 0.40 g to 7.06 ± 0.40 g).
Amplitudes of EPSCs evoked by primary afferent input were higher in neuropathic rats than those in sham rats
Response of first sensory synapses in superficial dorsal horn neurons (Sugiura et al. 1986; Basbaum et al. 2009) to peripheral sensory stimulation was studied by recording EPSCs, evoked by electrically stimulating the dorsal root entry zone, from superficial dorsal horn neurons in spinal slices. We first compared EPSCs in neuropathic and sham-operated rats evoked by primary afferent input elicited by the same stimulation intensity (0.2 ms duration and 0.8 mA). In general, EPSC amplitudes recorded from neuropathic rats (1045.44 ± 30.99 pA, n= 69) were significantly (P < 0.001) higher than those obtained from sham-operated rats (amplitude: 800.23 ± 74.05 pA; n= 31). The increased EPSC amplitude in neuropathic rats might result from alteration in any or a combination of the followings: (a) an increase of glutamate release from presynaptic terminals; (b) a decrease of glutamate uptake by glial cells (astrocytes) and/or neurons; (c) an increase in the function and/or number of postsynaptic glutamate receptors. In the following experiments, we specifically determined the contribution of presynaptic plasticity to the enhanced EPSC amplitude induced by nerve injury.
Glutamate release from the primary afferent central terminals in neuropathic rats was increased
To examine glutamate release in the first sensory synapse in the spinal dorsal horn, we first recorded mEPSCs in neuropathic rats and sham-operated rats in the presence of TTX (1 μm). Consistent with findings by others (Balasubramanyan et al. 2006; Takasusuki et al. 2007; Fukushima et al. 2011), we found that frequencies of mEPSCs in neuropathic rats (6.24 ± 0.85 Hz, n= 19) were significantly (P < 0.001) higher than those obtained from sham-operated rats (2.53 ± 0.36 Hz, n= 18) but amplitudes of mEPSCs in neuropathic (27.58 ± 1.39 pA, n= 19) and sham-operated (26.59 ± 1.49 pA, n= 18) rats were similar (Fig. 1A). These findings suggest an increase of glutamate release probability from presynaptic terminals in neuropathic rats. It is likely that synaptic input from the primary afferent fibres constitutes only a fraction of the total excitatory input to superficial dorsal horn neurons. The mEPSCs we recorded might well be a reflection of overall excitatory inputs from both the primary afferents and excitatory interneurons to the recorded neuron. To specifically address glutamate release from the primary afferents, a pair of EPSCs were evoked by a pair of electrical stimulating pulses (50 ms apart) applied to the spinal dorsal root. We measured the paired-pulse ratio (PPR) of EPSCs (i.e. the ratio of the second peak amplitude over the first peak amplitude induced by paired-pulse stimulation). Analysis of PPRs is a classic approach to determine the transmitter release probability from presynaptic terminals (Zucker, 1989; Foster & McNaughton, 1991; Manabe et al. 1993; Weng et al. 2007; Xu et al. 2008). A decrease of PPR indicates an increased probability of neurotransmitter release from presynaptic terminals. In contrast, an increase of PPRs indicates a decreased probability of neurotransmitter release from presynaptic terminals. We found that in comparison with PPRs of sham-operated rats (0.73 ± 0.02, n= 31), PPRs in neuropathic rats (0.63 ± 0.01, n= 69) were significantly (P < 0.001) reduced (Fig. 1B). Furthermore, we analysed the inverse squared coefficient of variation (CV−2) of the peak amplitudes of 10 evoked EPSCs. An increase of presynaptic release is expected to cause an increase in CV−2 (Bekkers & Stevens, 1990; Korn & Faber, 1991). We found that the CV−2 in neuropathic rats (1749.04 ± 192.6, n= 20) was significantly (P < 0.001) larger than that in sham-operated rats (496.90 ± 34.83, n= 20) (Fig. 1C). A decrease of the PPR and an increase of both the mEPSC frequency and CV−2 in neuropathic rats all indicate that an increase of glutamate release from the primary afferent terminals contributed to the enhanced EPSCs in neuropathic rats.
Figure 1. Glutamate release from the primary afferent terminals in neuropathic rats was increased in comparison with sham-operated rats.
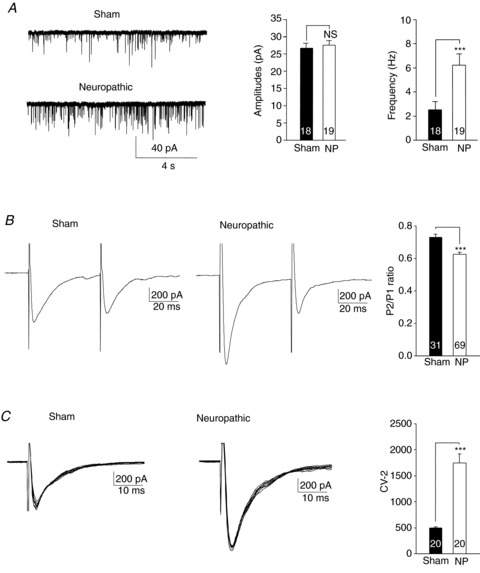
A, samples of mEPSC recordings recorded from sham-operated (Sham) and neuropathic rats. Bar graphs show comparisons of mean (+SEM) amplitudes (left) and frequencies (right) of mEPSCs between sham-operated and neuropathic rats. B, samples of EPSCs evoked by a pair of electrical pulses applied to the spinal dorsal root in a neuron from sham-operated rats and neuropathic rats. Bar graphs show the mean (+SEM) P2/P1 ratios in sham-operated and neuropathic rats. The ratio of the second peak amplitude (P2) over the first peak amplitude (P1) was lower in neuropathic rats than in sham-operated rats. C, samples of variability of 10 evoked EPSCs collected from a neuron of sham-operated rats and neuropathic rats. Bar graphs show the mean (+SEM) CV−2 values in sham-operated and neuropathic rats. The inverse squared coefficient of variation (CV−2) was larger in neuropathic rats than that in sham-operated rats. Number of neurons included in each group for the analysis is shown in each bar. * indicates comparison between sham-operated (Sham) and neuropathic (NP) rats. ***P < 0.001; NS, no statistical significance.
Blockade of presynaptic NMDARs reduced glutamate release from the presynaptic terminals in neuropathic rats
To investigate the role of presynaptic NMDARs in regulating glutamate release in neuropathic rat, we examined the effects induced by the selective NMDAR antagonist d-AP5 on non-NMDA mEPSCs and EPSCs evoked by stimulation of the spinal dorsal root. Bath application of d-AP5 (concentration in bath: 25 μm) significantly (P < 0.05) and reversibly reduced the mEPSC frequency from 7.37 ± 2.15 Hz to 5.71 ± 1.77 Hz (n= 5) but did not change the mEPSC amplitude in neurons recorded from the spinal L4 segment of neuropathic rats. Similarly, in neurons recorded from the spinal L5 segment of neuropathic rats, d-AP5 (25 μm) bath perfusion significantly (P < 0.05) decreased the mEPSC frequency from 7.70 ± 1.94 Hz to 5.66 ± 0.87 Hz (n= 5) but not the amplitude of mEPSCs. Percentages of the frequency reduction induced by d-AP5 in L4 spinal segment neurons (24.02 ± 2.83%) were similar (P= 0.59) to those of L5 spinal segment neurons (28.29 ± 4.95%). Thus, data from both the L4 and L5 spinal segments were combined and presented in Fig. 2A. In comparison, perfusion of d-AP5 (25 μm) did not alter the amplitude or the frequency of mEPSCs in neurons (n= 9) of spinal slices obtained from sham-operated animals (Fig. 2B).
Figure 2. Glutamate release from presynaptic terminals was enhanced by endogenous activation of presynaptic NMDARs in neuropathic rats but not in sham-operated rats.
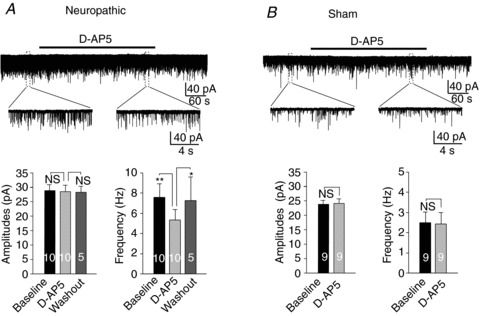
A, inhibition of NMDARs significantly and reversibly reduced the frequency of miniature EPSCs (mEPSCs) but did not change the amplitude of mEPSCs in neuropathic rats. B, inhibition of NMDARs did not significantly alter the mEPSC frequency or amplitude in sham-operated rats. Samples of mEPSC recordings before, during, and after washout of a NMDAR inhibitor (d-AP5, 25 μm) obtained from neuropathic and sham-operated rats are shown (top). The recordings before and during perfusion of d-AP5 were enlarged. Bar graphs show the mean (+SEM) frequencies and amplitudes of mEPSCs before, during, and after washout of d-AP5. *P < 0.05; **P < 0.01; NS, no statistical significance.
To specifically define the role of NMDARs of the primary afferents, we determined the effects of d-AP5 on a pair of EPSCs evoked by a pair of pulses (50 ms apart) of stimulation of the spinal dorsal root. Bath application of d-AP5 (25 μm) significantly and reversibly reduced the first peak amplitudes by 27.10 ± 2.62% (n= 9, P < 0.001) for L4 spinal segment neurons and by 28.2 ± 3.09% for L5 spinal segment neurons (n= 7, P < 0.001). This was accompanied by a significant increase of PPRs from 0.63 ± 0.02 to 0.73 ± 0.01 (n= 9, P < 0.001) for L4 spinal segment neurons and from 0.65 ± 0.03 to 0.76 ± 0.02 (n= 7, P < 0.001) for L5 spinal segment neurons. As the effects induced by d-AP5 on the first peak amplitude and PPR in L4 and L5 spinal segment neurons were similar (P > 0.79), combined data from both the L4 and L5 spinal segments were presented in Fig. 3A. Furthermore, in a subset of experiments, we also measured CV−2 values of the peak amplitudes of 10 evoked EPSCs before and after bath perfusion of d-AP5 (25 μm). The CV−2 was significantly reduced by d-AP5 from 1800.73 ± 589.50 to 229.18 ± 70.06 (n= 5, P < 0.05) for L4 spinal segment neurons and from 1652.48 ± 265.74 to 197.28 ± 28.73 (n= 5, P < 0.001) for L5 spinal segment neurons. The degree of changes induced by d-AP5 CV−2 in L4 and L5 spinal segment neurons were similar (P= 0.71). Data from L4 and L5 spinal segment neurons were pooled together and illustrated in Fig. 3B. Because d-AP5 had similar effects on L4 and L5 spinal segment neurons, data collected from L4 and L5 spinal segment neurons were combined for analysis in the rest of this paper. To further confirm the presynaptic action of d-AP5 under our experimental conditions, we recorded currents in superficial dorsal horn neurons elicited by exogenous glutamate applied through a puff electrode in 8 cells, we found that bath perfusion of d-AP5 (25 μm) did not change the currents elicited by exogenous glutamate (data not shown). Together, these results indicate that d-AP5 acted on presynaptic terminals and caused a reduced glutamate release from the primary afferents in neuropathic rats. In other words, endogenous activation of presynaptic NMDARs increased glutamate release from presynaptic terminals in the first sensory synapse in the spinal dorsal horn of neuropathic rats.
Figure 3. Endogenous activation of presynaptic NMDARs increased glutamate release from the presynaptic terminals in the first sensory synapse in the spinal dorsal horn of neuropathic rats but not in sham-operated rats.
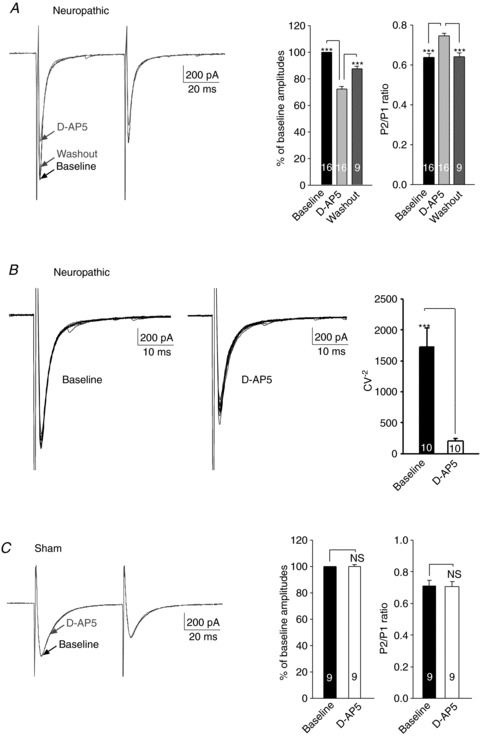
A, samples of EPSCs evoked by a pair of electrical pulses recorded from a neuron of neuropathic rats before (baseline), during and after washout of d-AP5 (25 μm). Bar graphs (right) show the mean (+SEM) amplitudes and P2/P1 ratios before, during, and after washout of d-AP5. Bath application of d-AP5 significantly and reversibly reduced the first peak amplitudes but increased the P2/P1 ratio. B, samples of variability of 10 evoked EPSCs collected from a neuron of neuropathic rats before (left) and during perfusion of d-AP5 (25 μm) are shown. The mean (+SEM) CV−2 values before and during perfusion of d-AP5 are shown in bar graphs (right). d-AP5 significantly increased the variability of evoked EPSCs. C, samples of EPSCs evoked by a pair of electrical pulses recorded from a neuron of sham-operated rats before (baseline) and during perfusion of d-AP5 (25 μm). Bar graphs (right) show the mean (+SEM) amplitudes and P2/P1 ratios before and during perfusion of d-AP5. Bath application of d-AP5 did not alter the first peak amplitudes or the P2/P1 ratio. ***P < 0.001; NS, no statistical significance.
In contrast, when we recorded afferent evoked EPSCs and PPRs in spinal slices taken from sham-operated rats, perfusion of d-AP5 (25 μm) did not change the EPSC amplitude or PPR in nine neurons tested (Fig. 3C). Together with the negative effects by d-AP5 on mEPSCs in slices from sham-operated rats, these data indicate that glutamate release from the primary afferents in the normal spinal dorsal horn is not regulated by endogenous activation of presynaptic NMDARs.
Exogenous NMDA increased glutamate release from the primary afferents in the spinal dorsal horn of neuropathic rats but not in sham-operated rats
To further confirm the role of presynaptic NMDARs and determine whether presynaptic NMDARs are functionally saturated by the endogenous agonist in neuropathic rats, we recorded a pair of EPSCs evoked by a pair of electrical pulses before and during perfusion of exogenous NMDA (50 μm) (Fig. 4A). In slices taken from neuropathic rats, addition of exogenous NMDA into the recording bath significantly and reversibly increased the peak amplitude of the first EPSCs by 23.42 ± 2.43% (n= 9, P < 0.001). Meanwhile, the PPR was decreased by exogenous NMDA from 0.57 ± 0.05 to 0.49 ± 0.04 (n= 9, P < 0.001). These data indicate that the function of presynaptic NMDARs in neuropathic rats was not saturated at baseline, and further confirm that activation of presynaptic NMDARs facilitated glutamate release from the primary afferents in the spinal dorsal horn of neuropathic rats.
Figure 4. Exogenous bath application of the NMDAR agonist NMDA enhanced glutamate release from the primary afferents in neuropathic rats but not in sham-operated rats.
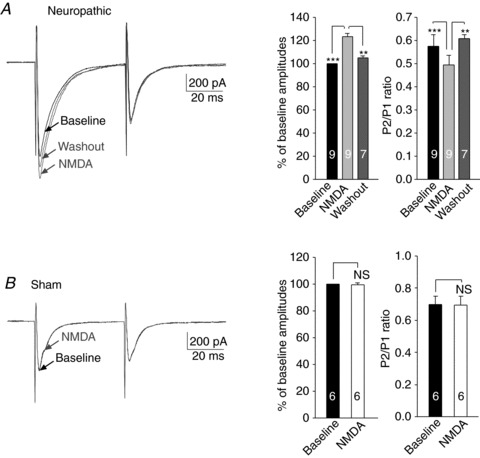
A, samples of EPSCs evoked by a pair of electrical pulses recorded from a neuron of neuropathic rats before (baseline), during and after washout of NMDA. Bar graphs (right) show the mean (+SEM) amplitudes and P2/P1 ratios before, during, and after washout of NMDA. Bath application of NMDA (50 μm) significantly and reversibly increased the first peak amplitudes but reduced the P2/P1 ratio. B, samples of EPSCs evoked by a pair of electrical pulses recorded from a neuron of sham-operated rats before (baseline) and during perfusion of NMDA (50 μm). Bar graphs (right) show the mean (+SEM) amplitudes and P2/P1 ratios before and during perfusion of NMDA. Bath application of NMDA (50 μm) did not significantly alter the first peak amplitudes or the P2/P1 ratio. **P < 0.01; ***P < 0.001; NS, no statistical significance.
We then, in spinal slices of sham-operated rats, perfused the NMDAR agonist NMDA (concentration in bath: 50 μm) after recording baseline afferent evoked EPSCs and PPRs in six neurons. Neither the amplitude nor the PPR was changed by exogenous NMDA (Fig. 4B). Thus, the lack of endogenous activation of presynaptic NMDARs in normal rats is not because ambient glutamate concentrations in sham-operated rats are lower than in neuropathic rats (Sung et al. 2003; Nie & Weng, 2010).
Exogenous d-serine increased glutamate release from the primary afferents in neuropathic rats but not in sham-operated rats
Activation of NMDARs requires not only glutamate or NMDA binding but also concurrent binding of glycine (or its endogenous ligand, d-serine) (Johnson & Ascher, 1987; Mothet et al. 2000) at the glycine site of NMDARs. It was reported that levels of NMDAR co-activator d-serine in the spinal dorsal horn are elevated under pathological pain conditions (Guo et al. 2006). It could be possible that ambient d-serine levels in normal conditions are not high enough to induce a sufficient number of activated NMDARs. In the following experiments, we recorded a pair of EPSCs evoked by a pair of electrical stimuli before and during perfusion of d-serine (200 μm) (Gaiarsa et al. 1990; Shuker et al. 1994; Li & Han, 2007) in spinal slices of sham-operated rats. We found that bath perfusion of d-serine (200 μm) plus NMDA (50 μm) did not change either the EPSC amplitude or PPR in neurons (n= 7) from sham-operated rats (Fig. 5A). These data indicate that low d-serine concentrations do not ascribe to the lack of presynaptic NMDAR-mediated glutamate release in sham-operated rats, and that the lack of NMDAR-mediated regulation of glutamate release in sham-operated rats results from the insufficient function and/or number of presynaptic NMDARs.
Figure 5. Exogenous application of d-serine plus NMDA increased glutamate release from the primary afferents in neuropathic rats but not in sham-operated rats.
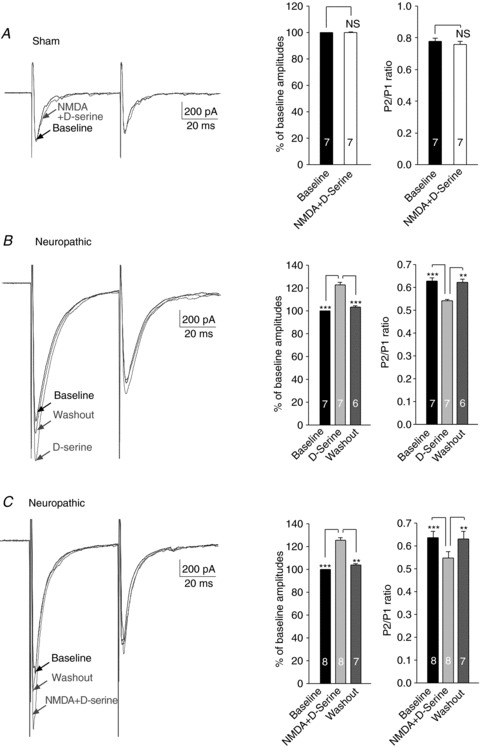
A, samples of EPSCs evoked by a pair of electrical pulses recorded from a neuron of sham-operated rats before (baseline) and during perfusion of d-serine (200 μm) plus NMDA (50 μm). Bar graphs (right) show the mean (+SEM) amplitudes and P2/P1 ratios before and during perfusion of d-serine plus NMDA. Bath application of d-serine plus NMDA did not significantly alter the first peak amplitudes or the P2/P1 ratio. B, samples of EPSCs evoked by a pair of electrical pulses recorded from a neuron of neuropathic rats before (baseline), during and after washout of d-serine (200 μm). Bar graphs (right) show the mean (+SEM) amplitudes and P2/P1 ratios before, during and after washout of d-serine. Bath application of d-serine significantly and reversibly enhanced the first peak amplitudes but reduced the P2/P1 ratio. C, samples of EPSCs evoked by a pair of electrical pulses recorded from a neuron of neuropathic rats before (baseline), during and after washout of d-serine (200 μm) plus NMDA (50 μm). Bar graphs (right) show the mean (+SEM) amplitudes and P2/P1 ratios before, during and after washout of d-serine. Bath application of d-serine plus NMDA significantly and reversibly enhanced the first peak amplitudes but reduced the P2/P1 ratio. **P < 0.01; ***P < 0.001.
In contrast, in spinal slices taken from neuropathic animals, d-serine (200 μm) significantly and reversibly increased the evoked EPSC amplitudes by 22.74 ± 2.00% (n= 7, P < 0.001), but reduced PPRs in neuropathic rats (Fig. 5B). In addition, concurrent application of both d-serine (200 μm) and NMDA (50 μm) produced similar changes in the evoked EPSC amplitudes and PPRs (Fig. 5C) as those induced by d-serine (50 μm) (Fig. 5B) or NMDA (50 μm) alone (Fig. 4A). These results indicate that, in neuropathic rats, presynaptic NMDARs are regulated by d-serine and the glycine site of NMDARs is not functionally saturated.
NR2B subunit was a dominant NMDAR at the primary afferent terminals in neuropathic rats
Most NMDARs in the central nervous system (CNS) are composed of NR1 and NR2 subunits forming a tetrameric complex of two NR1 and two NR2 subunits. NR2 receptors have different subtypes including NR2A, NR2B, NR2C and NR2D. Each subtype of NMDAR has different functional properties, and triggers different signalling pathways (Cull-Candy & Leszkiewicz, 2004). We then determined the contribution of the NR2B subunit to the composition of presynaptic NMDARs in neuropathic rats. In slices taken from neuropathic rats, we recorded a pair of EPSCs evoked by a pair of electrical stimuli before and during bath perfusion of the selective NR2B receptor antagonist Ro 25-6981 (1 μm) (Fischer et al. 1997; Mutel et al. 1998; Nie et al. 2010). Ro 25-6981 has more than 5000-fold greater selectivity for NR1/NR2B than for NR1/NR2A (Fischer et al. 1997). As shown in Fig. 6, perfusion of Ro 25-6981 significantly and reversibly reduced the amplitudes of the first EPSCs by 26.41 ± 3.303% (n= 6, P < 0.001), which was accompanied by a significant increase of PPRs. We also found that the percentage of reduction (26.41 ± 3.303%) induced by Ro 25-6981 was similar to those induced by d-AP5 (27.58 ± 1.94%), indicating that presynaptic NMDARs mediating glutamate release from the primary afferents in neuropathic rats are predominantly composed of NR2B subunits.
Figure 6. NR2B subunit was a dominant NMDAR at the presynaptic terminals in neuropathic rats.
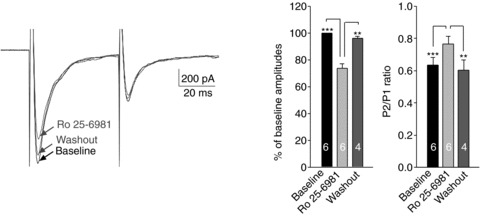
Original recordings show samples of EPSCs evoked by a pair of electrical pulses recorded from a neuron of neuropathic rats before (baseline), during and after washout of Ro 25-6981 (1 μm). Bar graphs (right) show the mean (+SEM) amplitudes and P2/P1 ratios before, during and after washout of Ro 25-6981. Bath application of Ro 25-6981 significantly and reversibly reduced the first peak amplitudes but increased the P2/P1 ratio. **P < 0.01; ***P < 0.001.
The function of presynapic NMDARs in neuropathic rats was dependent on activities of protein kinase C
It was reported previously that phosphorylation of NMDARs by protein kinase C (PKC) increases the function of NMDARs (Tingley et al. 1997; Lim et al. 2005; Kohno et al. 2008). We then tested whether the function of presynaptic NMDARs in neuropathic rats is regulated by PKC. We first determined the effects induced by the non-selective PKC inhibitor GF109203X on a pair of EPSCs evoked by a pair of electrical stimuli applied to the spinal dorsal root. In this set of experiments (all data in Fig. 7), a PKC inhibitor (PKCI 19–31, 5 μm) and a calcium chelator [1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA), 10 mm) were included in the pipette solution to preclude possible effects induced by GF109203X in the recorded postsynaptic neuron (Kohno et al. 2008) and signalling pathways activated by changes of intracellular Ca2+ levels in the recorded neuron (Kovalchuk et al. 2002; Gordon et al. 2005). After a pair of EPSCs evoked by a pair of electrical stimuli (50 ms apart) at baseline was recorded, GF109203X (4 μm) was perfused into the recording bath and a pair of EPSCs were recorded again. GF109203X significantly reduced the peak amplitude of the first EPSCs by 28.23 ± 1.62% (n= 23, P < 0.001) but increased the PPR from 0.63 ± 0.02 to 0.77 ± 0.02 (n= 23, P < 0.001) in neurons from neuropathic rats (Fig. 7). These data indicate that activation of PKC in presynaptic terminals increased glutamate release from the primary afferents in neuropathic rats.
Figure 7. The function of presynapic NMDARs in neuropathic rats was dependent on PKC activities.
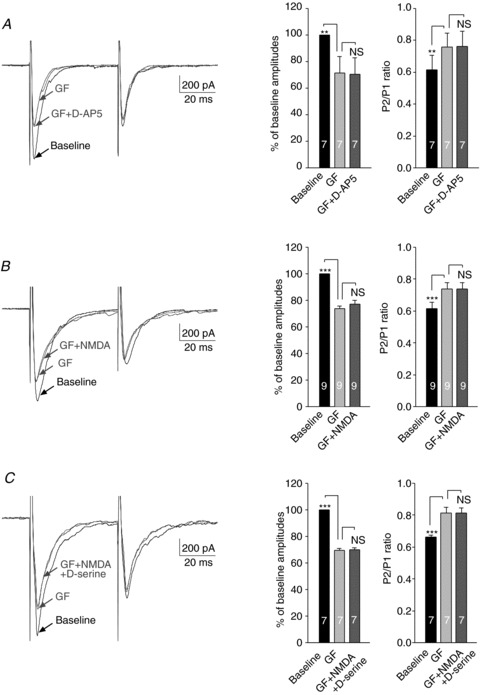
A, samples of EPSCs evoked by a pair of electrical pulses recorded from a neuron of neuropathic rats at baseline, during perfusion of a PKC inhibitor, GF109203X (GF, 4 μm), and then during addition of a NMDAR inhibitor, d-AP5 (25 μm) in the presence of GF109203X. Bar graphs (right) show the mean (+SEM) amplitudes and P2/P1 ratios at baseline, and during application of GF109203X, and then GF109203X plus d-AP5. Bath application of GF109203X significantly reduced the first peak amplitude but increased the P2/P1 ratio. Further addition of d-AP5 had no effects on the amplitude and P2/P1 ratio. B, samples of EPSCs evoked by a pair of electrical pulses recorded from a neuron of neuropathic rats at baseline, during perfusion of GF109203X (GF, 4 μm), and then during addition of NMDA (50 μm) in the presence of GF109203X. Bar graphs (right) show the mean (+SEM) amplitudes and P2/P1 ratios at baseline, and during application of GF109203X, and then GF109203X plus NMDA. NMDA had no effects on the amplitude and P2/P1 ratio in the presence of GF109203X. C, samples of EPSCs evoked by a pair of electrical pulses recorded from a neuron of neuropathic rats at baseline, during perfusion of GF109203X (GF, 4 μm), and then during addition of NMDA (50 μm) plus d-serine (200 μm) in the presence of GF109203X. Bar graphs (right) show the mean (+SEM) amplitudes and P2/P1 ratios at baseline, and during application of GF109203X, and then NMDA plus d-serine in the presence of GF109203X. NMDA plus d-serine had no effects on the amplitude and P2/P1 ratio in the presence of GF109203X. **P < 0.01; ***P < 0.001. NS, no statistical significance.
After the effects of GF109203X on the amplitude and PPR of evoked EPSCs were documented, we conducted the following experiments. In order to determine whether presynaptic NMDARs are the downstream effector of PKC in neuropathic rats, in the first subset of experiments, we examined the effects of d-AP5 on a pair of EPSCs in the presence of GF109203X. Bath application of d-AP5 did not alter either the amplitudes or the PPRs of evoked EPSCs in the presence of GF109203X in neuropathic rats (n= 7) (Fig. 7A). In other words, inhibition of PKC completely occluded the effects induced by d-AP5. This occlusion effect may result from direct suppressive effects induced by GF109203X on presynaptic NMDARs. Alternatively, inhibition of PKC by GF109203X may somehow reduce ambient glutamate concentrations, leading to deactivation of NMDARs. If this is the case, the enhancement induced by exogenous NMDA on the EPSCs in neuropathic rats demonstrated in Fig. 4A should be preserved in the presence of GF109203X. This possibility was excluded in the second subset of experiments. In these experiments, in the presence of GF109203X (4 μm), we further added exogenous NMDA (50 μm) into the recording bath and recorded the pair of evoked EPSCs again. We found that in the presence of GF109203X (4 μm), exogenous NMDA (50 μm) no longer altered the amplitude and PPR in neurons (n= 9) (Fig. 7B). Another possible mechanism underlying the suppressive effect induced by GF109203X on presynaptic NMDAR activities is that inhibition of PKC reduces levels of the endogenous agonist binding to the glycine site of NMDARs. To rule out such a possibility, in the third set of experiments, we added NMDA (50 μm) plus d-serine (200 μm) simultaneously into the recording bath in the presence of GF109203X (4 μm). As shown in Fig. 7C, concurrent bath application of exogenous NMDA and d-serine did not alter either the amplitudes or the PPRs of afferent evoked EPSCs in the presence of GF109203X. Taken together, these data indicated that the function of presynaptic NMDARs in neuropathic rats is dependent on the activation of PKC.
In comparison, perfusion of GF109203X (4 μm) did not alter the peak amplitude of the first EPSCs or the PPR recorded from neurons (n= 6) in slices of sham-operated rats (data not shown).
Protein expression of NR2B subunit in the L4 and L5 dorsal root ganglions and spinal dorsal horn ipsilateral to the injury site in neuropathic rats was upregulated
Finally, we conducted Western blot experiments to determine protein expression levels of the NR2B subunit in the spinal dorsal horn and dorsal root ganglions in neuropathic (n= 5) and sham-operated (n= 5) rats. As shown in Fig. 8, 7 days after ligation of the L5 spinal nerve, protein expression of the NR2B subunit was significantly increased by 95.97 ± 25.47% (P < 0.05) in the L4 spinal dorsal horns, and 106.46 ± 15.33% (P < 0.01) in the L5 spinal dorsal horns ipsilateral to the injury site in comparison with the sham-operated rats. At the same time, protein expression of the NR2B subunit in the L4 and L5 dorsal root ganglions ipsilateral to the injury site was also significantly increased by 140.81 ± 33.69% (P < 0.01) and 116.73 ± 12.40% (P < 0.01), respectively, in comparison with the sham-operated animals. The changes of NR2B subunit expression in the dorsal root ganglion and spinal dorsal horn at the L4 level were similar to those at the L5 level. These data support our conclusions that the lack of NMDAR-mediated regulation of glutamate release in sham-operated rats results from the insufficient function and/or number of presynaptic NMDARs.
Figure 8. Protein expression of NR2B subunit in the L4 and L5 dorsal root ganglions and spinal dorsal horn ipsilateral to the injury site in neuropathic rats was increased.
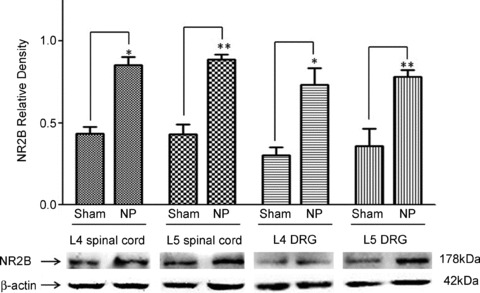
Samples of NR2B subunit expression in the spinal dorsal horn and dorsal root ganglion at the L4 and L5 levels in neuropathic (n= 5) and sham-operated (n= 5) rats are shown. Bath graphs show the mean (+SEM) relative density to β-actin in each group. DRG, dorsal root ganglion; *P < 0.05; **P < 0.01.
Discussion
Activation of NMDARs is a crucial mechanism underlying the development and maintenance of chronic pain, including neuropathic pain. Traditionally, the role of NMDARs in the pathogenesis of pain is ascribed to their activation and signalling cascades in postsynaptic neurons (Ren & Dubner, 2007; Salter & Pitcher, 2012). In this study, we, for the first time, identified endogenous activation of presynaptic NMDARs in the central terminals of primary afferents as a critical mechanism underlying the increased glutamatergic synaptic response in the first sensory synapse in the spinal dorsal horn of rats with neuropathic pain. Presynaptic NMDARs in neuropathic rats are mainly composed of NR2B receptors. This is consistent with and supported by an increase of NR2B subunit protein expression in both the dorsal root ganglion and spinal dorsal horn in neuropathic rats. The action of presynaptic NMDARs in neuropathic rats is enhanced by exogenous d-serine and/or NMDA and dependent on PKC activities. We demonstrated that glutamate release from the primary afferents in sham-operated rats is not regulated by presynaptic NMDARs. We also provide evidence that the lack of NMDAR-mediated regulation of glutamate release in sham-operated rats results from the insufficient function and/or number of presynaptic NMDARs.
Endogenous activation of presynaptic NMDARs as a novel mechanism underlying synaptic plasticity induced by nerve injury
The first nociceptive sensory synapse is the primary station for processing nociceptive information in the CNS. Glutamate is a major excitatory neurotransmitter released from the primary afferents in the spinal dorsal horn (De Biasi & Rustioni, 1988; Willis, 2002). In the neuroplasticity induced by nerve injury in the first nociceptive sensory synapse, changes in glutamate synaptic function are crucial to the development and maintenance of neuropathic pain (Salter & Pitcher, 2012). The function of glutamatergic synapses is governed by three key factors including the amount of glutamate release from presynaptic terminals, the rate at which glutamate is taken up by glutamate transporters, and the number and function of postsynaptic glutamate receptors (Danbolt, 2001). Glutamate release from primary afferent terminals in the spinal dorsal horn can be regulated by many receptors present presynaptically on the primary afferent terminals. For example, glutamate release from the primary afferents in the spinal dorsal horn is reduced upon activation of μ-opioid receptors (Heinke et al. 2011), α2 adrenoreceptors (Kawasaki et al. 2003), CB1 cannabinoid receptors (Lever & Malcangio, 2002), GABAA receptors (Willis, 2006), GABAB receptors (Ataka et al. 2000), and group II and group III metabotropic glutamate receptors (Gerber et al. 2000). On the other hand, activation of group I metabotropic glutamate receptors increases glutamate release from the central terminals of primary afferents (Song et al. 2009). Despite these extensive studies of the mechanism regulating glutamate release from primary afferent terminals, little is known about the role of presynaptic NMDARs in primary afferent terminals in pathological pain conditions. Our study reveals that endogenous activation of NMDARs in the central terminals of primary afferents in the spinal dorsal horn is critical to the increased glutamate release from primary afferent terminals triggered by peripheral sensory input in neuropathic rats. The suppression of glutamate release by the NMDAR antagonist d-AP5 found in this study may well contribute to the inhibitory effects of NMDA antagonists and PKC inhibitors on the genesis of neuropathic pain reported in numerous previous studies (Wang et al. 2004; Yajima et al. 2005; Brown & Krupp, 2006; Salter & Pitcher, 2012).
Our findings are reminiscent of a previous study reporting that activation of presynaptic NMDARs in morphine-tolerant neonatal rats increases glutamate release from the primary afferents in the spinal dorsal horn (Zeng et al. 2006). Increased glutamate release in forebrain areas in animal models of epilepsy is also related to the activation of presynaptic NMDARs (Yang et al. 2006; Graebenitz et al. 2010). Furthermore, activation of presynaptic NMDARs is involved in long-term potentiation (LTP) in the amygdala (Humeau et al. 2003; Samson & Pare, 2005), and long-term depression (LTD) in the neocortex (Sjöström et al. 2003) and cerebellum (Casado et al. 2002). Thus, endogenous activation of presynaptic NMDARs is a critical mechanism leading to synaptic plasticity induced by both pathological and physiological conditions.
Mechanisms underlying NMDAR-mediated enhancement of glutamate release in neuropathic rats
It is conceivable that activation of presynaptic NMDARs causes influx of Ca2+ into the central terminals of primary afferents and subsequent increase of glutamate release, as shown in hippocampal slices (McGuinness et al. 2010). The influx of Ca2+ could come directly from the opening of NMDARs (Glitsch & Marty, 1999) and/or from voltage-gated calcium channels in response to depolarization due to the opening of NMDARs (Awatramani et al. 2005; Christie & Jahr, 2008).
Prerequisites for the activation of NMDARs include binding of both glutamate (or NMDA) and glycine (or d-serine) to NMDARs and sufficient membrane depolarization to relieve Mg2+ blockade of the ion channels (Mayer et al. 1984; Oliet & Mothet, 2009). Glutamate binding to presynaptic NMDARs may come from three sources. Firstly, glutamate released from the presynaptic terminals may activate presynaptic NMDARs. This is supported by a recent hippocampal study showing that glutamate released from presynaptic terminals, triggered by the arrival of action potentials, can diffuse and bind to presynaptic NMDARs (autoreceptors), resulting in an increase of Ca2+ in the bouton of Schaffer collaterals (McGuinness et al. 2010) and an increased release of glutamate. Secondly, presynaptic NMDARs can be activated by glutamate released from astrocytes as shown in a recent report from studies of the hippocampal dentate gyrus (McGuinness et al. 2010). It remains to be further clarified if these two mechanisms occur in the spinal dorsal horn. Thirdly, NMDARs can be activated by ambient glutamate in extracellular space as demonstrated in forebrain slices (Herman & Jahr, 2007; Le Meur et al. 2007). Elevated ambient glutamate concentrations in neuropathic pain, caused by deficient glial glutamate uptake, would further increase the number of NMDARs activated. Our findings that exogenous application of NMDA increased evoked EPSC amplitudes but reduced EPSC PPRs provide direct evidence that presynaptic NMDARs can be activated by ambient agonists in the spinal dorsal horn. In this study, we also found that NR2B subunits are a predominant subtype in the primary afferent central terminals in neuropathic rats. In comparison with NR2A subunits, NR2B subunits have a higher affinity to glutamate (Erreger et al. 2007) and longer deactivation time (Monyer et al. 1994; Vicini et al. 1998). These special properties of NR2B subunits may yield a greater functional impact on the primary afferent central terminals in response to a given concentration of glutamate.
It is intriguing that the effects of presynaptic NMDARs shown in the current study and other studies are observed under normal extracellular Mg2+ concentrations (Zeng et al. 2006; Corlew et al. 2008). It was recently suggested that depolarization induced by the arrival of action potentials at the bouton removes Mg2+ block in NMDA channels (McGuinness et al. 2010). Although this may account for the effects of NMDAR inhibitors on EPSCs evoked by peripheral sensory input (Figs 3 and 6), it would not speak for the effects of d-AP5 on mEPSCs which were recorded when action potentials were blocked by TTX (Fig. 2). It is most likely that the mEPSCs recorded in this study are a reflection of spontaneous glutamate release from both the primary afferents and excitatory interneurons in the spinal dorsal horn. The resting potential in the primary afferent central terminals and excitatory interneuron terminals are unknown. Two possible mechanisms may take place. First, unblock of Mg2+ in NMDA channels may occurs if the resting potential in the primary afferent central terminals and excitatory interneuron terminals is less negative. Alternatively, depolarization of membrane potentials and removal of Mg2+ block in presynaptic terminals may be induced upon activation of AMPA and/or kainite receptors in the presynaptic terminals (Lee et al. 2002; Lu et al. 2002).
Mechanisms underlying the lack of presynaptic NMDAR-mediated regulation of glutamate release in normal rats
In sham-operated adult rats, we found that EPSCs evoked by the primary afferent input were not altered by selectively blocking NMDARs with d-AP5, indicating that glutamate release from the primary afferents is not endogenously regulated by NMDARs in normal adult rats. It could be possible that extracellular levels of the NMDAR agonist and/or coagonist (glycine and/or d-serine) in normal animals are not high enough to activate a sufficient number of NMDARs. This possibility has been ruled out by our results showing that bath application of NMDA alone or NMDA plus d-serine did not alter EPSCs evoked by the primary afferent input in sham-operated rats. Alternatively, the impact produced by the activity of NMDARs in the primary central terminals in normal animals is below a threshold needed to alter glutamate release. In other words the number of NMDARs, and/or the function of individual NMDARs, in the primary central terminals in normal animals is less than in those of neuropathic rats. This notion is supported by our findings that glutamate release regulated by presynaptic NMDARs in neuropathic rats is dependent on the PKC activity whereas glutamate release in normal rats is not altered by blocking PKC activity. In agreement with our findings, numerous studies have shown that the NMDAR function is increased upon phosphorylation by PKC (Tingley et al. 1997; Lim et al. 2005; Kohno et al. 2008). More specifically, activation of PKC enhances and prolongs the NMDA-evoked Ca2+ signals in the soma of the primary afferents (dorsal root ganglion neurons) (Castillo et al. 2011).
Function of presynaptic NMDARs in regulating glutamate release is synaptic specific, age- and function-dependent
The lack of regulation by NMDARs in the sham-operated adult rats in this study is in contrast with a previous report that exogenous application of NMDA into the recording bath reduces glutamate release from the primary afferent terminals in normal neonatal dorsal horn as measured by EPSCs in the spinal dorsal horn (Bardoni et al. 2004). It is noteworthy that the response to high doses of selective agonist in this study does not directly reveal the physiological role of presynaptic NMDARs when exposed to endogenous levels of glutamate. Nevertheless, the NMDAR number and function in the spinal dorsal horn alter significantly during development (Pattinson & Fitzgerald, 2004). Although the physiological role of NMDARs in the primary afferent central terminals in newborn rats remains to be determined, synaptic studies of other CNS areas show that glutamate release regulated by endogenous activation of presynaptic NMDARs in normal adult rats tends to be less marked or lost. For example, activation of presynaptic NMDARs enhances neurotransmitter release at synapses onto visual cortex pyramidal cells in mice before postnatal day 20, but has no apparent effect after postnatal day 23 (Corlew et al. 2007). The presynaptic effect of NMDA on glutamate release is observed in Purkinje cells recorded from juvenile but not from adult mice (Lonchamp et al. 2012). Similarly, endogenous activation of presynaptic NMDARs in the frontal cortex enhances GABA release in 12- to 15-day-old rats but not in 3-week-old rats (Pradhan et al. 2011). Furthermore, the activation of presynaptic NMDARs can increases glutamate release in some synapses (Berretta & Jones, 1996; Sjöström et al. 2003; Bender et al. 2006; Yang et al. 2006; Corlew et al. 2007; Brasier & Feldman, 2008) but decrease glutamate release in other synapses (Casado et al. 2002). Our current study extends this complexity by demonstrating that presynaptic NMDARs regulate glutamate release in the adult spinal dorsal horn after peripheral nerve injury but not in normal rats. This functional alteration induced by the nerve injury is accompanied by an increase of NR2B subunit protein expression in the dorsal root ganglion and spinal dorsal horn (Fig. 8) Hence, the function of presynaptic NMDARs in regulating glutamate release is synaptic specific and depends on the functional (physiological versus pathological) and developmental states (age).
In conclusion, this study reveals that suppression of the presynaptic NMDAR activity in the primary sensory afferents is an effective approach to attenuate the enhanced glutamatergic response in the spinal first sensory synapse induced by peripheral nerve injury, and presynaptic NMDARs might be a novel target for the development of analgesics.
Acknowledgments
This project was supported by the National Institute of Neurological Disorders and Stroke RO1 Grant NS-064289 to H.R. Weng.
Glossary
- mEPSCs
miniature EPSCs
- NMDAR
NMDA receptor
- PKC
protein kinase C
- PPR
paired-pulse ratio
- SNL
spinal nerve ligation injury
Author contributions
The experiments were performed at the University of Texas MD Anderson Cancer Center and the University of Georgia College of Pharmacy. X.Y., E.J. and M.G. participated in designing and performing the experiments, and data analysis. H.R.W. conceived of the study, participated in the design of the study and data analysis, and wrote the manuscript. All authors approved the final version of the manuscript.
References
- Ataka T, Kumamoto E, Shimoji K, Yoshimura M. Baclofen inhibits more effectively C-afferent than Aδ-afferent glutamatergic transmission in substantia gelatinosa neurons of adult rat spinal cord slices. Pain. 2000;86:273–282. doi: 10.1016/S0304-3959(00)00255-4. [DOI] [PubMed] [Google Scholar]
- Awatramani GB, Price GD, Trussell LO. Modulation of transmitter release by presynaptic resting potential and background calcium levels. Neuron. 2005;48:109–121. doi: 10.1016/j.neuron.2005.08.038. [DOI] [PubMed] [Google Scholar]
- Balasubramanyan S, Stemkowski PL, Stebbing MJ, Smith PA. Sciatic chronic constriction injury produces cell-type-specific changes in the electrophysiological properties of rat substantia gelatinosa neurons. J Neurophysiol. 2006;96:579–590. doi: 10.1152/jn.00087.2006. [DOI] [PubMed] [Google Scholar]
- Bardoni R, Torsney C, Tong CK, Prandini M, MacDermott AB. Presynaptic NMDA receptors modulate glutamate release from primary sensory neurons in rat spinal cord dorsal horn. J Neurosci. 2004;24:2774–2781. doi: 10.1523/JNEUROSCI.4637-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. 2009;139:267–284. doi: 10.1016/j.cell.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekkers JM, Stevens CF. Presynaptic mechanism for long-term potentiation in the hippocampus. Nature. 1990;346:724–729. doi: 10.1038/346724a0. [DOI] [PubMed] [Google Scholar]
- Bender VA, Bender KJ, Brasier DJ, Feldman DE. Two coincidence detectors for spike timing-dependent plasticity in somatosensory cortex. J Neurosci. 2006;26:4166–4177. doi: 10.1523/JNEUROSCI.0176-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berretta N, Jones RS. Tonic facilitation of glutamate release by presynaptic N-methyl-D-aspartate autoreceptors in the entorhinal cortex. Neuroscience. 1996;75:339–344. doi: 10.1016/0306-4522(96)00301-6. [DOI] [PubMed] [Google Scholar]
- Brasier DJ, Feldman DE. Synapse-specific expression of functional presynaptic NMDA receptors in rat somatosensory cortex. J Neurosci. 2008;28:2199–2211. doi: 10.1523/JNEUROSCI.3915-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DG, Krupp JJ. N-methyl-D-aspartate receptor (NMDA) antagonists as potential pain therapeutics. Curr Top Med Chem. 2006;6:749–770. doi: 10.2174/156802606777057571. [DOI] [PubMed] [Google Scholar]
- Casado M, Isope P, Ascher P. Involvement of presynaptic N-methyl-D-aspartate receptors in cerebellar long-term depression. Neuron. 2002;33:123–130. doi: 10.1016/s0896-6273(01)00568-2. [DOI] [PubMed] [Google Scholar]
- Castillo C, Norcini M, Baquero-Buitrago J, Levacic D, Medina R, Montoya-Gacharna JV, Blanck TJ, Dubois M, Recio-Pinto E. The N-methyl-D-aspartate-evoked cytoplasmic calcium increase in adult rat dorsal root ganglion neuronal somata was potentiated by substance P pretreatment in a protein kinase C-dependent manner. Neuroscience. 2011;177:308–320. doi: 10.1016/j.neuroscience.2010.12.040. [DOI] [PubMed] [Google Scholar]
- Chapman V, Dickenson AH. The combination of NMDA antagonism and morphine produces profound antinociception in the rat dorsal horn. Brain Res. 1992;573:321–323. doi: 10.1016/0006-8993(92)90780-d. [DOI] [PubMed] [Google Scholar]
- Chen Y, Balasubramanyan S, Lai AY, Todd KG, Smith PA. Effects of sciatic nerve axotomy on excitatory synaptic transmission in rat substantia gelatinosa. J Neurophysiol. 2009;102:3203–3215. doi: 10.1152/jn.00296.2009. [DOI] [PubMed] [Google Scholar]
- Christie JM, Jahr CE. Dendritic NMDA receptors activate axonal calcium channels. Neuron. 2008;60:298–307. doi: 10.1016/j.neuron.2008.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corlew R, Brasier DJ, Feldman DE, Philpot BD. Presynaptic NMDA receptors: newly appreciated roles in cortical synaptic function and plasticity. Neuroscientist. 2008;14:609–625. doi: 10.1177/1073858408322675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corlew R, Wang Y, Ghermazien H, Erisir A, Philpot BD. Developmental switch in the contribution of presynaptic and postsynaptic NMDA receptors to long-term depression. J Neurosci. 2007;27:9835–9845. doi: 10.1523/JNEUROSCI.5494-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cull-Candy SG, Leszkiewicz DN. Role of distinct NMDA receptor subtypes at central synapses. Sci STKE. 2004 doi: 10.1126/stke.2552004re16. 2004, re16. [DOI] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- De Biasi S, Rustioni A. Glutamate and substance P coexist in primary afferent terminals in the superficial laminae of spinal cord. Proc Natl Acad Sci U S A. 1988;85:7820–7824. doi: 10.1073/pnas.85.20.7820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolen S, Blake CB, Smith BN, Taylor BK. Peripheral nerve injury increases glutamate-evoked calcium mobilization in adult spinal cord neurons. Mol Pain. 2012;8:56. doi: 10.1186/1744-8069-8-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drdla R, Gassner M, Gingl E, Sandkuhler J. Induction of synaptic long-term potentiation after opioid withdrawal. Science. 2009;325:207–210. doi: 10.1126/science.1171759. [DOI] [PubMed] [Google Scholar]
- Erreger K, Geballe MT, Kristensen A, Chen PE, Hansen KB, Lee CJ, Yuan H, Le P, Lyuboslavsky PN, Micale N, Jorgensen L, Clausen RP, Wyllie DJ, Snyder JP, Traynelis SF. Subunit-specific agonist activity at NR2A-, NR2B-, NR2C-, and NR2D-containing N-methyl-D-aspartate glutamate receptors. Mol Pharmacol. 2007;72:907–920. doi: 10.1124/mol.107.037333. [DOI] [PubMed] [Google Scholar]
- Fischer G, Mutel V, Trube G, Malherbe P, Kew JN, Mohacsi E, Heitz MP, Kemp JA. Ro 25-6981, a highly potent and selective blocker of N-methyl-D-aspartate receptors containing the NR2B subunit. Characterization in vitro. J Pharmacol Exp Ther. 1997;283:1285–1292. [PubMed] [Google Scholar]
- Foster TC, McNaughton BL. Long-term enhancement of CA1 synaptic transmission is due to increased quantal size, not quantal content. Hippocampus. 1991;1:79–91. doi: 10.1002/hipo.450010108. [DOI] [PubMed] [Google Scholar]
- Fukushima T, Takasusuki T, Tomitori H, Hori Y. Possible involvement of syntaxin 1A downregulation in the late phase of allodynia induced by peripheral nerve injury. Neuroscience. 2011;175:344–357. doi: 10.1016/j.neuroscience.2010.11.049. [DOI] [PubMed] [Google Scholar]
- Gaiarsa JL, Corradetti R, Cherubini E, Ben-Ari Y. The allosteric glycine site of the N-methyl-D-aspartate receptor modulates GABAergic-mediated synaptic events in neonatal rat CA3 hippocampal neurons. Proc Natl Acad Sci U S A. 1990;87:343–346. doi: 10.1073/pnas.87.1.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber G, Zhong J, Youn DH, Randic M. Group II and group III metabotropic glutamate receptor agonists depress synaptic transmission in the rat spinal cord dorsal horn. Neuroscience. 2000;100:393–406. doi: 10.1016/s0306-4522(00)00269-4. [DOI] [PubMed] [Google Scholar]
- Glitsch M, Marty A. Presynaptic effects of NMDA in cerebellar Purkinje cells and interneurons. J Neurosci. 1999;19:511–519. doi: 10.1523/JNEUROSCI.19-02-00511.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graebenitz S, Lesting J, Sosulina L, Seidenbecher T, Pape HC. Alteration of NMDA receptor-mediated synaptic interactions in the lateral amygdala associated with seizure activity in a mouse model of chronic temporal lobe epilepsy. Epilepsia. 2010;51:1754–1762. doi: 10.1111/j.1528-1167.2010.02561.x. [DOI] [PubMed] [Google Scholar]
- Guo JD, Wang H, Zhang YQ, Zhao ZQ. Distinct effects of D-serine on spinal nociceptive responses in normal and carrageenan-injected rats. Biochem Biophys Res Commun. 2006;343:401–406. doi: 10.1016/j.bbrc.2006.02.156. [DOI] [PubMed] [Google Scholar]
- Gordon GR, Baimoukhametova DV, Hewitt SA, Rajapaksha WR, Fisher TE, Bains JS. Norepinephrine triggers release of glial ATP to increase postsynaptic efficacy. Nat Neurosci. 2005;8:1078–1086. doi: 10.1038/nn1498. [DOI] [PubMed] [Google Scholar]
- Heinke B, Gingl E, Sandkuhler J. Multiple targets of μ-opioid receptor-mediated presynaptic inhibition at primary afferent Aδ- and C-fibers. J Neurosci. 2011;31:1313–1322. doi: 10.1523/JNEUROSCI.4060-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman MA, Jahr CE. Extracellular glutamate concentration in hippocampal slice. J Neurosci. 2007;27:9736–9741. doi: 10.1523/JNEUROSCI.3009-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humeau Y, Shaban H, Bissiere S, Luthi A. Presynaptic induction of heterosynaptic associative plasticity in the mammalian brain. Nature. 2003;426:841–845. doi: 10.1038/nature02194. [DOI] [PubMed] [Google Scholar]
- Johnson JW, Ascher P. Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature. 1987;325:529–531. doi: 10.1038/325529a0. [DOI] [PubMed] [Google Scholar]
- Kawasaki Y, Kumamoto E, Furue H, Yoshimura M. α2 adrenoceptor-mediated presynaptic inhibition of primary afferent glutamatergic transmission in rat substantia gelatinosa neurons. Anesthesiology. 2003;98:682–689. doi: 10.1097/00000542-200303000-00016. [DOI] [PubMed] [Google Scholar]
- Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–364. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- Kohno T, Moore KA, Baba H, Woolf CJ. Peripheral nerve injury alters excitatory synaptic transmission in lamina II of the rat dorsal horn. J Physiol. 2003;548:131–138. doi: 10.1113/jphysiol.2002.036186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohno T, Wang H, Amaya F, Brenner GJ, Cheng JK, Ji RR, Woolf CJ. Bradykinin enhances AMPA and NMDA receptor activity in spinal cord dorsal horn neurons by activating multiple kinases to produce pain hypersensitivity. J Neurosci. 2008;28:4533–4540. doi: 10.1523/JNEUROSCI.5349-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn H, Faber DS. Quantal analysis and synaptic efficacy in the CNS. Trends Neurosci. 1991;14:439–445. doi: 10.1016/0166-2236(91)90042-s. [DOI] [PubMed] [Google Scholar]
- Kovalchuk Y, Hanse E, Kafitz KW, Konnerth A. Postsynaptic Induction of BDNF-Mediated Long-Term Potentiation. Science. 2002;295:1729–1734. doi: 10.1126/science.1067766. [DOI] [PubMed] [Google Scholar]
- Lee CJ, Bardoni R, Tong CK, Engelman HS, Joseph DJ, Magherini PC, MacDermott AB. Functional expression of AMPA receptors on central terminals of rat dorsal root ganglion neurons and presynaptic inhibition of glutamate release. Neuron. 2002;35:135–146. doi: 10.1016/s0896-6273(02)00729-8. [DOI] [PubMed] [Google Scholar]
- Le Meur K, Galante M, Angulo MC, Audinat E. Tonic activation of NMDA receptors by ambient glutamate of non-synaptic origin in the rat hippocampus. J Physiol. 2007;580:373–383. doi: 10.1113/jphysiol.2006.123570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lever IJ, Malcangio M. CB1 receptor antagonist SR141716A increases capsaicin-evoked release of substance P from the adult mouse spinal cord. Br J Pharmacol. 2002;135:21–24. doi: 10.1038/sj.bjp.0704506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, McRoberts JA, Ennes HS, Trevisani M, Nicoletti P, Mittal Y, Mayer EA. Experimental colitis modulates the functional properties of NMDA receptors in dorsal root ganglia neurons. Am J Physiol Gastrointest Liver Physiol. 2006;291:G219–G228. doi: 10.1152/ajpgi.00097.2006. [DOI] [PubMed] [Google Scholar]
- Li YH, Han TZ. Glycine binding sites of presynaptic NMDA receptors may tonically regulate glutamate release in the rat visual cortex. J Neurophysiol. 2007;97:817–823. doi: 10.1152/jn.00980.2006. [DOI] [PubMed] [Google Scholar]
- Lim G, Wang S, Zeng Q, Sung B, Yang L, Mao J. Expression of spinal NMDA receptor and PKCgamma after chronic morphine is regulated by spinal glucocorticoid receptor. J Neurosci. 2005;25:11145–11154. doi: 10.1523/JNEUROSCI.3768-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Mantyh PW, Basbaum AI. NMDA-receptor regulation of substance P release from primary afferent nociceptors. Nature. 1997;386:721–724. doi: 10.1038/386721a0. [DOI] [PubMed] [Google Scholar]
- Liu H, Wang H, Sheng M, Jan LY, Basbaum AI. Evidence for presynaptic N-methyl-D-aspartate autoreceptors in the spinal cord dorsal horn. Proc Natl Acad Sci U S A. 1994;91:8383–8387. doi: 10.1073/pnas.91.18.8383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonchamp E, Gambino F, Dupont JL, Doussau F, Valera A, Poulain B, Bossu JL. Pre and post synaptic NMDA effects targeting Purkinje cells in the mouse cerebellar cortex. PLoS One. 2012;7:e30180. doi: 10.1371/journal.pone.0030180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu CR, Hwang SJ, Phend KD, Rustioni A, Valtschanoff JG. Primary afferent terminals in spinal cord express presynaptic AMPA receptors. J Neurosci. 2002;22:9522–9529. doi: 10.1523/JNEUROSCI.22-21-09522.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutfy K, Cai SX, Woodward RM, Weber E. Antinociceptive effects of NMDA and non-NMDA receptor antagonists in the tail flick test in mice. Pain. 1997;70:31–40. doi: 10.1016/s0304-3959(96)03290-3. [DOI] [PubMed] [Google Scholar]
- Ma QP, Woolf CJ. Noxious stimuli induce an N-methyl-D-aspartate receptor-dependent hypersensitivity of the flexion withdrawal reflex to touch: implications for the treatment of mechanical allodynia. Pain. 1995;61:383–390. doi: 10.1016/0304-3959(94)00195-K. [DOI] [PubMed] [Google Scholar]
- McGuinness L, Taylor C, Taylor RD, Yau C, Langenhan T, Hart ML, Christian H, Tynan PW, Donnelly P, Emptage NJ. Presynaptic NMDARs in the hippocampus facilitate transmitter release at theta frequency. Neuron. 2010;68:1109–1127. doi: 10.1016/j.neuron.2010.11.023. [DOI] [PubMed] [Google Scholar]
- McRoberts JA, Ennes HS, Marvizon JC, Fanselow MS, Mayer EA, Vissel B. Selective knockdown of NMDA receptors in primary afferent neurons decreases pain during phase 2 of the formalin test. Neuroscience. 2010;172:474–482. doi: 10.1016/j.neuroscience.2010.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manabe T, Wyllie DJ, Perkel DJ, Nicoll RA. Modulation of synaptic transmission and long-term potentiation: effects on paired pulse facilitation and EPSC variance in the CA1 region of the hippocampus. J Neurophysiol. 1993;70:1451–1459. doi: 10.1152/jn.1993.70.4.1451. [DOI] [PubMed] [Google Scholar]
- Mayer ML, Westbrook GL, Guthrie PB. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature. 1984;309:261–263. doi: 10.1038/309261a0. [DOI] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- Mothet JP, Parent AT, Wolosker H, Brady RO, Jr, Linden DJ, Ferris CD, Rogawski MA, Snyder SH. D-Serine is an endogenous ligand for the glycine site of the N-methyl-D-aspartate receptor. Proc Natl Acad Sci U S A. 2000;97:4926–4931. doi: 10.1073/pnas.97.9.4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutel V, Buchy D, Klingelschmidt A, Messer J, Bleuel Z, Kemp JA, Richards JG. In vitro binding properties in rat brain of [3H]Ro 25-6981, a potent and selective antagonist of NMDA receptors containing NR2B subunits. J Neurochem. 1998;70:2147–2155. doi: 10.1046/j.1471-4159.1998.70052147.x. [DOI] [PubMed] [Google Scholar]
- Nakatsuka T, Tsuzuki K, Ling JX, Sonobe H, Gu JG. Distinct roles of P2X receptors in modulatiing glutamate release at different primary sensory synapses in rat spinal cord. J Neurophysiol. 2003;89:3243–3252. doi: 10.1152/jn.01172.2002. [DOI] [PubMed] [Google Scholar]
- Nie H, Weng HR. Glutamate transporters prevent excessive activation of NMDA receptors and extrasynaptic glutamate spillover in the spinal dorsal horn. J Neurophysiol. 2009;101:2041–2051. doi: 10.1152/jn.91138.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie H, Zhang H, Weng HR. Bidirectional neuron–glia interactions triggered by deficiency of glutamate uptake at spinal sensory synapses. J Neurophysiol. 2010;104:713–725. doi: 10.1152/jn.00282.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie H, Weng HR. Impaired glial glutamate uptake induces extrasynaptic glutamate spillover in the spinal sensory synapses of neuropathic rats. J Neurophysiol. 2010;103:2570–2580. doi: 10.1152/jn.00013.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliet SH, Mothet JP. Regulation of N-methyl-D-aspartate receptors by astrocytic D-serine. Neuroscience. 2009;158:275–283. doi: 10.1016/j.neuroscience.2008.01.071. [DOI] [PubMed] [Google Scholar]
- Pattinson D, Fitzgerald M. The neurobiology of infant pain: development of excitatory and inhibitory neurotransmission in the spinal dorsal horn. Reg Anesth Pain Med. 2004;29:36–44. doi: 10.1016/j.rapm.2003.10.018. [DOI] [PubMed] [Google Scholar]
- Pradhan A, Seena S, Pascoal C, Cassio F. Can metal nanoparticles be a threat to microbial decomposers of plant litter in streams. Microb Ecol. 2011;62:58–68. doi: 10.1007/s00248-011-9861-4. [DOI] [PubMed] [Google Scholar]
- Ren K, Dubner R. Pain facilitation and activity-dependent plasticity in pain modulatory circuitry: role of BDNF-TrkB signaling and NMDA receptors. Mol Neurobiol. 2007;35:224–235. doi: 10.1007/s12035-007-0028-8. [DOI] [PubMed] [Google Scholar]
- Salter MW, Pitcher GM. Dysregulated Src upregulation of NMDA receptor activity: a common link in chronic pain and schizophrenia. FEBS J. 2012;279:2–11. doi: 10.1111/j.1742-4658.2011.08390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samson RD, Pare D. Activity-dependent synaptic plasticity in the central nucleus of the amygdala. J Neurosci. 2005;25:1847–1855. doi: 10.1523/JNEUROSCI.3713-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuker MA, Bowser-Riley F, Davies SN. Possible NMDA antagonist properties of drugs that affect high pressure neurological syndrome. Br J Pharmacol. 1994;111:951–955. doi: 10.1111/j.1476-5381.1994.tb14831.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjöström PJ, Turrigiano GG, Nelson SB. Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron. 2003;39:641–654. doi: 10.1016/s0896-6273(03)00476-8. [DOI] [PubMed] [Google Scholar]
- Song JH, Park ES, Han SM, Han SR, Ahn DK, Youn DH. Signal transduction mechanisms underlying group I mGluR-mediated increase in frequency and amplitude of spontaneous EPSCs in the spinal trigeminal subnucleus oralis of the rat. Mol Pain. 2009;5:50. doi: 10.1186/1744-8069-5-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura Y, Lee CL, Perl ER. Central projections of identified, unmyelinated (C) afferent fibers innervating mammalian skin. Science. 1986;234:358–361. doi: 10.1126/science.3764416. [DOI] [PubMed] [Google Scholar]
- Sung B, Lim G, Mao J. Altered expression and uptake activity of spinal glutamate transporters after nerve injury contribute to the pathogenesis of neuropathic pain in rats. J Neurosci. 2003;23:2899–2910. doi: 10.1523/JNEUROSCI.23-07-02899.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasusuki T, Fujiwara T, Yamaguchi S, Fukushima T, Akagawa K, Hori Y. Enhancement of synaptic transmission and nociceptive behaviour in HPC-1/syntaxin 1A knockout mice following peripheral nerve injury. Eur J Neurosci. 2007;26:2179–2187. doi: 10.1111/j.1460-9568.2007.05830.x. [DOI] [PubMed] [Google Scholar]
- Tingley WG, Ehlers MD, Kameyama K, Doherty C, Ptak JB, Riley CT, Huganir RL. Characterization of protein kinase A and protein kinase C phosphorylation of the N-methyl-D-aspartate receptor NR1 subunit using phosphorylation site-specific antibodies. J Biol Chem. 1997;272:5157–5166. doi: 10.1074/jbc.272.8.5157. [DOI] [PubMed] [Google Scholar]
- Vicini S, Wang JF, Li JH, Zhu WJ, Wang YH, Luo JH, Wolfe BB, Grayson DR. Functional and pharmacological differences between recombinant N-methyl-D-aspartate receptors. J Neurophysiol. 1998;79:555–566. doi: 10.1152/jn.1998.79.2.555. [DOI] [PubMed] [Google Scholar]
- Wang S, Lim G, Zeng Q, Sung B, Ai Y, Guo G, Yang L, Mao J. Expression of central glucocorticoid receptors after peripheral nerve injury contributes to neuropathic pain behaviors in rats. J Neurosci. 2004;24:8595–8605. doi: 10.1523/JNEUROSCI.3058-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng HR, Chen JH, Cata JP. Inhibition of glutamate uptake in the spinal cord induces hyperalgesia and increased responses of spinal dorsal horn neurons to peripheral afferent stimulation. Neuroscience. 2006;138:1351–1360. doi: 10.1016/j.neuroscience.2005.11.061. [DOI] [PubMed] [Google Scholar]
- Weng HR, Chen JH, Pan ZZ, Nie H. Glial glutamate transporter 1 regulates the spatial and temporal coding of glutamatergic synaptic transmission in spinal lamina II neurons. Neuroscience. 2007;149:898–907. doi: 10.1016/j.neuroscience.2007.07.063. [DOI] [PubMed] [Google Scholar]
- Weng HR, Cordella JV, Dougherty PM. Changes in sensory processing in the spinal dorsal horn accompany vincristine-induced hyperalgesia and allodynia. Pain. 2003;103:131–138. doi: 10.1016/s0304-3959(02)00445-1. [DOI] [PubMed] [Google Scholar]
- Willis WD. Long-term potentiation in spinothalamic neurons. Brain Res Brain Res Rev. 2002;40:202–214. doi: 10.1016/s0165-0173(02)00202-3. [DOI] [PubMed] [Google Scholar]
- Willis WD. John Eccles’ studies of spinal cord presynaptic inhibition. Prog Neurobiol. 2006;78:189–214. doi: 10.1016/j.pneurobio.2006.02.007. [DOI] [PubMed] [Google Scholar]
- Wu LJ, Toyoda H, Zhao MG, Lee YS, Tang J, Ko SW, Jia YH, Shum FW, Zerbinatti CV, Bu G, Wei F, Xu TL, Muglia LJ, Chen ZF, Auberson YP, Kaang BK, Zhuo M. Upregulation of forebrain NMDA NR2B receptors contributes to behavioral sensitization after inflammation. J Neurosci. 2005;25:11107–11116. doi: 10.1523/JNEUROSCI.1678-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LJ, Zhuo M. Targeting the NMDA receptor subunit NR2B for the treatment of neuropathic pain. Neurotherapeutics. 2009;6:693–702. doi: 10.1016/j.nurt.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Wu LJ, Wang H, Zhang X, Vadakkan KI, Kim SS, Steenland HW, Zhuo M. Presynaptic and postsynaptic amplifications of neuropathic pain in the anterior cingulate cortex. J Neurosci. 2008;28:7445–7453. doi: 10.1523/JNEUROSCI.1812-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yajima Y, Narita M, Usui A, Kaneko C, Miyatake M, Yamaguchi T, Tamaki H, Wachi H, Seyama Y, Suzuki T. Direct evidence for the involvement of brain-derived neurotrophic factor in the development of a neuropathic pain-like state in mice. J Neurochem. 2005;93:584–594. doi: 10.1111/j.1471-4159.2005.03045.x. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Yaksh TL. Spinal pharmacology of thermal hyperesthesia induced by constriction injury of sciatic nerve. Excitatory amino acid antagonists. Pain. 1992;49:121–128. doi: 10.1016/0304-3959(92)90198-K. [DOI] [PubMed] [Google Scholar]
- Yang K, Takeuchi K, Wei F, Dubner R, Ren K. Activation of group I mGlu receptors contributes to facilitation of NMDA receptor membrane current in spinal dorsal horn neurons after hind paw inflammation in rats. Eur J Pharmacol. 2011;670:509–518. doi: 10.1016/j.ejphar.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Woodhall GL, Jones RS. Tonic facilitation of glutamate release by presynaptic NR2B-containing NMDA receptors is increased in the entorhinal cortex of chronically epileptic rats. J Neurosci. 2006;26:406–410. doi: 10.1523/JNEUROSCI.4413-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura M, Jessell TM. Primary afferent-evoked synaptic responses and slow potential generation in rat substantia gelatinosa neurons in vitro. J Neurophysiol. 1989;62:96–108. doi: 10.1152/jn.1989.62.1.96. [DOI] [PubMed] [Google Scholar]
- Yoshimura M, Nishi S. Blind patch-clamp recordings from substantia gelatinosa neurons in adult rat spinal cord slices: pharmacological properties of synaptic currents. Neuroscience. 1993;53:519–526. doi: 10.1016/0306-4522(93)90216-3. [DOI] [PubMed] [Google Scholar]
- Zeng J, Thomson LM, Aicher SA, Terman GW. Primary afferent NMDA receptors increase dorsal horn excitation and mediate opiate tolerance in neonatal rats. J Neurosci. 2006;26:12033–12042. doi: 10.1523/JNEUROSCI.2530-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS. Short-term synaptic plasticity. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]