

Abstract
BACKGROUND & AIMS
Intestinal microbes induce homeostatic mucosal immune responses, but can also cause inappropriate immune activation in genetically susceptible hosts. While immune responses to bacterial products have been studied extensively, little is known about how intestinal inflammation affects the function of commensal luminal microbes.
METHODS
Microarrays and real-time PCR were used to profile transcriptional changes in luminal bacteria from wild-type (WT) and IL-10−/− (KO) mice monoassociated with a non-pathogenic murine Escherichia coli isolate (NC101), which causes colitis in gnotobiotic KO mice. Colonic inflammation, innate and adaptive immune responses were measured in WT and KO mice monoassociated with mutant NC101 lacking selected upregulated genes and in KO mice co-colonized with mutant and parental NC101. Intracellular survival of bacteria within primary mouse macrophages and resultant TNF production was measured.
RESULTS
Significant upregulation of the stress response regulon, including the small heat shock proteins IbpA and IbpB that protect E. coli from oxidative stress, was observed in bacteria from KO mice with colitis compared to healthy WT controls. In KO mice, ibpAB expression resulted in reduced colonic histologic inflammation, secretion of IL-12/23p40 by colonic explant cultures, serologic reactivity to NC101 antigens, and IFNγ secretion by stimulated mesenteric lymph node cells. Infection of primary macrophages by bacteria expressing ibpAB was associated with decreased intracellular survival and attenuated TNF secretion.
CONCLUSIONS
Chronic intestinal inflammation causes functional alterations in gene expression of a commensal gut bacterium. Further studies of this component of the host-microbial dialogue may identify potential novel therapeutic targets to treat inflammatory bowel diseases.
Keywords: Inflammation, Bacteria, Gene Expression
INTRODUCTION
Relative to higher-order, multicellular organisms, microbes are unique in their ability to rapidly evolve with genetic adaptation to environmental conditions. Adaptation of non-pathogenic commensal microbes, such as those residing in the lumen of the gastrointestinal tract, occurs in response to host physiologic conditions. For instance, fucosylated glycans, mucus, and antigen-specific sIgA have been shown to shape luminal bacterial gene expression1–3. Although these studies indicate that the normal intestinal environment influences the composition and function of the gut microbiota, the impact of specific disease states on intestinal microbial communities is only now beginning to be explored.
A prevailing hypothesis is that inflammatory bowel diseases (IBD) are due to a combination of genetic and environmental factors, which predispose to dysregulated immune responses to the commensal intestinal bacteria4. Similar to human IBD, the intestinal microbiota is important in the pathogenesis of chronic, immune-mediated experimental colitis4. For instance, mice lacking the IL-10 gene (IL-10−/−) are healthy when raised in a germ-free (GF) environment, but rapidly develop chronic, bacterial antigen-specific T-cell-mediated colitis resembling IBD when transferred to specific-pathogen-free (SPF) conditions5, 6. The inflammation in human IBD and experimental colitis is also characterized by increased tissue levels of reactive oxygen (ROS) and nitrogen species7, 8.
While the intestinal microbiome is complex, preferential expansion of subsets of bacteria including E. coli is associated with Crohn’s disease9, 10. Certain commensal E. coli that adhere to and invade epithelial cells have been implicated in the development of ileal Crohn’s disease11. Similarly, in animal models, selective colonization (monoassociation) with a fecal murine isolate of non-pathogenic E. coli (NC101) for three weeks induces histological evidence of colitis in ex-GF IL-10−/− mice but not in wild-type (WT) controls12. This strain of E. coli exhibits many of the characteristics of adherent/invasive E. coli isolated from Crohn’s disease patients, including persistence within macrophages (unpublished data).
In addition to the expansion of select species of commensal bacteria such as E. coli, broad shifts in the composition of the gut microbial communities are also associated with intestinal inflammation in both human IBD and experimental colitis13. However, whether these changes are primary or secondary is unknown nor is much known about how pathologic inflammation affects the function of commensal intestinal microbes. We hypothesize that the unique adaptive capabilities of commensal gut microbes allow them to respond to intestinal inflammation by altering gene expression patterns that increase their survival and virulence. Here we provide evidence that a gut commensal strain, E. coli NC101, upregulates its stress response regulon in monoassociated IL-10−/− mice with colitis compared to healthy controls. Furthermore, we show using deletion mutants that expression of two components of the E. coli stress response regulon, ibpA and ibpB, is associated with decreased luminal growth, impaired intracellular macrophage survival, and diminished pro-inflammatory host immune responses. These findings suggest that chronic, immune-mediated intestinal inflammation upregulates a pathway in a commensal gut microbe that ultimately proves to be detrimental to bacterial growth and persistence within macrophages and, at the same time, attenuates the pro-inflammatory host response.
METHODS
Bacterial Strains, Lysates, and Growth Curves
The non-pathogenic murine E. coli strain designated NC101 was originally isolated from a randomly-chosen colony from the feces of WT mice raised in SPF conditions12. The Enterococcus faecalis (strain OG1RF) was originally obtained from a human oral isolate kindly provided by Dr. Mark Huycke. NC101ΔibpAB was constructed using λ-red recombinase and NC101ΔibpAB(pGEN-MCSibpAB) was constructed using standard molecular biology techniques (Supplementary Methods). Bacterial lysates were prepared from bacterial cultures grown in either LB or BHI media using mechanical disruption as described elsewhere12. For growth curves, LB or BHI was inoculated with an overnight bacterial culture and incubated at 37°C. The OD600 of the cultures was measured at the indicated time points.
Isolation of Bone Marrow-Derived Macrophages
The murine fibroblast cell line, L929, was used as a source of M-CSF and was kindly provided by Dr. Scott Plevy (UNC Chapel Hill, NC). Conditioned media from L929 cells, was made as previously described14. Bone marrow-derived macrophages (BMDM) were cultured from femurs and tibias of WT and IL-10−/− 129Sv/Ev mice as previously described15.
Mice
GF IL-10−/− and WT control (both on the 129S6/SvEV background) were originally derived in sterile conditions, by hysterectomy at the Gnotobiotic Laboratory (University of Wisconsin Madison). Mice maintained in germ-free conditions at the National Gnotobiotic Rodent Resource Center at UNC-Chapel Hill were monoassociated with bacteria by swabbing the snout and anus (experiments lasting >10 days) or by oral gavage of 1 × 109 CFU (experiments lasting ≤10 days) with an overnight bacterial culture. Inoculums for dual-association studies were prepared by mixing equal volumes of overnight cultures of NC101 and NC101ΔibpAB with same OD600 immediately prior to administration. Absence of isolator contamination was confirmed by Gram stain and culture of cecal contents on sheep blood agar or BHI plates under aerobic and anaerobic conditions. Animal use protocols were approved by the UNC-Chapel Hill Institutional Animal Care and Use Committees.
RNA Isolation From Cecal Contents
Approximately 300mg of freshly-harvested cecal or transverse colon contents were snap frozen in N2 (l) and stored at −80°C until ready for use. Frozen samples were thawed into 1mL of RNAProtect Bacteria Reagent (Qiagen) while vortexing, incubated at 25°C for 5 min, and bacterial RNA was isolated as described previously16.
Microarray Hybridization and Statistical Analysis
RNA samples were analyzed for purity and integrity on Agilent Bioanalyzer chips according to the manufacturer’s instructions (Agilent Technologies). All E. coli RNA samples were prepared for microarray hybridization according to the expression analysis technical manual from Affymetrix. Affymetrix E. coli Genome 2.0 arrays were washed and stained with the Fluidics Station 450. The arrays were scanned with the GeneChip Scanner 3000 7G Plus with Autoloader. Basic data analysis of the arrays was carried out with MAS 5.0 software to generate intensity values for each gene. Statistical analysis of microarray results was performed using GeneSpring 7.2 software. The list of normalized, differentially-regulated genes was narrowed to those genes with statistically significant differences when grouped by genetic characteristic (Welch t-test, p-value cutoff 0.05, multiple testing correction: Benjamini and Hochberg False Discovery Rate).
Transcription factor, including initiation factor σ, binding sites were identified in promoter elements of selected E. coli genes from the microarray results using the E. coli K-12 regulonDB 6.7 online database (http://regulondb.ccg.unam.mx/index.jsp)17.
Real-time PCR
Real-time oligonucleotide primers were designed using Applied Biosystems Primer Express 3.0 software based on E. coli MG1655 genomic DNA sequences published in Genbank (Table S4). First-strand cDNA was synthesized from 1μg total RNA using M-MLV Reverse Transcriptase (Invitrogen) per the manufacturer’s instructions. Two μL of the reaction were added to 3.75 pmol of each primer and 2× SYBR Green PCR Master Mix (Applied Biosystems) in a final volume of 12.5μL and analyzed in an ABI 7900HT thermocycler using the manufacturer’s universal thermocycling conditions: 95°C for 10 minutes then 40 cycles of 95°C for 15 sec and 60°C for 60 sec. The specificity of amplification was confirmed by melting curve analysis and agarose gel electrophoresis of PCR products. All reactions were run in triplicate. Fold-change was calculated using the ΔΔCt method relative to E. coli 16S rRNA.
Quantification of Luminal Bacteria
A small amount of cecal and mid-colonic luminal contents were collected into pre-weighed, sterile tubes. Assuming that 1g of contents has a volume of 1mL, the samples were serially diluted to 10−7, 10−8, and 10−9 in sterile phosphate-buffered saline (PBS). One-hundred μL of each dilution were plated onto BHI agar and incubated at 37°C for 24 hours under aerobic conditions. Results were expressed as colony forming units (CFU)/g of contents.
Histological Scoring
Sections of colon (proximal, transverse, distal) and cecum, were fixed in 10% neutral buffered formalin for 24–48 hrs. The fixed tissue was embedded in paraffin, sectioned and stained with hematoxylin and eosin (H&E). Individual sections were scored for severity of inflammation by a single, blinded individual using a validated scale of 0–412. Where indicated, a composite colonic inflammation score was obtained by summing the scores of four individual colonic sections (cecum, proximal, mid, and distal colon) from each animal.
Western Blots
Fifteen μg/lane of NC101 lysate prepared as described above were resolved on a 10% SDS/polyacrylamide gel, transferred to PVDF membrane, probed with 1:1000 dilution of mouse serum in 5% dry milk-TBST for 90 min, washed 3 times in TBST, incubated with 1:5000 dilution of sheep anti-mouse IgG-HRP in 5% dry milk-TBST for 60 min, washed 3 times, incubated with ECL-Plus (Amersham) for 5 min, and then exposed to X-ray film.
Colonic Tissue Fragment Cultures
Colonic tissue fragment cultures were prepared from the proximal colon as previously described5, 12. Briefly, the colonic tissue fragments were washed with PBS then RPMI containing 50μg/mL gentamicin, blotted dry, and weighed. Fragments were placed into 1mL of complete RPMI in 24-well plates, cut into pieces with scissors, and incubated for 20 hours at 37°C. Supernatants were collected and stored at −20°C.
Mesenteric Lymph Node Preparation and Culture
Mesenteric lymph nodes (MLN) were aseptically removed and single cell suspensions were prepared by gentle disruption between sterile, frosted glass microscope slides. Cells were washed and resuspended in RPMI with 10% FBS, 2mM L-glutamine, 1mM sodium pyruvate, 0.05mM 2-mercaptoethanol, and penicillin (100U/ml)-streptomycin (100μg/ml). 5 × 105 cells/well in quadruplicate were stimulated with either media control, or NC101 or E. faecalis lysates (5μg protein/mL) in 96-well plates (final volume of 0.2mL per well) for 72 hrs18. Culture supernatants were collected and stored at −20°C prior to cytokine quantification.
Gentamicin Protection Assays
Bacterial uptake and survival within macrophages was measured as described previously19. Briefly, approximately 10 bacteria/cell were added to BMDMs and incubated for 90 min and then treated for 90 min with media containing 100μg/mL gentamicin. Supernatants were removed and stored at −20°C for future cytokine analysis. In separate plates, after the initial gentamicin treatment and washing, 20μg gentamicin/mL was added and plates were incubated for an additional 23.5 hrs after which supernatants were removed and stored at −20°C for future cytokine analysis. Survival ratios were calculated by dividing the numberof bacteria detected after 24 hrs of gentamicin treatment by the number detected after 1.5 hrs of gentamicin treatment.
Cytokine Measurements
To measure amounts of TNF secreted by infected BMDM, IFN-γ secreted by stimulated MLN cells and IL-12/23 p40 secreted by colon tissue fragment cultures, we used commercially available monoclonal anti-mouse TNF (Cat.No. 555268), IFN-γ (Cat.No. 551866), and IL-12/23 p40 (Cat.No. 555165)- specific capture and detection reagents (BD Biosciences Pharmingen) in enzyme-linked immunosorbent assays (ELISAs) according to the manufacturer’s protocols with the following exceptions: 1) five washes were performed for all wash steps, and 2) for IL-12/23p40 ELISAs, we followed the TNF ELISA protocol, but used IL-12/23 capture and detection reagents. Cytokine levels were measured in duplicate supernatants and compared to standard curves generated using recombinant murine cytokines per the manufacturer’s instructions.
Statistical Analysis
Microarray data were analyzed as described above. All other data are presented as the mean ± SEM. P values were calculated using the unpaired, 2-tailed Student’s T-test.
RESULTS
The Intestinal Inflammatory Milieu Induces Stress Response Genes in a Commensal E. coli Strain
While the effects of bacterial products on intestinal immune responses are fairly well-characterized, the impact of inflammation on commensal bacterial function is poorly understood. We hypothesized that luminal bacteria adapt to chronic, immune-mediated colitis by altering the expression of genes in a manner that promotes bacterial survival in a hostile environment. To test this hypothesis, we monoassociated GF WT or IL-10−/− mice with the non-pathogenic, commensal murine strain E. coli NC101, which was previously shown to cause a Th1/Th17-mediated colitis in IL-10−/−, but not WT, mice20. Consistent with prior results, IL-10−/−, but not WT, mice had moderate histologic cecal inflammation 3 and 7 weeks post-colonization (Figure 1A). Similarly, innate immune responses, measured by spontaneous secretion of IL-12/23p40 from colonic explant cultures, were increased in IL-10−/− mice compared to WT mice colonized with NC101 (Figure 1B). Together, these data confirm that NC101 induces moderate chronic inflammation in monoassociated IL-10−/−, but not WT, mice.
Figure 1.
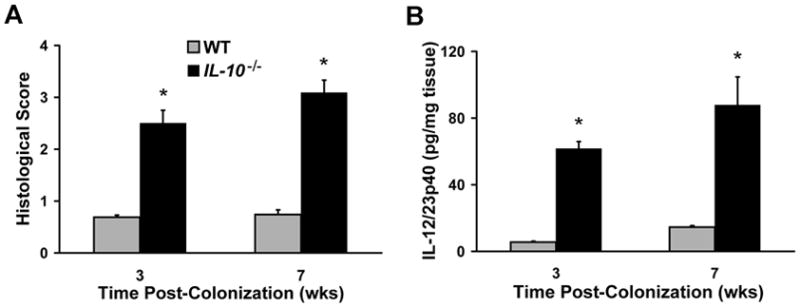
Spontaneous intestinal inflammation develops in E. coli NC101-monoassociated IL-10−/− mice. WT and IL-10−/− mice were monoassociated with E. coli NC101 for 3 and 7 weeks. Histological inflammation in the cecum (A) and spontaneous secretion of IL-12/23p40 from proximal colon explant cultures (B) was measured. Data are means + SEM, n=5 mice/group,*p<0.05 relative to WT.
We next performed transcriptional profiling of luminal bacteria isolated from the ceca from colitic IL-10−/− and healthy WT mice monoassociated with NC101 for 7 weeks using microarrays. Sixty-four genes were upregulated ≥1.5-fold and 150 genes were downregulated ≥2-fold in NC101 from IL-10−/− mice with colitis compared to NC101 from WT mice (Tables S1 & S2). In general, genes encoding enzymes of amino group and butanoate metabolism were differentially regulated and TCA cycle-associated genes were downregulated (Table S3). Interestingly, 27% of the identified upregulated genes are involved in E. coli stress responses and many of the genes are transcribed by alternative σ factors previously identified to control the E. coli stress response regulon (Table 1). Real-time PCR results of selected upregulated genes confirmed the microarray results (Figure S1). These data suggest that E. coli NC101 responds to chronic intestinal inflammation, in part, by upregulating stress response genes. For example, acid tolerance genes (gadAB, hdeAB, yhiX, evgA) are upregulated although pH of cecal contents is no different (data not shown). Furthermore, some of the most highly upregulated genes (ibpB, ibpA, oxyS) have been shown to protect bacteria from ROS21–24, which are increased in intestinal tissue of patients with IBD and animals with experimental colitis7, 8.
Table 1.
Selected genes upregulated in E. coli NC101 during experimental colitis.
| Gene | Fold Increase | Description | Alternative σ |
|---|---|---|---|
|
| |||
| ibpB | 7.3 | Heat shock protein | 32,54 |
| glpA | 6.9 | Glycerol-3-phosphate dehydrogenase, subunit A | |
| cspH | 6.2 | Cold shock-like protein | |
| glpC | 5.3 | Glycerol-3-phosphate dehydrogenase, subunit C | |
| gadA | 5.1 | Glutamate decarboxylase beta, acid tolerance | 38 |
| ibpA | 4.7 | Heat shock protein | 32 |
| gadB | 4.4 | Glutamate decarboxylase beta, acid tolerance | 38 |
| oxyS | 4.4 | Global regulatory RNA, oxidative damage | |
| ycfR | 4.0 | Hypothetical protein, biofilm formation | |
| ydgF | 3.6 | Hypothetical protein, putative chaperone | |
| hdeB | 3.2 | Hypothetical protein, acid resistance chaperone | 38 |
| glpD | 3.1 | Glycerol-3-phosphate dehydrogenase, subunit D | |
| evgA | 2.6 | Positive transcription regulator of gadE | |
| yhiX | 2.3 | Transcriptional regulator of gadE, gadB | 38 |
| inaA | 1.8 | pH-inducible protein involved in stress response | |
| rpoH | 1.6 | RNA polymerase sigma-32 factor | 38,54 |
| hslV | 1.5 | ATP-dependent protease; heat shock protein | 32 |
Expression of Genes Encoding the Small Heat Shock Proteins IbpAB in E. coli Decreases Bacterial Survival in vivo and Attenuates Experimental Colitis
Assuming bacteria upregulate stress responses during intestinal inflammation to survive, we hypothesized that deletion of specific stress response genes from bacteria may decrease their survival and virulence in the inflammatory milieu resulting in attenuated intestinal inflammation. Specifically, since ibpA and ibpB (ibpAB) are among the most highly upregulated stress response genes and have been shown to protect E. coli from ROS, we predicted that these genes enhance the survival and virulence of NC101 in the intestine of IL-10−/− mice with colitis. To test this hypothesis, we constructed a mutant strain of NC101 lacking ibpAB (NC101ΔibpAB) and measured growth rates and indicators of chronic, immune-mediated colitis in monoassociated WT and IL-10−/− mice. Interestingly, while in vitro growth of NC101 and NC101ΔibpAB was similar, luminal concentrations of NC101ΔibpAB were significantly greater than NC101 in monoassociated WT and IL-10−/− mice (Figure 2). We confirmed the absence of ibpAB expression in cecal bacteria from NC101ΔibpAB-monoassociated IL-10−/− mice using real-time PCR (Figure S2). These findings suggest that, contrary to our original hypothesis, expression of ibpAB in NC101 limits its growth in vivo independent of intestinal inflammation.
Figure 2.
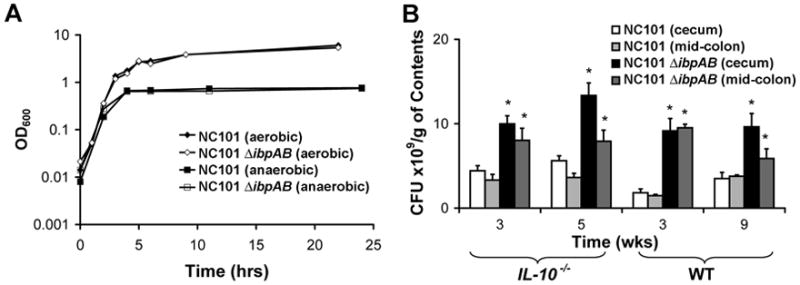
Survival of NC101 is attenuated relative to NC101ΔibpAB in vivo, but not in vitro. (A) NC101 or NC101ΔibpAB were grown in LB under aerobic or anaerobic conditions and bacterial density was measured using OD600. Data are presented as the mean+/−SEM, n=3 cultures/strain. (B) Luminal bacterial concentrations in monoassociated WT and IL-10−/− mice. Data are means + SEM, n=5–6 mice/group, *p<0.05 relative to NC101-monoassociated mice.
To determine whether in vivo survival differences between the two strains are associated with differences in their colitogenic potential in IL-10−/− mice, we quantified colonic inflammation in the two groups of mice. Increased bacterial density correlated with higher cumulative colitis scores in IL-10−/− mice monoassociated with NC101ΔibpAB compared to NC101 (Figures 3A&B).
Figure 3.

Expression of E. coli ibpAB is associated with decreased inflammatory responses in NC101-monoassociated IL-10−/− and WT mice. (A) Composite blinded histological inflammation scores of 4 colonic segments from IL-10−/− or WT mice. (B) Representative photomicrographs of cecum and mid-colon from IL-10−/− mice monoassociated for 5 weeks, 20x magnification. (C) Western blots of NC101 lysate using sera from 13 IL-10−/− mice monoassociated with the indicated bacteria for 5 weeks. (D) Spontaneous secretion of IL-12/23p40 in culture supernatants from proximal colon explants. (E) IFN-γ secretion by unseparated MLN cells stimulated ex vivo with NC101 lysate. Data are means + SEM, n=5–6 mice/group, *p<0.05 relative to NC101-monoassociated mice.
Since others have demonstrated that enhanced humoral immune responses to microbial, including E. coli, antigens in patients with Crohn’s disease correlates with a more aggressive disease course25, we questioned whether increased inflammation in NC101ΔibpAB- compared to NC101-monoassociated IL-10−/− mice was associated with increased levels of anti-E. coli antibodies. Indeed we found increased numbers and intensities of immunoreactive bands on Western blots of NC101 lysate probed with sera from NC101ΔibpABNC101-monoassociated IL-10−/− mice (Figure 3C).
IL-12 and IL-23 are produced by macrophages and dendritic cells when microbial products bind to Toll-like receptors (TLRs), and promote the differentiation or expansion of effector Th1 and Th17 cells, respectively, both of which participate in the development of chronic colitis26, 27. To assess the role of mucosal innate immune responses in potentiating colitis in NC101ΔibpAB-monoassociated IL-10−/− mice, we measured spontaneous secretion of IL-12/23p40 in colonic explant cultures. Consistent with increased histologic inflammation, colonic explants from NC101ΔibpAB-monoassociated IL-10−/− mice secreted more IL-12/23p40 than those from NC101-monoassociated mice suggesting activation of innate immune pathways (Figure 3D).
Since IL-12/23p40 effectively links the innate and the adaptive immune systems, we wanted to determine whether the increased production of IL-12/23p40 observed in NC101ΔibpAB-monoassociated mice is associated with enhanced bacterial antigen-specific adaptive immune responses in these mice. To test this, we measured IFN- production by NC101 lysate-stimulated mesenteric lymph node (MLN) cells obtained from NC101ΔibpAB- and NC101-monoassociated WT and IL-10−/− mice. Stimulated MLN cells from IL-10−/− mice colonized with NC101ΔibpAB secreted more IFN- compared to those from mice colonized with NC101 for both 3 and 5 weeks (Figure 3E). There were no significant differences in IFN-γ secretion by MLN cells from the two groups of animals when stimulated with lysates from an unrelated Gram positive bacterium, Enterococcus faecalis, and the amounts of secreted IFN-γ were negligible (data not shown). Importantly, in IL-10−/− mice monoassociated with a complemented strain of NC101ΔibpAB [NC101ΔibpAB(pGEN-MCSibpAB)] for 3 weeks, luminal bacterial concentrations, histologic inflammation, and IFN-γ secretion by stimulated MLN cells were similar to results obtained in NC101-monoassociated IL-10−/− mice (Figure S3). Expression of ibpAB in luminal E. coli from the NC101ΔibpAB(pGEN-MCSibpAB)-monoassociated mice was confirmed by real-time PCR (Figure S2). Taken together, these results suggest that, contrary to our original hypothesis, expression of ibpAB in E. coli is detrimental to luminal bacterial survival and attenuates innate, and adaptive mucosal immune responses in both IL-10−/− and WT mice. However, the mechanisms by which this occurs is unclear.
Decreased E. coli Survival Associated with ibpAB Expression Does Not Correlate with Reduced Histologic Inflammation at Early Time Points
Since our initial observations indicated that ibpAB expression in NC101 is associated with decreased bacterial growth and attenuated mucosal inflammation at later time points (≥3 weeks), we hypothesized that reduced mucosal inflammatory responses are due to impaired luminal bacterial survival in NC101-monoassociated IL-10−/− mice rather than a direct effect of ibpAB on the host immune system. To test this, we dual-associated IL-10−/− mice with equivalent numbers of NC101 and NC101ΔibpAB to determine whether host inflammatory responses correlated with the proportion of luminal NC101ΔibpAB or total bacteria. At 5 weeks, there were similar numbers of luminal bacteria in NC101ΔibpAB-monoassociated and dual-associated mice, and 82 ± 4.6% of cecal bacteria in dual-associated mice were NC101ΔibpAB (Figure S4). Histologic inflammation and IL-12/23p40 secretion from colonic explants were equivalent in the two groups of mice suggesting that host inflammatory responses correlate better with total bacteria number than with proportion of ibpAB-expressing bacteria (Figure S4). However, since the vast majority of bacteria in the dual-associated mice were NC101ΔibpAB, we cannot exclude the possibility that ibpAB expression may directly influence host immune responses independent of luminal bacteria numbers.
Next, we monoassociated IL-10−/− mice with NC101 or NC101ΔibpAB for shorter times (3 and 10 days) to determine whether differences in luminal bacterial concentrations at these early time points correlate with differences in the degree of mucosal inflammation as they had at later time points. NC101ΔibpAB grew to higher densities than NC101 in cecal and transverse colon contents as early as 3 days after colonization, and this difference persisted through five weeks in cecal contents, but decreased in magnitude in transverse colon contents (Figures 4A and B). However, at 3 days, the difference in IL-12/23p40 secretion from colonic explants, a measure of early inflammatory responses, and IFN- production by NC101 lysate-stimulated MLN cells from the two groups of animals were still relatively small, but statistically significant (Figures 4C & D). Moreover, there was no difference in histologic inflammation at 3 and 10 days (Figure 4E). Thus, peak differences in luminal bacterial concentrations between NC101 and NC101ΔibpAB occurred soon after monoassociation, at a time when differences in measures of inflammation were minimal. Though changes in inflammation may lag alterations in luminal bacterial concentrations, these data suggest that the disparity in inflammation noted between NC101- and NC101ΔibpAB-monoassociated IL-10−/− mice may not be entirely dependent on differences in luminal bacterial concentrations of the two strains.
Figure 4.
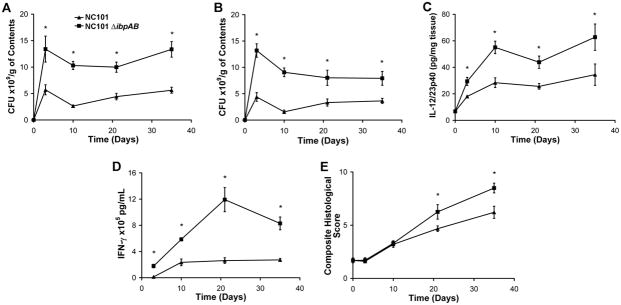
Attenuated pro-inflammatory responses in IL-10−/− mice monoassociated with NC101 compared to NC101ΔibpAB are not proportional to differences in luminal bacterial concentrations at early time points. Luminal bacterial concentrations in the cecum (A) or mid-colon (B) from IL-10−/− mice monoassociated with NC101 or NC101ΔibpAB. (C) Spontaneous IL-12/23p40 secretion in proximal colon explant cultures. (D) IFN-γ secretion by unseparated MLN cells stimulated ex vivo with NC101 lysate. (E) Blinded composite histological inflammation scores of 4 colonic segments. Data are means +/− SEM, n=5–6 mice/group, *p<0.05 relative to NC101-monoassociated mice.
IbpAB Expression is Associated with Impaired Survival of E. coli NC101 in Murine Macrophages
Since decreased growth of luminal NC101 compared to NC101ΔibpAB did not completely correlate with decreased measures of inflammation, we wanted to determine whether ibpAB expression affected bacterial clearance by innate immune cells. Others have shown that adherent, invasive E. coli strains isolated from CD patients survive and replicate within the phagolysosome of murine macrophages and that intracellular persistence in macrophages is associated with constitutive secretion of TNF-α, a pro-inflammatory cytokine important in the pathogenesis of CD28. We therefore hypothesized that ibpAB expression may reduce intestinal inflammation in part by decreasing survival of NC101 within mucosal macrophages resulting in decreased TNF secretion. To test this hypothesis, we performed gentamicin protection assays using murine bone marrow-derived macrophages (BMDM) to determine whether NC101 was more vulnerable to killing within macrophages compared to NC101ΔibpAB, and if so, whether this correlated with decreased production of TNF by macrophages. We found that expression of ibpAB influenced initial uptake of NC101 in IL-10−/−, but not WT, macrophages by reducing the number of internalized bacteria at 1 hr (Figure 5A). However, once internalized, NC101ΔibpAB survived better in macrophages from both mouse backgrounds at 24 hrs than did NC101 (Figure 5A & B). Furthermore, while there was no significant difference in the survival ratio of NC101ΔibpAB between WT and IL-10−/− macrophages, NC101 had a significantly lower survival ratio in IL-10−/− versus WT macrophages. These data indicate that ibpAB expression impairs intracellular survival of NC101 in macrophages, and that this effect is more pronounced in the absence of IL-10. In addition, while NC101-infected WT and IL-10−/− macrophages secreted more TNF during the first 3 hours of infection, they produced significantly less TNF from 3–24 hours compared to NC101ΔibpAB-infected macrophages (Figure 5C). Thus, decreased long-term intracellular survival of NC101 in macrophages compared to NC101ΔibpAB correlates with reduced secretion of TNF at later time-points in vitro. Whether this effect occurs in intestinal lamina propria macrophages in vivo and contributes to the decreased histologic inflammation noted in NC101-monoassociated IL-10−/− mice remains to be determined.
Figure 5.
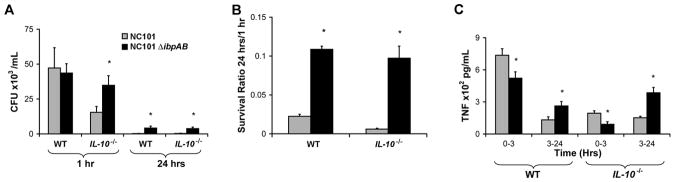
Expression of ibpAB by E. coli NC101 is associated with attenuated survival within macrophages and decreased TNF secretion. (A) Intracellular survival of NC101 and NC101ΔibpAB in WT and IL-10−/− bone marrow-derived macrophages. (B) Survival ratios (24h/1h) of intracellular bacteria. (C) TNF secretion by NC101- or NC101ΔibpAB-infected macrophages. Data are means + SEM, 3 wells/condition, representative of results from 3 different mice, *p<0.05 relative to NC101.
DISCUSSION
To the best of our knowledge, this is the first published study of commensal bacterial gene regulation in response to chronic, T-cell-mediated intestinal inflammation. We demonstrated that the E. coli stress response regulon, including the small heat shock proteins IbpAB, is upregulated in the IL-10−/− mouse model of IBD. Contrary to our initial hypothesis, ibpAB expression inhibits NC101 growth within the intestinal lumen and survival within primary macrophages. This is evident from our studies using ibpAB-deficient E. coli NC101, which showed enhanced luminal bacterial growth and inflammatory host responses in vivo, the ability of ibpAB-deficient E. coli NC101 to outcompete parental NC101 in dual-association studies, and increased bacterial survival within macrophages and elevated TNF secretion in vitro. The reasons why bacteria would upregulate genes in response to inflammation that decrease their survival in vivo are currently unclear.
The paradox could be explained by limitations of our experimental model. First, we are studying E. coli in an un-natural, monoassociated system without the influences of other commensal microbes, which could affect E. coli gene responses to intestinal inflammation. Second, we have only studied gene expression levels in luminal bacteria whereas the results from mucosally-associated bacteria may be quite different. Third, we inferred that ibpAB expression in luminal E. coli is detrimental to the bacterium because its complete absence resulted in increased growth. However, there could be a range of ibpAB expression levels, below which fitness of the bacterium is inversely proportional to expression and above which fitness is proportional to expression. Fourth, the complete absence of IbpAB may cause alterations in expression of other genes, which in turn results in the observed phenotype. However, we detected no differences in mRNA levels of genes adjacent to ibpAB in NC101ΔibpAB compared to NC101 (data not shown).
While the functions of ibpAB in cultured bacteria are beginning to be elucidated, little is known about their role in E. coli within the intestinal lumen. Others have shown that IbpAB promote bacterial survival in vitro by functioning as molecular chaperones in conditions of heat shock, enhancing resistance to oxidative stress, and facilitating biofilm formation23, 29, 30. For example, oxidized proteins accumulate in ibpAB-deficient E. coli exposed to copper ions, suggesting that ibpAB protects proteins from copper-induced oxidative damage23. However, based on our results, we propose that E. coli ibpAB function as negative survival factors in the colon, though the mechanisms by which this occurs are unclear. We speculate that IbpAB preserve the function of proteins in certain metabolic pathways that are less-efficient at extracting energy from dietary substances. This is supported by evidence that ibpAB can protect metabolic enzymes from oxidative damage22.
Although we have shown that ibpAB are upregulated in luminal E.coli isolated from animals with experimental colitis compared to healthy controls, the host-derived stimuli and their cognate bacterial pathways that enhance ibpAB expression are unknown. In vitro studies suggest that both ibpA and ibpB can be transcribed as an operon from a common promoter that binds the alternative σ factor, σ32, whereas ibpB also can be independently transcribed from a separate promoter located between the two genes, which binds σ54, a different alternative σ factor, 31. Because we noted that both ibpA and ibpB were upregulated during intestinal inflammation, the major stimulus is probably acting via σ32. However, since the core temperature of the mice is probably minimally changed by colitis (though we did not measure this), we doubt heat is causing increased ibpAB expression. More likely, the inflamed intestine produces factors, such as reactive oxygen species, which cause unfolded proteins to accumulate in the cytoplasm of luminal bacteria resulting in upregulation of σ32-dependent ibpAB expression. Indeed, others have shown that reactive oxygen species are increased in intestinal inflammation and that ibpAB protects E. coli from oxidative damage7, 8, 21. Additional studies to identify the factors in the inflamed intestine that induce ibpAB in luminal E. coli are needed.
In summary, we show that a commensal E. coli strain upregulates a broad set of stress response genes, particularly ibpAB, in response to intestinal inflammation in monoassociated IL-10−/− mice. Furthermore, contrary to our original hypothesis, we provide evidence that ibpAB expression in E. coli is associated with reduced survival of luminal and intra-macrophage bacteria and increased host pro-inflammatory immune responses. These data suggest that commensal intestinal microbes are not passive bystanders during intestinal inflammation, but dynamically react to host-derived factors, even if it ultimately proves detrimental to bacterial survival in the gut. Further studies of this important host-microbial dialogue may provide clues to the pathogenesis of human IBD and potentially offer novel therapeutic targets.
Supplementary Material
Acknowledgments
We thank Dr. Virginia Miller for her expert advice and critical review of the manuscript and Drs. M. Chelsea Lane and Kim Walker for their technical help creating the mutant and complemented strains of E. coli NC101. We acknowledge Dr. Michael Vernon at the UNC Functional Genomics Core for performing the microarray experiments, and the Gnotobiotic, Histology, and Immunotechnology Cores at the UNC Center for Gastrointestinal Biology and Disease (supported by NIH P30DK34987). This work was funded by grants from the UNC Holderness Distinguished Medical Scholars Program (L.G.P.), NIH K08DK087896 (J.J.H.), Crohn’s and Colitis Foundation of America (CCFA) Career Development Award (J.J.H.), Center for Gastrointestinal Biology and Disease Pilot Feasibility Grant to J.J.H. (NIH P30DK34987, Robert S. Sandler, PI), NIH R01DK53347(RBS), NIH P40RR18803(RBS), CCFA Gnotobiotic Animal Facility(RBS), and NIH/NCI 1U54CA121852-01A1 National Center for the Multiscale Analysis of Genomic and Cellular Networks(Y.A.L.).
Abbreviations
- BHI
brain heart infusion
- BMDM
bone marrow-derived macrophages
- CD
Crohn’s disease
- ELISA
enzyme-linked immunosorbent assay
- FBS
fetal bovine serum
- GF
germ free
- IBD
inflammatory bowel diseases
- LB
Luria-Bertani
- MLN
mesenteric lymph node
- PCR
polymerase chain reaction
- SPF
specific pathogen free
- TBS
Tris-buffered saline
- WT
wild-type
Footnotes
Author Contributions:
L.G.P., study concept and design; data acquisition, analysis, and interpretation; drafting of manuscript; obtained funding
T.F., study concept and design; data acquisition, analysis, and interpretation; critical revision of manuscript
S.T., study concept and design, data acquisition, analysis, and interpretation, critical revision of manuscript
Y.L., data analysis and interpretation
Y.A.L., data analysis and interpretation; obtained funding; critical revision of manuscript
R.B.S., study concept and design; critical revision of manuscript for important intellectual content; study supervision
J.J.H., study concept and design; data acquisition, analysis, and interpretation; drafting of manuscript; obtained funding; critical revision of manuscript for important intellectual content; study supervision
Disclosure Statement: The authors have no conflicts of interest to disclose.
References
- 1.Hooper LV, Xu J, Falk PG, et al. A molecular sensor that allows a gut commensal to control its nutrient foundation in a competitive ecosystem. Proc Natl Acad Sci U S A. 1999;96:9833–8. doi: 10.1073/pnas.96.17.9833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peterson DA, McNulty NP, Guruge JL, et al. IgA response to symbiotic bacteria as a mediator of gut homeostasis. Cell Host Microbe. 2007;2:328–39. doi: 10.1016/j.chom.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 3.Sonnenburg JL, Xu J, Leip DD, et al. Glycan foraging in vivo by an intestine-adapted bacterial symbiont. Science. 2005;307:1955–9. doi: 10.1126/science.1109051. [DOI] [PubMed] [Google Scholar]
- 4.Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology. 2008;134:577–94. doi: 10.1053/j.gastro.2007.11.059. [DOI] [PubMed] [Google Scholar]
- 5.Sellon RK, Tonkonogy S, Schultz M, et al. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun. 1998;66:5224–31. doi: 10.1128/iai.66.11.5224-5231.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mombaerts P, Mizoguchi E, Grusby MJ, et al. Spontaneous development of inflammatory bowel disease in T cell receptor mutant mice. Cell. 1993;75:274–82. doi: 10.1016/0092-8674(93)80069-q. [DOI] [PubMed] [Google Scholar]
- 7.Carroll IM, Andrus JM, Bruno-Barcena JM, et al. Anti-inflammatory properties of Lactobacillus gasseri expressing manganese superoxide dismutase using the interleukin 10-deficient mouse model of colitis. Am J Physiol Gastrointest Liver Physiol. 2007;293:G729–38. doi: 10.1152/ajpgi.00132.2007. [DOI] [PubMed] [Google Scholar]
- 8.Keshavarzian A, Banan A, Farhadi A, et al. Increases in free radicals and cytoskeletal protein oxidation and nitration in the colon of patients with inflammatory bowel disease. Gut. 2003;52:720–8. doi: 10.1136/gut.52.5.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kotlowski R, Bernstein CN, Sepehri S, et al. High prevalence of Escherichia coli belonging to the B2+D phylogenetic group in inflammatory bowel disease. Gut. 2007;56:669–75. doi: 10.1136/gut.2006.099796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frank DN, St Amand AL, Feldman RA, et al. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A. 2007;104:13780–5. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Darfeuille-Michaud A, Neut C, Barnich N, et al. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn’s disease. Gastroenterology. 1998;115:1405–13. doi: 10.1016/s0016-5085(98)70019-8. [DOI] [PubMed] [Google Scholar]
- 12.Kim SC, Tonkonogy SL, Albright CA, et al. Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology. 2005;128:891–906. doi: 10.1053/j.gastro.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 13.Hansen JJ, Gulati A, Sartor RB. The role of mucosal immunity and host genetics in defining intestinal commensal bacteria. Curr Opin Gastroenterol. 2010:26. doi: 10.1097/MOG.0b013e32833f1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stanley ER, Heard PM. Factors regulating macrophage production and growth. Purification and some properties of the colony stimulating factor from medium conditioned by mouse L cells. J Biol Chem. 1977;252:4305–12. [PubMed] [Google Scholar]
- 15.Lutz MB, Kukutsch N, Ogilvie AL, et al. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods. 1999;223:77–92. doi: 10.1016/s0022-1759(98)00204-x. [DOI] [PubMed] [Google Scholar]
- 16.Rey FE, Faith JJ, Bain J, et al. Dissecting the in vivo metabolic potential of two human gut acetogens. J Biol Chem. 2010;285:22082–90. doi: 10.1074/jbc.M110.117713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gama-Castro S, Jimenez-Jacinto V, Peralta-Gil M, et al. RegulonDB (version 6. 0): gene regulation model of Escherichia coli K-12 beyond transcription, active (experimental) annotated promoters and Textpresso navigation. Nucleic Acids Res. 2008;36:D120–4. doi: 10.1093/nar/gkm994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoentjen F, Tonkonogy SL, Liu B, et al. Adoptive transfer of nontransgenic mesenteric lymph node cells induces colitis in athymic HLA-B27 transgenic nude rats. 2006;143:474. doi: 10.1111/j.1365-2249.2006.03013.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Darfeuille-Michaud A, Boudeau J, Bulois P, et al. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology. 2004;127:412–21. doi: 10.1053/j.gastro.2004.04.061. [DOI] [PubMed] [Google Scholar]
- 20.Kim SC, Tonkonogy SL, Karrasch T, et al. Dual-association of gnotobiotic IL-10−/ − mice with 2 nonpathogenic commensal bacteria induces aggressive pancolitis. Inflamm Bowel Dis. 2007;13:1457–66. doi: 10.1002/ibd.20246. [DOI] [PubMed] [Google Scholar]
- 21.Kitagawa M, Matsumura Y, Tsuchido T. Small heat shock proteins, IbpA and IbpB, are involved in resistances to heat and superoxide stresses in Escherichia coli. FEMS Microbiol Lett. 2000;184:165–71. doi: 10.1111/j.1574-6968.2000.tb09009.x. [DOI] [PubMed] [Google Scholar]
- 22.Kitagawa M, Miyakawa M, Matsumura Y, et al. Escherichia coli small heat shock proteins, IbpA and IbpB, protect enzymes from inactivation by heat and oxidants. Eur J Biochem. 2002;269:2907–17. doi: 10.1046/j.1432-1033.2002.02958.x. [DOI] [PubMed] [Google Scholar]
- 23.Matuszewska E, Kwiatkowska J, Kuczynska-Wisnik D, et al. Escherichia coli heat-shock proteins IbpA/B are involved in resistance to oxidative stress induced by copper. Microbiology. 2008;154:1739–47. doi: 10.1099/mic.0.2007/014696-0. [DOI] [PubMed] [Google Scholar]
- 24.Altuvia S, Weinstein-Fischer D, Zhang A, et al. A small, stable RNA induced by oxidative stress: role as a pleiotropic regulator and antimutator. Cell. 1997;90:43–53. doi: 10.1016/s0092-8674(00)80312-8. [DOI] [PubMed] [Google Scholar]
- 25.Arnott ID, Landers CJ, Nimmo EJ, et al. Sero-reactivity to microbial components in Crohn’s disease is associated with disease severity and progression, but not NOD2/CARD15 genotype. Am J Gastroenterol. 2004;99:2376–84. doi: 10.1111/j.1572-0241.2004.40417.x. [DOI] [PubMed] [Google Scholar]
- 26.Yen D, Cheung J, Scheerens H, et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest. 2006;116:1310–6. doi: 10.1172/JCI21404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davidson NJ, Hudak SA, Lesley RE, et al. IL-12, but not IFN-gamma, plays a major role in sustaining the chronic phase of colitis in IL-10-deficient mice. J Immunol. 1998;161:3143–9. [PubMed] [Google Scholar]
- 28.Glasser AL, Boudeau J, Barnich N, et al. Adherent invasive Escherichia coli strains from patients with Crohn’s disease survive and replicate within macrophages without inducing host cell death. Infect Immun. 2001;69:5529–37. doi: 10.1128/IAI.69.9.5529-5537.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matuszewska M, Kuczynska-Wisnik D, Laskowska E, et al. The small heat shock protein IbpA of Escherichia coli cooperates with IbpB in stabilization of thermally aggregated proteins in a disaggregation competent state. J Biol Chem. 2005;280:12292–8. doi: 10.1074/jbc.M412706200. [DOI] [PubMed] [Google Scholar]
- 30.Kuczynska-Wisnik D, Matuszewska E, Laskowska E. Escherichia coli heat-shock proteins IbpA and IbpB affect biofilm formation by influencing the level of extracellular indole. Microbiology. 156:148–57. doi: 10.1099/mic.0.032334-0. [DOI] [PubMed] [Google Scholar]
- 31.Kuczynska-Wisnik D, Laskowska E, Taylor A. Transcription of the ibpB heat-shock gene is under control of sigma(32)- and sigma(54)-promoters, a third regulon of heat-shock response. Biochem Biophys Res Commun. 2001;284:57–64. doi: 10.1006/bbrc.2001.4926. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.