

Significance
Mechanical forces transmitted between a cell and its surrounding extracellular matrix determine functions like proliferation or differentiation, and drive processes in development, tumorigenesis, and wound healing. However, the molecules involved in this force transmission remain unclear. Here we show that forces exerted by cells are transmitted to the extracellular matrix through α-actinin molecules via the transmembrane protein integrins. Furthermore, this transmission enables the growth and maturation of adhesion sites to the matrix, and takes place in competition with another molecule submitted to force, talin. This force regulation mechanism may help us understand the role of force in different biological scenarios.
Abstract
Focal adhesions are mechanosensitive elements that enable mechanical communication between cells and the extracellular matrix. Here, we demonstrate a major mechanosensitive pathway in which α-actinin triggers adhesion maturation by linking integrins to actin in nascent adhesions. We show that depletion of the focal adhesion protein α-actinin enhances force generation in initial adhesions on fibronectin, but impairs mechanotransduction in a subsequent step, preventing adhesion maturation. Expression of an α-actinin fragment containing the integrin binding domain, however, dramatically reduces force generation in depleted cells. This behavior can be explained by a competition between talin (which mediates initial adhesion and force generation) and α-actinin for integrin binding. Indeed, we show in an in vitro assay that talin and α-actinin compete for binding to β3 integrins, but cooperate in binding to β1 integrins. Consistently, we find opposite effects of α-actinin depletion and expression of mutants on substrates that bind β3 integrins (fibronectin and vitronectin) versus substrates that only bind β1 integrins (collagen). We thus suggest that nascent adhesions composed of β3 integrins are initially linked to the actin cytoskeleton by talin, and then α-actinin competes with talin to bind β3 integrins. Force transmitted through α-actinin then triggers adhesion maturation. Once adhesions have matured, α-actinin recruitment correlates with force generation, suggesting that α-actinin is the main link transmitting force between integrins and the cytoskeleton in mature adhesions. Such a multistep process enables cells to adjust forces on matrices, unveiling a role of α-actinin that is different from its well-studied function as an actin cross-linker.
Mechanical stimulus-response pathways are essential in establishing the proper physical communication between a cell and its environment. This role is exemplified not only by the wide-ranging effects of external mechanical signals, such as substrate rigidity or applied forces (1–3), but also by the impairment of cellular function that results from inhibiting internal force generation in cells by molecular motors (4–6). To understand this mechanical signaling, the study of cell adhesion and spreading has proven to be a particularly powerful tool. Indeed, cell spreading showcases the transition from an isolated cell in suspension to full mechanochemical contact with the extracellular matrix (ECM) in a short period. Furthermore, as the first mechanical events in adhesion, the steps occurring during spreading are necessarily upstream of the longer-term mechanically controlled events in development, cancer, wound healing, and other processes (2, 7, 8).
After the initial rapid spreading of fibroblasts on fibronectin, myosin-generated contractility is necessary to develop adhesion sites (3, 9). However, to generate contractile forces on a substrate, the cytoskeleton must be linked to the ECM through integrins. Integrins initially link to actin through talin, which activates integrin binding to matrix (10) and is subjected to forces that aid initial adhesion site formation (11, 12). Stretching of talin also exposes buried binding sites to the focal adhesion protein vinculin (13), providing a mechanotransduction mechanism by which actomyosin forces applied to talin could trigger initial adhesion formation. After this initial talin-dependent phase, however, the molecules and steps necessary to transmit forces from the cytoskeleton to integrins, and thereby trigger further adhesion maturation, remain undefined.
A particularly interesting candidate for force transmission is α-actinin. Like talin, α-actinin can bind to both actin and integrins (14–16) and is a prominent component in mature adhesions. Certainly, although it has mainly been studied as an actin cross-linker because it is an antiparallel dimer (17), α-actinin has been shown to be required for focal adhesions to both mature (18) and attach to actin filaments (19). Furthermore, exogenous fragments of α-actinin containing either the actin- or the integrin-binding domains can disassemble stress fibers or focal adhesions, respectively (20, 21). Thus, α-actinin has a role in the maturation of adhesions, which could involve force transmission. Moreover, as α-actinin and talin bind to overlapping domains in integrin β-tails (22, 23), this role is most likely in competition or cooperation with talin. Indeed, here we report that α-actinin competes with talin for the binding to β3 tails, and then transmits the cytoskeletal forces that trigger mechanotransduction, adhesion maturation, and dynamic force regulation.
Results
Binding of α-Actinin to Actin Enables Adhesion Maturation.
We first analyzed the role of α-actinin in adhesion formation by knocking down the levels of α-actinin using shRNA. Mouse embryonic fibroblasts were transfected with a plasmid containing an shRNA with either a nontargeting (NT) sequence or a sequence targeting α-actinin 4. Four days after transfection, this process resulted in a 94% decrease in total α-actinin levels (Fig. 1A), showing that α-actinin 4 was knocked down and that the level of the other α-actinin isoform present in fibroblasts (α-actinin 1) was small. Furthermore, even though in some systems different localizations of both α-actinin isoforms have been reported (24, 25), both isoforms behaved in the same way in our early spreading system (Fig. S1 Aand B). Thus, we find that this shRNA transfection causes the loss of over 90% of the endogenous α-actinin.
Fig. 1.
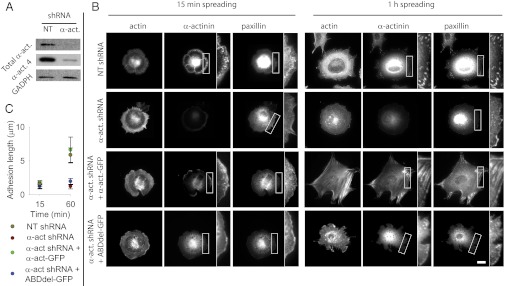
Binding of α-actinin to actin enables adhesion maturation. (A) Western blot showing levels of total α-actinin and α-actinin 4 in cells transfected with a shRNA with a NT or α-actinin 4 targeting (α-act) sequence. (B) Cells transfected with NT or α-actinin shRNA and rescued with FL or ABDdel α-actinin-GFP stained after 15 min or 1 h for F-actin, α-actinin, and paxillin. In rescue cells, α-actinin image is the corresponding GFP construct. Expanded views amplify the areas marked with a white rectangle. (Scale bar, 20 µm.) (C) Quantification of paxillin adhesion length for the different conditions at the 15-min and 1-h time points. Both NT-transfected cells and α-actinin depleted cells rescued with FL α-actinin-GFP experienced significant increases in adhesion size with time (P < 0.01), whereas α-actinin depleted cells either not rescued or rescued with ABDdel-GFP did not (n ≥ 6 cells measured on 2 different days).
We then plated cells on fibronectin-coated coverslips, fixed them, and stained for α-actinin, f-actin, and the focal adhesion protein paxillin. After 15 min of spreading, α-actinin in control (NT-transfected) cells first assembled as a rim in the lamellipodium (Fig. 1B). In this rim, small nascent adhesions containing paxillin and α-actinin could be observed, but neither stress fibers nor large focal adhesions had formed (Fig. 1B). After 1 h of spreading, cells transitioned from a rounded isotropic to a polarized shape, where α-actinin colocalized with paxillin at focal adhesions forming the ends of actin stress fibers. However, α-actinin–depleted cells remained in an isotropic shape even 1 h after spreading, and focal adhesions and stress fibers did not form. Thus, α-actinin was required to develop focal adhesions and stress fibers, as previously reported (18). To understand if α-actinin performed this role in adhesions by mechanically connecting them to the cytoskeleton, we rescued depleted cells with either full-length (FL) α-actinin 1-GFP or a construct where the actin binding domain (ABD) had been deleted (ABDdel). In early spreading cells, both FL-rescued and ABDdel-rescued cells developed a rim of GFP staining that colocalized with paxillin adhesions. However, at 1 h, only FL α-actinin was capable of maturing into focal adhesions as in control cells (Fig. 1B). Indeed, the ABDdel construct still localized at the rim of the lamellipodium, but did not mature. To quantify these results, we measured the length of paxillin adhesions (Fig. 1C). Confirming our observations, adhesion length increased over threefold from the 15 min to the 1-h time point in control cells and α-actinin–depleted cells rescued with FL α-actinin-GFP. In contrast, adhesion length did not increase during the same period in cells depleted of α-actinin or rescued with ABDdel-GFP.
To check the dynamics of this process, we monitored the spreading of cells rescued with either FL or ABDdel α-actinin-GFP using total internal reflection fluorescence (TIRF) time-lapse microscopy (Fig. S2). This technique allowed us to monitor only the bottom layer of the cell, directly in contact with the ECM, and thus responsible for adhesion formation. Time-lapse sequences confirmed that the initial lamellipodial rim of FL α-actinin quickly transitioned into elongated adhesion sites extending rearward into the cell (Fig. S2 and Movie S1). In contrast, the rim of ABDdel α-actinin did not coalesce into mature adhesion sites (Fig. S2 and Movie S2). Even though it also showed a diffuse cytoplasmic distribution, ABDdel α-actinin still localized specifically to initial adhesions at the lamellipodium. Thus, this finding suggests that the absence of a connection to actin prevented adhesion maturation and cell polarization.
Force Transmitted Through α-Actinin Triggers Adhesion Maturation and Mechanosensing.
We next analyzed whether α-actinin triggered adhesion maturation by transmitting forces between the actomyosin cytoskeleton and the ECM. To do this, we used a magnetic tweezers device to apply high pulsatile forces of 1 nN [a force magnitude characteristic of maturing adhesions (26)] to fibronectin-coated magnetic beads attached to cells (Fig. 2A). The diameter of the beads (3 μm) was sufficient to allow cells to form paxillin-rich mature adhesions around the beads (Fig. S3A). It has been shown that the application of forces to cell-ECM adhesions results in their reinforcement (27) and maturation (3). Consistently, in NT cells there was a progressive decrease in the force-induced displacements as a function of time, showing that the cell detected the applied force and responded to it by reinforcing the adhesion (Fig. 2B). However, there was no decrease in the oscillation of beads on α-actinin–depleted cells as a function of time and, thus, they did not reinforce (Fig. 2B). Reinforcement was rescued by FL α-actinin-GFP (although a little less robustly than in control cells) but not by the ABDdel construct (Fig. 2C). This finding showed that the bridging to actin was crucial. Thus, α-actinin was necessary for transmitting forces from the cytoskeleton to adhesion sites, enabling mechanotransduction, reinforcement, and the subsequent maturation of focal adhesions.
Fig. 2.

Force transmitted through α-actinin triggers adhesion maturation and mechanosensing. (A) Magnetic tweezers assay (from left to right). Fibronectin-coated 3-µm magnetic beads are deposited on a substrate where cells are allowed to spread. As the cells spread, they contact and adhere to the beads, and start transporting them with the rearward moving actin cytoskeleton (black arrow). At this point, a magnetic tip is placed close to the bead and a 1-nN force pulsating at 1 Hz is applied (orange arrow). The pulsation of the bead in response to the force is then monitored. (B) Example bead traces (Left) and average relative stiffness (Right) as a function of time of fibronectin-coated beads submitted to a 1-nN force pulsating at 1 Hz attached to cells transfected with NT or α-actinin shRNA. The sample traces show the oscillation of the beads as a function of time in response to the applied force (which begins at the 20-s time point). Relative stiffness values represent bead stiffness (applied force/observed movement) normalized by initial stiffness. Thus, a constant value of 1 would indicate that stiffness is constant (no reinforcement), whereas values higher than one indicate reinforcement (reduced bead pulsation) from initial movement. (C) Example bead traces and average relative stiffness for cells transfected with α-actinin shRNA and rescued with FL or ABDdel α-actinin-GFP. (P < 0.05 for both B and C, n ≥ 26 beads from ≥ 11 cells measured on 2 different days).
α-Actinin and Its Fragments Mediate Force Generation and Release.
If forces from the cytoskeleton were transmitted to adhesion sites by α-actinin, then blocking this link should cause a decrease in cell-ECM traction forces. To measure traction forces, cells were plated on flexible fibronectin-coated polydimethylsiloxane pillars. As the cells spread, they adhered and deflected the pillars by a distance that was directly proportional to the applied force (Fig. 3A). As expected, α-actinin–depleted cells rescued with FL α-actinin-GFP exerted the same forces as NT cells. In contrast, ABDdel-rescued cells exerted reduced forces (Fig. 3 B and C). This result indicated that α-actinin–actin interactions contributed to force generation. However, cells depleted of α-actinin, but not rescued with any mutant, exerted higher forces than all other cells (Fig. 3 B and C). Thus, the presence of α-actinin itself impaired force generation. This effect of α-actinin could not be merely a result of interactions with actin, because the ABDdel construct dramatically reduced contractility in depleted cells without restoring actin binding. Alternatively, α-actinin fragments could be binding integrins and blocking binding and force transmission by another molecule. To test for this possibility, we prepared a construct (SR12-GFP) containing only the integrin binding domains of α-actinin, the spectrin-like repeats (SR) 1 and 2. These repeats do not contain binding domains for other focal adhesion components, such as vinculin (29) or zyxin (30) (Fig. 3D). Similarly to ABDdel-GFP, SR12-GFP colocalized with paxillin in nascent adhesions, but did not rescue focal adhesion formation (Fig. S4A) or mechanotransduction (Fig. S4 B and C) in α-actinin–depleted cells. In the pillar force assay, if integrin binding by α-actinin was sufficient to block force generation, rescue of α-actinin depleted cells with SR12-GFP or ABDdel-GFP should lead to the same reduced forces, as was indeed observed (Fig. 3 B and C). Thus, cells expressing α-actinin’s SR1 and SR2 domains pulled with lower forces than controls, whereas cells depleted of α-actinin pulled with higher forces than controls. Forces in depleted cells were also maintained longer and were less dynamic than in controls (Fig. 3E, and Movies S3 and S4). A possible explanation for these findings was thus that α-actinin normally competed with an early integrin–actin link during adhesion maturation.
Fig. 3.
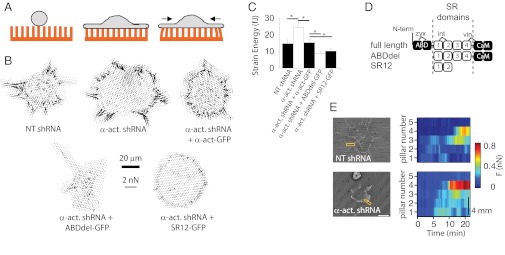
α-actinin mediates force generation and release through interaction with integrins. (A) Flexible pillar assay (from left to right). Cells are deposited on an array of fibronectin-coated flexible poly(dimethylsiloxane) pillars of 1-µm diameter. As the cells spread, they attach and exert contractile forces (black arrows), deflecting the pillars in proportion to the applied force. (B) Vector plots with arrows depicting the magnitude and direction of forces exerted on 1-µm pillar arrays. Cells were transfected with either NT or α-actinin shRNA and rescued with FL, ABDdel, or SR12 α-actinin-GFP. (Scale bar, 20 µm; force scale bar indicates the length of a force arrow of 2 nN.) (C) Corresponding quantification of average forces (strain energy) exerted by cells. *P < 0.05, n ≥ 14 cells measured on ≥ 2 different days. (D) Diagram of α-actinin constructs used showing the ABD, SR1–SR4, and calmodulin-like domain (CaM). Approximate binding domains to integrin-β talis (int), zyxin (zyx), and vinculin (vin) are shown. GFP was at the C terminus. (E, Left) Examples of cells on pillars transfected with NT or α-actinin shRNA. (Scale bar, 20 μm.) (Right) Forces as a function of time exerted by the row of pillars marked in yellow in the left image (see also Movies S3 and S4). Each horizontal color sequence represents an individual pillar, with the lowest pillar number being closest to the cell center.
α-Actinin and Talin Compete for Binding to Integrin β3 Tails.
Talin may be this early integrin–actin link competing with α-actinin during adhesion maturation, because it was necessary for spreading, force generation on the ECM, and mechanosensing at the earliest stages of adhesion (11, 31), and its binding domain in integrin β-tails overlaps with that of α-actinin (22, 23). To explore whether talin and α-actinin could compete for the binding to integrin β-tails, we first analyzed the distribution of α-actinin, talin, and β1 and β3 integrins (the two main fibronectin receptors (27, 32)). After 15 min of spreading, initial adhesions showed colocalization between talin, α-actinin, and β3 integrins (Fig. 4 A and B). In contrast, β1 integrins did not clearly localize to initial adhesions (Fig. 4 C and D), confirming the predominant role of β3 in early adhesion steps (12, 33). After 1 h of spreading, both integrins colocalized with α-actinin and talin in mature focal adhesions as expected (Fig. 4 A–D). This colocalization was confirmed by calculating Pearson correlation coefficients between talin and α-actinin and both integrins (34). Pearson coefficients were high (0.6–0.8) in all cases except between α-actinin and β1 integrins in initial adhesions, confirming our observations (Fig. 4E). Correlation coefficients were also slightly higher between integrins and talin than between integrins and α-actinin (Fig. 4E), probably reflecting that α-actinin also localizes to actin filaments outside adhesions. In α-actinin–depleted cells, talin not only colocalized with remaining α-actinin, but maintained a distribution strikingly similar to the characteristic lamellipodial rim observed in α-actinin (Fig. S5). Using an immunoprecipitation approach, α-actinin also coprecipitated with β3 integrins, much more strongly in control cells than in α-actinin–depleted cells (Fig. 4F). This coprecipitated α-actinin was observed at the expected α-actinin molecular weight (100 KDa) but more strongly at a lower band at 80 kDa, suggesting that some α-actinin was degraded during the immunoprecipitation procedure. This same 80-kDa α-actinin band was observed when coprecipitated with β1 integrins (Fig. 4G), although the expected undegraded band at 100 kDa was not clear. Thus, α-actinin fragments coprecipitated with β1 integrins, although we could not confirm that full-length α-actinin did. However, β1 integrin–α-actinin interactions have been clearly observed in vitro (28, 35, 36). Thus, α-actinin binds to β3 integrins in vivo, and colocalizes with β3 integrins and talin in nascent adhesions, enabling a potential competition.
Fig. 4.

α-Actinin and talin compete for binding to β3 integrins. (A) Localization of α-actinin, talin, and β3 integrins in cells spread for 15 min or 1 h. Merged images show α-actinin in green, talin in red, and integrins in blue. White areas denote colocalization of all three proteins. Expanded views amplify the areas marked with a red rectangle. (B) fluorescence intensity profiles (arbitrary units) across the red dotted lines shown in the corresponding zoomed view. Profiles of α-actinin, talin, and β3 integrins are shown respectively in green, red, and blue. (C and D) Same as A and B with β1 instead of β3 integrins. (Scale bars in A and C, 20 µm.) Colocalization of β1 integrins with talin and α-actinin was not clearly observable after 15 min. After 1 h, colocalization was clear for large focal adhesions (black arrow in D) but not for smaller adhesions (peaks to the right of black arrow). (E) Pearson coefficients quantifying the colocalization between talin and α-actinin to β3 and β1 integrins in staining images (1 = perfect overlap, 0 = unrelated distributions). Colocalizations with integrins were higher for talin than α-actinin (P < 0.001). For α-actinin, colocalization at 15-min spreading with β1 integrins was significantly lower than at 1 h (P < 0.01). n ≥ 6 cells measured on two diferent days. (F and G) Pull-down with β3 antibody (F) or β1 antibody (G) of lysates from cells transfected with NT or α-actinin shRNA. Lanes labeled as total show the lysate before pull-down (8× dilution), whereas lanes labeled as pull-down show the pulled-down proteins. (H) Talin head and α-actinin SR12 fragment were incubated with beads coated with β1, β3, and scrambled β3 (β3SCR) integrin cytoplasmic tails. The amount of talin head and SR12 (1 μM) bound to integrins was then assessed by protein staining after a pull-down assay. (I) Talin head (1 μM) was incubated with beads coated with β1 or β3 tails (Upper and Lower, respectively) either in the absence or in the presence (5 μM) of SR12. Amounts of talin head and SR12 were then assessed as in (H). Quantifications (to the right of each blot) show the average amounts of talin head in arbitrary units (AU), where the value for the case with no SR12 (−) is set to 1 (n ≥ 3).
To test directly if talin and α-actinin competed for binding to β-integrin tails, we conducted an in vitro competition assay. We first purified the cytoplasmic domains of β1 and β3 integrins, and the integrin binding domains of talin [talin head (37)] and α-actinin [the SR12 domain (28)]. We then bound the integrin tails to Ni-NTA beads through a 6xHis-tag, incubated the beads with talin head and α-actinin-SR12, and used Western blotting to evaluate their binding after washing. Using this assay, we initially confirmed the specificity of the interaction by checking that the talin head and α-actinin-SR12 bound to β1 or β3 integrin-coated beads, but not to beads coated with a mutant of the β3 with a scrambled amino acid sequence (Fig. 4H). We then observed that the presence of α-actinin-SR12 impaired the binding of talin to β3 integrin tails, whereas it enhanced talin binding to the β1 integrin tail (Fig. 4I). Because the β3 integrin is required for early adhesion formation (12, 33) and the β1 integrin is involved in more mature adhesions (38), this difference indicates that α-actinin would inhibit talin binding to early adhesions but may enhance binding in mature adhesions.
Initial Adhesion Formation Is Enhanced Upon α-Actinin Depletion.
Our data seem thus to suggest a model in which talin first mediates binding and activation of β3 integrins (10) and the subsequent force generation and reinforcement of initial adhesions (11, 12). Then, α-actinin competes with talin to bind β3 integrins, transmit forces, and trigger maturation (Figs. 1 and 2). This model predicts that the initial talin-mediated step of adhesion formation should be enhanced after depleting α-actinin because of reduced competition. To test this theory, we placed 0.5-μm beads in contact with the lamellipodium of cells with an optical trap, and observed the ability of cells to adhere to the beads and pull them out of the trap (Fig. S3). In contrast to the 3-μm beads used earlier for the magnetic tweezers assay, these beads were too small to support adhesion maturation without external forces (Fig. S3A), and were thus suited to test only initial adhesion formation. In earlier results, we showed that talin depletion drastically reduced the ability of cells to pull beads out of the optical trap (31). In contrast and as predicted, α-actinin–depleted cells pulled the beads more rapidly (Fig. S3 C and D) and with more persistent linkages that resisted stalling (Fig. S3E). After normalizing by actin rearward flow (which was slightly but nonsignificantly higher in α-actinin–depleted cells) (Fig. S3 F and G), pulling speeds in α-actinin–depleted cells remained strikingly higher (Fig. S3H). Thus, the difference observed was not caused by changes in rearward speed [or in talin expression levels, which remained unaffected (Fig. S1C)].
α-Actinin and Not Talin Correlates with Force Generation in Mature Adhesions.
To further test this model, we used TIRF time-lapse microscopy to analyze the spreading on fibronectin of cells cotransfected with α-actinin-GFP and talin-mCherry. This process showed that maturing adhesions progressively recruited both talin and α-actinin, with α-actinin recruitment being apparently faster (Fig. 5 A–C and Movie S5). Thus, competition for β3 binding could explain the incorporation of α-actinin, although this would not lead to a substitution of talin but rather to stable fractions of α-actinin and talin-bound β3 tails. As adhesions matured, incorporation of β1 integrins may further help recruiting both talin and α-actinin in a cooperative manner. Regardless of this, however, if adhesion maturation is triggered by forces transmitted by α-actinin and not talin, then in mature adhesions α-actinin and not talin should regulate forces on the matrix. To test this hypothesis, we analyzed the forces generated over time by cells cotransfected with α-actinin-GFP and talin-mCherry and plated on fibronectin-coated 1-μm pillars. When attached to pillars larger than 0.5 μm, cells organize their adhesions around each pillar, thus allowing the study of adhesion dynamics and force generation for each individual pillar (39). After adhering to pillars, cells indeed localized both talin and α-actinin in circles around each pillar (Fig. 5D). However, whereas the intensity of talin fluorescence around each pillar remained fairly constant, that of α-actinin was highly dynamic (Fig. 5 E and F and Movie S6). Furthermore, the oscillations in fluorescence intensity of α-actinin showed a moderate correlation with force generation on the underlying pillars (Figs. 5 F and G), suggesting that α-actinin could regulate force transmission. The correlation between force generation and talin dynamics was significantly lower (Fig. 5 F and G). To check if the constant intensity of talin-mCherry could be because of an artifact, we repeated the experiment with GFP-talin. GFP-talin localized identically to talin-mCherry for cells spread on fibronectin-coated glass (Fig. S6A) and in most cases also showed a constant intensity on pillars uncorrelated to pillar force (Fig. S6 B–D). The constant talin fluorescence levels, however, may still hide small scale oscillations correlated with force. To discard this, we specifically searched for pillars that didn’t have constant talin levels, and showed visible force and talin fluorescence fluctuations. These fluctuations were not correlated (Fig. S6 E–G). Thus, force generation in mature adhesions better correlated with the recruitment of α-actinin than talin.
Fig. 5.
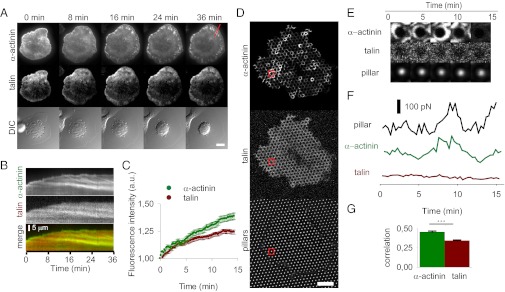
Dynamics of talin and α-actinin. (A) Time sequence of cells cotransfected with α-actinin-GFP and talin-mCherry. DIC, differential interference contrast. (Scale bar, 20 µm.) (See also Movie S5). (B) Kymograph corresponding to red line in A showing the formation of α-actinin and talin adhesions (green and red, respectively, in merged image) as a function of time. (C) Quantification of relative increase in talin and α-actinin fluorescence with maturation. The fluorescence of both molecules was arbitrarily set to 1 at the beginning of the measurement. Differences were significant (n = 87 adhesions in four cells measured on 2 different days, P < 0.001). (D) Confocal image showing a cell transfected with α-actinin-GFP and talin-mCherry plated on a 1-µm pillar array. Fluorescence GFP and mCherry images and bright-field pillar image are shown. (Scale bar, 10 µm.) (See also Movie S6). (E) Time sequence of GFP and mCherry fluorescence intensity and pillar position corresponding to the pillar marked in red in D. (F) Corresponding traces of pillar force, GFP, and mCherry fluorescence. Fluorescence intensity is in arbitrary units. (G) Average correlation between α-actinin and force traces (green) and between talin and force traces (red). A correlation of 1 would indicate that both traces follow the exact same trend, and a correlation of 0 would indicate two completely unrelated traces. n = 110 pillars from three cells measured on 2 different days, ***P < 0.001.
Cell Response to α-Actinin Depletion Depends on Integrin Specificity of the Substrate.
Another prediction of our model is that the different interactions between α-actinin and talin with β3 integrins (competition) and with β1 integrins (cooperation and perhaps weaker interaction in vivo) should lead to opposite effects of α-actinin depletion on substrates binding to β3 or β1 integrins. To test this hypothesis, we analyzed cell force generation on pillars coated with either vitronectin or collagen I, matrices which bind respectively to β3 and β1 integrins (32, 40). On vitronectin-coated pillars we obtained the same results as on fibronectin (Fig. 3 B and C). Namely, the depletion of α-actinin resulted in increased forces, which decreased to control levels upon rescue with FL α-actinin, and were reduced even further upon rescue with SR12-GFP (Fig. 6 A and B). On collagen-coated pillars, as predicted, we found the opposite results: in α-actinin–depleted cells, force generation was decreased. Rescue of depleted cells by FL α-actinin increased force generation, but rescue with SR12-GFP did not (Fig. 6 C and D). This substrate-specific behavior strongly suggests direct force transmission between integrins and α-actinin in vivo, and further supports our competition/cooperation model. Indeed, on collagen, α-actinin–depleted cells would show decreased force generation because of decreased cooperation between α-actinin and talin for β1 binding. Alternatively, if interactions of α-actinin with β1 integrin were less relevant than with β3 (as suggested by the immunoprecipitation results), decreased forces on collagen could simply be a result of decreased actin cross-linking. The only partial rescue of force generation in α-actinin 4 depleted cells by FL α-actinin 1 may hint at a specific role of α-actinin 4. Indeed, such a specific role of α-actinin 4 in stress fiber cross-linking has been suggested (25).
Fig. 6.

Role of α-actinin in cell contractility depends on matrix coating. (A) Vector plots with arrows depicting the magnitude and direction of forces exerted on 1-µm pillar arrays coated with vitronectin. Cells were transfected with either NT or α-actinin shRNA and rescued with FL or SR12 α-actinin-GFP. (Scale bar, 20 µm; force scale bar indicates the length of a force arrow of 2 nN.) (B) Corresponding quantification of average forces (strain energy) exerted by cells. (C and D) Same results on collagen I-coated pillars. *P < 0.05, n ≥ 11 cells measured on 2 different days.
Alternative Hypotheses.
Actin cross-linking is one of the main functions of α-actinin (17, 18), and could potentially explain some of our phenotypes. Indeed, a myosin IIA mutant (GFP-MIIA-N93K) that constitutively cross-links actin (5, 41) but has no motor activity (42) was shown previously to rescue the orientation and elongation of maturing adhesions in α-actinin–depleted cells (18). To evaluate this result, we rescued actin cross-linking in α-actinin–depleted cells with GFP-MIIA-N93K. However, whereas we did observe that rescued cells had longer and more centripetally oriented adhesions, the large focal adhesions present in control cells were not restored (Fig. S7 A and B). Similarly, GFP-MIIA-N93K rescue did not change force generation of depleted cells (Fig. S7 C and D), nor did it rescue reinforcement (Fig. S7 E and F). Thus, restoring actin cross-linking is not sufficient to enable focal adhesion formation, normal force generation, or mechanotransduction.
Besides actin cross-linking, overexpression of myosin light chain 2 (MLC2) has also been suggested to explain the increased contractility on collagen of cells from a stable α-actinin 4 knock-down line (43). We observed an opposite effect on collagen (Fig. 6 C and D), and we did not observe an overexpression of MLC2 (Fig. S1C). This finding suggests that high contractility and MLC2 expression could be a compensatory mechanism of the stable knock-down cell line. Furthermore, this knock-down cell line had unmodified levels of total α-actinin (unlike with our shRNA approach) (Fig. 1), likely masking any effect of competition or cooperation. Thus, neither actin cross-linking nor MLC2 expression could explain our results. This finding is therefore consistent with the substrate-specific role of α-actinin in force generation (Fig. 6), which may not be explained by interactions of α-actinin with binding partners like actin or myosin. In conclusion, only effects on substrate-specific molecules like integrins could account for our results. These effects should be given by α-actinin–integrin interactions and not by indirect effects on integrin expression, because integrin expression levels on the membrane before spreading were not affected by α-actinin depletion or the different α-actinin mutants studied (Fig. S8).
Discussion
Here, we show that α-actinin competes with talin for the binding to the tails of β3 integrins, and cooperates for the binding to β1 integrins (Fig. 4). A structural alignment model had already suggested this cooperation for β1 binding (28), whereas the competition for β3 binding might arise from the sequence differences between β3 and β1 cytoplasmic tails (40%) (22, 28). These interactions can explain why α-actinin impairs the talin- and β3-dependent formation of nascent adhesions (Fig. S3) and also the opposite effects of α-actinin on the generation of force on collagen vs. fibronectin/vitronectin substrates (Figs. 3 and 6). Once bound to integrins, α-actinin appears to transmit force from the cytoskeleton to integrins (Fig. 3), triggering mechanotransduction (Fig. 2) and adhesion maturation (Fig. 1). As adhesions are formed, α-actinin correlates with force generation (Fig. 5). We thus show how α-actinin transmits force to initial adhesions, and catalyzes adhesion maturation. This mechanism also explains earlier findings showing that α-actinin is required for adhesion maturation (18), and that fragments of α-actinin can impair focal adhesion formation (20, 21).
Although the role and relevance of the actin cross-linking properties of α-actinin are well studied (17), here we find that actin cross-linking does not rescue focal adhesion formation, force generation, or mechanotransduction in α-actinin–depleted cells (Fig. S7). Even though it has been previously suggested (43), changes in MLC expression are not consistent with our results either (Fig. S1). Other alternative hypotheses could be envisioned: α-actinin changes could be acting through effects on stress fibers (19), signaling through tyrosine phosphorylation (44), or by interacting with myosin and impairing force generation, for example. However, these mechanisms do not depend on the ECM, and therefore cannot explain the opposite effects of α-actinin depletion on collagen versus fibronectin or vitronectin matrices (Figs. 3 and 6). The finding that expressing α-actinin’s integrin binding domain dramatically reduced force generation on fibronectin in depleted cells without restoring actin binding, stress fibers, or focal adhesions (Figs. 1 and 3) further discards any explanation based merely on actin interactions. Thus, only interactions between α-actinin and matrix-binding molecules like integrins can explain our data. For example, α-actinin could have an opposite role in the activation of β1 vs. β3 integrins. However, talin and kindlin appear to be sufficient for activating integrins (10, 37, 45), and any impairment of activation should result in a decrease not only in forces but also in spreading and adhesion on the corresponding substrate, which we did not observe. Indirect effects of α-actinin depletion on integrin expression were also discarded (Fig. S8). The simplest substrate-specific mechanism that can account for our results is therefore the observed competition between talin and α-actinin for binding to β3 integrins, and cooperation (or perhaps weak or no interaction) in binding to β1 integrins.
Based on this competition/cooperation mechanism, we propose the following model. First, the early molecular mechanical contacts between the cell and the ECM are formed in a process that can be measured with the small probes and low forces characteristic of optical tweezers. In this step on fibronectin, talin forms an initial link between β3 integrins and the cytoskeleton that results in forces on the ECM (12). Talin is required to activate β3 integrins (10) for binding to the ECM, and its stretching by force can readily lead to mechanotransduction events such as vinculin binding (13) and initial adhesion reinforcement. This step appears to be inhibited by α-actinin, because it will compete with talin for binding to β3 integrins but it will not activate integrins, impairing integrin-fibronectin binding and, therefore, the formation of a nascent adhesion. Once the early adhesion forms, the normal turnover of talin (46) could result in replacement by either another talin or a competing α-actinin molecule. This process leads to a progressive buildup of α-actinin molecules in adhesions, reaching an eventual equilibrium between integrin-bound α-actinin and talin molecules. At this point, α-actinin can effectively connect actin to integrins (which have previously been activated by talin) and transmit forces to the ECM. This force transmission leads to another mechanotransduction step (measurable with larger probes and the higher forces exerted by magnetic tweezers) resulting in adhesion maturation. In mature adhesions, further from the leading edge and where β1 integrins predominate (38), talin and α-actinin could then strengthen the adhesion by possibly cooperating in binding to β1. Regarding the fraction of α-actinin molecules likely bound to integrins, both termini of α-actinin have been recently found in average to localize closer to actin than to integrins in the z axis (47), an expected result because in the α-actinin antiparallel dimer both termini are right next to an actin binding domain. This result suggests that the fraction of α-actinin involved in integrin binding is the minor fraction of the total α-actinin.
In α-actinin–depleted cells, talin would still mediate initial force generation, but it would not be replaced by α-actinin, leading to a high-force state without stress fibers or focal adhesions that does not further mature. Interestingly, this phenotype of α-actinin–depleted cells confirms that, even if there is often a correlation, force generation does not require stress fibers and focal adhesions. Indeed, high forces in spreading cells are generated early and coinciding with the beginning of the P2 spreading phase, when focal adhesions are not yet formed (48–50). Therefore, a contractile actin cytoskeleton can generate high forces with only early adhesions. Even if talin is known to mediate this initial force generation, however, it remains unclear how forces are transmitted in the absence of α-actinin. Indeed, we did not observe a correlation between talin and force generation (Fig. 5) and talin domains are two orders of magnitude softer than α-actinin’s (16), suggesting that their stretching may not be very effective at transmitting forces. Supporting this theory, stretching cycles of single talin molecules (39) have a time scale which does not coincide with that of force generation in small adhesions (51). Thus, although talin stretching could provide the initial mechanosensing event leading to the formation of initial adhesions, molecules recruited downstream of this event (currently unidentified) could be responsible for the transmission of forces. In any case, in the absence of α-actinin this force transmission would not lead to adhesion maturation.
In the maturation step, the properties of α-actinin as a docking platform put it in an ideal position to serve as a connector to the actin cytoskeleton, where it binds more tightly than talin (52). First, α-actinin could trigger mechanotransduction and subsequent adhesion maturation by transmitting forces to several binding partners, including integrins themselves, vinculin (53), zyxin (30), zyxin binding proteins such as p130Cas (54) or Ena/VASP proteins (55), and others. Second, it could adjust this force transmission through mechanisms such as the regulation of its integrin binding (56) or actin cross-linking (57) by phosphoinositides. Third, α-actinin could promote adhesion dynamics and force release by serving as a docking platform for FAK (44, 58, 59) or upon cleavage by calpain (60). Thus, although the specific mechanisms remain to be elucidated, these interactions could allow α-actinin to increase force generation through increased actin cross-linking and integrin binding. This process would then result in the dynamic force generation enabled by α-actinin (Fig. 3E), and supports the correlation observed between force generation and α-actinin recruitment (Fig. 5).
In summary, we establish that α-actinin competes with talin for binding to β3 integrin. We suggest that α-actinin enters nascent adhesions through this competition, and then triggers adhesion maturation, dynamic force generation, and force release by connecting integrins to the cytoskeleton. Once the two mechanotransduction steps led by talin and α-actinin have established both initial and mature adhesions, the cell is in place for the essential biomechanical cell–ECM interplay that determines downstream processes in motility, development, and cancer. Given the integration of multiple signals and the various steps that cells require to develop adhesions for biological functions, nonlinear mechanisms, such as the one unveiled here, are likely to occur in many motile phenomena.
Materials and Methods
Spreading Experiments.
For spreading experiments, cells were trypsinized (0.05% Trypsin-EDTA) and resuspended in Ringer buffer solution (150 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 20 mM Hepes, and 2 g/L glucose, pH 7.4). After allowing the cells to recover for 30 min at 37 °C, cells were then plated on coverslips silanized with 1,1,1,3,3,3,-hexamethyldisilazane (Sigma) and coated with 10 µg/mL fibronectin (Roche) for 2 h at 37 °C. For immunofluorescence microscopy, cells were then fixed with 4% (vol/vol) paraformaldehyde, permeabilized with 0.1% Triton X-100, and labeled first with primary antibodies (1 h at room temperature) and then Alexa-conjugated IgG secondary antibodies (1 h at room temperature; Invitrogen). Actin was stained with phalloidin-Texas red (Invitrogen) together with primary antibodies. Integrin β3 and talin localization were assessed by transfecting cells with integrin β3-GFP, GFP-talin, and talin-mCherry (not with antibodies). Fluorescence, differential interference contrast, and TIRF images were taken using an Olympus IX81 fluorescence microscope with a 60×, N.A. 1.45 objective. To quantify colocalization between proteins, the Pearson correlation coefficient between images was calculated using the JaCoP plugin in ImageJ (34) after removing image background. Only the cell periphery where adhesions form was used for the analysis.
Magnetic and Optical Tweezer Measurements.
Carboxylated 3-µm magnetic beads (Invitrogen) and silica beads (Kisker Biotech), and avidinated 0.5-µm silica beads (Bangs laboratories) were coated with a mixture of biotinylated pentameric FN7-10 and biotinylated BSA as previously described (27). FN7-10 (61) is a four-domain segment of fibronectin responsible for cell binding and containing the RGD and PHSRN motifs. Reinforcement was measured with magnetic tweezers as previously described (27). Briefly, magnetic beads in cell lamellipodia were observed for 20 s and then submitted to a 1-Hz 1-nN pulsation. Reinforcement was calculated as bead stiffness (applied force/pulsation amplitude) normalized by initial stiffness. For optical tweezer measurements, 0.5-µm beads were captured with a 100-pN/µm laser trap setup (Axiovert 100TV; Zeiss), brought into contact with the cell (1–2 μm from the leading edge), and tracked while the cell established adhesion and attempted to displace them from the trap. In the same cells, the movement of cytoplasmic markers near the beads was also tracked to estimate the speed of rearward flow.
Pillar Measurements.
Preparation of pillar arrays (6-µm height, 1-µm diameter, 2-µm center to center, k =1.87 nN/µm) coated with 10 μg/mL of fibronectin (Invitrogen), collagen I (Roche Applied Science), or vitronectin (Invitrogen) and measurement of forces exerted by cells on them was done as previously described (9). Briefly, multiple-particle tracking software (62) was used to calculate the deflection d of pillars from the initial undeflected array. Pillar forces were then calculated as kd, and the strain energy exerted by the cell on the pillars was calculated as the sum of 1/2 kd2 for all pillars in the cell periphery. Correlations between pillar force and GFP and mCherry fluorescence intensities (Fig. 5) were computed by detrending the pillar force and fluorescence traces (obtained with a 60× objective in an Olympus Fluoview FV500 laser-scanning confocal microscope) and then calculating the normalized cross-correlation function between them.
Western Blotting and Pull-Down Assays.
Cells were rinsed once with PBS and lysed with RIPA buffer or Nonidet P-40 buffer (for immunoprecipitation). For imminoprecipitation, lysates were then incubated for 1 h with rabbit polyclonal anti-β3 or anti-β1 Ab (Millipore) and 1 h with protein A/G slurry (Santa Cruz), and washed three times. Samples were then combined with 4× loading buffer, boiled for 5 min at 100 °C and loaded onto 4–20% gradient Tris-glycine gels (Lonza). Protein was then transferred to an Optitran reinforced nitrocellulose membrane (Whatman), which was blocked with 5% (wt/vol) dry milk-Tris buffered saline (TBS) and incubated with primary antibodies overnight at 4 °C. The membrane was then incubated with secondary anti-rabbit or anti-mouse–HRP Abs (Amersham Biosciences), or Clean-Blot IP Detection Reagent (Thermo Scientific). HRP signal was detected with ECL Western blotting reagents (Amersham Biosciences) on Hyblot CL film (Denville Scientific). As the β3 Ab did not recognize denatured β3, for β3 immunoprecipitation cells were also transfected with β3-GFP, and EGFP Ab was used to quantify pull-down. For in vitro pull-down assays, 6xHis-tagged β1A, β3, and β3SCR integrin cytoplasmic tails were expressed as described previously (63) from cDNAs obtained as a gift from the Ginsberg laboratory (San Diego, CA). His-tagged talin head (aa 196–405) was a gift of the Critchley laboratory (Leicester, United Kingdom). The His-tag in the expressed talin head protein was cleaved before experiments. The SR12 construct was cloned from the GFP vector used for cell transfection into the pGEX-6P-1 vector, and then the expressed GST-protein was purified. Pull-down assay was then conducted by first binding the integrin tails to Ni-NTA magnetic beads (Qiagen), and then mixing with SR12-GST or talin head as described previously (63). After washing beads, Western blotting was used to detect binding of talin head (H-300 polyclonal talin antibody; Santa Cruz Biotechnology) and SR12-GST (GST monoclonal antibody; Millipore). Signal quantification was done with ImageJ software.
Cells, Constructs, and Reagents.
Mouse embryonic fibroblasts were cultured in DMEM supplemented with 10% (vol/vol) FBS. Transfection was carried out using the Amaxa Nucleofector System (Lonza), using 106 cells per reaction and 5–6 µg DNA. shRNA pLKO.1-puro vectors with a nontargeting sequence (CAACAAGATGAAGAGCACCAA) and a sequence targeting mouse α-actinin 4 (CCTCTCTTTCTCAGTCTTGTA) were from Sigma. Cells were transfected 4 d before measurements and selected using 1.5 µg/mL puromycin (Sigma) 6 h after transfection. Antibodies used for immunostaining and Western blotting were polyclonal rabbit antibodies against α-actinin 4 (Alexis Biochemicals), total myosin light chain (MRCL3/MRCL2/MYL9; Santa Cruz), MLC2 (Cell Signaling) monoclonal mouse antibodies against total α-actinin (US Biological; clone O.T.02), GADPH (Santa Cruz Antibodies; clone sc-32233), paxillin (BD transduction laboratories; clone 349), talin (Sigma; clone 8D4), and EGFP (Clontech; 632569) and monoclonal rat antibody against activated β1 integrin (BD Biosciences; clone 9EG7). To check integrin expression levels on the membrane, we used function-blocking monoclonal antibodies against α5β1 (Millipore; clone BMB5 in rat), β3 (Beckton Dickinson; clone 2C9.G2 in Armenian hamster), and Trinitrophenol (Beckton Dickinson; clone A19-3 in Armenian hamster). Human α-actinin 1-GFP (50) and integrin β3-GFP (64) were described previously. ABDdel and SR12 constructs were mutated from α-actinin-GFP using the Quikchange mutagenesis kit (Stratagene). In ABDdel, the actin binding domain (amino acids 30–253) was deleted, and SR12 contained only the first 29 amino acids of the protein, domains SR1 and SR2 (amino acids 267–507), and the last 5 amino acids. Talin-mCherry was prepared from GFP-talin, which was described previously (11). GFP-MIIA-N93K was a gift of Miguel Vicente-Manzanares (Universidad Autónoma de Madrid, Madrid, Spain) (18).
Statistical Analysis.
Statistical comparisons were done with two-tailed Student t tests when two cases were compared and with ANOVA tests when more comparisons were done. When data did not meet normality criteria, Mann–Whitney rank sum tests were performed instead. All data shown are mean ± SEM.
Supplementary Material
Acknowledgments
We thank L. Chew, S. Moore, X. Zhang, S. Ghassemi, G. Meacci, and D. Navajas for technical support and useful discussions, and the members of the M.P.S. laboratory. This work was funded by the National Institutes of Health through RO1 Grant EB001480 (to M.P.S.), the National Institutes of Health Roadmap for Medical Research (PN2 EY016586); Spanish Ministry of Economy and Competitiveness (Grant BFU2011-23111); a Career Integration Grant within the 7th European Community Framework Programme (PCIG10-GA-2011-303848); a Marie Curie International Outgoing Fellowship within the 7th European Community Framework Programme (PIOF-GA-2008-219401, to P.R.-C.); a National Institutes of Health award (to A.d.R.); and a Ramon y Cajal award (to A.d.R.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1220723110/-/DCSupplemental.
References
- 1.Paszek MJ, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8(3):241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 2.Mammoto T, Ingber DE. Mechanical control of tissue and organ development. Development. 2010;137(9):1407–1420. doi: 10.1242/dev.024166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Riveline D, et al. Focal contacts as mechanosensors: Externally applied local mechanical force induces growth of focal contacts by an mDia1-dependent and ROCK-independent mechanism. J Cell Biol. 2001;153(6):1175–1186. doi: 10.1083/jcb.153.6.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cai YF, Sheetz MP. Force propagation across cells: Mechanical coherence of dynamic cytoskeletons. Curr Opin Cell Biol. 2009;21(1):47–50. doi: 10.1016/j.ceb.2009.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vicente-Manzanares M, Zareno J, Whitmore L, Choi CK, Horwitz AF. Regulation of protrusion, adhesion dynamics, and polarity by myosins IIA and IIB in migrating cells. J Cell Biol. 2007;176(5):573–580. doi: 10.1083/jcb.200612043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galbraith CG, Yamada KM, Sheetz MP. The relationship between force and focal complex development. J Cell Biol. 2002;159(4):695–705. doi: 10.1083/jcb.200204153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jaalouk DE, Lammerding J. Mechanotransduction gone awry. Nat Rev Mol Cell Biol. 2009;10(1):63–73. doi: 10.1038/nrm2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fenteany G, Janmey PA, Stossel TP. Signaling pathways and cell mechanics involved in wound closure by epithelial cell sheets. Curr Biol. 2000;10(14):831–838. doi: 10.1016/s0960-9822(00)00579-0. [DOI] [PubMed] [Google Scholar]
- 9.Cai YF, et al. Nonmuscle myosin IIA-dependent force inhibits cell spreading and drives F-actin flow. Biophys J. 2006;91(10):3907–3920. doi: 10.1529/biophysj.106.084806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bouaouina M, Lad Y, Calderwood DA. The N-terminal domains of talin cooperate with the phosphotyrosine binding-like domain to activate beta1 and beta3 integrins. J Biol Chem. 2008;283(10):6118–6125. doi: 10.1074/jbc.M709527200. [DOI] [PubMed] [Google Scholar]
- 11.Zhang X, et al. Talin depletion reveals independence of initial cell spreading from integrin activation and traction. Nat Cell Biol. 2008;10(9):1062–1068. doi: 10.1038/ncb1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang GY, Giannone G, Critchley DR, Fukumoto E, Sheetz MP. Two-piconewton slip bond between fibronectin and the cytoskeleton depends on talin. Nature. 2003;424(6946):334–337. doi: 10.1038/nature01805. [DOI] [PubMed] [Google Scholar]
- 13.del Rio A, et al. Stretching single talin rod molecules activates vinculin binding. Science. 2009;323(5914):638–641. doi: 10.1126/science.1162912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pavalko FM, LaRoche SM. Activation of human neutrophils induces an interaction between the integrin beta 2-subunit (CD18) and the actin binding protein alpha-actinin. J Immunol. 1993;151(7):3795–3807. [PubMed] [Google Scholar]
- 15.Pavalko FM, Otey CA, Simon KO, Burridge K. Alpha-actinin: A direct link between actin and integrins. Biochem Soc Trans. 1991;19(4):1065–1069. doi: 10.1042/bst0191065. [DOI] [PubMed] [Google Scholar]
- 16.Roca-Cusachs P, Iskratsch T, Sheetz MP. Finding the weakest link: Exploring integrin-mediated mechanical molecular pathways. J Cell Sci. 2012;125(Pt 13):3025–3038. doi: 10.1242/jcs.095794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sjöblom B, Salmazo A, Djinović-Carugo K. Alpha-actinin structure and regulation. Cell Mol Life Sci. 2008;65(17):2688–2701. doi: 10.1007/s00018-008-8080-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choi CK, et al. Actin and alpha-actinin orchestrate the assembly and maturation of nascent adhesions in a myosin II motor-independent manner. Nat Cell Biol. 2008;10(9):1039–1050. doi: 10.1038/ncb1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rajfur Z, Roy P, Otey C, Romer L, Jacobson K. Dissecting the link between stress fibres and focal adhesions by CALI with EGFP fusion proteins. Nat Cell Biol. 2002;4(4):286–293. doi: 10.1038/ncb772. [DOI] [PubMed] [Google Scholar]
- 20.Pavalko FM, Burridge K. Disruption of the actin cytoskeleton after microinjection of proteolytic fragments of alpha-actinin. J Cell Biol. 1991;114(3):481–491. doi: 10.1083/jcb.114.3.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Triplett JW, Pavalko FM. Disruption of alpha-actinin-integrin interactions at focal adhesions renders osteoblasts susceptible to apoptosis. Am J Physiol Cell Physiol. 2006;291(5):C909–C921. doi: 10.1152/ajpcell.00113.2006. [DOI] [PubMed] [Google Scholar]
- 22.Legate KR, Fässler R. Mechanisms that regulate adaptor binding to beta-integrin cytoplasmic tails. J Cell Sci. 2009;122(Pt 2):187–198. doi: 10.1242/jcs.041624. [DOI] [PubMed] [Google Scholar]
- 23.Harburger DS, Calderwood DA. Integrin signalling at a glance. J Cell Sci. 2009;122(Pt 2):159–163. doi: 10.1242/jcs.018093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Araki N, Hatae T, Yamada T, Hirohashi S. Actinin-4 is preferentially involved in circular ruffling and macropinocytosis in mouse macrophages: Analysis by fluorescence ratio imaging. J Cell Sci. 2000;113(Pt 18):3329–3340. doi: 10.1242/jcs.113.18.3329. [DOI] [PubMed] [Google Scholar]
- 25.Honda K, et al. Actinin-4, a novel actin-bundling protein associated with cell motility and cancer invasion. J Cell Biol. 1998;140(6):1383–1393. doi: 10.1083/jcb.140.6.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Balaban NQ, et al. Force and focal adhesion assembly: A close relationship studied using elastic micropatterned substrates. Nat Cell Biol. 2001;3(5):466–472. doi: 10.1038/35074532. [DOI] [PubMed] [Google Scholar]
- 27.Roca-Cusachs P, Gauthier NC, Del Rio A, Sheetz MP. Clustering of α(5)β(1) integrins determines adhesion strength whereas α(v)β(3) and talin enable mechanotransduction. Proc Natl Acad Sci USA. 2009;106(38):16245–16250. doi: 10.1073/pnas.0902818106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kelly DF, Taylor KA. Identification of the beta1-integrin binding site on alpha-actinin by cryoelectron microscopy. J Struct Biol. 2005;149(3):290–302. doi: 10.1016/j.jsb.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 29.McGregor A, Blanchard AD, Rowe AJ, Critchley DR. Identification of the vinculin-binding site in the cytoskeletal protein alpha-actinin. Biochem J. 1994;301(Pt 1):225–233. doi: 10.1042/bj3010225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crawford AW, Michelsen JW, Beckerle MC. An interaction between zyxin and alpha-actinin. J Cell Biol. 1992;116(6):1381–1393. doi: 10.1083/jcb.116.6.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giannone G, Jiang G, Sutton DH, Critchley DR, Sheetz MP. Talin1 is critical for force-dependent reinforcement of initial integrin-cytoskeleton bonds but not tyrosine kinase activation. J Cell Biol. 2003;163(2):409–419. doi: 10.1083/jcb.200302001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hynes RO. Integrins: Bidirectional, allosteric signaling machines. Cell. 2002;110(6):673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 33.Cluzel C, et al. The mechanisms and dynamics of (alpha)v(beta)3 integrin clustering in living cells. J Cell Biol. 2005;171(2):383–392. doi: 10.1083/jcb.200503017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bolte S, Cordelières FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224(Pt 3):213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- 35.Otey CA, Pavalko FM, Burridge K. An interaction between alpha-actinin and the beta 1 integrin subunit in vitro. J Cell Biol. 1990;111(2):721–729. doi: 10.1083/jcb.111.2.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Otey CA, Vasquez GB, Burridge K, Erickson BW. Mapping of the alpha-actinin binding site within the beta 1 integrin cytoplasmic domain. J Biol Chem. 1993;268(28):21193–21197. [PubMed] [Google Scholar]
- 37.Calderwood DA, et al. The Talin head domain binds to integrin beta subunit cytoplasmic tails and regulates integrin activation. J Biol Chem. 1999;274(40):28071–28074. doi: 10.1074/jbc.274.40.28071. [DOI] [PubMed] [Google Scholar]
- 38.Zamir E, et al. Dynamics and segregation of cell-matrix adhesions in cultured fibroblasts. Nat Cell Biol. 2000;2(4):191–196. doi: 10.1038/35008607. [DOI] [PubMed] [Google Scholar]
- 39.Ghassemi S, et al. Cells test substrate rigidity by local contractions on submicrometer pillars. Proc Natl Acad Sci USA. 2012;109(14):5328–5333. doi: 10.1073/pnas.1119886109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Felding-Habermann B, Cheresh DA. Vitronectin and its receptors. Curr Opin Cell Biol. 1993;5(5):864–868. doi: 10.1016/0955-0674(93)90036-p. [DOI] [PubMed] [Google Scholar]
- 41.Hu AH, Wang F, Sellers JR. Mutations in human nonmuscle myosin IIA found in patients with May-Hegglin anomaly and Fechtner syndrome result in impaired enzymatic function. J Biol Chem. 2002;277(48):46512–46517. doi: 10.1074/jbc.M208506200. [DOI] [PubMed] [Google Scholar]
- 42.Kim KY, Kovács M, Kawamoto S, Sellers JR, Adelstein RS. Disease-associated mutations and alternative splicing alter the enzymatic and motile activity of nonmuscle myosins II-B and II-C. J Biol Chem. 2005;280(24):22769–22775. doi: 10.1074/jbc.M503488200. [DOI] [PubMed] [Google Scholar]
- 43.Shao H, Wang JH, Pollak MR, Wells A. α-Actinin-4 is essential for maintaining the spreading, motility and contractility of fibroblasts. PLoS ONE. 2010;5(11):e13921. doi: 10.1371/journal.pone.0013921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Izaguirre G, et al. The cytoskeletal/non-muscle isoform of alpha-actinin is phosphorylated on its actin-binding domain by the focal adhesion kinase. J Biol Chem. 2001;276(31):28676–28685. doi: 10.1074/jbc.M101678200. [DOI] [PubMed] [Google Scholar]
- 45.Moser M, Nieswandt B, Ussar S, Pozgajova M, Fässler R. Kindlin-3 is essential for integrin activation and platelet aggregation. Nat Med. 2008;14(3):325–330. doi: 10.1038/nm1722. [DOI] [PubMed] [Google Scholar]
- 46.Lele TP, Thodeti CK, Pendse J, Ingber DE. Investigating complexity of protein-protein interactions in focal adhesions. Biochem Biophys Res Commun. 2008;369(3):929–934. doi: 10.1016/j.bbrc.2008.02.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kanchanawong P, et al. Nanoscale architecture of integrin-based cell adhesions. Nature. 2010;468(7323):580–584. doi: 10.1038/nature09621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cai YF, et al. Cytoskeletal coherence requires myosin-IIA contractility. J Cell Sci. 2010;123(Pt 3):413–423. doi: 10.1242/jcs.058297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dubin-Thaler BJ, et al. Quantification of cell edge velocities and traction forces reveals distinct motility modules during cell spreading. PLoS ONE. 2008;3(11):e3735. doi: 10.1371/journal.pone.0003735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Giannone G, et al. Periodic lamellipodial contractions correlate with rearward actin waves. Cell. 2004;116(3):431–443. doi: 10.1016/s0092-8674(04)00058-3. [DOI] [PubMed] [Google Scholar]
- 51.Margadant F, et al. Mechanotransduction in vivo by repeated talin stretch-relaxation events depends upon vinculin. PLoS Biol. 2011;9(12):e1001223. doi: 10.1371/journal.pbio.1001223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hu K, Ji L, Applegate KT, Danuser G, Waterman-Storer CM. Differential transmission of actin motion within focal adhesions. Science. 2007;315(5808):111–115. doi: 10.1126/science.1135085. [DOI] [PubMed] [Google Scholar]
- 53.Wachsstock DH, Wilkins JA, Lin S. Specific interaction of vinculin with alpha-actinin. Biochem Biophys Res Commun. 1987;146(2):554–560. doi: 10.1016/0006-291x(87)90564-x. [DOI] [PubMed] [Google Scholar]
- 54.Yi JS, et al. Members of the Zyxin family of LIM proteins interact with members of the p130Cas family of signal transducers. J Biol Chem. 2002;277(11):9580–9589. doi: 10.1074/jbc.M106922200. [DOI] [PubMed] [Google Scholar]
- 55.Hoffman LM, et al. Genetic ablation of zyxin causes Mena/VASP mislocalization, increased motility, and deficits in actin remodeling. J Cell Biol. 2006;172(5):771–782. doi: 10.1083/jcb.200512115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Greenwood JA, Theibert AB, Prestwich GD, Murphy-Ullrich JE. Restructuring of focal adhesion plaques by PI 3-kinase. Regulation by PtdIns (3,4,5)-p(3) binding to alpha-actinin. J Cell Biol. 2000;150(3):627–642. doi: 10.1083/jcb.150.3.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fraley TS, et al. Phosphoinositide binding inhibits alpha-actinin bundling activity. J Biol Chem. 2003;278(26):24039–24045. doi: 10.1074/jbc.M213288200. [DOI] [PubMed] [Google Scholar]
- 58.Zhang ZY, Lin SY, Neel BG, Haimovich B. Phosphorylated alpha-actinin and protein-tyrosine phosphatase 1B coregulate the disassembly of the focal adhesion kinase x Src complex and promote cell migration. J Biol Chem. 2006;281(3):1746–1754. doi: 10.1074/jbc.M509590200. [DOI] [PubMed] [Google Scholar]
- 59.von Wichert G, Haimovich B, Feng GS, Sheetz MP. Force-dependent integrin-cytoskeleton linkage formation requires downregulation of focal complex dynamics by Shp2. EMBO J. 2003;22(19):5023–5035. doi: 10.1093/emboj/cdg492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sprague CR, Fraley TS, Jang HS, Lal S, Greenwood JA. Phosphoinositide binding to the substrate regulates susceptibility to proteolysis by calpain. J Biol Chem. 2008;283(14):9217–9223. doi: 10.1074/jbc.M707436200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Coussen F, Choquet D, Sheetz MP, Erickson HP. Trimers of the fibronectin cell adhesion domain localize to actin filament bundles and undergo rearward translocation. J Cell Sci. 2002;115(Pt 12):2581–2590. doi: 10.1242/jcs.115.12.2581. [DOI] [PubMed] [Google Scholar]
- 62.du Roure O, et al. Force mapping in epithelial cell migration. Proc Natl Acad Sci USA. 2005;102(7):2390–2395. doi: 10.1073/pnas.0408482102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lad Y, Harburger DS, Calderwood DA. Integrin cytoskeletal interactions. Methods Enzymol. 2007;426:69–84. doi: 10.1016/S0076-6879(07)26004-5. [DOI] [PubMed] [Google Scholar]
- 64.Plançon S, Morel-Kopp MC, Schaffner-Reckinger E, Chen P, Kieffer N. Green fluorescent protein (GFP) tagged to the cytoplasmic tail of alphaIIb or beta3 allows the expression of a fully functional integrin alphaIIb(beta3): Effect of beta3GFP on alphaIIb(beta3) ligand binding. Biochem J. 2001;357(Pt 2):529–536. doi: 10.1042/0264-6021:3570529. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.