

Abstract
BACKGROUND & AIMS
The soluble hematopoietin receptor Epstein–Barr virus–induced protein (EBI)-3 is an immune regulator that has been associated with the pathogenesis of inflammatory bowel disease. However, the concept that EBI3 is part of an interleukin (IL)-27 heterodimer that mediates chronic inflammatory and auto-immune diseases has been challenged by the description of IL-35, a bioactive cytokine comprising EBI3 and IL-12 p35. We investigated the roles of IL-27 and IL-35 in chronic inflammation of the intestine.
METHODS
We analyzed EBI3-deficient mice and IL-27p28 – deficient mice with spontaneous or T-cell transfer-induced colitis and compared outcomes with wild-type mice (controls). We constructed vectors that express EBI3 covalently linked to the IL-12p35 chain (recombinant [r]IL-35).
RESULTS
Intestines of EBI3-deficient mice had increased pathologic features of colitis, compared with IL-27p28 – deficient or control mice; they also had shorter survival times, indicating that IL-35, rather than IL-27, protects the intestine from immune responses in mice. The mucosa of EBI3-deficient mice accumulated subsets of activated CD4+ T cells that produced T-helper (Th)1 and Th17 cytokines. Adoptive transfer of these T cells induced colitis in RAG-deficient mice. The rIL-35 significantly reduced the development of several forms of experimental colitis and reduced levels of markers of Th1 and Th17 cells.
CONCLUSIONS
IL-35 controls the development of T-cell– dependent colitis in mice. It might be developed as a therapeutic target for patients with chronic intestinal inflammation.
Keywords: IBD, Mouse Model, Immune Regulation, Suppression
The etiology of inflammatory bowel disease (IBD) still remains incompletely understood, but it is generally agreed that a complex interplay between genetic, environmental, and immunologic factors contributes to disease initiation and progression.1 Numerous studies in animal models clearly identified members of the family of interleukin (IL)-12–related cytokines as central mediators of mucosal inflammation.2 In both animal models and human Crohn’s disease there is evidence for dominance of a polarized T-helper 1 (TH1) pathway that depends on increased mucosal expression of IL-12 (p40:p35), which is the key T-cell differentiation factor driving cellular immune responses characterized by the production of proinflammatory cytokines interferon (IFN)-γ and tumor necrosis factor-α.3 Consistently, targeting IL-12 may be effective in treating CD and such strategies currently are evaluated.4 However, previous concepts on the role of T-helper cells in inflammatory and autoimmune diseases have been challenged by the recent description of the highly proinflammatory TH17 subset characterized by production of IL-17 and IL-22.5 IL-23 (p40:p19), a cytokine sharing the p40 subunit with IL-12, is essential for maintenance of the TH17 phenotype and is associated with disease in colitis models.6 Taken together with the discovery of the protective Arg381Gln polymorphism in the IL-23R gene in Crohn’s disease, these findings strongly suggest a major role of this cytokine in IBD.
More recently, computational studies identified IL-27, a heterodimer composed of Epstein–Barr virus–induced protein (EBI)-3, and an IL-27p28 subunit that binds to a receptor formed by the WSX-1 and gp130 chains.7 Initially, IL-27 was described as a TH1-promoting factor synergizing with IL-12 to drive proliferation and IFN-γ production of naive but not memory CD4+ T cells. However, subsequent studies elucidated that the biological functions of IL-27 are more complex, and a role for IL-27 as a negative regulator of TH2 and particularly TH17 responses emerged.8 IL-27 exerts these functions by inducing IL-10 production in T cells and directly blocking ROR-γT production in a signal transducer and activator of transcription (STAT)1-dependent manner.9 In addition, we and others have identified EBI3 as a regulator of innate immune cells such as granulocytes and macrophages.10,11 Such immunoregulatory potential of EBI3 is implicated further by the recent discovery that EBI3 is expressed in Foxp3+ CD25+ Treg and constitutes with the IL-12p35 chain the novel cytokine IL-35.12–14 IL-35 is induced in CD4+ CD25+ Treg on contact with CD4+CD25− effector cells and contributes to their suppressive activity in vitro and in vivo.15,16 Furthermore, rIL-35 suppressed the proliferation of CD4+ effector cells and inhibited TH17 polarization. In vivo, recombinant (r)IL-35 was shown to dramatically inhibit the onset of collagen-induced arthritis and, in contrast to rIL-27, even was able to modulate already established disease, suggesting that IL-35 administration could be a potential therapeutic strategy in inflammation and autoimmunity.17 However, the lack of knowledge about the IL-35 receptor, current limitations in its detection, and the fact that EBI3−/− mice lack with IL-27 and IL-35, 2 cytokines with largely redundant biological functions, is currently limiting the understanding of its precise biological function. This is reflected by the data on the role of IL-27 and IL-35 in the context of IBD, in which studies exist showing that IL-27R deficiency protects from colitis in IL-10−/− mice,18 whereas the same strain was shown to be affected in dextrane sodium sulfate (DSS) colitis.11,19 On the other hand, EBI3 deficiency does not lead to changes in intestinal pathology in trinitrobenzene sulfonic acid colitis, but is protective in oxazolone-induced colitis.20
Here, we analyzed the differential role of IL-27 and IL-35 for the development of intestinal pathology by direct comparison of EBI3 and IL-27p28 – deficient mice in models of T-cell– dependent colitis. Although EBI3-deficient mice developed early severe intestinal disease, IL-27p28−/− mice were phenotypically similar to wild-type mice, suggesting that IL-35 rather than IL-27 is a critical factor limiting intestinal inflammation in these models. Furthermore, administration of a single-chain IL-35 fusion-protein led to suppression of colitis activity, indicating that IL-35 is able to suppress pathologic intestinal immune responses in vivo.
Material and Methods
Mice
EBI3-deficient mice were described previously.20 UbC-luc mice were obtained from Caliper Lifesciences (Mainz, Germany). For generation of IL-27p28 – deficient mice clones containing the IL-27 genomic locus were derived from a mouse BAC library as described previously.21 IL-27p28 – deficient C57BL/6x129 F1 ES-cell clones were injected into C57BL6/J blastocysts to generate chimeras. Male chimeras were mated with female C57BL6/J mice to generate F1 breeders. Both strains were back-crossed to the C57BL/6 background for at least 10 times. Some mice were back-crossed to a RAG1-knockout background. To generate IL-27/IL-35– deficient mice with conditional ablation of STAT3 in myeloid cells, EBI3−/− and p28−/− mice were mated with LysmCre mice obtained from Jackson Laboratories (C56BL/6-background). Littermate Cre-negative wild-type or STAT3 heterozygous mutants were used as controls. Induction of DSS and trinitrobenzene sulfonic acid colitis was performed as previously described.22 All mice used in experiments were bred and maintained in microisolator cages and all mouse procedures were performed using committee-approved protocols.
Primary Cells
Murine lamina propria mononuclear cells (LPMCs) were isolated as previously described.22 Briefly, colons were mechanically dissected into small pieces and intestinal epithelial cells were removed by incubation in 5 mmol/L ethylenediaminetet-raacetic acid. Remaining tissue was digested using collagenase D, DNaseI, and DispaseII (all Roche Diagnostics, Mannheim, Germany). Digested tissue was passed through a 40-μm cell strainer, and the remaining cellular content was separated from debris using a 40%/80% Percoll gradient. In some experiments, CD4+ T cells from mesenteric lymph nodes (MLN) were isolated by MACS separation (Miltenyi-Biotec, Bergisch-Gladbach, Germany) to a purity degree of more than 95%, as evaluated by FACS.
Histologic Scoring of Inflammation
Inflammation was graded semiquantitatively on a scale from 0 to 6 in a blinded fashion. Two subscores grading the degree of inflammatory cell infiltrations (0 –3) and tissue damage (0 –3) were determined, resulting in a combined score ranging from 0 (no changes) to 6 (widespread cellular infiltrations and extensive tissue damage). For grading infiltration of inflammatory cells, an infrequent presence of inflammatory cells in the lamina propria was classified as 0; increased numbers of inflammatory cells, including neutrophils, as 1; submucosal presence of inflammatory cell clusters as 2; and a score of 3 was applied for transmural cell infiltrations. For grading of epithelial damage, a normal mucosal structure was classified as 0, isolated focal epithelial damage was counted as 1, the presence of mucosal erosions/ulcerations was counted as 2, and a score of 3 was given if extensive mucosal damage and extension through deeper structures of the bowel wall was present.
Mouse Endoscopic Procedures
Colitis activity was monitored with a high-resolution video endoscopic system (Karl Storz, Tuttlingen, Germany) at indicated time points in anesthetized mice. Endoscopic scoring of colitis activity was based on the evaluation of mucosal translucency, vascularity, granularity, fibrin deposition, and stool consistency as previously described.22
Measurement of Cytokines
For measurement of cytokines in supernatants from cell preparations or full-thickness organ cultures we used the Flow-Cytomix (Ebioscience, Frankfurt, Germany) according to the manufacturer’s instructions using a FACSCantoII (Becton Dickinson, Heidelberg, Germany). IL-27 levels were determined by enzyme-linked immunosorbent assay (Ebioscience).
Analysis of Gene Expression
Total RNA was extracted with RNeasy columns (Qiagen, Hilden, Germany) including DNaseI digestion. RNA was reverse-transcribed with the QuantiTect Reverse Transcription Kit (Qiagen) using random hexamers. Quantitative polymerase chain reaction (PCR) analysis was performed using Quantitect Primer assays from Qiagen in a CFX96 system (Biorad). Relative differences between samples were calculated with the Pfaffl-model-based Rest2009 software using HPRT as the reference gene.23
IL-35 Expression Vector
For construction of IL-35, complementary DNAs encoding for EBI3 and p35 were cloned from lipopolysaccharide-stimulated splenocytes by reverse-transcription PCR. For cloning of single-chain IL-35, fragments encoding EBI3, followed by a (GlyGlyGlySer)4 linker and the mature coding sequence of p35 were generated by PCR and cloned in a expression plasmid containing regulatory regions from the ubiquitous EF1α promoter and HTLV enhancer. To ensure efficient secretion, the EBI3 leader sequence was replaced by the signal peptide of IgGκ. Plasmid DNA was isolated with Qiagen Plasmid Gigakits including endotoxin removal. To further ensure endotoxin removal, DNA was treated with the Miraclean endotoxin removal Kit (MirusBio, Madison, Wisconsin). For in vivo gene transfer, 100 μg of IL-35 expression vector or empty control vector were complexed with 16 μL in vivo jetPEI reagent according to the manufacturer’s instructions (Polyplus, Illkirch, France) and intraperitoneally injected in a final volume of 600 μL.
Statistical Analysis
Data are expressed as means of individual determinations ± standard deviations. Statistical analysis was performed using the unpaired Student t test or for survival analysis with a log-rank test (*P < .05; **P <.01; ***P <.001).
Results
IL-27 and IL-35 Subunits Are Up-Regulated in Chronic Colitis
IBD is characterized by an inflammatory response with production of proinflammatory cytokines including IL-12 and IL-23.4 We and others previously have shown that IL-27 and IL-35 subunits are produced in macrophages and dendritic cells upon Toll-like receptor stimulation, however, little is known about the role of these IL-12 family members in mucosal inflammation of the gut.24,25 Therefore, we analyzed in an initial series of studies their colonic expression in wild-type mice and mice with STAT3 deficiency in myeloid cells that spontaneously develop colitis.26 First, we isolated intestinal epithelial cells and LPMCs from colons of wild-type mice and analyzed the expression of transcripts of IL-12, IL-23, IL-27, and IL-35 by quantitative PCR. Marked expression of EBI3 p35 and p19 was detected in intestinal epithelial cells, whereas little or no expression of IL-27 p28 and IL-12/IL-23 p40 was noted in these cells (Figure 1A). In contrast, in LPMCs the expression of the earlier-described transcripts was below the detection limit of the assay (not shown). However, in 5-month-old mice with established enterocolitis transcripts of the analyzed targets including EBI3, p28, and p35 were increased, consistent with a potential regulatory role of IL-27 and IL-35 (Figure 1B). Consistently, expression of the IL-27 heterodimer was augmented significantly in supernatants of LPMCs from colitic mice as compared with controls (Figure 1C).
Figure 1.
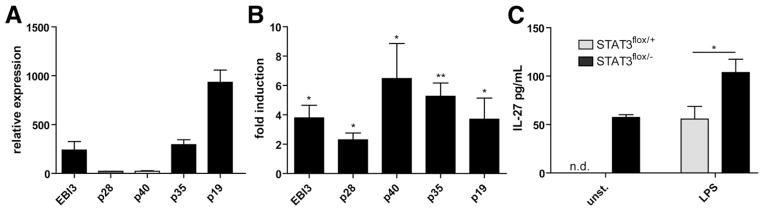
Transcripts of IL-27 and IL-35 subunits are up-regulated in spontaneous colitis. (A) Constitutive levels of transcripts of IL-12–related cytokines (EBI3, p28, p35, p40, p19) in RNA from colonic epithelial cells of 10-week-old C57BL/6 mice were determined by quantitative PCR (N = 5 mice). (B) Quantitative PCR analysis with LPMC RNA from 5-month-old LysMcre/STAT3fl/− with enterocolitis and controls. Fold induction in STAT3flox/− relative to STAT3flox/− mice is shown. (C) Isolated LPMCs (2 × 106) from STAT3-deficient and control mice were left unstimulated or were stimulated with 1 μg/mL lipopolysaccharide (LPS). Twenty-four hours later supernatants were collected and analyzed per IL-27–specific enzyme-linked immunosorbent assay. Data represent mean ± standard error of the mean of 5 mice/group. *P < .05; **P < .01.
Early Onset of Severe Enterocolitis in EBI3 but Not p28-Deficient Mice
To analyze the relative contribution of IL-27 and IL-35 in the development of colitis, we created a novel mouse strain with a targeted deletion of the IL-27p28 gene (Figure 2A). The last 4 of 5 exons of the endogenous gene were replaced by in-frame insertion of the reporter gene LacZ, leading to mice that did not produce any detectable p28 protein in vivo after injection of lipopoly-saccharide or in vitro in stimulated bone marrow derived dendritic cells (BMDC) (Figure 2B). IL-27p28−/− mice developed normally and had no gross or histologic abnormalities in various organs including liver, kidney, spleen, colon, small intestine, lung, and heart (not shown).
Figure 2.

EBI3 deficiency but not p28 deficiency protects mice from colitis. (A) Targeting strategy for generation of IL-27p28 – deficient mice. Mice were genotyped using PCR with tail DNA using a common primer upstream of exon 2 and a genotype-specific reverse primer within the p28 gene (wild-type allele) or the LacZ region (targeted allele), respectively (right). (B) Serum IL-27p28 protein levels of wild-type or targeted mice 24 hours after intraperitoneal injection of 10 mg/kg lipopolysaccharide (LPS) (left) or isolated bone marrow derived dendritic cells (BMDC) (right). IL-27p28 levels were determined by enzyme-linked immunosorbent assay. One representative experiment of 2 is shown. N = 4 –5/group. (C) Survival analysis of EBI3−/− or IL-27p28−/− mice or controls with conditional ablation of STAT3 in myeloid cells (N = 7–10/group). (D) Weight analysis of EBI3- and p28-deficient RAG−/− mice adoptively transferred with 0.5 × 106 CD4+ CD25− T cells (N = 9 –13/group). (E) Survival analysis of EBI3−/− or IL-27p28−/− mice or controls after oral treatment with 3% DSS at indicated time points (n = 8 –12/group). *P < .05; **P < .01; ***P < .001.
Next, EBI3 or IL-27p28 deficiency was bred into mice with conditional ablation of STAT3 in myeloid cells.26 Both EBI3 and IL-27p28 STAT3 double-mutant strains were born at predicted Mendelian ratios. However, at 5– 6 weeks of age, EBI3/STAT3 mice started to lose weight and showed pronounced signs of intestinal inflammation (diarrhea, rectal prolapse) and none of these mice survived more than 13–14 weeks (Figure 2C). In contrast, IL-27p28/STAT3-deficient mice did not show such early lethality and were phenotypically indistinguishable from controls. To confirm the observed phenotype in another model of chronic colitis, we crossed EBI3−/− and p28−/− mice into the Rag1−/− background and adoptively transferred EBI3−/− /Rag1−/−, p28−/− /Rag1−/−, and Rag1−/− control mice with purified colitogenic CD4+ CD25− T cells to induce chronic colitis. As shown in Figure 2D, EBI3−/− mice developed significantly more wasting disease earlier than p28−/− and control mice. Further survival analysis of EBI3−/− and IL-27p28−/− mice in the DSS colitis model showed that EBI3 deficiency is again associated with increased mortality, suggesting a pivotal role of this molecule for protective mucosal responses during colitis (Figure 2E).
Next, we analyzed intestinal pathology in the different STAT3 mutant strains. When colitis severity was analyzed in 5-week-old mice by colonoscopy, we observed diarrhea, massive thickening of the colon wall, and fibrin depositions in EBI3/STAT3-deficient mice, whereas IL-27p28/STAT3-deficient and control mice still had normal mucosal surfaces at this age (Figure 3A). Consistently, in vivo imaging of myeloperoxidase activity by injection of the bioluminescent agent luminol revealed that EBI3 deficiency is associated with marked intestinal accumulation of inflammatory cells (Figure 3B). Moreover, assessment of colonic cross-sections of these mice showed profound changes in the normal gut architecture characterized particularly by elongated crypts, gland loss, and massive mononuclear cell infiltrates (Figure 3C). In contrast, these changes were not seen in p28/STAT3-deficient mice and controls. To further characterize the contribution of IL-27p28 to colitis development in this model, we analyzed 6-month-old p28/STAT3-deficient mice by endoscopy and histology. At this age these mice had established full-blown enterocolitis similar to controls (Figure 3D and E). Compared with p28−/− Rag1−/− and controls, EBI3−/− Rag1−/− mice developed faster colitis after transfer of CD4+ CD25− T cells (Figure 4A–C). Transfer colitis is associated with proliferation and intestinal accumulation of engrafted cells. We therefore assessed the effects of EBI3/p28 deficiency on the proliferation capacity of these cells by FACS. In fact, we observed vigorous accumulation and splenic expansion of transferred T cells in EBI3−/− Rag1−/− mice (Figure 4C and D). As shown by in vivo imaging after transfer of bioluminescent donor cells, these cells strongly accumulated in the gut in the absence of EBI3 but not p28, suggesting that EBI3 is a critical regulator of colitogenic T-cell responses.
Figure 3.
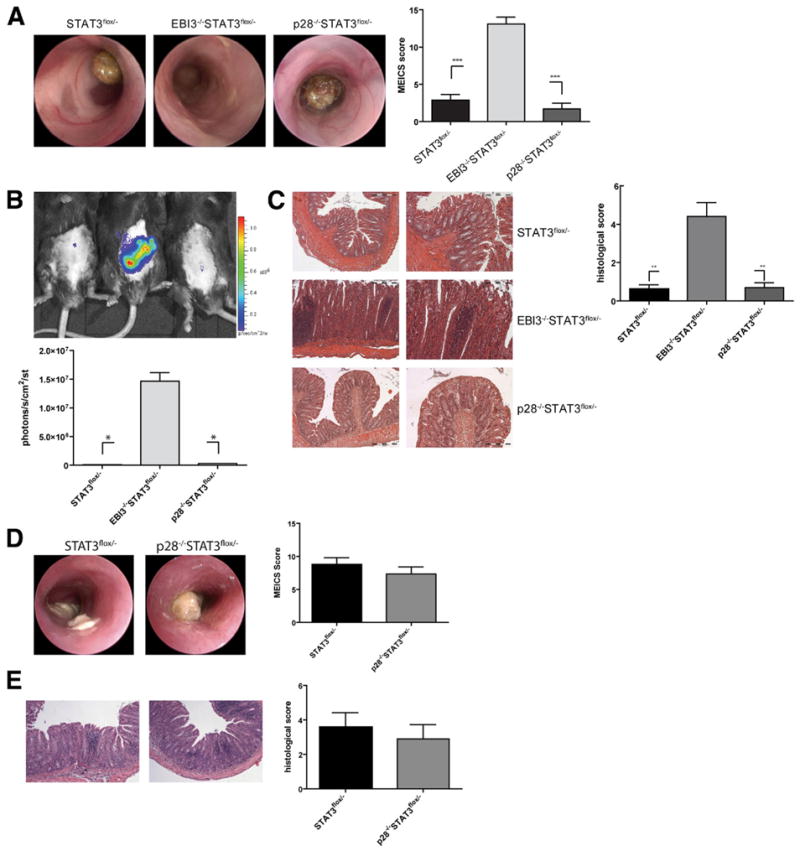
Severe enterocolitis in EBI3- but not IL-27p28-deficient STAT3fl/− mice. (A) Mice were subjected to colonoscopy at 5 weeks of age. The mean value ± standard error of the mean of the MEICS score in the groups are indicated. N = 6 –10/group. (B) Mice received an intraperitoneal injection of 200 mg/kg luminol in phosphate-buffered saline for bioluminescence imaging of myeloperoxidase activity. Ten minutes later the mice were imaged. (C) Cross-sections from mid-colons taken at 6 weeks of age were stained with H&E. Histopathology was scored as described in the Materials and Methods section. Mean value ± standard error of the mean (N = 6 –10 mice/group). (D) Colonoscopy and (E) histopathologic analysis of mice at 6 months of age. The mean values ± standard error of the mean of the murine endoscopic score of colitis severity (MEICS) score and the histologic score in groups are indicated. No significant differences between the groups were noted (N = 5/group). *P < .05; **P < .01; ***P < .001.
Figure 4.

EBI3 deficiency is associated with increased transfer colitis. Purified splenic CD4+CD25− T cells (0.5 × 105) were injected intraperitoneally into mice. (A) Colonoscopy and (B) histopathologic analysis of mice 3 weeks after adoptive transfer. The mean values ± standard error of the mean of the MEICS score and the histologic score in the groups are indicated. N = 7/group. (C) CD4+ T cells in spleens were quantified by FACS. *P < .05; **P <.01. (D) CD4+ CD25− T cells from UbC-luc mice were used for adoptive transfer. Mice were injected intraperitoneally with 100 mg/kg D-luciferin and imaged 10 minutes later.
Increased Production of Inflammatory Mediators in EBI3/STAT3 Mice
Similarly to IBD in human beings, it previously has been shown that T cells in LysMCreSTAT3 mice produce augmented amounts of proinflammatory cytokines.26,27 Specifically, T cells in LysMCreSTAT3 mice displayed signs of TH1 polarization characterized by increased production of the signature cytokine IFN-γ, and this process has been strongly implicated in enterocolitis development in these mice.26,28 To analyze intestinal T-cell polarization and cytokine responses in the absence of EBI3 in this model, we isolated LPMCs from 5-week-old EBI3/STAT3 or control mice and analyzed the expression of inflammation-associated genes by quantitative PCR. In line with high numbers of infiltrating T-cell CD3 transcripts, chemokines and cytokine receptors associated with TH1 and TH17 immune cell activation were increased in LPMCs from EBI3/STAT3 mice compared with controls (Figure 5A). Moreover, consistent with severe intestinal inflammation in these mice, a markedly increased presence of transcripts of TH1 and TH17 signature cytokines was observed. We next isolated LPMCs from EBI3/STAT3, p28/STAT3, and control mice and analyzed their cytokine-expression profile in vitro in response to anti-CD3/anti-CD28 stimulation. Isolated LPMCs from EBI3/STAT3 mice produced significantly higher amounts of proinflammatory cytokines including IL-17, IL-6, tumor necrosis factor-α, and granulocyte-macrophage colony–stimulating factor compared with LPMCs from p28/STAT3 mice (Figure 5B). Because IL-27 initially was described as a TH1-inducing factor and TH1 deficiency has been suggested to be responsible for protection of IL-27R−/− mice in DSS colitis,19 we next analyzed the expression of IFN-γ. Interestingly, there was a highly significant increase in the production of IFN-γ in EBI3/STAT3-deficient mice, indicating that TH1 responses contribute to intestinal pathology in this model.
Figure 5.

Highly increased expression of proinflammatory mediators in EBI3-deficient STAT3flox/− mice. (A) Total RNA from LPMCs of 5-week-old STAT3flox/− and EBI3−/− STAT3flox/− mice was isolated, reverse transcribed, and quantitative PCR was performed. Fold induction in EBI3−/− STAT3flox/− relative to STAT3flox/− mice is shown. 1 × 106 LPMC of 5-week-old (B) and (C) or MLN of 6-month-old (D) mice were stimulated with aCD3/aCD28, 1 μg/mL LPS, or 1 μM CPG1668 under nonpolarizing conditions. Forty-eight hours later supernatants were collected, and cytokines were measured as described in the Materials and Methods section. (E) Intracellular FACS analysis of MLN cells of 6-month-old mice. Data are representative of 2 independent experiments with 3–5 mice/group. *P < .05; **P < .01. LPS, lipopolysaccharide.
Previous studies have shown that IL-27 and IL-35 exert their immunoregulatory functions in part by inducing IL-10.8,17 However, we found no reduction in T-cell– derived IL-10 levels in EBI3/STAT3 mice, suggesting that lack of IL-10 is not primarily responsible for the development of enterocolitis in these mice (Figure 5B). This was supported further by the finding that IL-10 production by LPMCs after stimulation with the Toll-like receptor-9 ligand CPG1668 is strikingly higher in EBI/STAT3 mice than in controls (Figure 5C). Although less affected than EBI3/STAT3 mice at a younger age, p28/STAT3 mice developed severe intestinal pathology similarly to control STAT3 mice at several months of age that was characterized by the intestinal accumulation of ROR-γt and Tbet-positive T cells, which may account for the production of proinflammatory IFN-γ, IL-17, and IL-6 cytokines (Figure 5D and E).
T Cells From EBI/STAT3 Transfer Enterocolitis to Rag1−/− Mice
Our data were consistent with the idea that EBI3 deficiency in colitis leads to intestinal accumulation of highly activated and potentially colitogenic T-cell populations. Because EBI3-related cytokines have been implicated in the modulation of both T cells and accessory cells,29 we crossed EBI3/STAT3 mice with lymphopenic Rag1−/− mice. Colonoscopic, macroscopic, and histologic evaluation of these mice did not show evidence of colitis at 12 weeks of age when Rag-proficient mice of this strain already suffered from severe disease. Further analysis showed that these mice as controls do not develop colitis at 10 month of age (not shown), underlining the important role of adaptive immunity for disease manifestation (Figure 6A). To further analyze the importance of T cells for the observed intestinal phenotype, we next evaluated whether T-helper cells from EBI3/STAT3 mice can adoptively transfer disease to Rag1−/− mice. Therefore, we isolated CD4+ T cells from MLN of 6-week-old littermate controls, STAT3, and EBI3/STAT3 mice by MACS and injected them into Rag1−/− mice. As shown in Figure 6B and C, T cells from 6-week-old STAT3 mice without established colitis induced only mild disease. In contrast, mice reconstituted with MLN T cells from 6-week-old colitic EBI3/STAT3 mice progressively developed marked colitis and wasting disease, indicating that EBI3 deficiency in STAT3 mutant mice leads to the generation of highly colitogenic T-cell populations that are capable of adoptively transferring disease to immunodeficient hosts. Similarly to control mice, MLN CD4+ T cells from p28/STAT3 mice were only able to transfer disease when isolated from several-month-old animals with established colitis (Figure 6D).
Figure 6.

T cells from EBI3−/− STAT3flox/− mice transfer enterocolitis to immunodeficient mice. (A) Conditional EBI3/STAT3-deficient and STAT3-deficient mice were crossed with Rag1−/− mice. Representative photomicrographs of H&E-stained midcolon sections and endoscopic images of mice at 3 months of age are shown. CD4+ T cells of the indicated groups were isolated from MLN of (B and C) 6-week-old mice or (D) 6-month-old mice by MACS. Cells (1 × 106) were injected intraperitoneally into Rag1−/− mice. Mice of indicated groups were subjected to colonoscopy or histopathologic analysis at 7 weeks of age. The mean endoscopic and histopathologic scores ± standard error of the mean are shown (N = 4 –5/group). *P < .05.
IL-35 Inhibits Development of Colitis
Previous studies showed that IL-35 administration inhibits the development of collagen-induced arthritis and ceases progression of established collagen-induced arthritis by suppressing TH1 and TH17 responses in vivo.14,17 Based on these studies and our finding that EBI3 deficiency rather than lack of IL-27 may lead to increased gut pathology prompted us to explore the potential of IL-35 to suppress colitis. Therefore, we constructed expression vectors for a single-chain IL-35 fusion protein by covalently linking EBI to the IL-12p35 chain (Figure 7A) and determined whether IL-35 through administration of this expression vector contributed to the attenuation or exacerbation of DSS colitis. Therefore, C56BL/6 mice that were treated orally with DSS received repeated injections of IL-35 or control vector complexed in a polyethylenimine-based gene delivery solution (Figure 7A). Remarkably, IL-35 gene administration was able to significantly inhibit inflammation-dependent wasting disease compared with controls (Figure 7B) in both preventive and therapeutic settings. In addition, in contrast to IL-35–treated mice, control vector–treated mice developed marked colitis, as determined by endoscopic and histopathologic criteria (Figure 7C). Similar efficacy was shown in the trinitrobenzene sulfonic acid colitis model (Figure 7F), in which rIL-35 treatment resulted in a significant reduction of weight loss compared with control mice.
Figure 7.

IL-35 therapy protects from colitis. (A) Schematic of the cloned IL-35 construct. Serum IL-35Fc concentrations of injected mice as determined by enzyme-linked immunosorbent assay. (B) Mice were treated orally with 2.5% DSS (preventive treatment) or 3% (therapeutic treatment) for 7 days. At indicated time points (arrows) mice were injected intraperitoneally with control or IL-35 constructs as described in the Materials and Methods section. Weight analysis during colitis of IL-35– or mock-treated mice. (C) Mice were subjected to colonoscopy at day 10 after starting DSS treatment. (D) Cross-sections from colons of animals killed on day 11 were stained with H&E. (E) Full-thickness colon organ cultures were obtained using a 3-mm dermal punch and incubated in culture medium. Forty-eight hours later supernatants were collected and cytokines were measured. In addition, colonic RNA was isolated and messenger RNA expression of the T-helper cell lineage-specific transcription factors ROR-γt, GATA3, tbet, and FoxP3 was determined by quantitative PCR. (F) Mice with trinitrobenzene sulfonic acid colitis were treated with rIL-35 expression vector at the indicated time points. On day 3 colons were removed for analysis. Data represent mean values ± standard error of the mean of 5 mice/group. **P < .01; *P < .05.
In the next series of studies, we analyzed if ameliorated disease in IL-35–treated mice is associated with changes in the intestinal production of cytokines. Analysis of colonic organ cultures revealed that consistent with their improved clinical outcome, IL-35–treated mice produced less IFN-γ, IL-6, and IL-17 cytokines in the mucosa (Figure 7D). Finally, in contrast to GATA3 and FoxP3, expression levels of the TH1- and TH17-associated transcription factors T-bet and ROR-γt were suppressed significantly in IL-35–treated mice compared with controls (Figure 7E), suggesting that IL-35 is able to actively suppress TH1- and TH17-dependent inflammatory responses in the gut and may play an important role in protective intestinal immune responses.
Discussion
EBI3 associates with p28 to form IL-27 or with IL-12p35 to form IL-35.7,13 Both cytokines may have immunosuppressive functions and particularly IL-35 has been implicated in the suppressive capacity of Treg. In addition to its immunoregulatory functions, IL-27 may act as an initiator of proinflammatory TH1-type responses and inhibits the transforming growth factor-β– dependent development of Foxp3+ iTregs.9 However, although studies have shown their up-regulation in the inflamed IBD mucosa,30,31 the precise biological function of the EBI3-related cytokines in intestinal inflammation remains largely unclear. Here, we provide the outcome of a direct comparison of EBI3−/− (lacking both IL-27 and IL-35) and IL-27p28−/− (lacking IL-27 only) mice in experimental models of colitis. In contrast to p28−/− mice, we found that EBI3−/− mice are highly susceptible to spontaneous colitis caused by STAT3 deficiency in myeloid cells and developed disease significantly earlier than p28−/− mice. In addition, EBI3 deficiency led to early onset of transfer colitis. Despite the fact that the Th1-promoting factor IL-27 is lacking in these mice, pathology in EBI3−/− mice in the absence of IL-35 was associated with an increase of both Th1 and Th17 responses. In addition, overexpression of an IL-35 fusion protein was sufficient to attenuate the development of mucosal damage, suggesting that the anti-inflammatory properties of IL-35 may be a key regulatory factor in IBD.
There is a great body of evidence that intestinal inflammation is characterized by enhanced TH1 and TH17 responses against antigens of the commensal gut flora.6,32 Previous studies in mice have shown a differential role of IL-27 in colitis depending on the experimental context. Honda et al19 showed that IL-27R– deficient mice are significantly protected from DSS-induced colitis presumably owing to a reduced TH1 response in these mice. In line with this, decreased CD4+ T-cell IFN-γ production has been implicated in the delayed onset of colitis in IL-27R−/− mice crossed to IL-10−/− mice.18
In contrast, another study in the DSS model, in which intestinal damage was induced by high-dose DSS treatment, revealed that IL-27/IL-27R signaling protects from fatal TH17-mediated immunopathology,11 suggesting a dual function of IL-27 as a mediator and suppressor of mucosal cytokine responses in acute colitis. Therefore, we analyzed in an initial series of studies whether IL-27 deficiency plays a regulatory role in models of chronic colitis by taking advantage of newly generated p28-knockout mice. Although IL-27 is up-regulated in this model, p28−/− STAT3fl/− mice in the present study developed a similar intestinal pathology and survival as wild-type STAT3fl/− mice for up to 6 months of age, indicating that proinflammatory and anti-inflammatory functions of IL-27 might counterbalance each other. In line with this apparent complexity of the immunologic functions of IL-27, a very recent study showed that IL-27Rα −/− CD45RbHigh T cells promote only reduced transfer colitis.33 Although IL-27 is a well-known inhibitor of assumedly colitogenic Th17 cells and is an inducer of Tr1 cells, this report clearly suggests that under certain in vivo conditions a lack of IL-27R signaling in T cells instead leads to their increased conversion to anti-inflammatory Tregs.33 We have not seen such protection from transfer colitis, when naive T cells were transferred into IL-27p28−/−/Rag mice, indicating that IL-27 could have important effects on non–T cells in this model. In addition, IL-27p28 recently was described to form a novel heterodimeric cytokine with Cytokine-like factor (CLF)-1. Thus, it remains possible that IL-27/IL-27Rα–independent effects may be one explanation for the contrasting features of IL-27 in different mouse models of IBD. However, EBI3−/− mice developed early, more serious colitis in the models that we have analyzed, showing that EBI3 independently of IL-27 plays a crucial role in suppression of intestinal inflammation.
EBI3 initially was reported to be synthesized by Epstein–Barr virus-transformed B cells. Although some cells such as activated macrophages and dendritic cells produce EBI3 and p28 to secrete the IL-27 heterodimer,7,24 other cell types including CD4+ T cells, intestinal epithelial cells, syncytiotrophoblasts, and tumor cells have been reported to produce EBI3 that apparently do not co-express the p28 chain.12 Recently, IL-35, a novel heterodimer of EBI3 and IL-12p35, has been described. Functional studies revealed that IL-35 is produced by Treg rather than effector CD4+ T cells and contributes to their maximal regulatory function.13,14 In line with the observation that EBI3−/− Treg were not able to suppress effector T-cell responses in vitro and transfer colitis in vivo,13 colitis in EBI3−/− STAT3fl/− mice was characterized by strong infiltrations with activated T cells that were, when adoptively transferred, able to induce disease in Rag1−/− mice. Subsequent analysis showed that TH1 and TH17 cytokines are suppressed in the absence of EBI3 in STAT3flox/− mice, suggesting that IL-35 controls T-cell activation in colitis in vivo. However, because EBI3−/− mice lack both IL-27 and IL-35 pathways the phenotype of these mice can be a mixed effect on both pathways. The discovery of the cellular receptor of IL-35 certainly would help to precisely dissect its role in the immune system.
To provide further evidence that lack of IL-35 is eventually critically important for colitis activity, we constructed an EBI3-p35 fusion protein and ectopically expressed IL-35 in the course of colitis. Remarkably, rIL-35 was able to significantly improve inflammation, and ameliorated disease in IL-35–treated mice is associated with blunted intestinal production of proinflammatory cytokines. Furthermore, expression levels of the TH1- and TH17-associated transcription factors T-bet and ROR-γt were suppressed during treatment with rIL-35, suggesting that IL-35 is able to actively balance immune responses in the gut. This observation is consistent with previous studies reporting that IL-35 is sufficient to inhibit collagen-induced arthritis.14,17
In conclusion, our results indicate that EBI3, as part of the immunosuppressive IL-35 cytokine, plays an important role in the regulation of intestinal immune responses by controlling overwhelming T-cell activation. Modulating the intestinal levels of this cytokine may be relevant in patients with IBD.
Acknowledgments
The authors thank I. Tubbe, K. Cappel, D. Huse, C. Lindner, and A. Taut for excellent technical assistance.
Funding: Supported by the Collaborative Research Center (grants SFB490 and SFB796 of the Deutsche Forschungsgemeinschaft to S.W. and M.F.N.).
Abbreviations used in this paper
- DSS
dextrane sodium sulfate
- EBI
Epstein–Barr virus–induced protein
- IBD
inflammatory bowel disease
- IFN
interferon
- IL
interleukin
- LPMC
lamina propria mononuclear cell
- MLN
mesenteric lymph nodes
- PCR
polymerase chain reaction
- rIL
recombinant interleukin
Footnotes
Conflicts of interest: The authors disclose no conflicts
References
- 1.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427– 434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 2.Wirtz S, Neurath MF. Mouse models of inflammatory bowel disease. Adv Drug Deliv Rev. 2007;59:1073–1083. doi: 10.1016/j.addr.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 3.Magram J, Connaughton SE, Warrier RR, et al. IL-12-deficient mice are defective in IFN gamma production and type 1 cytokine responses. Immunity. 1996;4:471– 481. doi: 10.1016/s1074-7613(00)80413-6. [DOI] [PubMed] [Google Scholar]
- 4.Mannon PJ, Fuss IJ, Mayer L, et al. Anti-interleukin-12 antibody for active Crohn’s disease. N Engl J Med. 2004;351:2069–2079. doi: 10.1056/NEJMoa033402. [DOI] [PubMed] [Google Scholar]
- 5.Veldhoen M, Hocking RJ, Atkins CJ, et al. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 6.Ahern PP, Schiering C, Buonocore S, et al. Interleukin-23 drives intestinal inflammation through direct activity on T cells. Immunity. 2010;33:279–288. doi: 10.1016/j.immuni.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pflanz S, Timans JC, Cheung J, et al. IL-27, a heterodimeric cytokine composed of EBI3 and p28 protein, induces proliferation of naive CD4(+) T cells. Immunity. 2002;16:779–790. doi: 10.1016/s1074-7613(02)00324-2. [DOI] [PubMed] [Google Scholar]
- 8.Stumhofer JS, Silver JS, Laurence A, et al. Interleukins 27 and 6 induce STAT3-mediated T cell production of interleukin 10. Nat Immunol. 2007;8:1363–1371. doi: 10.1038/ni1537. [DOI] [PubMed] [Google Scholar]
- 9.Neufert C, Becker C, Wirtz S, et al. IL-27 controls the development of inducible regulatory T cells and Th17 cells via differential effects on STAT1. Eur J Immunol. 2007;37:1809–1816. doi: 10.1002/eji.200636896. [DOI] [PubMed] [Google Scholar]
- 10.Wirtz S, Tubbe I, Galle PR, et al. Protection from lethal septic peritonitis by neutralizing the biological function of interleukin 27. J Exp Med. 2006;203:1875–1881. doi: 10.1084/jem.20060471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Troy AE, Zaph C, Du Y, et al. IL-27 regulates homeostasis of the intestinal CD4+ effector T cell pool and limits intestinal inflammation in a murine model of colitis. J Immunol. 2009;183:2037–2044. doi: 10.4049/jimmunol.0802918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chaudhry A, Rudra D, Treuting P, et al. CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science. 2009;326:986–991. doi: 10.1126/science.1172702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Collison LW, Workman CJ, Kuo TT, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. 2007;450:566–569. doi: 10.1038/nature06306. [DOI] [PubMed] [Google Scholar]
- 14.Niedbala W, Wei XQ, Cai B, et al. IL-35 is a novel cytokine with therapeutic effects against collagen-induced arthritis through the expansion of regulatory T cells and suppression of Th17 cells. Eur J Immunol. 2007;37:3021–3029. doi: 10.1002/eji.200737810. [DOI] [PubMed] [Google Scholar]
- 15.Collison LW, Pillai MR, Chaturvedi V, et al. Regulatory T cell suppression is potentiated by target T cells in a cell contact, IL-35- and IL-10-dependent manner. J Immunol. 2009;182:6121–6128. doi: 10.4049/jimmunol.0803646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Collison LW, Chaturvedi V, Henderson AL, et al. IL-35-mediated induction of a potent regulatory T cell population. Nat Immunol. 2010;11:1093–1101. doi: 10.1038/ni.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kochetkova I, Golden S, Holderness K, et al. IL-35 stimulation of CD39+ regulatory T cells confers protection against collagen II-induced arthritis via the production of IL-10. J Immunol. 2010;184:7144–7153. doi: 10.4049/jimmunol.0902739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Villarino AV, Artis D, Bezbradica JS, et al. IL-27R deficiency delays the onset of colitis and protects from helminth-induced pathology in a model of chronic IBD. Int Immunol. 2008;20:739–752. doi: 10.1093/intimm/dxn032. [DOI] [PubMed] [Google Scholar]
- 19.Honda K, Nakamura K, Matsui N, et al. T helper 1-inducing property of IL-27/WSX-1 signaling is required for the induction of experimental colitis. Inflamm Bowel Dis. 2005;11:1044–1052. doi: 10.1097/01.mib.0000191611.05466.1f. [DOI] [PubMed] [Google Scholar]
- 20.Nieuwenhuis EE, Neurath MF, Corazza N, et al. Disruption of T helper 2-immune responses in Epstein-Barr virus-induced gene 3-deficient mice. Proc Natl Acad Sci U S A. 2002;99:16951–16956. doi: 10.1073/pnas.252648899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Valenzuela DM, Murphy AJ, Frendewey D, et al. High-throughput engineering of the mouse genome coupled with high-resolution expression analysis. Nat Biotechnol. 2003;21:652– 659. doi: 10.1038/nbt822. [DOI] [PubMed] [Google Scholar]
- 22.Wirtz S, Neufert C, Weigmann B, et al. Chemically induced mouse models of intestinal inflammation. Nat Protoc. 2007;2:541–546. doi: 10.1038/nprot.2007.41. [DOI] [PubMed] [Google Scholar]
- 23.Pfaffl MW, Horgan GW, Dempfle L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002;30:e36. doi: 10.1093/nar/30.9.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wirtz S, Becker C, Fantini MC, et al. EBV-induced gene 3 transcription is induced by TLR signaling in primary dendritic cells via NF-kappa B activation. J Immunol. 2005;174:2814–2824. doi: 10.4049/jimmunol.174.5.2814. [DOI] [PubMed] [Google Scholar]
- 25.Molle C, Goldman M, Goriely S. Critical role of the IFN-stimulated gene factor 3 complex in TLR-mediated IL-27p28 gene expression revealing a two-step activation process. J Immunol. 2010;184:1784–92. doi: 10.4049/jimmunol.0902005. [DOI] [PubMed] [Google Scholar]
- 26.Takeda K, Clausen BE, Kaisho T, et al. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 1999;10:39– 49. doi: 10.1016/s1074-7613(00)80005-9. [DOI] [PubMed] [Google Scholar]
- 27.Kobayashi M, Kweon MN, Kuwata H, et al. Toll-like receptor-dependent production of IL-12p40 causes chronic enterocolitis in myeloid cell-specific Stat3-deficient mice. J Clin Invest. 2003;111:1297–1308. doi: 10.1172/JCI17085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheng F, Wang HW, Cuenca A, et al. A critical role for Stat3 signaling in immune tolerance. Immunity. 2003;19:425– 436. doi: 10.1016/s1074-7613(03)00232-2. [DOI] [PubMed] [Google Scholar]
- 29.Yoshida H, Miyazaki Y. Regulation of immune responses by interleukin-27. Immunol Rev. 2008;226:234–247. doi: 10.1111/j.1600-065X.2008.00710.x. [DOI] [PubMed] [Google Scholar]
- 30.Omata F, Birkenbach M, Matsuzaki S, et al. The expression of IL-12 p40 and its homologue, Epstein-Barr virus-induced gene 3, in inflammatory bowel disease. Inflamm Bowel Dis. 2001;7:215–220. doi: 10.1097/00054725-200108000-00006. [DOI] [PubMed] [Google Scholar]
- 31.Larousserie F, Pflanz S, Coulomb-L’Hermine A, et al. Expression of IL-27 in human Th1-associated granulomatous diseases. J Pathol. 2004;202:164–171. doi: 10.1002/path.1508. [DOI] [PubMed] [Google Scholar]
- 32.Fina D, Sarra M, Fantini MC, et al. Regulation of gut inflammation and Th17 cell response by interleukin-21. Gastroenterology. 2008;134:1038–1048. doi: 10.1053/j.gastro.2008.01.041. [DOI] [PubMed] [Google Scholar]
- 33.Cox JH, Kljavin NM, Ramamoorthi N, et al. IL-27 promotes T cell-dependent colitis through multiple mechanisms. J Exp Med. 2011;208:115–123. doi: 10.1084/jem.20100410. [DOI] [PMC free article] [PubMed] [Google Scholar]