

Abstract
Glucocorticosteroid hormones, including prednisone and dexamethasone (Dex), have been used to treat lymphoid malignancies for many years because they readily induce apoptosis in immature lymphocytes lacking Bcl-2. However, elevated expression of the anti-apoptotic protein Bcl-2 inhibits apoptosis and contributes to glucocorticoid resistance. Using the Bcl-2-negative WEHI7.2 lymphoma line as an experimental model, we found that Dex not only induces apoptosis but also induces autophagy. The caspase inhibitor Z-VAD-fmk inhibited apoptosis but not autophagy in Dex-treated cells. Bcl-2 overexpression inhibited Dex-induced apoptosis even more potently than Z-VAD-fmk and, contrary to previous reports, Bcl-2 neither interacted with Beclin-1 nor inhibited autophagy. Rather, Bcl-2 overexpression facilitated detection of Dex-induced autophagy by both steady state methods and flux measurements, ostensibly due to apoptosis inhibition. Autophagy contributed to prolonged survival of Bcl-2-positive lymphoma cells following Dex treatment, as survival was reduced when autophagy was inhibited by 3-methyladenine. These findings emphasize the important interplay between apoptosis and autophagy and suggest a novel mechanism by which Bcl-2, which is frequently elevated in lymphoid malignancies, contributes to glucocorticoid resistance and survival of lymphoma cells.
Key words: apoptosis, autophagy, lymphocyte, lymphoma, dexamethasone, glucocorticoid, glucocorticosteroid, Bcl-2
Introduction
Glucocorticosteroid hormones have a wide range of physiological actions and play critical roles in development and metabolism. Glucocorticoids are particularly important in the immune system, especially in the regulation of immune system development and homeostasis. In the thymus glucocorticoids have both positive and negative actions.1 At physiological concentrations glucocorticoids promote the survival and proliferation of immature T cells by upregulating cytokine receptors; but at pharmacological concentrations glucocorticoids induce apoptosis. Glucocorticoid-induced apoptosis is mediated through the glucocorticoid receptor, a ligand regulated transcription factor, and involves induction of a variety of genes that contribute to cell death, including the gene encoding the pro-apoptotic protein Bim.2,3 In addition to apoptosis-related genes, gene expression profiling has uncovered glucocorticoid regulation of genes involved in cellular metabolism, including genes that regulate glucose homeostasis and respond to ER stress.2,4-7 This is not surprising since it has been known for over forty years that glucocorticoids inhibit glucose uptake and glycolysis in thymocytes.8,9 These well-documented effects on lymphocyte metabolism suggested to us that glucocorticoids might induce macroautophagy (hereafter referred to as autophagy), as well as apoptosis.
Metabolic stress, induced by nutrient deprivation and growth factor withdrawal, induces autophagy. Autophagy is a highly conserved process involved in protein degradation and maintenance of cellular homeostasis in yeast, plants and mammals.10,11 Through this process, cells are able to remain viable during periods of metabolic stress by using their own proteins and organelles as substrates for energy production, although sustained autophagy ultimately leads to cell death. Hence, autophagy is often referred to as Type II cell death, with apoptosis referred to as Type I cell death.12 Like apoptosis, autophagy is a genetically programmed process and the genes encoding autophagy are highly conserved from yeast to mammals.10,11 An energy dependent multi-step process, autophagy begins with the formation of a double membrane structure, known as the autophagosome, thought to be derived from the endoplasmic reticulum. Autophagosomes sequester organelles and cytoplasmic materials, ultimately fusing with lysosomes to form autolysosomes. Lysosomal hydrolases then degrade the intracellular material for energy.13
Many highly conserved genes are involved in mediating autophagy, including Beclin 1 (yeast homologue Atg 6) and the microtubule-associated protein 1 light chain 3 (LC3, yeast homologue Atg 8).14 Following synthesis, LC3 is converted into a proteolytically processed form, LC3 I, which is cytoplasmic in location. During the process of autophagy, LC3 I is modified by conjugation to phosphatidylethanolamine, giving rise to LC3 II.15 LC3 II plays an essential role in autophagosome formation, associating with the inner and outer membrane of the autophagosome. Increased levels of LC3 II are indicative of the extent of autophagosome formation in the cell; therefore LC3 II is commonly used as a marker of autophagy.16,17
Because of the known glucocorticoid-mediated metabolic effects in lymphocytes, the present study was undertaken to determine if glucocorticoids induce autophagy. At present, the only indication that steroid hormones induce autophagy comes from work in lower organisms, including Anguilla rostrata (North American eels) and in the fat body of insects.18,19 Also, hydrocortisone has been reported to stimulate an autophagic process in newborn rat hepatocytes.20 In the present study, we mainly employed the WEHI7.2 murine T cell lymphoma line. The anti-apoptotic protein Bcl-2 is virtually absent in WEHI7.2 cells; hence, these cells readily undergo apoptosis following exposure to the glucocorticoid hormone dexamethasone (Dex) and represent a good experimental system for investigating glucocorticoid-induced apoptosis.2 Moreover, Tome et al,21 have shown that glucocorticoid treatment inhibits glucose uptake and represses glycolysis in WEHI7.2 cells, reflecting similar metabolic alterations observed in primary thymocytes.8,9 As reported here, we discovered that Dex induces autophagy in WEHI7.2 cells, based on multiple complementary detection methods. Moreover, although it was reported previously that Bcl-2 inhibits autophagy,22,23 we found that detection of Dex-induced autophagy is enhanced by Bcl-2 overexpression, ostensibly due to inhibition of Dex-induced apoptosis. These findings emphasize the important interplay between apoptosis and autophagy and have important implications regarding the therapeutic actions of glucocorticoids in the wide variety of lymphoid malignancies associated with elevated Bcl-2 expression.
Results
Experiments reported here were performed not only in WEHI7.2 cells lacking Bcl-2 (either wild type WEHI7.2 cells or stable control transfectants), but also in stable WEHI7.2 transfectants that overexpress Bcl-2. Although the Bcl-2-positive and -negative clones employed in these experiments have already been extensively characterized,24 the resistance of Bcl-2-positive clones to apoptosis is highly relevant to the present study and is therefore documented in Figure 1. In these experiments, Bcl-2-negative and -positive cells were treated with Dex for various periods of time and then analyzed by flow cytometry to determine the percentage of cells undergoing apoptosis, based on sub-G1 DNA content (Fig. 1A). The findings confirm that Dex induces apoptosis in Bcl-2-negative cells, readily detected at 24–30 hr after Dex addition, and that Bcl-2 overexpression inhibits Dex-induced apoptosis. Notably, Bcl-2 preserved cell survival for up to 72 hr following Dex addition. Extended time points were not tested in Bcl-2-negative cells due to the large amount of cell death that ensues after Dex treatment for more than 24–30 hr. The anti-apoptotic activity of Bcl-2 in this experimental system was further confirmed by measuring the activation of caspase-3 (Fig. 1B).
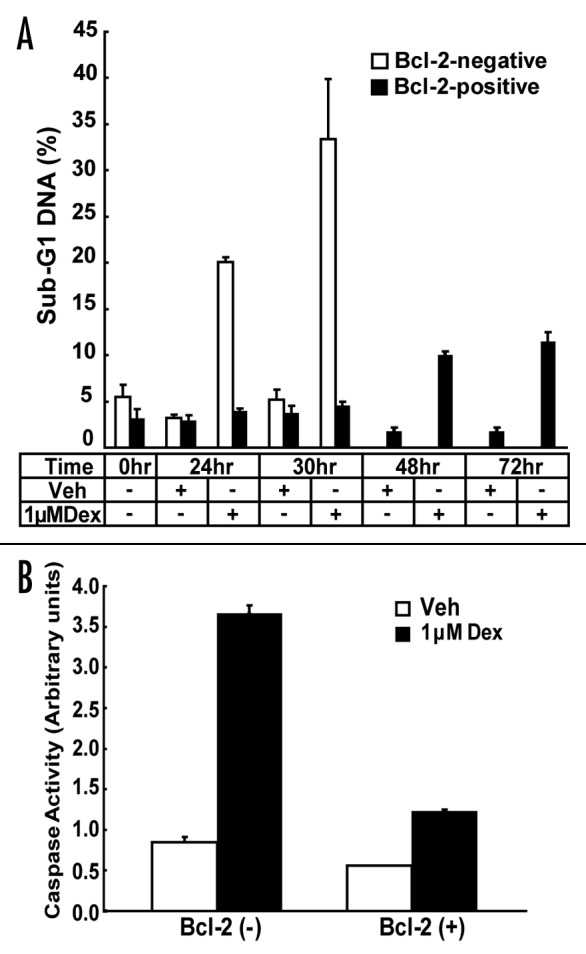
Figure 1. Bcl-2 inhibits Dex-induced apoptosis in WEHI7.2 cells. (A) The percentage of propidium iodide-stained cells with sub-G1 DNA content, indicative of apoptosis, following Dex treatment was measured by flow cytometry and represented in the bar graph (mean ± SE; n = 5). Bcl-2-negative and -positive cells were treated with vehicle or 1 μM Dex for up to 30 hr and Bcl-2-positive cells were treated for up to 72 hr. (B) Caspase 3 activity was measured following 1 μM Dex treatment for 24 hr in Bcl-2-negative and -positive cells (mean ± SE; n = 2).
The conversion of 18 kD LC3 I to the 16 kD LC3 II correlates with autophagy induction.15,17 Therefore, LC3 immunoblotting was used to test if Dex induces autophagy in WEHI7.2 cells. Serum reduction was employed as a positive control, since serum reduction is well known to trigger autophagy.25 Only LC3 I was detected when untreated cells were cultured in 10% serum (either Bcl-2-negative or -positive) (Fig. 2A, lanes 5 and 6), whereas both LC3 I and II were detected when serum was reduced (Fig. 2A, lanes 1 and 2). Dex treatment for 24 hr did not appear to elevate levels of LC3 II in either Bcl-2-negative or -positive cells cultured in 10% serum (Fig. 2A, lanes 7 and 8), although a discernable elevation of LC3 II was detected when serum-deprived cells were treated for the same time period with Dex (Fig. 2A, lanes 3 and 4). These findings suggested that Dex does not induce autophagy when WEHI7.2 cells are treated for only 24 hr with Dex under optimal culture conditions (i.e., 10% serum). However, when Bcl-2-positive cells were treated with Dex for longer periods of time, conversion of LC3 I to LC3 II was readily detected (Fig. 2B). The induction of autophagy in Bcl-2-positive cells was dose-related, being readily detected after 48 hr treatment with either 0.1 or 1 μM Dex, but not lower concentrations of Dex (Fig. 2C). Bcl-2-negative cells were not treated for longer than 24 hr with Dex because of the marked degree of cell death that ensues following more prolonged Dex treatment.
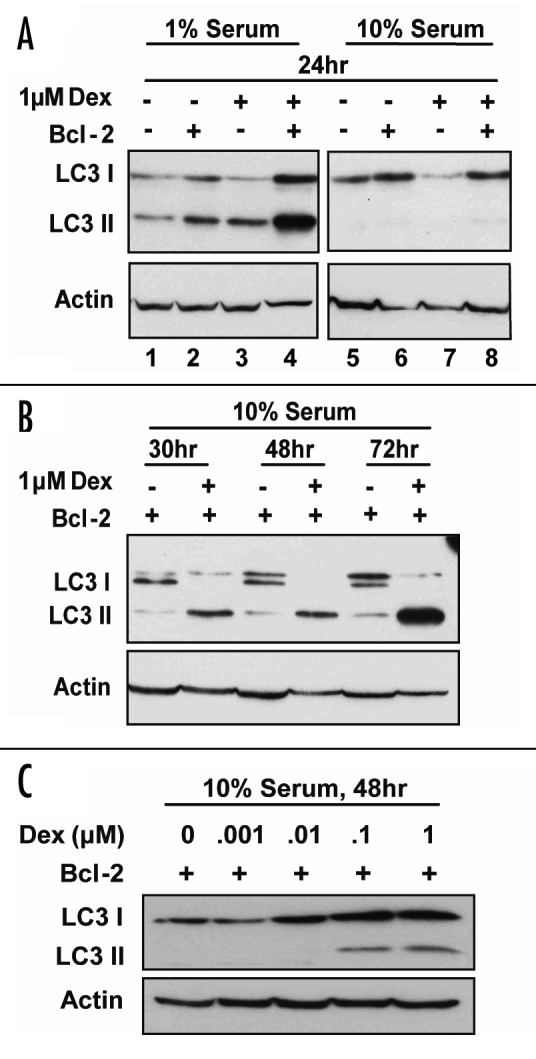
Figure 2. Dex induces LC3 II accumulation in Bcl-2-positive cells. Autophagy was detected by immunoblotting based on conversion of LC3 I to LC3 II. Shown are immunoblots representative of multiple experiments. (A) Bcl-2-positive and -negative cells were treated with either vehicle or 1 μM Dex for 24 hr under normal culture conditions (10% serum) or under low serum conditions (1% serum). Results confirm that serum deprivation induces autophagy, but suggest that Dex treatment for 24 hr may not induce autophagy. (B) Bcl-2-positive cells were treated with vehicle or 1 μM Dex for 30 to 72 hr and immunoblots readily detect conversion of LC3 I to LC3 II, indicating that prolonged treatment of Bcl-2-positive cells with Dex induces autophagy. (C) Bcl-2-positive cells were treated for 48 hr with vehicle or 0.001 to 1 μM Dex, indicating that 0.1 or 1 μM Dex induces autophagy but that lower concentrations of Dex do not.
The immunoblotting method employed in Figure 2, which does not include use of lysosomal inhibitors, constitutes a steady state measurement.16 To confirm that Dex induces autophagy in Bcl-2-positive cells, two types of flux measurements were also employed. The first was to measure the effect of Dex treatment on the degradation rate of 14C-valine-labeled proteins, since autophagy is the major mechanism for the degradation of long-lived proteins in cells.16,17 There was a significant increase in protein degradation following Dex treatment for 30, 48 and 72 hr (p < 0.01), supporting the conclusion that Dex induces autophagy in Bcl-2-positive cells (Fig. 3A). The second flux measurement involved time-lapse imaging of GFP-LC3 to monitor the formation of autophagosomes and subsequent degradation of the LC3 portion of the chimera. Bcl-2-positive cells that had been transiently transfected with GFP-LC3 were treated with 1 μM Dex and time-lapse images were collected at 30 min intervals over a 20 hr period. Selected frames tracking two representative cells throughout the entire movie are shown in Figure 3B and the full time course is included as a movie in supplemental material. Note that lymphocytes adhere only loosely to poly-L-lysine-coated -coverslips and hence the cellular movement observed in this movie is to be expected. Because LC3 I is cytoplasmic, GFP-LC3 was initially observed in a diffuse cytoplasmic pattern. Over time following Dex treatment, GFP-LC3 took on a punctate appearance, corresponding to the incorporation of LC3 in autophagosome membranes, evident in the selected movie panels shown in Figure 3B. Examination of the online movie provides additional information, as follows. First, the coalescence of GFP-LC3 into a punctate pattern is evident as early as 4 hr after Dex addition and further increases over time. Also, in the final movie frames, GFP is seen to switch back from punctate to diffuse, suggesting that the LC3 was degraded, releasing free GFP back into the cytoplasm and thus bringing the autophagic process to completion (for review of autophagic flux process see ref. 16).
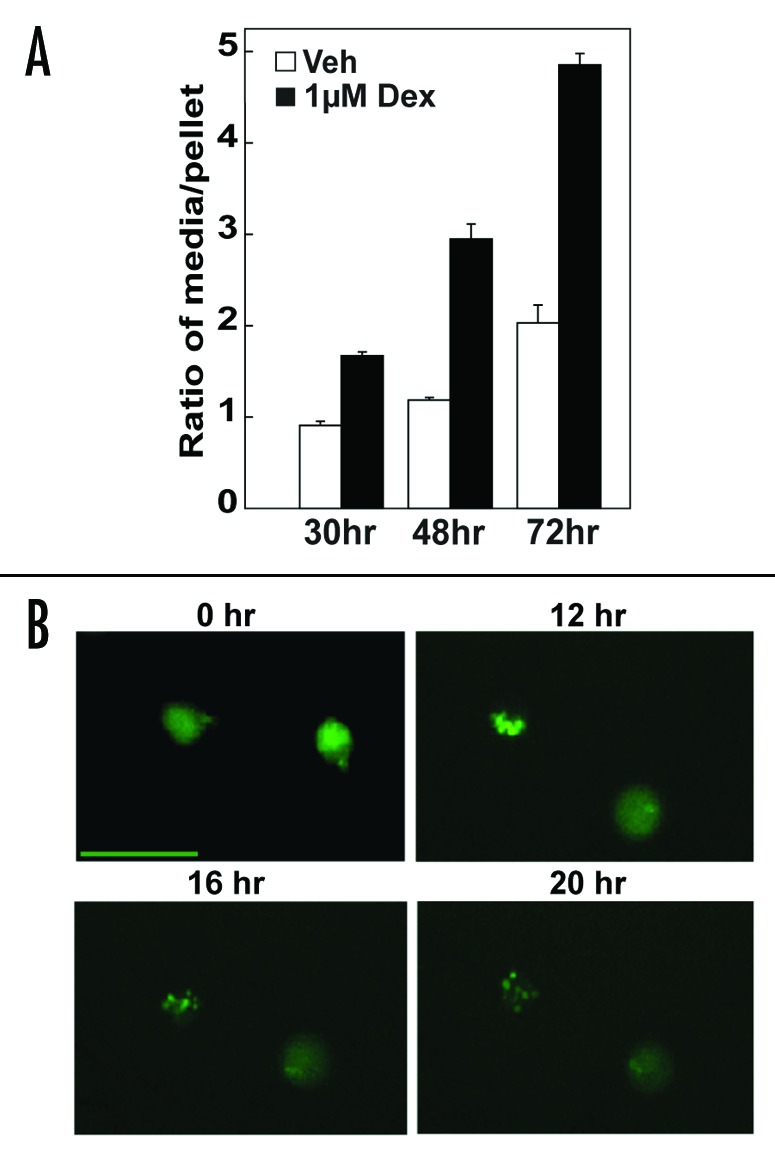
Figure 3. Flux measurements confirm the induction of autophagy by Dex in Bcl-2-positive cells. (A) Induction of protein degradation by Dex in Bcl-2-positive cells. Cells were labeled with 14C-valine for 24 hr, washed and then treated with vehicle or 1 μM Dex for the indicated times. Trichloroacetic acid precipitates (pellet) and supernatants (media) were dissolved and the amount of protein degradation was calculated as the ratio of cpm from the media to the cpm of the pellet. There was a significant (p < 0.01) increase in protein degradation in the Dex treated versus the vehicle treated samples at each time point. Symbols represent mean ± SE, n = 3 (B) GFP-LC3 accumulation in autophagosomes induced by Dex in Bcl-2-positive cells. Cells were transfected with GFP-LC3 and conversion of GFP-LC3 from a diffuse to punctate pattern, indicative of autophagosome formation, was recorded by time-lapse microscopy at 30 min intervals over a 20 hr period following the addition of 1 μM Dex. Images taken at the indicated times are representative of findings in two separate experiments. Scale bar is 20 microns.
Based on these findings, we conclude that Dex induces autophagy in Bcl-2-positive cells. It had been suggested previously that Bcl-2 inhibits autophagy by binding Beclin-1.22 However, in co-immunoprecipitation experiments we did not detect an interaction of Beclin-1 with Bcl-2, although as a positive control we were able to detect co-immunoprecipitation of the inositol 1,4,5-trisphosphate receptor (IP3R) with Bcl-2, as described previously in multiple laboratories (Fig. 4A).24,26,27 Overall, these findings are consistent with recent reports indicating that Bcl-2 does not impede autophagy, but rather enables cells to undergo autophagy by inhibiting apoptosis.28,29 Moreover, consistent with these reports, we find that autophagy contributes to the survival of Bcl-2-positive cells when treated with Dex. Thus, Bcl-2-positive WEHI7.2 cells remain viable for up to 72 hr when treated with Dex, but die when co-treated with 3-methyladenine (3-MA), a known autophagy inhibitor (Fig. 4B).

Figure 4. Contribution of autophagy to survival of Bcl-2-positive cells treated with Dex. (A) Failure to detect an interaction of Beclin-1 with Bcl-2. Bcl-2 immunoprecipitates from Bcl-2-negative and -positive WEHI7.2 cells were investigated by immunoblotting using either antibody to Beclin-1 or antibody to IP3R. The results, confirmed in two separate experiments, indicate that IP3R but not Beclin-1 co-immunoprecipitates with Bcl-2. (B) Decreased viability of Bcl-2-positive cells following treatment with 3-MA and Dex. Bcl-2-positive cells were treated with 1 μM Dex, 1 mM 3-MA plus vehicle or 1 mM 3-MA plus 1 μM Dex (mean ± SE, n = 3).
Having established that Dex induces autophagy in cells that are protected from undergoing apoptosis by Bcl-2, we next turned to the question of whether or not Bcl-2 is required for autophagy induction by Dex. Our initial examination of LC3 by immunoblotting did not detect autophagy induction in either Bcl-2-negative or -positive cells treated with Dex for 24 hr (Fig. 2A) and testing Bcl-2-negative cells after longer exposure to Dex was impractical due to the rapid onset of apoptotic cell death. However, rapid degradation of LC3 II by lysosomal proteases following the fusion of autophagosomes with lysosomes could conceivably limit autophagy detection by LC3 immunoblotting. Therefore, a series of immunoblotting experiments were performed using the lysosomal protease inhibitors pepstatin A and E64d to prevent LC3 II degradation.16,30 In the course of these experiments we discovered that the transfer of LC3 II to the membrane during the immunoblotting procedure was markedly affected by the pH of the transfer buffer, leading to a technical modification that significantly improved LC3 detection (Fig. 5A). The pH of Towbin transfer buffer generally employed in our laboratory for the investigation of many different proteins is 8.3, close to the isoelectric point of LC3 which ranges from 8.73 to 9.14 according to LC3 isoform. In a direct comparison, much more LC3 II was detected on immunoblotting of Bcl-2-positive cells when transfer buffer at pH 11.0 was employed versus transfer buffer at pH 8.3 (Fig. 5A). This particular experiment was performed in Bcl-2-positive cells. Note that Dex-induced LC3 II elevation was not detected at 24 hr when LC3 protein transfer was less than optimal (i.e., transfer buffer pH 8.3), even with the use of lysosomal protease inhibitors, but was detected using improved transfer conditions (i.e., transfer buffer pH 11.0) (Fig. 5A). Therefore, subsequent -experiments primarily designed to test for autophagy induction by 24 hr of Dex treatment in Bcl-2-negative versus Bcl-2-positive cells employed only pH 11.0 transfer buffer. As shown in Figure 5B, Dex-induced LC3 II elevation was detected in both Bcl-2-negative and -positive cells when lysosomal protease inhibitors were used to stabilize LC3 II, but a significant elevation of LC3 II was not detected when either Bcl-2-negative or -positive cells were treated with Dex in the absence of lysosomal protease inhibitors. Also, the addition of lysosomal protease inhibitors markedly enhanced LC3 II detection (compare lane 3 versus lane 9 in Fig. 5B). Thus, we conclude that a combination of optimal protein transfer conditions plus treating cells with lysosomal protease inhibitors increases the sensitivity of the LC3 immunoblotting assay, enabling detection of LC3 I to LC3 II conversion not only at an earlier time point but also in Bcl-2-negative cells. Moreover, we conclude that Bcl-2 expression is not necessary for Dex to induce autophagy, but that it enables autophagy detection over a prolonged duration of Dex treatment by inhibiting apoptosis.

Figure 5. Autophagy detection in both Bcl-2-negative and -positive cells following Dex treatment in presence of lysosomal protease inhibitors. (A) Bcl-2-positive cells were treated with vehicle or 1 μM Dex for the indicated periods of time in the absence (lanes 1 and 8) or presence (all other lanes) of lysosomal protease inhibitors (5 μg/ml E64d and 10 μg/ml pepstatin A). Cell extracts were subjected to SDS-PAGE and transfered to PDVF membranes using either CAPS or Towbin transfer buffer. LC3 II detection was markedly enhanced both by use of lysosomal protease inhibitors and use of CAPS transfer buffer, confirming LC3 II elevation in Dex-treated cells. (B) The effect of lysosomal protease inhibitors on LC3 II detection in Bcl-2-negative and -positive cells treated with either vehicle or 1 μM Dex for 24 hr. Since lysosomal protease inhibitors are dissolved in ethanol or DMSO (see Methods) potential effects of these vehicles were controlled for by inclusion of a lane corresponding to untreated cells (lanes 2 and 5) or cells treated with ethanol and DMSO at concentrations corresponding to that used in treating cells with protease inhibitors (lanes 8 and 11). Also, LC3 Standard protein demarcates the migration of LC3 I on immunoblots (lane 1). Dex-induced elevation of LC3 II was not detected in the absence of protease inhibitors (lanes 3, 4, 6 and 7), but was detected in both Bcl-2-negative and -positive cells when protease inhibitors were included (lanes 9, 10, 12 and 13).
To further explore the inter-relationship of apoptosis and autophagy, the effect of the caspase inhibitor Z-VAD-fmk on autophagy induction by Dex was tested in Bcl-2-negative cells. Previous studies in this laboratory confirmed that Z-VAD-fmk inhibits apoptosis induction in Dex-treated WEHI7.2 cells.31 Therefore, in the current experiment caspase activation was monitored by immunoblotting to detect caspase-mediated cleavage of poly-ADP-ribose polymerase (PARP), a well-documented caspase target. Treatment of WEHI7.2 cells with 1 μM Dex for 24 hr induced considerable cell death (44% viability compared to 90% viability for vehicle-treated cells) accompanied by PARP cleavage (Fig. 6, lane 4). Co-treatment with Z-VAD-fmk significantly inhibited cell death and PARP cleavage in cells treated with Dex for 24 hr (84% viability compared to 90% viability for vehicle-treated cells) (Fig. 6, lane 7). Analysis of LC3 revealed significant conversion of LC3 I to LC3 II in cells treated with Dex for 24 hr in the absence of Z-VAD-fmk (Fig. 6, lane 4), confirming that Dex induces autophagy in Bcl-2-negative cells. Moreover, the level of Dex-induced LC3 II accumulation appeared to be increased by co-treatment with Z-VAD-fmk, suggesting that autophagy detection is enhanced by apoptosis inhibition (Fig. 6, lane 7). But treatment with Z-VAD-fmk was not nearly as effective at inhibiting apoptosis as Bcl-2 overexpression and hence was unable to preserve the viability of Dex-treated cells for prolonged periods of time. Hence, virtually all Bcl-2-negative cells were dead following Dex treatment for 48 hr, even in the presence of Z-VAD-fmk.
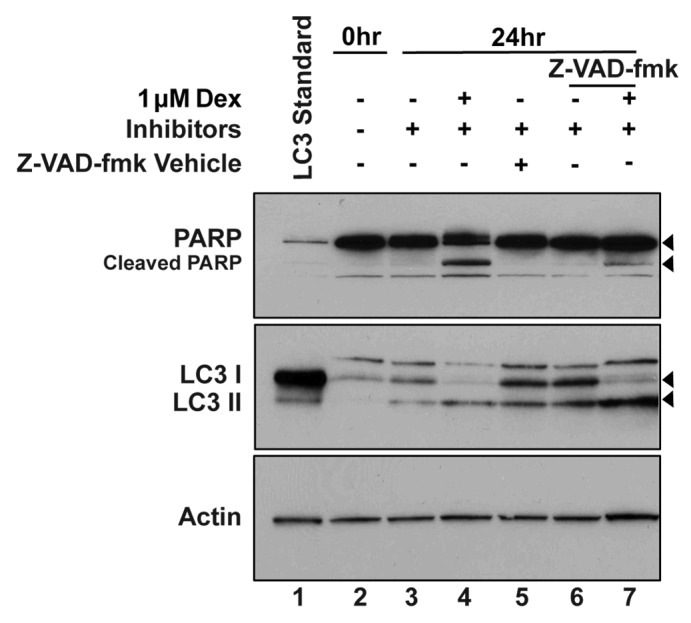
Figure 6. Effect of apoptosis inhibition on autophagy induction by Dex in Bcl-2-negative cells. Bcl-2-negative cells were treated with vehicle or 1 μM Dex and lysosomal protease inhibitors as indicated. In addition, a portion of the cells were co-treated with Z-VAD-fmk. Caspase activity was monitored according to PARP cleavage detected by immunoblotting. Dex-induced PARP cleavage was inhibited by Z-VAD-fmk, which enhanced accumulation of LC3 II in Dex-treated cells.
Detection of autophagosomes by electron microscopy (EM) is a hallmark of autophagy.16 Although it was impractical in this study to examine a sufficient number of cells by EM to reach quantitatively significant conclusions, EM provided qualitative information supporting the conclusion that Dex induces autophagy (Fig. 7). Autophagosomes were rarely detected in vehicle-treated Bcl-2-negative cells (Fig. 7A, Part i and ii) and vehicle-treated Bcl-2-positive cells (Fig. 7B, Part i and ii). Autophagosomes were detected in a small proportion of Dex-treated Bcl-2-negative cells, with a representative example shown in Figure 7A, Part iv. Chromatin condensation indicative of apoptosis was only detected in Bcl-2-negative cells following Dex treatment (Fig. 7A, Part iii) and was not detected in Bcl-2-positive cells (Fig. 7B). Bcl-2-positive cells afforded a better opportunity for a more detailed examination by EM because of their more prolonged survival compared to Bcl-2-negative cells in the face of Dex treatment. In these cells, autophagic vacuoles, including both autophagosomes and autolysosomes, were detected by EM at 30, 48 and 72 hr after Dex treatment (Fig. 7B, Part iii–vi). Moreover, autophagosomes appeared to take up a progressively greater proportion of the cytoplasmic space with time (Fig. 7B). Representative images illustrate three vacuoles with degraded intracellular components at 30 hr with Dex (Fig. 7B, Part iii and iv). Also, both an autophagosome and an autolysosome are illustrated in the Dex-treated Bcl-2-positive cells (Fig. 7B, Part v). Remarkably, at 72 hr of Dex treatment autophagic vacuoles occupy much of the cell (Fig. 7B, Part vi).

Figure 7. Electron microscopic detection of autophagic vesicles in Dex-treated cells. Bcl-2-negative cells (A) and Bcl-2-positive cells (B) were treated with either vehicle or 1 μM Dex for the indicated time periods. Shown are representative images at the following magnifications (A): (Part i) 3,600X, (Part ii–iv) 5,400X; (B): (Part i) 4,000X, (Part ii and iii) 6,000X, (Part iv) 30,000X, (Part v) 15,000X, (Part vi) 8,000X. In (Part v), * indicates an autophagosome and ** indicates an autolysosome.
Discussion
Here we report that the glucocorticosteroid hormone Dex induces autophagy in the murine T cell lymphoma line WEHI7.2. Following current recommendations, autophagy was documented by both steady state methods and flux measurements.16 Autophagy induction was detected in both Bcl-2-negative and Bcl-2-positive cells, although autophagy detection was clearly enhanced in the Bcl-2 overexpressing cells. In Bcl-2-negative cells, Dex induced both apoptosis and autophagy. In this case, apoptosis overwhelmed any obvious protective effect that autophagy might provide. But apoptosis was markedly inhibited by Bcl-2 overexpression, making it much easier to detect autophagy in Bcl-2-positive cells than in Bcl-2-negative cells. Moreover, autophagy appears to contribute significantly to the prolonged survival of Bcl-2-positive cells following Dex treatment, as treating these cells with the autophagy inhibitor 3-MA triggered cell death.
The observation that Dex induces autophagy may appear unexpected in view of earlier reports indicating that rapamycin, a known autophagy inducer, enhances apoptosis induction by Dex.32-34 At first this may seem paradoxical, as rapamycin is reported to induce autophagy through inhibition of mTOR.35 However, mTOR also reduces expression of a number of anti-apoptotic proteins, including Bcl-2 and Mcl-1.33,34 Thus, we speculate that rapamycin may possibly enhance Dex-induced apoptosis by downregulating expression of anti-apoptotic proteins.
Also, finding that both serum withdrawal and Dex treatment induce autophagy in Bcl-2-positive cells appears to be in conflict with reports that Bcl-2 inhibits autophagy by binding to Beclin-1.22 As reported in this earlier study, under nutrient rich conditions Bcl-2 binds to Beclin-1 and thereby inhibits autophagy, but following nutrient starvation Bcl-2 dissociates from Beclin-1.22 Also, consistent with the concept that Bcl-2 is capable of inhibiting autophagy, it has recently been reported that dissociation of Bcl-2 from Beclin-1 by the Bcl-2 antagonist ABT737 triggers autophagy.36 Because of these reports, we entertained the concept that perhaps Dex induces autophagy in Bcl-2-positive cells by causing dissociation of Bcl-2 from Beclin-1; however, Beclin-1 does not co-immunoprecipitate with Bcl-2 even in untreated WEHI7.2 cells. Notably, we found that Bcl-2-positive lymphocytes also undergo autophagy in response to serum withdrawal. Moreover, our findings are consistent with recent evidence that Bcl-2, by inhibiting apoptosis, enables autophagy to occur when cells are subjected to metabolic stress, the DNA damaging agent etoposide or the kinase inhibitor staurosporine.29,37
A variety of assays were employed to detect autophagy in the present study. As discussed in a recent review no one single assay is perfect; hence, the use of a combination of assays is advisable.16 The finding that Dex induces autophagy was supported by each of the assays performed herein. Through experience gained in conducting these studies we found that increasing the pH of the transfer buffer above the isoelectric point of LC3 markedly enhanced LC3 detection by immunoblotting. Initially we did not detect autophagy in Bcl-2-negative cells until this technical modification was made.
Although it is clear from the present study that Dex induces autophagy, the mechanism is yet to be elucidated. Through gene expression profiling in the CEM T cell leukemia line4 and the WEHI7.2 T cell lymphoma line,2 it is known that glucocorticoids repress the expression of genes encoding most of the enzymes involved in glycolysis. Consistent with these gene expression changes, metabolic profiles generated in WEHI7.2 cells by NMR spectroscopy indicate that Dex induces a generalized downregulation of glycolysis rather than a block at a single step in this complex process.21 These findings, coupled with earlier evidence that glucocorticoids inhibit lymphocyte glucose uptake,8,9,21 suggest that glucocorticoid-induced autophagy may be a consequence of metabolic stress, although formal proof of this hypothesis will require further investigation.
The discovery that Dex induces autophagy has important clinical implications. The glucocorticoid hormones prednisone and dexamethasone are commonly used to treat lymphoid malignancies, stemming from the 1940’s when increased adrenal activity was observed to induce involution of the thymus38 and glucocorticoid administration was found to induce regression of lymphoid tumors in mice and humans.39 Several decades later it was discovered that the negative effect of glucocorticoids on lymphocyte survival was due to induction of apoptosis.40 Interestingly, it was reported in the same time period that an autophagic pathway is active in untreated normal human lymphocytes and chronic lymphocytic leukemia cells that accounts for up to one-third of the cells overall protein degradation.41 Now we report for the first time that glucocorticosteroid hormones induce autophagy in lymphoma cells and that autophagy induction is particularly evident when Bcl-2 is overexpressed. Bcl-2 is frequently overexpressed in lymphoid malignancies, causing resistance to glucocorticoid-induced apoptosis.42,43 Our observation that Dex induces autophagy in Bcl-2 overexpressing WEHI7.2 cells and that autophagy supports prolonged survival of Bcl-2 overexpressing cells treated with Dex is consistent with recent evidence that autophagy maintains the survival of metabolically stressed cancer cells when apoptosis is blocked by Bcl-2.29 Thus, it is likely that autophagy contributes to the survival of Bcl-2-positive lymphoma and leukemia cells in patients treated with glucocorticoids. If so, it may prove useful to target autophagy to enhance the therapy of lymphoid malignancies.
Methods
Reagents
Dexamethasone was purchased from Sigma and 14C-valine from GE Healthcare. E64d and Caspase Inhibitor I (Z-VAD-fmk) were purchased from BioMol and pepstatin A from Roche. All other chemicals were from Amresco.
Cell culture
WEHI7.2 cells were cultured in Dulbecco’s Modified Eagle’s Medium supplemented with 2 mM L-glutamine, 0.1 mM non-essential amino acids and 10% bovine calf serum. As described previously24 cells were stably transfected with an expression vector encoding full-length human Bcl-2, followed by isolation of individual clones on soft agar. Control cells, which were transfected with an empty expression vector, are Bcl-2-negative. Bcl-2 is readily detected by immunoblotting in cells transfected with the Bcl-2 expression vector; therefore these cells are referred to as Bcl-2-positive. Dex was dissolved in 100% ethanol vehicle and WEHI7.2 cells were treated with either vehicle or concentrations of Dex ranging from 0.001 μM–1 μM, for up to 72 hr. Where indicated cells were cultured in the presence of lysosomal inhibitors E64d at 5 ug/ml and pepstatin A at 10 ug/ml, which were added at the same time as either ethanol vehicle or Dex. E64d was dissolved in 100% ethanol and pepstatin A was dissolved in dimethyl sulfoxide (DMSO).
Immunoblotting
Cells were lysed in buffer containing 50 mM Tris pH 7.5, 1% Triton X-100, 0.1% (w/v) sodium dodecyl sulfate, 150 mM NaCl, 200 mM DTT and a complete Mini protease inhibitor cocktail (Roche Applied Science) for 30 min at 4°C. Cell lysates containing ~50 μg of protein were separated by either 15% or 4–20% gradient SDS-PAGE and transferred in either Towbin transfer buffer (25 mM Tris, 192 mM glycine and 20% methanol, pH 8.3) or CAPS transfer buffer (10 mM CAPS (3-cyclohexylamino-1 propanesulfonic acid), 10% methanol and NaOH to pH 11.0) to PVDF for 1–1.5 hr at 25–100 volts. An extract from serum starved Neuro 2A cells (Axxora, NT-0911-2) was used as an LC3 Standard.
Anti-LC3 antibody from MBL International Corporation (#PD012) was used in initial experiments (Fig. 2) but following its discontinuation anti-LC3 antibody from Novus Biologicals (#NB1002331) was used in all subsequent experiments. Anti-actin antibody was from Sigma-Aldrich. Bands on immunoblots were visualized by adding ECL or ECL plus (GE Healthcare).
Immunoprecipitation
Co-immunoprecipitation procedures were described previously.24 The following antibodies were used: anti-human Bcl-2 (BD Biosciences), anti-IP3R type 1 (Calbiochem) and anti-Beclin 1 (BD Biosciences).
GFP-LC3 expression and time-lapse imaging
Bcl-2-positive cells were transiently transfected by electroporation with the pEGFP-C1-LC3 plasmid, kindly provided by Tamotsu Yoshimori and Noboru Mizushima15 using previously described electroporation conditions.44 After incubation for 1 hr, 1 μM Dex was added to the cells which were then incubated in poly-L-lysine coated MatTek cultureware plates for 1 hr at 37°C, allowing the cells to adhere. Images were taken using an inverted Leica DMI 6000 fluorescence microscope, equipped with an environmental chamber set at 37°C and 7% CO2. Fluorescence images were acquired every 30 min over a period of 20 hr, controlled by MetaMorph (Vers. 7.04), using a 20X/0.4 CORR HCX PL FLUOTAR objective.
Protein degradation assay
The rate of protein degradation was measured as previously described.45 Bcl-2-positive cells were labeled for 24 hr in 30 ml of DMEM containing a concentration of 0.2 μCi per ml 14C-valine. Cells were washed with PBS three times to remove unincorporated 14C-valine. The cells were further incubated in DMEM without 14C-valine for 1 hr and then treated with 1 μM Dex or vehicle for 30, 48 or 72 hr. To precipitate cellular proteins 10% trichloroacetic acid was added and the cells were incubated at 4°C for 10 min and centrifuged. Trichloroacetic acid precipitates and supernatants were dissolved and radioactivity was counted in a liquid scintillation counter (Beckman LS6000IC). The amount of protein degradation at each time point was calculated as the ratio of the cpm from the media to the cpm from the pellet.
Caspase 3 activity
Caspase 3 activity was measured using an assay based on the enzymatic cleavage of the fluorogenic caspase-3 substrate Ac-DEVD-AFC (Calbiochem). Cell lysates were harvested and lysed in 60 μL buffer (150 mmol/L NaCl, 10 mmol/L HEPES (pH 7.4) and 1% CHAPS). Then caspase-3 activity was measured in black plastic well plates containing 100 μg protein from cell lysates, 1 μg Ac-DEVD-AFC and reaction buffer (50 mmol/L Tris-HCl (pH 7.2), 100 mmol/L KCl, 10% sucrose, 0.1% CHAPS, 10 mmol/L DTT) to make a total volume of 100 μL/well. After incubation for 1 hr at 37°C, fluorescence emission (excitation 365–380 nm and emission 430–460 nm) was measured on a Victor 3 plate reader (Perkin-Elmer) and expressed as arbitrary relative fluorescent units.
Electron microscopy
Cells were fixed for 2 hr at room temperature in a 0.05 M phosphate-buffered solution (pH 7.4) containing 2.5% glutaraldehyde, 2% paraformaldehyde and 4% sucrose and then postfixed in 1% osmium tetroxide for 1 hr. Samples were then dehydrated in ascending concentration of ethanol and embedded in Epon 812. Ultrathin sections were stained with 2% uranyl acetate in 50% methanol and lead citrate, and then examined in a JEOL 1200 EX electron microscope (Japan).
Flow cytometry
Flow cytometry was performed to detect and quantify apoptotic cells. Cells were stained with propidium iodide for 30 min, run on a EPICS-XL MCL and analyzed with Winlist as previously described.2
Supplementary Material
Acknowledgements
We thank Tamotsu Yoshimori and Noboru Mizushima for providing the LC3 cDNA and we also thank Hisashi Fujioka for help with the electron microscopy. We thank Vivian Gama and Jose Gomez in Shigemi Matsuyama’s laboratory for help with the caspase assay. This work was supported by NIH grants RO1 CA42755 and CA85804 (to CWD) and T32 HL007147 (to SS). This research utilized the Confocal Microscopy Core Facility and Flow Cytometry Core Facility of the Comprehensive Cancer Center of Case Western Reserve University and University Hospitals of Cleveland (P30 CA43703-12).
Note
Supplementary materials can be found at: http://www.landesbioscience.com/journals/autophagy/article/5920
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/5920
References
- 1.Jondal M, Pazirandeh A, Okret S. Different roles for glucocorticoids in thymocyte homeostasis? Trends Immunol. 2004;25:595–600. doi: 10.1016/j.it.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 2.Wang Z, Malone MH, He H, McColl KS, Distelhorst CW. Microarray analysis uncovers the induction of the proapoptotic BH3-only protein Bim in multiple models of glucocorticoid-induced apoptosis. J Biol Chem. 2003;278:23861–7. doi: 10.1074/jbc.M301843200. [DOI] [PubMed] [Google Scholar]
- 3.Zhang L, Insel PA. The pro-apoptotic protein Bim is a convergence point for cAMP/protein kinase A- and glucocorticoid-promoted apoptosis of lymphoid cells. J Biol Chem. 2004;279:20858–65. doi: 10.1074/jbc.M310643200. [DOI] [PubMed] [Google Scholar]
- 4.Tonko M, Ausserlechner MJ, Bernhard D, Helmberg A, Kofler R. Gene expression profiles of proliferating vs. G1/G0 arrested human leukemia cells suggest a mechanism for glucocorticoid-induced apoptosis. FASEB J. 2001;15:693–9. doi: 10.1096/fj.00-0327com. [DOI] [PubMed] [Google Scholar]
- 5.Yoshida NL, Miyashita T, U M, Yamada M, Reed JC, Sugita Y, et al. Analysis of gene expression patterns during glucocorticoid-induced apoptosis using oligonucleotide arrays. Biochem Biophys Res Commun. 2002;293:1254–61. doi: 10.1016/S0006-291X(02)00361-3. [DOI] [PubMed] [Google Scholar]
- 6.Wang Z, Malone MH, Thomenius MJ, Zhong F, Xu F, Distelhorst CW. Dexamethasone-induced gene 2 (dig2) is a novel pro-survival stress gene induced rapidly by diverse apoptotic signals. J Biol Chem. 2003;278:27053–8. doi: 10.1074/jbc.M303723200. [DOI] [PubMed] [Google Scholar]
- 7.Medh RD, Webb MS, Miller AL, Johnson BH, Fofanov Y, Li T, et al. Gene expression profile of human lymphoid CEM cells sensitive and resistant to glucocorticoid-evoked apoptosis. Genomics. 2003;81:543–55. doi: 10.1016/S0888-7543(03)00045-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kattwinkel J, Munck A. Activities in vitro of glucocorticoids and related steroids on glucose uptake by rat thymus cell suspensions. Endocrinology. 1966;79:387–90. doi: 10.1210/endo-79-2-387. [DOI] [PubMed] [Google Scholar]
- 9.Young DA. Glucocorticoid action on rat thymus cells. Interrelationships between carbohydrate, protein, and adenine nucleotide metabolism and cortisol effects on these functions in vitro. J Biol Chem. 1969;244:2210–7. [PubMed] [Google Scholar]
- 10.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–77. doi: 10.1016/S1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 11.Lum JJ, DeBerardinis RJ, Thompson CB. Autophagy in metazoans: cell survival in the land of plenty. Nat Rev Mol Cell Biol. 2005;6:439–48. doi: 10.1038/nrm1660. [DOI] [PubMed] [Google Scholar]
- 12.Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 2004;23:2891–906. doi: 10.1038/sj.onc.1207521. [DOI] [PubMed] [Google Scholar]
- 13.Mizushima N, Ohsumi Y, Yoshimori T. Autophagosome formation in mammalian cells. Cell Struct Funct. 2002;27:421–9. doi: 10.1247/csf.27.421. [DOI] [PubMed] [Google Scholar]
- 14.Edinger AL, Thompson CB. Defective autophagy leads to cancer. Cancer Cell. 2003;4:422–4. doi: 10.1016/S1535-6108(03)00306-4. [DOI] [PubMed] [Google Scholar]
- 15.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–8. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–75. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mizushima N. Methods for monitoring autophagy. Int J Biochem Cell Biol. 2004;36:2491–502. doi: 10.1016/j.biocel.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 18.Bhattacharyya TK, Butler DG. Ultrastructure of the adrenocortical homologue in dexamethasone-treated eels. J Anat. 1980;130:323–32. [PMC free article] [PubMed] [Google Scholar]
- 19.Rusten TE, Lindmo K, Juhász G, Sass M, Seglen PO, Brech A, et al. Programmed autophagy in the Drosophila fat body is induced by ecdysone through regulation of the PI3K pathway. Dev Cell. 2004;7:179–92. doi: 10.1016/j.devcel.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 20.Kalamidas SA, Kotoulas OB. Glycogen autophagy in newborn rat hepatocytes. Histol Histopathol. 2000;15:1011–8. doi: 10.14670/HH-15.1011. [DOI] [PubMed] [Google Scholar]
- 21.Tome ME, Lutz NW, Briehl MM. Overexpression of catalase or Bcl-2 alters glucose and energy metabolism concomitant with dexamethasone resistance. Biochim Biophys Acta. 2004;1693:57–72. doi: 10.1016/j.bbamcr.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 22.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–39. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 23.Criollo A, Maiuri MC, Tasdemir E, Vitale I, Fiebig AA, Andrews D, et al. Regulation of autophagy by the inositol trisphosphate receptor. Cell Death Differ. 2007;14:1029–39. doi: 10.1038/sj.cdd.4402099. [DOI] [PubMed] [Google Scholar]
- 24.Chen R, Valencia I, Zhong F, McColl KS, Roderick HL, Bootman MD, et al. Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. J Cell Biol. 2004;166:193–203. doi: 10.1083/jcb.200309146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–11. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oakes SA, Scorrano L, Opferman JT, Bassik MC, Nishino M, Pozzan T, et al. Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum. Proc Natl Acad Sci U S A. 2005;102:105–10. doi: 10.1073/pnas.0408352102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu L, Kong D, Zhu L, Zhu W, Andrews DW, Kuo TH. Suppression of IP3-mediated calcium release and apoptosis by Bcl-2 involves the participation of protein phosphatase 1. Mol Cell Biochem. 2007;295:153–65. doi: 10.1007/s11010-006-9285-5. [DOI] [PubMed] [Google Scholar]
- 28.Xue LY, Chiu SM, Azizuddin K, Joseph S, Oleinick NL. The death of human cancer cells following photodynamic therapy: apoptosis competence is necessary for Bcl-2 protection but not for induction of autophagy. Photochem Photobiol. 2007;83:1016–23. doi: 10.1111/j.1751-1097.2007.00159.x. [DOI] [PubMed] [Google Scholar]
- 29.Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10:51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mizushima N, Yoshimori T. How to Interpret LC3 Immunoblotting. Autophagy. 2007;3:542–5. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- 31.McColl KS, He H, Zhong H, Whitacre CM, Berger NA, Distelhorst CW. Apoptosis induction by the glucocorticoid hormone dexamethasone and the calcium-ATPase inhibitor thapsigargin involves Bc1-2 regulated caspase activation. Mol Cell Endocrinol. 1998;139:229–38. doi: 10.1016/S0303-7207(98)00051-3. [DOI] [PubMed] [Google Scholar]
- 32.Strömberg T, Dimberg A, Hammarberg A, Carlson K, Osterborg A, Nilsson K, et al. Rapamycin sensitizes multiple myeloma cells to apoptosis induced by dexamethasone. Blood. 2004;103:3138–47. doi: 10.1182/blood-2003-05-1543. [DOI] [PubMed] [Google Scholar]
- 33.Yan H, Frost P, Shi Y, Hoang B, Sharma S, Fisher M, et al. Mechanism by which mammalian target of rapamycin inhibitors sensitize multiple myeloma cells to dexamethasone-induced apoptosis. Cancer Res. 2006;66:2305–13. doi: 10.1158/0008-5472.CAN-05-2447. [DOI] [PubMed] [Google Scholar]
- 34.Wei G, Twomey D, Lamb J, Schlis K, Agarwal J, Stam RW, et al. Gene expression-based chemical genomics identifies rapamycin as a modulator of MCL1 and glucocorticoid resistance. Cancer Cell. 2006;10:331–42. doi: 10.1016/j.ccr.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 35.Takeuchi H, Kondo Y, Fujiwara K, Kanzawa T, Aoki H, Mills GB, et al. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res. 2005;65:3336–46. doi: 10.1158/0008-5472.CAN-04-3640. [DOI] [PubMed] [Google Scholar]
- 36.Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007;26:2527–39. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, et al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. 2004;6:1221–8. doi: 10.1038/ncb1192. [DOI] [PubMed] [Google Scholar]
- 38.Dougherty TF, White A. Effect of pituitary adrenotropic hormone on lymphoid tissue. Proc Soc Exp Biol Med. 1943;53:132–3. [Google Scholar]
- 39.Pearson OH, Eliel LP, Rawson RW, Dobriner K, Rhoads CP. Adrenocorticotropic hormone- and cortisone-induced regression of lymphoid tumors in man; a preliminary report. Cancer. 1949;2:943–5. doi: 10.1002/1097-0142(194911)2:6<943::AID-CNCR2820020602>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 40.Wyllie AH. Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature. 1980;284:555–6. doi: 10.1038/284555a0. [DOI] [PubMed] [Google Scholar]
- 41.Seglen PO, Munthe-Kaas AC, Dybedal MA. Amino acid control of protein degradation in normal and leukemic human lymphocytes. Exp Cell Res. 1984;155:121–8. doi: 10.1016/0014-4827(84)90773-0. [DOI] [PubMed] [Google Scholar]
- 42.Haarman EG, Kaspers GJ, Pieters R, van Zantwijk CH, Broekema GJ, Hählen K, et al. BCL-2 expression in childhood leukemia versus spontaneous apoptosis, drug induced apoptosis, and in vitro drug resistance. Adv Exp Med Biol. 1999;457:325–33. doi: 10.1007/978-1-4615-4811-9_35. [DOI] [PubMed] [Google Scholar]
- 43.Reed JC, Miyashita T, Krajewski S, Takayama S, Aime-Sempe C, Kitada S, et al. Bcl-2 family proteins and the regulation of programmed cell death in leukemia and lymphoma. Cancer Treat Res. 1996;84:31–72. doi: 10.1007/978-1-4613-1261-1_3. [DOI] [PubMed] [Google Scholar]
- 44.Wang Z, Rong YP, Malone MH, Davis MC, Zhong F, Distelhorst CW. Thioredoxin-interacting protein (txnip) is a glucocorticoid-regulated primary response gene involved in mediating glucocorticoid-induced apoptosis. Oncogene. 2006;25:1903–13. doi: 10.1038/sj.onc.1209218. [DOI] [PubMed] [Google Scholar]
- 45.Høyer-Hansen M, Bastholm L, Mathiasen IS, Elling F, Jäättelä M. Vitamin D analog EB1089 triggers dramatic lysosomal changes and Beclin 1-mediated autophagic cell death. Cell Death Differ. 2005;12:1297–309. doi: 10.1038/sj.cdd.4401651. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.