

Abstract
Purpose
To assess prognostic roles of various KRAS oncogene mutations in colorectal cancer, BRAF mutation status must be controlled for because BRAF mutation is associated with poor prognosis, and almost all BRAF mutants are present among KRAS-wild-type tumors. Taking into account experimental data supporting a greater oncogenic effect of codon 12 mutations compared to codon 13 mutations, we hypothesized that KRAS codon 12 mutated colorectal cancers might behave more aggressively than KRAS-wild-type tumors and codon 13 mutants.
Experimental design
Utilizing molecular pathological epidemiology database of 1261 rectal and colon cancers, we examined clinical outcome and tumor biomarkers of KRAS codon 12 and 13 mutations in 1075 BRAF-wild-type cancers (i.e., controlling for BRAF status). Cox proportional hazards model was used to compute mortality hazard ratio (HR), adjusting for potential confounders, including stage, PIK3CA mutations, microsatellite instability, CpG island methylator phenotype, and LINE-1 methylation.
Results
Compared to patients with KRAS-wild-type/BRAF-wild-type cancers (N=635), those with KRAS codon 12 mutations (N=332) experienced significantly higher colorectal cancer-specific mortality [log-rank P=0.0001; multivariate HR=1.30; 95% confidence interval (CI), 1.02–1.67; P=0.037], whereas KRAS codon 13 mutated cases (N=108) were not significantly associated with prognosis. Among the seven most common KRAS mutations, c.35G>T (p.G12V; N=93) was associated with significantly higher colorectal cancer-specific mortality (log-rank P=0.0007; multivariate HR=2.00, 95% CI, 1.38–2.90, P=0.0003) compared to KRAS-wild-type/BRAF-wild-type cases.
Conclusions
KRAS codon 12 mutations (in particular, c.35G>T), but not codon 13 mutations, are associated with inferior survival in BRAF-wild-type colorectal cancer. Our data highlight the importance of accurate molecular characterization in colorectal cancer.
Keywords: colon cancer, genetics, oncogenic, molecular diagnostics, personalized medicine, RAF, RAS
INTRODUCTION
Colorectal cancer develops through a multistep carcinogenic process with an accumulation of epigenetic and genetic changes, including KRAS mutation. Approximately 40% of colorectal cancers harbor KRAS mutations, and 90% of those mutations occur in codons 12 and 13 (1–3). In contrast to the widely-accepted predictive role of KRAS mutation in identifying resistance to anti-EGFR therapy (3–8) the prognostic role of KRAS mutation in colorectal cancer remains uncertain (9–14). Recently, the differential biological effect of various KRAS mutations in colorectal cancer was brought to light by data from De Rook et al. (15), showing that the c.38G>A (p.G13D) mutation was associated with benefit from cetuximab whereas KRAS codon 12 mutations were associated with resistance to cetuximab among chemotherapy-refractory colorectal cancer patients. A search of the literature to-date reveals that several studies (16–21) have compared the prognostic roles of KRAS codon 12 mutations with those of codon 13. Nonetheless, there is a lack of agreement as to the prognostic difference between KRAS codon 12 and codon 13 mutations in colorectal cancer (Table 1).
Table 1.
Studies on prognostic significance of KRAS codon 12 and 13 mutations in colorectal cancer
| Ref. | Authors (year) | No. of hospitals | Sample size | Tumor location | Disease stage | No. of KRAS mutants
|
BRAF data | Multivariate HR (95% CI) (vs. KRAS-wild-type as a referent, unless otherwise specified) | Notes | |
|---|---|---|---|---|---|---|---|---|---|---|
| codon 12 (No. of events) | codon 13 (No. of events) | |||||||||
| 16 | Samowitz et al. (2000) | Many | 1413 | Colon | I–IV | 353 (-) | 100 (-) | No | Cancer-specific survival codon 12, 1.0 (0.8 – 1.2) codon 13, 1.4 (0.95 – 2.0) c.35G>A, 1.1 (0.8 – 1.5) c.35G>T, 0.8 (0.5 – 1.2) c.38G>A, 1.4 (0.95 – 2.0) |
Adjusted by age and stage. |
| 17 | Andreyev et al. (2001) (Meta-analysis) | <35 | 2832 | Colon & rectum | I–IV | <900 (<395) | <297 (<139) | No | Overall survival c.34G>A, 1.20 (0.86 – 1.70) c.34G>T, 1.26 (0.93 – 1.62) c.35G>A, 0.94 (0.79 – 1.11) c.35G>C, 1.35 (0.98 – 1.87) c.35G>T, 1.29 (1.08 – 1.55) c.38G>A, 0.93 (0.78 – 1.12) |
Adjusted by age, stage and center. |
| 18 | Bazan et al. (2002) | 1 | 160 | Colon & rectum | I–IV | 40 (21) | 34 (28) | No | Cancer-specific survival codon 13, 1.93 (1.17 – 3.18) |
Covariates were location, stage, surgical resection, nodal metastasis, tumor growth pattern, lymphovascular invasion, lymphocytic infiltration, DNA aneuploidy status and synthesis-phase fraction status. |
| 19 | Roth et al. (2010) | Many | 1299 | Colon | II–III | 372 (-) | 102 (-) | No | Relapse-free survival c.34G>A, 0.99 (0.46 – 2.09) c.34G>T, 1.40 (0.89 – 2.21) c.35G>A, 0.98 (0.72 – 1.34) c.35G>C, 0.97 (0.48 – 1.95) c.35G>T, 1.09 (0.76 – 1.57) c.38G>A, 0.99 (0.68 – 1.44) |
Adjusted by treatment arm and stage. |
| 20 | Zlobec et al. (2010) | 2 | 392 | Colon & rectum | I–III | 71 (-) | 27 (-) | No | Cancer-specific survival c.35G>A, HR=0.82 (P=0.044) |
Covariates were tumor depth, nodal metastasis and MSI. |
| 21 | Yokota et al. (2011) | 1 | 229 | Colon & rectum | Advanced & recurrent | 53 (-) | 26 (-) | Yes | Univariate HR for overall survival (vs. KRAS-wild-type/BRAF-wild-type as a referent) (no multivariate HR provided) codon 12, 1.28 (0.74 – 2.19) codon 13, 2.03 (1.10 – 3.74) |
Multivariate analysis included both BRAF mutants and BRAF-wild-type tumors. Covariates were age, sex, performance status, BRAF, pathological type, number of metastasis and presence of metastasis (liver, lung and peritoneum). |
| Imamura et al. (Current study) | Many | 1261(1075 BRAF-wild-type tumors) | Colon & rectum | I–IV | 332 (119) | 108 (31) | Yes | Cancer-specific survival (vs. KRAS-wild-type/BRAF-wild-type as a referent) codon 12, 1.29 (1.01 – 1.65) codon 13, 0.86 (0.58 – 1.27) c.34G>A, 1.00 (0.42 – 2.34) c.34G>C, 3.21 (1.22 – 8.45) c.34G>T, 1.52 (0.90 – 2.58) c.35G>A, 1.09 (0.78 – 1.53) c.35G>C, 0.55 (0.24 – 1.28) c.35G>T, 1.94 (1.34 – 2.80) c.38G>A, 0.88 (0.59 – 1.30) |
Adjusted by stage; covariates were age, sex, year of diagnosis, tumor location, tumor differentiation, family history of colorectal cancer in any first degree relative, MSI, CpG island methylator phenotype, PIK3CA, and LINE-1 methylation. | |
CI, confidence interval; HR, hazard ratio; MSI, microsatellite instability.
Of note, little attention has been given to the confounding effect of BRAF mutation on the relationship between KRAS mutation and clinical outcome in colorectal cancer. Almost all BRAF-mutated colorectal cancers are present within the group of KRAS-wild-type cancers. Compared to BRAF-wild-type cases, BRAF mutation has been associated with poorer prognosis in several studies (10, 11, 19, 21–23), hence, it is impossible to clarify the exact prognostic roles of KRAS mutations in colorectal cancer without controlling for BRAF mutation. Importantly, none of the previous large studies (with a sample size of N≥300) (16, 17, 19, 20) controlled for the potential confounding effect of BRAF mutation, while only one smaller study (N=229) assessed BRAF status (21) (Table 1). One way of controlling for BRAF mutation is to examine the prognostic significance of KRAS mutation in BRAF-wild-type colorectal cancers. Considering experimental data (24, 25) supporting a greater oncogenic effect of KRAS codon 12 mutations than codon 13 mutations, we hypothesized that KRAS codon 12 mutated colorectal cancer might behave more aggressively than codon 13 mutants and KRAS-wild-type tumors.
We therefore tested this hypothesis by conducting a study on the prognostic roles of KRAS codon 12 and 13 mutations utilizing 1261 colorectal cancers within two U.S. nationwide prospective cohort studies, in which there were 1075 BRAF-wild-type cancers. Because our molecular pathological epidemiology (26–28) database included tumor molecular variables including microsatellite instability (MSI), CpG island methylator phenotype (CIMP), BRAF and PIK3CA mutations, and LINE-1 methylation, we could evaluate the prognostic role of KRAS codon 12 and 13 mutations independent of other potential molecular confounders. Our findings raise a possible need for tumor subtyping based on specific KRAS and BRAF oncogene mutations in colorectal cancer.
MATERIALS AND METHODS
Study population
We utilized the database of two U.S. nationwide prospective cohort studies, the Nurses’ Health Study (N=121,701 women followed since 1976) and the Health Professionals Follow-up Study (N=51,529 men followed since 1986) (29). Every two years, cohort participants have been sent follow-up questionnaires to identify newly diagnosed cancers in themselves and their first degree relatives. We collected paraffin-embedded tissue blocks from hospitals where patients underwent colorectal cancer resections (29). We collected diagnostic biopsy specimens for rectal cancer patients who received preoperative treatment, in order to avoid artifacts or bias introduced by treatment. Hematoxylin and eosin (HE) stained tissue sections from all colorectal cancer cases were reviewed by a pathologist (S.O.) unaware of other data. A subset of cases (N=172) were reviewed by another pathologist (T.M.), and the concordance between the two observers was 0.96 (κ =0.72; P<0.0001), indicating substantial agreement. The tumor differentiation was categorized as well-moderate vs. poor (>50% vs. ≤50% gland formation). Initially, 1261 colorectal cancer cases diagnosed up to 2006 were included based on the availability of tumor tissue, sequencing data for both KRAS and BRAF, and survival data (Table 2). Treatment data were not available in this study. In our current study, BRAF-mutated cancers (N=181) were excluded in order to assess the prognostic role of various KRAS mutations in a pool of BRAF-wild-type cases. Tumors harboring mutations in both codons 12 and 13 (N=5) of KRAS were excluded, resulting in a final total of 1075 BRAF-wild-type cases as our survival analysis study base (Figure 1, Supplementary Table 1). Patients were observed until death or January 1, 2011, whichever came first. Death of a participant was confirmed by searching the National Death Index. Informed consent was obtained from all study subjects. This study was approved by the Human Subjects Committees at Harvard School of Public Health and Brigham and Women’s Hospital.
Table 2.
Clinical, pathological and molecular characteristics according to KRAS mutation status
| Clinical, pathological or molecular feature | Total
|
KRAS-wild-type
|
P (wild-type vs. all mutants together) |
KRAS mutant
|
|||
|---|---|---|---|---|---|---|---|
| No. (%) | No. (%) | Mutation in either codon 12 or codon 13 only
|
Mutations in both codon 12 and codon 13
|
||||
| Codon 12
|
P (codon 12 vs. codon 13) | Codon 13
|
|||||
| No. (%) | No. (%) | No. (%) | |||||
| Total No. of patients | 1261 | 810 | 335 | 110 | 6 | ||
| Sex | 0.0049 | 0.39 | |||||
| Male | 568 (45%) | 341 (42%) | 164 (49%) | 59 (54%) | 4 (67%) | ||
| Female | 693 (55%) | 469 (58%) | 171 (51%) | 51 (46%) | 2 (33%) | ||
| Mean age (years) ± SD | 68.5 ± 8.7 | 68.2 ± 8.7 | 0.18 | 69.4 ± 8.6 | 0.044 | 67.4 ± 9.1 | 68.7 ± 5.2 |
| Year of diagnosis | 0.48 | 0.0079 | |||||
| Prior to 1995 | 396 (31%) | 246 (30%) | 105 (31%) | 41 (37%) | 4 (67%) | ||
| 1995 – 1999 | 405 (32%) | 260 (32%) | 100 (30%) | 44 (40%) | 1 (17%) | ||
| 2000 – 2006 | 460 (36%) | 304 (38%) | 130 (39%) | 25 (23%) | 1 (17%) | ||
| Family history of colorectal cancer in first degree relative(s) | 0.35 | 0.95 | |||||
| Absent | 1020 (81%) | 649 (80%) | 275 (82%) | 90 (82%) | 6 (100%) | ||
| Present | 241 (19%) | 161 (20%) | 60 (18%) | 20 (18%) | 0 | ||
| Tumor location | <0.0001 | 0.68 | |||||
| Cecum | 210 (17%) | 100 (12%) | 82 (25%) | 27 (25%) | 1 (17%) | ||
| Ascending to transverse colon | 374 (30%) | 260 (33%) | 80 (24%) | 32 (29%) | 2 (33%) | ||
| Splenic flexure to sigmoid colon | 388 (31%) | 254 (32%) | 104 (31%) | 29 (26%) | 1 (17%) | ||
| Rectum | 276 (22%) | 187 (23%) | 65 (20%) | 22 (20%) | 2 (33%) | ||
| Disease stage | 0.026 | 0.72 | |||||
| I | 300 (24%) | 199 (25%) | 80 (24%) | 20 (18%) | 1 (17%) | ||
| II | 356 (28%) | 248 (31%) | 77 (23%) | 30 (27%) | 1 (17%) | ||
| III | 324 (26%) | 195 (24%) | 95 (28%) | 30 (27%) | 4 (67%) | ||
| IV | 166 (13%) | 95 (12%) | 52 (16%) | 19 (17%) | 0 | ||
| Unknown | 115 (9.1%) | 73 (9.0%) | 31 (9.3%) | 11 (10%) | 0 | ||
| Tumor differentiation | 0.0018 | 0.29 | |||||
| Well to moderate | 1132 (90%) | 711 (88%) | 315 (95%) | 101 (92%) | 5 (83%) | ||
| Poor | 122 (9.7%) | 94 (12%) | 18 (5.4%) | 9 (8.2%) | 1 (17%) | ||
| MSI status | <0.0001 | 0.86 | |||||
| MSI-low/MSS | 1047 (85%) | 625 (79%) | 315 (95%) | 102 (94%) | 5 (83%) | ||
| MSI-high | 190 (15%) | 166 (21%) | 17 (5.1%) | 6 (5.6%) | 1 (17%) | ||
| CIMP status | <0.0001 | 0.40 | |||||
| CIMP-0 | 518 (44%) | 335 (45%) | 140 (44%) | 39 (37%) | 4 (67%) | ||
| CIMP-low | 456 (39%) | 242 (32%) | 154 (49%) | 59 (56%) | 1 (17%) | ||
| CIMP-high | 203 (17%) | 174 (23%) | 21 (6.7%) | 7 (6.7%) | 1 (17%) | ||
| BRAF mutation status | <0.0001 | 0.43 | |||||
| Wild-type | 1080 (86%) | 635 (78%) | 332 (99%) | 108 (98%) | 5 (83%) | ||
| Mutant | 181 (14%) | 175 (22%) | 3 (0.9%) | 2 (1.8%) | 1 (17%) | ||
| PIK3CA mutation status | <0.0001 | 0.48 | |||||
| Wild-type | 972 (84%) | 656 (88%) | 240 (77%) | 72 (73%) | 4 (67%) | ||
| Mutant | 188 (16%) | 88 (12%) | 72 (23%) | 26 (26%) | 2 (33%) | ||
| Mean LINE-1 methylation level (%) ± SD | 62.7 ± 9.4 | 62.8 ± 9.7 | 0.43 | 62.8 ± 9.1 | 0.15 | 61.3 ± 8.3 | 61.8 ± 9.0 |
(%) indicates the proportion of cases with a specific clinical, pathologic or molecular feature among each KRAS mutation status group. A P-value for significance was adjusted for multiple hypothesis testing to P=0.05/24=0.0021. Thus, a P-value between 0.05 and 0.0021 should be regarded as of borderline significance.
CIMP, CpG island methylator phenotype; MSI, microsatellite instability; MSS, microsatellite stable; SD, standard deviation.
Figure 1.
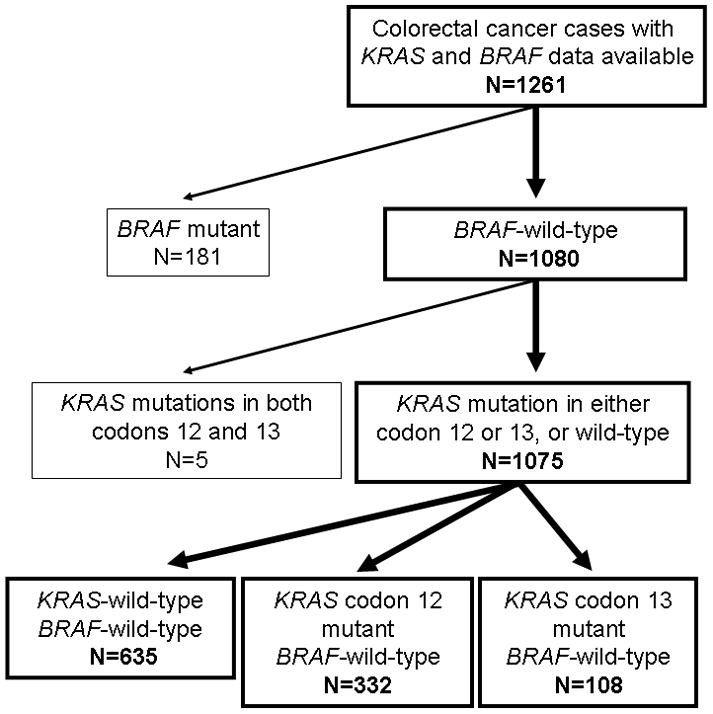
Flow chart of the current study. BRAF-mutated cases (N=181) were excluded from survival analysis to assess a prognostic role of KRAS mutation in BRAF-wild-type tumors. In addition, cases with KRAS mutations in both codons 12 and 13 (N=5) were excluded, in order to assess a prognostic effect of KRAS codon 12 mutations separately from that of KRAS codon 13 mutations.
Sequencing of KRAS, BRAF and PIK3CA, and microsatellite instability (MSI) analysis
Resected tissue was fixed, processed and stored according to standard protocols by hospitals across the U.S., where study participants had undergone surgery. Retrieved paraffin-embedded tissue blocks were stored at room temperature before use. Tissue for analysis was macrodissected from 10 micron sections on glass slides while guided by a HE-stained section with tumor areas marked by a pathologist (S.O.). Accordingly, we extracted DNA from tumor tissue enriched with neoplastic cells, without adjacent normal tissue. DNA was stored at −20 degrees centigrade before use. We performed PCR and Pyrosequencing targeted for KRAS (codons 12 and 13) (30), BRAF (codon 600) (31) and PIK3CA (exons 9 and 20) as previously described (32). Pyrosequencing technology has been shown to reliably detect KRAS mutation with 100% analytic sensitivity and specificity, even when the proportion of mutant alleles is as low as 10% (30). We dissected tumor-only areas, maintaining neoplastic cellularity of at least 30%. Assuming no laboratory error, both positive and negative predictive values are estimated to be 100% MSI analysis was performed using 10 microsatellite markers (D2S123, D5S346, D17S250, BAT25, BAT26, BAT40, D18S55, D18S56, D18S67 and D18S487) (2). MSI-high was defined as instability in ≥30% of the markers, and MSI-low/microsatellite stability (MSS) as instability in 0–29% of the markers.
Methylation analyses for CpG islands and LINE-1
Using validated bisulfite DNA treatment and real-time PCR (MethyLight), we quantified DNA methylation in eight CIMP-specific promoters [CACNA1G, CDKN2A (p16), CRABP1, IGF2, MLH1, NEUROG1, RUNX3 and SOCS1] (2, 33). CIMP-high was defined as the presence of ≥6/8 methylated promoters, CIMP-low as 1/8 to 5/8 methylated promoters, and CIMP-0 as the absence (0/8) of methylated promoters according to the previously established criteria (2). In order to accurately quantify differences in relatively high LINE-1 methylation levels, we used Pyrosequencing as previously described (34, 35).
Statistical analysis
All statistical analyses were performed by using SAS (Version 9.2, SAS Institute, Cary, NC). All P-values were two-sided. For our main hypothesis on the prognostic significance of KRAS codon 12 mutation among BRAF-wild-type cases, a P-value for significance was set at P=0.05. When we performed multiple hypothesis testing on specific KRAS mutations (the seven most common mutations), the P-value for significance was adjusted by Bonferroni correction to P=0.007 (=0.05/7). When we performed multiple hypothesis testing on associations or interactions between KRAS mutations (codon 12 or 13) and other covariates, a P-value for significance was adjusted by Bonferroni correction to P=0.0021 (=0.05/24). For categorical data, the chi-square test was performed. To compare mean age and mean LINE-1 methylation level, the t-test assuming equal variances was performed.
The Kaplan-Meier method was used to assess survival time distribution, and log-rank test was used. For analyses of colorectal cancer-specific mortality, deaths as a result of other causes were censored. To control for confounding, we used multivariate Cox proportional hazards regression models. A multivariate model initially included sex, age at diagnosis (continuous), year of diagnosis (continuous), family history of colorectal cancer in any first-degree relative (present vs. absent), tumor location (colon vs. rectum), tumor differentiation (well to moderate vs. poor), MSI (high vs. low/MSS), CIMP (high vs. low/0), PIK3CA and LINE-1 methylation (continuous). To avoid overfitting and residual confounding, disease stage (I, IIA, IIB–C, IIIA, IIIB, IIIC, IV or unknown) was used as a stratifying variable using the “strata” option in the SAS “proc phreg” command. A backward stepwise elimination was performed with P=0.20, as a threshold to avoid overfitting. For cases with missing information in any of the categorical covariates [tumor location (0.5%), tumor differentiation (0.7%), MSI (2.0%), CIMP (7.2%) and PIK3CA (8.5%)], we included those cases in the majority category of a given covariate to avoid overfitting. We confirmed that excluding cases with missing information in any of the covariates did not substantially alter results (data not shown). The proportionality of hazards assumption was satisfied by evaluating time-dependent variables, which were the cross products of the KRAS indicator variables (codon 12 mutant and codon 13 mutant; vs. KRAS-wild-type/BRAF-wild-type) and survival time (all P values >0.14 for colorectal cancer-specific mortality and overall mortality). We also tested for potential interaction between KRAS mutation status and each of the other covariates (including sex, age at diagnosis, family history of colorectal cancer, tumor location, disease stage, tumor differentiation, MSI, CIMP, PIK3CA, and LINE-1 methylation). An interaction was assessed by the Wald test on the cross product of each of the KRAS indicator variables and another variable of interest (without data-missing cases) in a multivariate Cox model.
RESULTS
KRAS mutation status in colorectal cancer
Among 1261 patients with incident colorectal cancer in the two U.S. nationwide prospective cohort studies, we detected KRAS codon 12 and/or 13 mutations in 451 (36%) patients; 335 in codon 12 only, 110 in codon 13 only, and six in both codons 12 and 13. For KRAS codon 12 mutations, we identified 161 cases with c.35G>A (p.G12D, codon 12 GGT>GAT), 95 cases with c.35G>T (p.G12V, codon 12 GGT>GTT), 44 cases with c.34G>T (p.G12C, codon 12 GGT>TGT), 20 cases with c.35G>C (p.G12A, codon 12 GGT>GCT), 12 cases with c.34G>A (p.G12S, codon 12 GGT>AGT), eight cases with c.34G>C (p.G12R, codon 12 GGT>CGT) and one case with c.35_36delinsCA (p.G12A, codon 12 GGT>GCA). In codon 13, we identified 110 cases with c.38G>A (p.G13D, codon 13 GGC>GAC), three cases with c.37G>T (p.G13C, codon 13 GGC>TGC), two cases with c.38G>T (p.G13V, codon 13 GGC>GTC) and one case with c.37G>C (p.G13R, codon 13 GGC>CGC).
Table 2 summarizes the baseline characteristics of study subjects (N=1261) according to KRAS mutation status. There was no significant difference in any of the features examined between KRAS codon 12 and codon 13 mutants. Supplementary Table 1 summarizes the baseline characteristics of BRAF-wild-type cases (N=1075) which were used for subsequent survival analyses.
KRAS mutation status and patient survival in BRAF-wild-type cases
When assessing the prognostic effect of KRAS mutation, it is necessary to consider a confounding effect of BRAF mutation, because BRAF mutation is associated with poorer prognosis (10, 11, 19, 21–23) and inversely associated with KRAS mutation (Table 2). In order to assess prognostic roles of various KRAS mutations, independent of BRAF mutation status, we used BRAF-wild-type tumors only, and compared KRAS-mutated BRAF-wild-type cases to KRAS-wild-type/BRAF-wild-type cases. Thus, among the 1261 patients with KRAS and BRAF data, 181 BRAF-mutated cases were excluded (Figure 1). Within the remaining 1080 cases, five cases with KRAS mutations in both codons 12 and 13 were excluded in order to analyze the prognostic effect of KRAS codon 12 mutation separately from that of codon 13 mutation. As a result, a total of 1075 BRAF-wild-type cases were used for survival analyses (Figure 1, Supplementary Table 1). There were 512 deaths, including 299 colorectal cancer-specific deaths, during a median follow-up of 11.7 years (interquartile range, 8.3–16.1 years) for censored cases.
The 5-year colorectal cancer-specific survival probabilities were 81.5% for patients with KRAS-wild-type/BRAF-wild-type, 68.8% for those with KRAS codon 12 mutations (with wild-type BRAF), and 75.3% for those with KRAS codon 13 mutations (with wild-type BRAF; Figure 2 Panels A–B). Compared to patients with KRAS-wild-type/BRAF-wild-type cancers, those with KRAS codon 12 mutations experienced a significant increase in colorectal cancer-specific mortality in Kaplan-Meier analysis (log-rank P=0.0001) and in Cox regression analysis [univariate hazard ratio (HR)=1.68, 95% confidence interval (CI), 1.32–2.14, P<0.0001; multivariate HR=1.30, 95% CI, 1.02–1.67, P=0.037; Table 3]. In contrast, compared to KRAS-wild-type/BRAF-wild-type cases, patients with KRAS codon 13 mutations did not experience any significant reduction in survival (Table 3).
Figure 2.
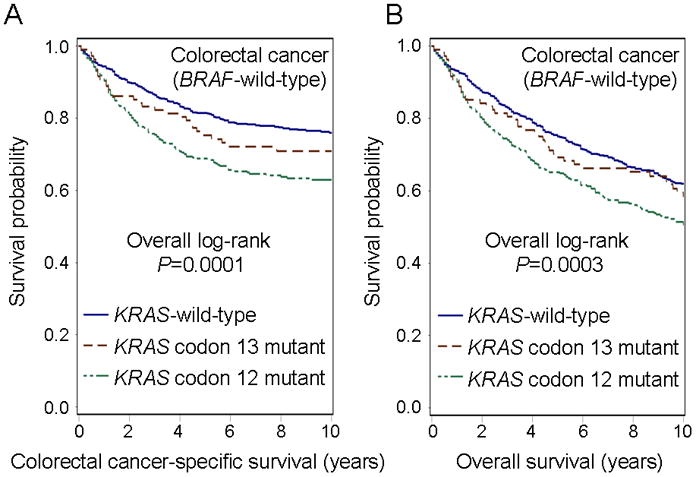
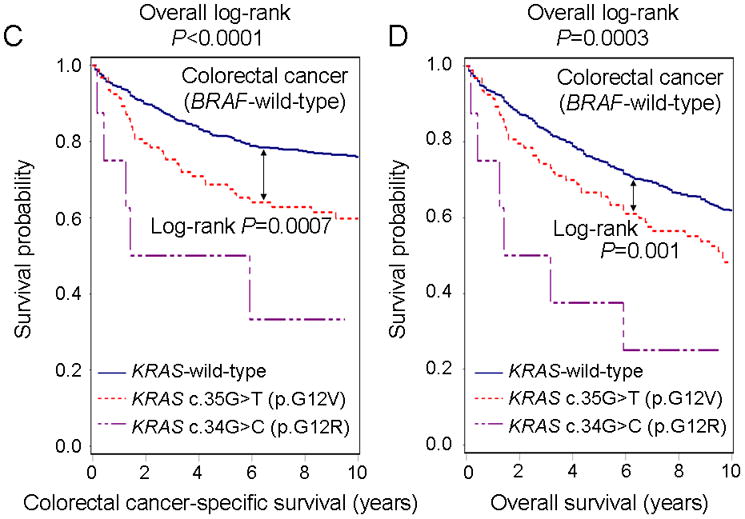
Kaplan-Meier curves of BRAF-wild-type colorectal cancer patients according to KRAS mutation status. KRAS-mutated BRAF-wild-type cases were compared to KRAS-wild-type/BRAF-wild-type cases to assess a prognostic role of KRAS mutation independent of BRAF mutation status. Table indicates the number of patients who were alive and at risk of death at each time point after diagnosis of colorectal cancer. (A) Colorectal cancer-specific survival according to KRAS codon 12 or 13 mutation status. (B) Overall survival according to KRAS codon 12 or 13 mutation status. (C) Colorectal cancer-specific survival according to KRAS c.35G>T (p.G12V) or c.34G>C (p.G12R) mutation status. (D) Overall survival according to KRAS c.35G>T (p.G12V) or c.34G>C (p.G12R) mutation status.
Table 3.
Colorectal cancer patient mortality according to KRAS mutation status in 1075 BRAF-wild-type cases
| KRAS | BRAF | Total N | Colorectal cancer-specific mortality
|
Overall mortality
|
||||
|---|---|---|---|---|---|---|---|---|
| No. of events | Univariate HR (95% CI) | Multivariate HR (95% CI) | No. of events | Univariate HR (95% CI) | Multivariate HR (95% CI) | |||
| Wild-type | Wild-type | 635 | 149 | 1 (referent) | 1 (referent) | 275 | 1 (referent) | 1 (referent) |
| All codon 12 mutants | Wild-type | 332 | 119 | 1.68 (1.32 – 2.14) P<0.0001 |
1.30 (1.02 – 1.67) P=0.037 |
183 | 1.46 (1.21 – 1.77) P<0.0001 |
1.24 (1.02 – 1.51) P=0.029 |
| All codon 13 mutants | Wild-type | 108 | 31 | 1.25 (0.85 – 1.84) NS |
0.86 (0.58 – 1.27) NS |
54 | 1.17 (0.87 – 1.57) NS |
0.96 (0.71 – 1.30) NS |
We tested the specific study hypothesis on the prognostic role of KRAS codon 12 mutations, among BRAF-wild-type cases. Thus, a P-value for significance was set at P=0.05.
The multivariate, stage-stratified Cox regression model initially included age, sex, year of diagnosis, tumor location, tumor differentiation, family history of colorectal cancer, microsatellite instability, CpG island methylator phenotype, PIK3CA and LINE-1 methylation. A backward stepwise elimination with a threshold of P=0.20 was used to select variables in the final model.
CI, confidence interval; HR, hazard ratio; NS, not significant.
Among the seven most common KRAS codon 12 and 13 mutations, c.35G>T (p.G12V; N=93) was associated with significantly higher colorectal cancer-specific mortality (log-rank P=0.0007; multivariate HR=2.00, 95% CI, 1.38–2.90, P=0.0003) compared to KRAS-wild-type/BRAF-wild-type (Figure 2 Panels C–D, Table 4). In addition, c.34G>C (p.G12R; N=8) was associated with higher colorectal cancer-specific mortality (univariate HR=4.22, 95% CI, 1.72–10.4, P=0.0017; multivariate HR=3.39, 95% CI, 1.28–9.00, P=0.014) compared to KRAS-wild-type/BRAF-wild-type. Nonetheless, due to the lower statistical power, as well as multiple hypothesis testing (requiring an adjusted stringent significance level at P=0.05/7=0.007), our findings from mutation-specific survival analyses require validation in independent datasets. Analyses excluding the cases with unknown disease stage (N=110), yielded similar results (Supplementary Table 2).
Table 4.
Colorectal cancer patient mortality among the seven most common KRAS codon 12–13 mutations in 1075 BRAF-wild-type cases
| KRAS | BRAF | Total N | Colorectal cancer-specific mortality
|
Overall mortality
|
||||
|---|---|---|---|---|---|---|---|---|
| No. of events | Univariate HR (95% CI) | Multivariate HR (95% CI) | No. of events | Univariate HR (95% CI) | Multivariate HR (95% CI) | |||
| Wild-type | Wild-type | 635 | 149 | 1 (referent) | 1 (referent) | 275 | 1 (referent) | 1 (referent) |
| c.34G>A (p.G12S) | Wild-type | 12 | 6 | 2.61 (1.15 – 5.91) P=0.022 |
1.03 (0.44 – 2.44) NS |
7 | 1.63 (0.77 – 3.47) NS |
0.96 (0.44 – 2.08) NS |
| c.34G>C (p.G12R) | Wild-type | 8 | 5 | 4.22 (1.72 – 10.4) P=0.0017 |
3.39 (1.28 – 9.00) P=0.014 |
6 | 2.79 (1.24 – 6.31) P=0.014 |
3.19 (1.32 – 7.69) P=0.010 |
| c.34G>T (p.G12C) | Wild-type | 44 | 16 | 1.70 (1.01 – 2.85) P=0.045 |
1.56 (0.92 – 2.65) NS |
25 | 1.48 (0.98 – 2.24) NS |
1.42 (0.93 – 2.17) NS |
| c.35G>A (p.G12D) | Wild-type | 155 | 49 | 1.47 (1.07 – 2.04) P=0.019 |
1.08 (0.76 – 1.51) NS |
79 | 1.38 (1.07 – 1.77) P=0.013 |
1.16 (0.89 – 1.51) NS |
| c.35G>C (p.G12A) | Wild-type | 19 | 6 | 1.36 (0.60 – 3.08) NS |
0.56 (0.24 – 1.30) NS |
9 | 1.03 (0.53 – 2.01) NS |
0.60 (0.31 – 1.19) NS |
| c.35G>T (p.G12V) | Wild-type | 93 | 37 |
1.84 (1.28 – 2.64) P=0.0010 |
2.00 (1.38 – 2.90) P=0.0003 |
57 |
1.61 (1.21 – 2.14) P=0.0012 |
1.54 (1.15 – 2.07) P=0.0042 |
| c.38G>A (p.G13D) | Wild-type | 103 | 31 | 1.33 (0.90 – 1.96) NS |
0.88 (0.59 – 1.30) NS |
52 | 1.21 (0.90 – 1.62) NS |
0.98 (0.72 – 1.35) NS |
The multivariate Cox regression model included the same set of covariates selected as in Table 3. A P-value for significance was adjusted for multiple hypothesis testing to P=0.05/7=0.007. Thus, a P-value between 0.05 and 0.007 should be regarded as of borderline significance. CI, confidence interval; HR, hazard ratio; NS, not significant.
In order to assess the impact of confounding by BRAF mutation, we repeated the above survival analyses including BRAF-mutated cases, most of which were included in the KRAS wild-type group. A total case number for this additional analysis was 1255, including 180 BRAF-mutated cases. Compared to KRAS-wild-type cases, KRAS codon 12 mutations were not significantly associated with colorectal cancer-specific mortality in multivariate analysis and the HR effect estimate was substantially attenuated (Supplementary Table 3). Among the seven most common KRAS codon 12 and 13 mutations, compared to KRAS-wild-type cases, the HR effect estimates for c.34G>C (p.G12R) and c.35G>T (p.G12V) mutations were considerably attenuated.
KRAS mutation status and mortality in strata of other variables
As exploratory analyses, we examined the prognostic association of KRAS codon 12 and 13 mutation status among BRAF-wild-type tumors in various strata, including disease stage, sex, age, family history of colorectal cancer, tumor location, tumor differentiation, MSI, CIMP, PIK3CA, and LINE-1 methylation. We did not observe considerable or significant modifying effect by any of these variables on KRAS codon 12 or 13 mutation [all Pinteraction>0.02; given multiple hypothesis testing, a statistical significance level was adjusted to Pinteraction=0.0021)].
DISCUSSION
We conducted this study to assess whether KRAS codon 12 mutated tumors represent a more aggressive subtype as compared to either KRAS codon 13 mutated tumors or KRAS-wild-type tumors, within a group of BRAF-wild-type tumors (i.e., controlling for BRAF mutation status). We showed that KRAS codon 12 mutations, but not codon 13 mutations, were associated with significantly higher mortality compared to KRAS-wild-type/BRAF-wild-type cases. In particular, c.35G>T (p.G12V) mutation was associated with the highest colorectal cancer-specific mortality (multivariate HR=2.00, 95% CI, 1.38–2.90, P=0.0003). Our data are consistent with previous laboratory studies (24, 25) suggesting that the presence of a mutation in KRAS codon 12 confers substantially greater oncogenic potential as compared to codon 13 mutation. Our data are also consistent with a recent study that showed that KRAS codon 12 mutations, but not codon 13 mutations, conferred resistance to cetuximab in advanced colorectal cancer (15).
Detection of somatic molecular aberrations and tumor molecular classification are increasingly important in colorectal cancer (36–39). We used Pyrosequencing technology, which has been shown to be more sensitive than Sanger sequencing in detecting KRAS mutations in paraffin-embedded archival tissue (30, 40, 41). Pyrosequencing is a sensitive sequencing assay and can reliably detect mutant alleles of low abundance (10% mutant) among wild-type alleles, which is a common situation in solid tumors (30, 40, 41).
To the best of our knowledge based on the literature search in Pubmed, this is the first study to address the prognostic difference between KRAS codon 12 and codon 13 mutations in more than 1000 of BRAF-wild-type colorectal cancers (i.e., controlling for BRAF mutation status). Although several previous studies have distinguished between the prognostic associations of KRAS mutations in codon 12 and codon 13 (Table 1) (16–21), none of the large studies (with a sample size N≥300) (16, 17, 19, 20) controlled for BRAF mutation status in their analyses, and the results are conflicting. Given the consistent significant negative prognostic impact of BRAF mutations on patient survival (10, 11, 19, 21–23), and its association with KRAS-wild-type tumors, the presence of patients harboring BRAF-mutated tumors within a KRAS-wild-type control group would attenuate any negative prognostic effect associated with KRAS mutation status. Therefore, BRAF mutation status must be controlled to assess the precise oncogenic effect of KRAS mutation status. A simple way of controlling for BRAF mutation status is to examine the prognostic role of KRAS mutation in BRAF-wild-type tumors. Indeed, BRAF mutation confounded and attenuated the negative prognostic effects of KRAS mutations in this study.
Regulation of RAS involves binding of GTP, which activates the protein. Activation of RAS enables high affinity interactions with downstream effectors such as RAF-MAPK and PI3K. Subsequently, slow intrinsic GTPase activity leads to RAS functional inactivation. This on and off switch regulation is tightly controlled by ARHGAP (Rho-GTPase activating proteins) and RAPGEF (Rap guanine-nucleotide exchange factors) (42). Interestingly, RAS mutants are resistant to ARHGAP-mediated GTPase activation, leading to elevated cellular levels of RAS-GTP (42). Guerrero and colleagues (24) found that KRAS codon 12 mutation, by altering the threshold for induction of apoptosis, confers a more aggressive tumor phenotype than codon 13 mutation. This suggests that codon 12 mutation results in greater resistance to ARHGAP-mediated GTPase activation than codon 13 mutation (24). Consequently, codon 12 mutated RAS theoretically remains in an active GTP-bound state longer than codon 13 mutated or wild-type RAS. Experimental data suggest that, among the many different KRAS codon 12 mutations, c.34G>C (p.G12R) and c.35G>T (p.G12V) mutations confer more potent transforming ability than other KRAS mutations including c.34G>A (p.G12S), c.34G>T (p.G12C), c.35G>A (p.G12D) and c.35G>C (p.G12A) (43). Moreover, the GTPase activity of c.34G>C (p.G12R) and c.35G>T (p.G12V) mutants is lower than that of other KRAS mutations (25, 44). These experimental data are consistent with our observations that KRAS codon 12 mutation, especially c.34G>C (p.G12R) and c.35G>T (p.G12V), might be associated with more aggressive tumor behavior. Our findings underscore the importance of our awareness that different mutations (even in a single gene) may contribute to different tumor characteristics and support the unique tumor principle (45–47).
Limitations of the current study include the lack of data on cancer treatment. Chemotherapeutic and surgical interventions have a significant impact on disease progression in metastatic colorectal cancer. We cannot exclude the possibility that there may have been an imbalance in the use of therapeutic interventions between subgroups. KRAS mutation status has recently become an important biomarker when selecting chemotherapeutic agents for colorectal cancer therapy (6–8, 48). Given that we could not control for use of EGFR inhibitors, such as cetuximab and panitumumab, bias may have arisen through selective use of these agents within the study group. Nonetheless, it seems unlikely that chemotherapy use or regimen differed substantially by tumor KRAS mutation status, since a vast majority of cases were diagnosed in 1990’s to early 2000’s, before 2006, when KRAS mutation emerged as a predictive biomarker in stage IV colorectal cancer. In addition, our molecular data were not available for patients or clinicians for treatment decision making. Another weakness of this study is the absence of data on cancer recurrence, and, as a result, disease-free survival was not an available outcome measurement in these cohorts. Because the median survival of metastatic colorectal cancer patients was 10 to 12 months during the time period of this study (49), we believe that colorectal cancer-specific survival is a reasonably robust surrogate for cancer-specific outcomes. In fact, disease-free survival has been shown to be highly correlated with overall survival (50).
Strengths of the current study include the use of data from two U.S. nationwide prospective cohort studies. Information on disease staging, family history of cancer, and other clinicopathologic and tumor molecular data was prospectively integrated into the molecular pathological epidemiology database (26–28). Cohort participants who were diagnosed with colorectal cancer were presented and treated at hospitals throughout the United States, and thus more representative colorectal cancers in the general U.S. population than are patients in single or few academic medical centers. Finally, by virtue of our molecular pathological epidemiology (26–28) database, we assessed the effects of KRAS codon 12 and 13 mutations independent of various clinicopathologic features and other critical molecular events such as BRAF and PIK3CA mutations, MSI, CIMP and LINE-1 hypomethylation, all of which have been associated with colorectal cancer prognosis (2, 34).
In conclusion, our study of over 1000 colorectal cancers has shown that KRAS codon 12 mutation (in particular, c.35G>T, p.G12V), but not codon 13 mutation, is associated with worse prognosis in BRAF-wild-type colorectal cancers. Different mutations in a single gene may have distinct biological effects and clinical implications (47). Since, controlling for BRAF status, KRAS codon 12 mutations contribute to poor prognosis in colorectal cancer, it might be prudent to control for mutations in BRAF and KRAS in the study arms of future clinical trials.
Supplementary Material
Translational Relevance.
To assess prognostic roles of KRAS mutations in colorectal cancer, BRAF mutation status must be controlled because BRAF-mutated cancers are associated with poorer prognosis than BRAF-wild-type cases, and almost all BRAF mutants are present among KRAS-wild-type tumors. However, no previous large study (with a sample size of N≥300) has controlled for the effect of BRAF mutation. We examined the prognostic roles of various KRAS codon 12 and 13 mutations in 1075 BRAF-wild-type colorectal cancer cases with available clinical data, adequate follow-up, and other important molecular data relevant to colorectal cancer. This study, including over 1000 BRAF-wild-type colorectal cancers, shows that KRAS codon 12 mutations (in particular, c.35G>T), but not codon 13 mutations, are associated with inferior survival independent of clinical, pathological and molecular features of colorectal cancer. Our data suggest the potential need to evaluate both BRAF mutation status and specific KRAS mutation status as prognostic biomarkers for colorectal cancer.
Acknowledgments
Grant Support
This work was supported by the National Institute of Health [P01 CA87969 (to S.E. Hankinson), P01 CA55075 (to W.C. Willett), P50 CA127003 (to C.S.F.), and R01 CA151993 (to S.O.)]; the Bennett Family Fund for Targeted Therapies Research; and the Entertainment Industry Foundation through National Colorectal Cancer Research Alliance. TM was supported by a fellowship grant from the Japan Society for Promotion of Science. PL was supported by Frank Knox Memorial Fellowship from Harvard University. The content is solely the responsibility of the authors and does not necessarily represent the official views of NCI or NIH. Funding agencies did not have any role in the design of the study; the collection, analysis, or interpretation of the data; the decision to submit the manuscript for publication; or the writing of the manuscript.
We deeply thank hospitals and pathology departments throughout the U.S. for generously providing us with tissue specimens. In addition, we would like to thank the participants and staff of the Nurses’ Health Study and the Health Professionals Follow-Up Study, for their valuable contributions as well as the U.S. state cancer registries for their help.
Footnotes
Conflict of interest: The authors have declared no conflicts of interest.
References
- 1.Janakiraman M, Vakiani E, Zeng Z, Pratilas CA, Taylor BS, Chitale D, et al. Genomic and biological characterization of exon 4 KRAS mutations in human cancer. Cancer Res. 2010;70:5901–11. doi: 10.1158/0008-5472.CAN-10-0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nosho K, Irahara N, Shima K, Kure S, Kirkner GJ, Schernhammer ES, et al. Comprehensive biostatistical analysis of CpG island methylator phenotype in colorectal cancer using a large population-based sample. PLoS One. 2008;3:e3698. doi: 10.1371/journal.pone.0003698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roock WD, Vriendt VD, Normanno N, Ciardiello F, Tejpar S. KRAS, BRAF, PIK3CA, and PTEN mutations: implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol. 2011;12:594–603. doi: 10.1016/S1470-2045(10)70209-6. [DOI] [PubMed] [Google Scholar]
- 4.Allegra CJ, Jessup JM, Somerfield MR, Hamilton SR, Hammond EH, Hayes DF, et al. American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncol. 2009;27:2091–6. doi: 10.1200/JCO.2009.21.9170. [DOI] [PubMed] [Google Scholar]
- 5.Jimeno A, Messersmith WA, Hirsch FR, Franklin WA, Eckhardt SG. KRAS mutations and sensitivity to epidermal growth factor receptor inhibitors in colorectal cancer: practical application of patient selection. J Clin Oncol. 2009;27:1130–6. doi: 10.1200/JCO.2008.19.8168. [DOI] [PubMed] [Google Scholar]
- 6.Van Cutsem E, Kohne CH, Lang I, Folprecht G, Nowacki MP, Cascinu S, et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol. 2011;29:2011–9. doi: 10.1200/JCO.2010.33.5091. [DOI] [PubMed] [Google Scholar]
- 7.Douillard JY, Siena S, Cassidy J, Tabernero J, Burkes R, Barugel M, et al. Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. J Clin Oncol. 2010;28:4697–705. doi: 10.1200/JCO.2009.27.4860. [DOI] [PubMed] [Google Scholar]
- 8.Bokemeyer C, Bondarenko I, Makhson A, Hartmann JT, Aparicio J, de Braud F, et al. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J Clin Oncol. 2009;27:663–71. doi: 10.1200/JCO.2008.20.8397. [DOI] [PubMed] [Google Scholar]
- 9.Zlobec I, Bihl MP, Schwarb H, Terracciano L, Lugli A. Clinicopathological and protein characterization of BRAF- and K-RAS-mutated colorectal cancer and implications for prognosis. Int J Cancer. 2010;127:367–80. doi: 10.1002/ijc.25042. [DOI] [PubMed] [Google Scholar]
- 10.Farina-Sarasqueta A, van Lijnschoten G, Moerland E, Creemers GJ, Lemmens VE, Rutten HJ, et al. The BRAF V600E mutation is an independent prognostic factor for survival in stage II and stage III colon cancer patients. Ann Oncol. 2010;21:2396–402. doi: 10.1093/annonc/mdq258. [DOI] [PubMed] [Google Scholar]
- 11.Richman SD, Seymour MT, Chambers P, Elliott F, Daly CL, Meade AM, et al. KRAS and BRAF mutations in advanced colorectal cancer are associated with poor prognosis but do not preclude benefit from oxaliplatin or irinotecan: results from the MRC FOCUS trial. J Clin Oncol. 2009;27:5931–7. doi: 10.1200/JCO.2009.22.4295. [DOI] [PubMed] [Google Scholar]
- 12.Samowitz WS, Curtin K, Wolff RK, Tripp SR, Caan BJ, Slattery ML. Microsatellite instability and survival in rectal cancer. Cancer Causes Control. 2009;20:1763–8. doi: 10.1007/s10552-009-9410-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barault L, Charon-Barra C, Jooste V, de la Vega MF, Martin L, Roignot P, et al. Hypermethylator phenotype in sporadic colon cancer: study on a population-based series of 582 cases. Cancer Res. 2008;68:8541–6. doi: 10.1158/0008-5472.CAN-08-1171. [DOI] [PubMed] [Google Scholar]
- 14.Ogino S, Meyerhardt JA, Irahara N, Niedzwiecki D, Hollis D, Saltz LB, et al. KRAS mutation in stage III colon cancer and clinical outcome following intergroup trial CALGB 89803. Clin Cancer Res. 2009;15:7322–9. doi: 10.1158/1078-0432.CCR-09-1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Roock W, Jonker DJ, Di Nicolantonio F, Sartore-Bianchi A, Tu D, Siena S, et al. Association of KRAS p. G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA. 2010;304:1812–20. doi: 10.1001/jama.2010.1535. [DOI] [PubMed] [Google Scholar]
- 16.Samowitz WS, Curtin K, Schaffer D, Robertson M, Leppert M, Slattery ML. Relationship of Ki-ras mutations in colon cancers to tumor location, stage, and survival: a population-based study. Cancer Epidemiol Biomarkers Prev. 2000;9:1193–7. [PubMed] [Google Scholar]
- 17.Andreyev HJ, Norman AR, Cunningham D, Oates J, Dix BR, Iacopetta BJ, et al. Kirsten ras mutations in patients with colorectal cancer: the ‘RASCAL II’ study. Br J Cancer. 2001;85:692–6. doi: 10.1054/bjoc.2001.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bazan V, Migliavacca M, Zanna I, Tubiolo C, Grassi N, Latteri MA, et al. Specific codon 13 K-ras mutations are predictive of clinical outcome in colorectal cancer patients, whereas codon 12 K-ras mutations are associated with mucinous histotype. Ann Oncol. 2002;13:1438–46. doi: 10.1093/annonc/mdf226. [DOI] [PubMed] [Google Scholar]
- 19.Roth AD, Tejpar S, Delorenzi M, Yan P, Fiocca R, Klingbiel D, et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol. 2010;28:466–74. doi: 10.1200/JCO.2009.23.3452. [DOI] [PubMed] [Google Scholar]
- 20.Zlobec I, Kovac M, Erzberger P, Molinari F, Bihl MP, Rufle A, et al. Combined analysis of specific KRAS mutation, BRAF and microsatellite instability identifies prognostic subgroups of sporadic and hereditary colorectal cancer. Int J Cancer. 2010;127:2569–75. doi: 10.1002/ijc.25265. [DOI] [PubMed] [Google Scholar]
- 21.Yokota T, Ura T, Shibata N, Takahari D, Shitara K, Nomura M, et al. BRAF mutation is a powerful prognostic factor in advanced and recurrent colorectal cancer. Br J Cancer. 2011;104:856–62. doi: 10.1038/bjc.2011.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Samowitz WS, Sweeney C, Herrick J, Albertsen H, Levin TR, Murtaugh MA, et al. Poor survival associated with the BRAF V600E mutation in microsatellite-stable colon cancers. Cancer Res. 2005;65:6063–9. doi: 10.1158/0008-5472.CAN-05-0404. [DOI] [PubMed] [Google Scholar]
- 23.French AJ, Sargent DJ, Burgart LJ, Foster NR, Kabat BF, Goldberg R, et al. Prognostic significance of defective mismatch repair and BRAF V600E in patients with colon cancer. Clin Cancer Res. 2008;14:3408–15. doi: 10.1158/1078-0432.CCR-07-1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guerrero S, Casanova I, Farre L, Mazo A, Capella G, Mangues R. K-ras codon 12 mutation induces higher level of resistance to apoptosis and predisposition to anchorage-independent growth than codon 13 mutation or proto-oncogene overexpression. Cancer Res. 2000;60:6750–6. [PubMed] [Google Scholar]
- 25.Bollag G, McCormick F. Intrinsic and GTPase-activating protein-stimulated Ras GTPase assays. Methods Enzymol. 1995;255:161–70. doi: 10.1016/s0076-6879(95)55020-8. [DOI] [PubMed] [Google Scholar]
- 26.Ogino S, Chan AT, Fuchs CS, Giovannucci E. Molecular pathological epidemiology of colorectal neoplasia: an emerging transdisciplinary and interdisciplinary field. Gut. 2011;60:397–411. doi: 10.1136/gut.2010.217182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ogino S, Galon J, Fuchs CS, Dranoff G. Cancer immunology--analysis of host and tumor factors for personalized medicine. Nat Rev Clin Oncol. 2011;8:711–9. doi: 10.1038/nrclinonc.2011.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ogino S, Stampfer M. Lifestyle factors and microsatellite instability in colorectal cancer: The evolving field of molecular pathological epidemiology. J Natl Cancer Inst. 2010;102:365–367. doi: 10.1093/jnci/djq031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morikawa T, Kuchiba A, Yamauchi M, Meyerhardt JA, Shima K, Nosho K, et al. Association of CTNNB1 (beta-catenin) alterations, body mass index, and physical activity with survival in patients with colorectal cancer. JAMA. 2011;305:1685–94. doi: 10.1001/jama.2011.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ogino S, Kawasaki T, Brahmandam M, Yan L, Cantor M, Namgyal C, et al. Sensitive sequencing method for KRAS mutation detection by Pyrosequencing. J Mol Diagn. 2005;7:413–21. doi: 10.1016/S1525-1578(10)60571-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ogino S, Kawasaki T, Kirkner GJ, Loda M, Fuchs CS. CpG island methylator phenotype-low (CIMP-low) in colorectal cancer: possible associations with male sex and KRAS mutations. J Mol Diagn. 2006;8:582–8. doi: 10.2353/jmoldx.2006.060082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liao X, Morikawa T, Lochhead P, Imamura Y, Kuchiba A, Yamauchi M, et al. Prognostic role of PIK3CA mutation in colorectal cancer: cohort study and literature review. Clin Cancer Res. 2012;18:2257–68. doi: 10.1158/1078-0432.CCR-11-2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38:787–93. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- 34.Ogino S, Nosho K, Kirkner GJ, Kawasaki T, Chan AT, Schernhammer ES, et al. A cohort study of tumoral LINE–1 hypomethylation and prognosis in colon cancer. J Natl Cancer Inst. 2008;100:1734–8. doi: 10.1093/jnci/djn359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Irahara N, Nosho K, Baba Y, Shima K, Lindeman NI, Hazra A, et al. Precision of pyrosequencing assay to measure LINE-1 methylation in colon cancer, normal colonic mucosa, and peripheral blood cells. J Mol Diagn. 2010;12:177–83. doi: 10.2353/jmoldx.2010.090106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tanaka M, Chang P, Li Y, Li D, Overman M, Maru DM, et al. Association of CHFR promoter methylation with disease recurrence in locally advanced colon cancer. Clin Cancer Res. 2011;17:4531–40. doi: 10.1158/1078-0432.CCR-10-0763. [DOI] [PubMed] [Google Scholar]
- 37.Dahlin AM, Palmqvist R, Henriksson ML, Jacobsson M, Eklof V, Rutegard J, et al. The role of the CpG island methylator phenotype in colorectal cancer prognosis depends on microsatellite instability screening status. Clin Cancer Res. 2010;16:1845–55. doi: 10.1158/1078-0432.CCR-09-2594. [DOI] [PubMed] [Google Scholar]
- 38.Yagi K, Akagi K, Hayashi H, Nagae G, Tsuji S, Isagawa T, et al. Three DNA methylation epigenotypes in human colorectal cancer. Clin Cancer Res. 2010;16:21–33. doi: 10.1158/1078-0432.CCR-09-2006. [DOI] [PubMed] [Google Scholar]
- 39.Balaguer F, Moreira L, Lozano JJ, Link A, Ramirez G, Shen Y, et al. Colorectal cancers with microsatellite instability display unique miRNA profiles. Clin Cancer Res. 2011;17:6239–49. doi: 10.1158/1078-0432.CCR-11-1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weichert W, Schewe C, Lehmann A, Sers C, Denkert C, Budczies J, et al. KRAS genotyping of paraffin-embedded colorectal cancer tissue in routine diagnostics: comparison of methods and impact of histology. J Mol Diagn. 2010;12:35–42. doi: 10.2353/jmoldx.2010.090079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsiatis AC, Norris-Kirby A, Rich RG, Hafez MJ, Gocke CD, Eshleman JR, et al. Comparison of Sanger sequencing, pyrosequencing, and melting curve analysis for the detection of KRAS mutations: diagnostic and clinical implications. J Mol Diagn. 2010;12:425–32. doi: 10.2353/jmoldx.2010.090188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol. 2008;9:517–31. doi: 10.1038/nrm2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Seeburg PH, Colby WW, Capon DJ, Goeddel DV, Levinson AD. Biological properties of human c-Ha-ras1 genes mutated at codon 12. Nature. 1984;312:71–5. doi: 10.1038/312071a0. [DOI] [PubMed] [Google Scholar]
- 44.Al-Mulla F, Milner-White EJ, Going JJ, Birnie GD. Structural differences between valine-12 and aspartate-12 Ras proteins may modify carcinoma aggression. J Pathol. 1999;187:433–8. doi: 10.1002/(SICI)1096-9896(199903)187:4<433::AID-PATH273>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 45.Ogino S, Goel A. Molecular classification and correlates in colorectal cancer. J Mol Diagn. 2008;10:13–27. doi: 10.2353/jmoldx.2008.070082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ogino S, Giovannucci E. Lifestyle factors and colorectal cancer microsatellite instability: molecular pathological epidemiology science, based on unique tumour principle. J Epidemiol. 2012 doi: 10.1093/ije/dys076. in press (published online) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ogino S, Fuchs CS, Giovannucci E. How many tumor molecular subtype? Implications of unique tumor principle in personalized medicine and molecular diagnostics. Expert Rev Mol Diagn. 2012 doi: 10.1586/erm.12.46. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moosmann N, von Weikersthal LF, Vehling-Kaiser U, Stauch M, Hass HG, Dietzfelbinger H, et al. Cetuximab plus capecitabine and irinotecan compared with cetuximab plus capecitabine and oxaliplatin as first-line treatment for patients with metastatic colorectal cancer: AIO KRK-0104--a randomized trial of the German AIO CRC study group. J Clin Oncol. 2011;29:1050–8. doi: 10.1200/JCO.2010.31.1936. [DOI] [PubMed] [Google Scholar]
- 49.Ogino S, Nosho K, Irahara N, Shima K, Baba Y, Kirkner GJ, et al. Prognostic significance and molecular associations of 18q loss of heterozygosity: a cohort study of microsatellite stable colorectal cancers. J Clin Oncol. 2009;27:4591–8. doi: 10.1200/JCO.2009.22.8858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sargent DJ, Wieand HS, Haller DG, Gray R, Benedetti JK, Buyse M, et al. Disease-free survival versus overall survival as a primary end point for adjuvant colon cancer studies: individual patient data from 20,898 patients on 18 randomized trials. J Clin Oncol. 2005;23:8664–70. doi: 10.1200/JCO.2005.01.6071. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.