

Abstract
Aminophospholipid (APL) trafficking across the plasma membrane is a key event in cell activation, apoptosis, and aging and is required for clearance of dying cells and coagulation. Currently the phospholipid molecular species externalized are unknown. Using a lipidomic method, we show that thrombin, collagen, or ionophore-activated human platelets externalize two phosphatidylserines (PSs) and five phosphatidylethanolamines (PEs). Four percent of the total cellular PE/PS pool (∼300 ng/2 × 108 cells, thrombin), is externalized via calcium mobilization and protease-activated receptors-1 and -4, and 48% is contained in microparticles. Apoptosis and energy depletion (aging) externalized the same APLs in a calcium-dependent manner, and all stimuli externalized oxidized phospholipids, termed hydroxyeicosatetraenoic acid-PEs. Transmembrane protein-16F (TMEM-16F), the protein mutated in Scott syndrome, was required for PE/PS externalization during thrombin activation and energy depletion, but not apoptosis. Platelet-specific APLs optimally supported tissue factor-dependent coagulation in human plasma, vs. APL with longer or shorter fatty acyl chains. This finding demonstrates fatty acids as molecular determinants of APL that regulate hemostasis. Thus, the molecular species of externalized APL during platelet activation, apoptosis, and energy depletion were characterized, and their ability to support coagulation revealed. The findings have therapeutic implications for bleeding disorders and transfusion therapy. The assay could be applied to other cell events characterized by APL externalization, including cell division and vesiculation.
Plasma membrane phospholipids are organized with phosphatidylcholine (PC) in the outer leaflet, and the aminophospholipids (APLs), phosphatidylserine (PS), and phosphatidylethanolamine (PE) facing the cytoplasm (1). Loss of asymmetry is a key feature of apoptosis, cell aging, clearance, and coagulation (2). Externalization of APLs by platelets allows coagulation factors to bind and form enzyme/cofactor complexes and aids complement binding (3). Loss of this ability to externalize APLs is characteristic of the rare bleeding disorder Scott syndrome (4). To date, APL trafficking has been measured using indirect approaches including (i) fluorescent PLs added to cells (5), and (ii) annexin V-FITC flow cytometry. This protein recognizes APL on the platelet surface and is used as a nonquantitative probe (6–8). In recent years it has been described as detecting PS exposure, although several annexins also bind PE (6, 9–13). Other proteins that indirectly measure APL exposure include lactadherin and cinnamycin, reported as specific PS or PE indicators, respectively (14–16).
PE and PS are families of lipids that differ according to fatty acyl composition, including both saturated (sn1) and unsaturated (sn2) species such as palmitic (16:0), linoleic (18:1), arachidonic (20:4), and docosahexanoic acids (22:6). For PE, sn1 bond can be acyl, alkyl ether, or vinyl ether (plasmalogen). Currently the specific molecules of APLs that externalize during cell activation or apoptosis are unknown. This information could lead to mechanistic insights into their specific role(s) and reveal functional groups involved in APL-dependent events. Herein we describe characterization and quantitation of APL trafficking across the platelet plasma membrane during activation, apoptosis, and energy depletion (model for aging), using mass spectrometry. Lastly, the APL species exposed by platelets were examined for their activity in supporting tissue factor-dependent thrombin generation in human plasma.
Results
Quantitative Assay for Surface Exposed Aminophospholipids.
EZ-Link Sulfo-NHS-Biotin (SNB; Thermo Fisher Scientific) is a cell-impermeable reagent that biotinylates primary amines and is extensively used for proteomics (17). Similarly, external facing amine headgroups of PE and PS can be derivatized using this label. These lipids are then extracted and analyzed using LC/MS/MS (Figs. S1–S3). A series of biotinylated APL standards were synthesized, and characterized (SI Materials and Methods). Assay optimization used liposomes of defined APL composition, varying time, SNB concentration, lipid extraction, and chromatographic separation. Thus, a quantitative MS method that specifically measures external APL was developed (SI Materials and Methods).
Platelets Externalize Multiple Species of PS and PE on Activation.
Platelet activation by thrombin (0.2 U/mL) or ionophore (10 µM) caused significant PS and PE externalization, with seven molecular species detected (Figs. 1 and 2 A–D). The predominance of 20:4 species reflects its abundance in platelet APL. Collagen-dependent (10 μg/mL) externalization was lower than thrombin, with ionophore highest (Fig. 2 A and B). By pooling data from seven donors, mean values for APL on unstimulated platelets were 34 ± 8 ng PS and 97 ± 33 ng PE/2 × 108 cells (mean ± SEM). After 30 min thrombin, this externalization increased to 215 ± 57 ng PS and 508 ± 152 ng PE/2 × 108 cells. Genetically unrelated donors showed a 10-fold difference between highest and lowest levels. Derivatizing total platelet APLs using the cell-permeable analog, NHS-biotin (NB), demonstrated ∼3% of total PE or PS externalized in response to thrombin (Fig. 2 C and D). Selective protease-activated receptor (PAR) agonists, TFLLR-NH2 (PAR1) or AY-NH2 (PAR4), activated APL externalization (Fig. S3C). Thrombin (0.2 U/mL) activated within 2 min, was with levels elevating for up to 1 h and remaining stable for up to 3 h, and was dose dependent (Figs. 2 E and F and 3 A and B).
Fig. 1.

Structures of APL externalized by human platelets.
Fig. 2.
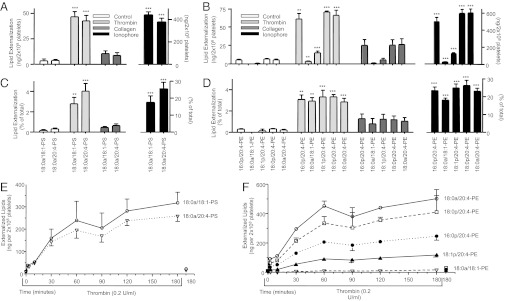
Several APL molecular species are externalized by human platelets in response to agonists and are dose-dependent to thrombin. (A and B) Platelets were activated using thrombin (0.2 U/mL), collagen (10 µg/mL), or ionophore (A23187, 10 µM) at 37° for 30 min, before biotinylation, lipid extraction, and LC/MS/MS analysis as in Materials and Methods. (C and D) Total APL in each sample was determined by biotinylation using NB, as described in Materials and Methods, and used to calculate the relative proportion externalized on platelet activation. (E and F) Platelets were activated by thrombin (0.2 U/mL) for varying times, as shown, before biotinylation, lipid extraction, and LC/MS/MS analysis as in Materials and Methods (n = 3; mean ± SEM; data representative of three independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. control, using ANOVA and Bonferroni post hoc test).
Fig. 3.
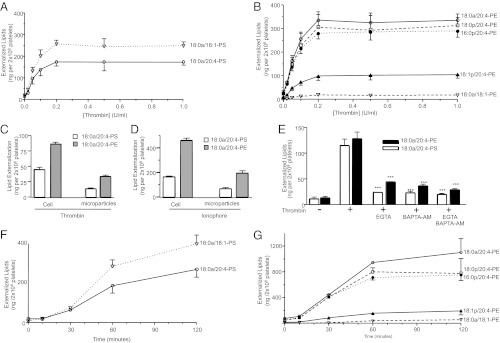
Externalization of APL is time dependent, associated with microparticles, requires calcium, and occurs during apoptosis. (A and B) Platelets were activated with varying amounts of thrombin for 120 min, before biotinylation, lipid extraction, and LC/MS/MS analysis as in Materials and Methods. (C and D) Platelets were activated using thrombin (0.2 U/mL, 30 min), then centrifuged as described in Materials and Methods. Supernatant (microparticles) and pellets were separately analyzed for surface exposed APLs. (E) Platelets were activated using thrombin (0.2 U/mL, 30 min) after 10 min preincubation with Ca2+ chelators (EGTA, 1mM and/or BAPTA-AM, 10 μM). (F and G) Platelets were activated using 1 μM ABT-737 for varying times before lipid extraction and analysis. For all experiments, n = 3; mean ± SEM; data representative of three independent donors. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. control, using ANOVA and Bonferroni post hoc test.
Platelet Microparticles Contain External Facing APLs, and Externalization Requires Calcium Influx.
Although the majority of the externalized APLs were present in the platelets, a significant proportion remained in the supernatant, presumably associated with microparticles, with the microparticle external APL composition reflecting the pattern observed in platelets (Fig. 3 C and D). APL externalization was blocked by chelation of extra- (EGTA) or intracellular Ca2+ 1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester) (BAPTA-AM) (Fig. 3E). Potential roles for several other signaling pathways were excluded using pharmacological inhibitors, including p38 MAP kinase (SB203580), MAPK/ERK kinase 1 (PD98059), phospholipase C (U-73112), Src-family tyrosine kinase (PP2), PI3 kinase (wortmannin), or protein kinase C (GF-109203X) (Fig. S3 D and E).
Platelet APL Exposure During Apoptosis, Energy Depletion, and in Scott Syndrome.
Platelets become annexin V positive during apoptosis and aging (18–20). Recent studies on Scott syndrome identified a critical role for a unique protein, transmembrane protein-16F (TMEM-16F), in APL externalization (21, 22). Here, APL externalized during “aging,” apoptosis, and in Scott syndrome are compared. For apoptosis, ABT-737, that targets B-cell lymphoma 2 (Bcl-2) and B-cell lymphoma-extra large (Bcl-xL), was used (Fig. 3 F and G) (23). To mimic aging-dependent changes, thought due to energy depletion, rotenone, an inhibitor of complex I, that causes loss of cellular ATP, in glucose-free buffer was used (24, 25). The pattern and magnitude of APL externalization was similar to thrombin, with the same predominant species externalized (Figs. 2 E and F, 3 F and G, and 4 A and B). Both rotenone and ABT-737–dependent externalization were inhibited by EGTA/BAPTA-AM, indicating requirement for calcium (Fig. 4 C and D). Only the ABT-737 response was blocked by the caspase inhibitor Q-VD-OPH (Fig. 4D). In contrast, in the absence of TMEM-16F, thrombin, and rotenone were unable to stimulate robust APL externalization, although the apoptosis response was still largely intact (Fig. 4 E and F). This finding indicates that energy depletion and thrombin activation of APL externalization is distinct from apoptosis, although calcium is required for all.
Fig. 4.
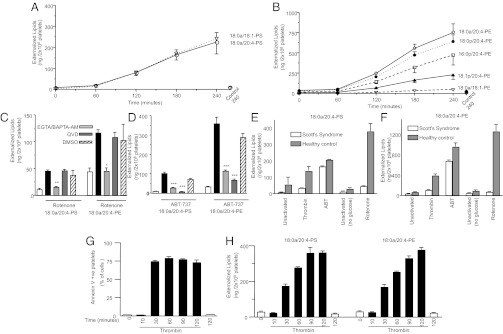
(i) Energy depletion causes similar APL externalization to thrombin, (ii) energy depletion and apoptosis APL exposure require calcium mobilization, (iii) TMEM-16F is required for APL externalization in response to thrombin, or energy depletion, but not apoptosis, and (iv) annexin V-FITC binding vs. biotinylation of APL reveals different kinetics. (A and B) Platelets were activated using 10 μM rotenone for varying times before lipid extraction and analysis. (C and D) Platelets were activated with 10 μM rotenone for 180 min, or with 1 μM ABT-737 for 60 min, after preincubation with inhibitors (Q-VD-OPH, caspase inhibitor, 10 μM), EGTA (1 mM), BAPTA-AM (10 μM), then surface APL analyzed as described in Materials and Methods. (E and F) Scott syndrome or healthy control platelets were activated with 0.2 U/mL thrombin for 30 min, 1 μM ABT-737 for 60 min, or 10 μM rotenone (in the absence of glucose) for 180 min, then surface APL analyzed as described in Materials and Methods. (G and H) Annexin V-FITC binding was determined using flow cytometry, or APL exposure measured using biotinylation, as described in Materials and Methods, after activation using 0.2 U/mL thrombin. For all experiments, n = 3; mean ± SEM; data representative of three independent donors. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. rotenone or thrombin (+DMSO) samples, as appropriate, using ANOVA and Bonferroni post hoc test.
Comparison of Annexin V Binding with MS Analysis of APL Externalization.
Annexin V binding kinetics saturated at much earlier time points than biotinylation (Fig. 4 G and H). Once cells become coated in annexin V-FITC, steric hindrance may prevent additional molecules of PE and PC binding this large protein.
Externalization of Hydroxyeicosatetraenoic Acid-PEs During Platelet Activation, Apoptosis, and Aging.
We recently demonstrated that platelets generate and externalize the unique procoagulant 12-HETE-PEs (hydroxyeicosatetraenoic acid-PEs) on thrombin activation, and we show here that they are externalized on apoptosis or energy depletion (Fig. 5 A and B) (26).
Fig. 5.
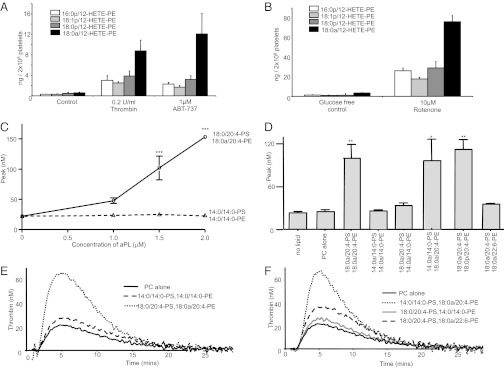
Thrombin, apoptosis, and energy depletion cause externalization of HETE-PEs, and tissue factor-dependent thrombin generation requires specific fatty acids on PE, but not PS. (A and B) Externalized HETE-PEs were determined on platelets activated using thrombin (30 min), ABT-737 (60 min) or rotenone/glucose-free (3 h), before biotinylation and analysis as described in Materials and Methods. Biotinylated 18:0a/12-HETE-PE was used as primary standard for quantitation. (C) Tissue factor (1 pM)-dependent thrombin generation was determined in human plasma using liposomes containing 4 μM PLs (with APL varying from 0 to 2 μM) of varying fatty acid composition, as described in Materials and Methods. (D) Tissue factor-dependent thrombin generation was determined using liposomes containing 1.5 µM APL (in a total of 4 µM PL), as described in Materials and Methods. APL composition is varied according to the labels on the figure, but PS:PE is maintained at 1:2.7 throughout. Representative experiments are shown in E and F. For all experiments, n = 3; mean ± SEM; data from three independent experiments. ***P < 0.001, **P < 0.01, *P < 0.05 vs. PC alone control, using a two-way ANOVA and Bonferroni post hoc test.
Composition of Fatty-Acyl Chains in PE Is Critical for Thrombin Generation.
The role of APL headgroups in facilitating coagulation at the plasma membrane of activated platelets is well known; however, whether the fatty acids attached to APL can also regulate this phenomenon is unknown (27). To address this idea, liposomes were generated that contain the amount of APLs found to be externalized, at a physiological platelet concentration. They included those found in platelets (18:0a/20:4-PS and -PE), shorter chain forms (di-14:0-PS and -PE), a longer Ω-3 lipid (18:0a/22:6-PE) and a plasmalogen also externalized by platelets (18:0p/20:4-PE), with 18:0a/18:0-PC as carrier lipid. Thus, liposome composition and concentration was designed to mimic physiological lipid concentrations of activated platelets in plasma. The liposomes were added to pooled human plasma and coagulation initiated using tissue factor.
For 18:0/20:4-PS and -PE, dose-dependent thrombin generation was observed (Fig. 5C). In contrast, di-14:0-PS and -PE together did not support coagulation (Fig. 5C). At 1.5 μM APL, 18:0a/20:4-PE effectively supported thrombin generation with 14:0a/14:0-PS, as well as 18:0a/20:4-PE/PS together (Fig. 5 D–F). In contrast, 18:0a/20:4-PS was inactive when using 14:0a/14:0-PE (Fig. 5 D–F). This data indicates that the acyl-chain length of PE, but not PS, is an essential determinant of the ability of APL to support coagulation. Similarly, a longer chain PE containing docosahexanoic (DHA) acid at sn2 was also associated with reduced coagulation, whereas a plasmalogen with 20:4 at sn2 was similar to its corresponding acyl derivative (Fig. 5 D and F).
Discussion
Herein, the molecular species of APLs that appear on the surface of activated human platelets were identified and quantitated (Figs. 1–4). Then, a critical role for PE fatty acyl chain length in regulating coagulation was revealed (Fig. 5). The findings change our understanding of how distinct functional groups of APL regulate a key physiological process, raising questions regarding their mechanisms of interaction with coagulation factors. Up to now, APL externalization measurements were neither quantitative nor informed on headgroup or fatty acid composition (6, 9–13). Thus, this study represents a significant step forward, providing detailed insight through demonstrating amount, percentage total APL pool externalized, specific APL species expressed under different conditions (agonist types and amounts, and time courses), and receptors and intracellular signaling pathways involved (Figs. 2–4 and SI Results). The ratio of total platelet PS to PE (∼1:1.9) was similar to previous reports in platelets, measured using older techniques (Fig. 2 C and D) (28, 29). Our observations are consistent with those from previous studies using annexin V (although kinetics differ); for example, stimulation by multiple agonists, the presence of APLs in microparticles, and the requirement for Ca2+ mobilization for their exposure (Figs. 2–4) (30–32). APL externalization showed significant variation, indicating heterogeneity among genetically unrelated donors. Whether this finding translates into different levels of coagulation factor activities is unknown.
Platelet activation, apoptosis, and aging are all important for regulating normal clotting, controlling platelet viability/number, and extending the life span of platelets ex vivo for transfusion. Thus, knowing which APLs are exposed may lead to important insights into platelet biology and has implications for cardiovascular disease, acquired and congenital bleeding disorders, disorders of platelet number (both low and raised), and transfusion medicine. Profiling APL exposure during thrombin activation, apoptosis, or ATP depletion demonstrated the same species externalized regardless of stimulus (Fig. 2–4). Thus, clearance pathways and coagulation reactions would not distinguish between platelets activated by different pathways. In contrast to thrombin or rotenone stimulation, TMEM-16F is not required for apoptosis-dependent APL exposure (Fig. 4). This recently discovered protein is considered a regulator of calcium-dependent scramblase and is mutated in Scott syndrome (21, 22). The existence of PS/PE externalization pathways independent of TMEM-16F may explain why people with Scott syndrome have a relatively mild bleeding pattern and are not predisposed to malignancy.
The ability of APLs to interact with clotting factors is well known. Specifically, tissue factor/factor VIIa, factor VIIIa/IXa (tenase complex), and factor Va/Xa (prothrombinase complex) interact with surface APL in a Ca2+-dependent manner, enhancing enzymatic activity (27, 33, 34). This interaction occurs through multiple γ-carboxylated glutamic acids (Gla domains), and the mechanisms by which these interactions occur have been described at an atomic level (35). Each Gla domain is thought to have one specific interaction with the carboxylate on the headgroup of PS and approximately six interactions with phosphates on any PL except PC. This idea means that PS is required for coagulation to take place but PE can enhance the rate of these reactions (27). To date, the interaction between APL and coagulation factors was assumed to involve the headgroup only, and a role for fatty acid functional groups was unknown. Here, fatty acid chain length is shown to regulate the ability of PE to support coagulation, with platelet-specific PEs showing optimum activity (Fig. 5). In agreement, lyso-PE (which contains no sn2 fatty acid) was previously shown to not support activity of the tenase complex in PS liposomes (27). Our finding that short chain PE poorly supports coagulation whereas short chain PS supports coagulation normally, suggests that these molecules interact differentially with coagulation factors through their Gla domains. Further studies are required to elucidate the precise mechanism. Omega-3 fatty acids are thought to increase bleeding time and suppress thrombosis through COX-1–dependent generation of bioactive lipids that have platelet inhibitory actions (Fig. 5D) (36, 37). Our data indicate that DHA-containing PE is less able to support thrombin generation, indicating a unique mechanism for the anti-coagulant actions of this lipid, unrelated to eicosanoid generation.
In summary, we characterized the specific endogenous phospholipid molecular species that cross the membrane of activated platelets. This finding led us to demonstrate a critical role for APL fatty acyl groups in regulating coagulation factor activity and has therapeutic implications for our understanding of the functions of APL in hemostasis and platelet transfusion. The approach could be applied to the study of APL externalization in other cell types, to provide insight into how these molecules regulate additional critical cellular events, including vesiculation, phagocytosis, and cell division.
Materials and Methods
Materials.
EZ-Link SNB, rotenone, and Q-VD-OPH were from R&D Systems. NB and HPLC grade solvents were obtained from Thermo Fisher Scientific. Corn trypsin inhibitor was from Haematologic Technologies. Phospholipid standards are from Avanti Polar Lipids. Inhibitors of intracellular signaling pathways were from Calbiochem. Unless otherwise stated, other reagents are from Sigma-Aldrich. ABT737 was a gift from Abbott Laboratories.
Platelet Isolation and Activation.
Whole blood was obtained from healthy volunteers free of nonsteroidal antiinflammatory drugs for ≥14 d. Ethical permission was obtained from the School of Medicine Research Ethics Committee, Cardiff University, and was with informed consent. Ethical approval for Scott syndrome samples was from the South East Wales Research Ethics Committee. Washed human platelets were isolated as previously described and maintained at 22 °C in calcium-free Tyrode’s buffer (38). Platelets (2 × 108 cells/mL) were prewarmed for 10 min at 37 °C in the presence of 1 mM CaCl2 with/without inhibitors, before addition of stimuli. Inhibitors were preincubated with platelets for 10 min before stimulation. EGTA/BAPTA-AM was used in calcium-free buffer.
Biotinylation and Extraction of APL.
For external APL derivatization, platelets (200 µL of 2 × 108/mL) or liposomes (4 μM) were incubated with 1.5 mg/mL (3.4 mM) SNB for 10 min at 22 °C, followed by addition of 50 mM lysine for a 10 min at 22 °C. For total cellular APLs, cells were incubated with NB (Thermo Fisher Scientific): one-tenth volume of 20 mM NB in DMSO was added to the cells and incubated at 22 °C for 30 min. Biotinylated APLs were extracted as described below. Results are expressed as surface lipid (SNB-derivatized) as a percentage of total lipid (NB-derivatized) for each molecular species. In some experiments, cells were centrifuged at 900 × g for 10 min to pellet intact platelets from microparticles. Biotinylated lipids were extracted by a modified Bligh and Dyer method (39). Samples (400 µL in Tyrode’s buffer) were added to 1.5 mL of chloroform and methanol (1:2) with internal standards (10 ng each of di-14:0-PE-B and di-14:0-PS-B) and vortexed. A further 0.5 mL CHCl3 was added, followed by 0.5 mL water with vortexing. The sample was centrifuged for 3 min at 895 × g and the lower CHCl3 phase recovered, dried, dissolved in methanol, and stored at −80 °C.
Reverse Phase LC/MS/MS of Biotinylated Phospholipids.
Lipid extracts were separated by reverse phase HPLC using an Ascentis C18, 5-µm particle size, 150 × 2.1-mm column (Sigma-Aldrich) with a mobile phase of methanol, 0.2% ammonium acetate at 400 μL/min, with MS detection on a 4000 Q-Trap (AB Sciex). Biotinylated PS and PE lipids (PS-B, PE-B) were quantitated using standard curves. Multiple reaction monitoring transitions and MS settings are given in SI Materials and Methods.
Measurement of Thrombin Generation by Calibrated Automated Thrombography.
Platelet poor plasma was obtained from human blood [drawn into 20 µg/mL corn trypsin inhibitor and 4% (wt/vol) sodium citrate] by two rounds of centrifugation (1,500 × g for 15 min) and filtration (0.22-µm filter). Trigger solution was prepared by dilution of liposomes in Hepes buffer, 0.5% BSA, and addition of tissue factor (Innovin, Dade). As control, trigger solution with tissue factor and no liposomes was used. Plasma (80 µL) was added in triplicate to wells of a 96-well plate followed by trigger solution (20 µL). For each sample set, a separate calibrator well, containing thrombin calibrator (α2-macroglobulin-thrombin instead of liposomes) was used. The 96-well plate was warmed and placed into a Fluoroskan Ascent Reader (Thermo Electron) and automated addition of fluorogenic substrate for thrombin initiated (Z-Gly-Gly-Arg). The final concentrations were 4 µM liposomes, 1 pM tissue factor, and 5 mM fluorogenic substrate. The thrombin concentration reported represents the maximum present during the assay (40).
Statistics.
Data are representative of at least three separate donors, with samples run in triplicate for each experiment, and expressed as mean ± SEM. Statistical significance was assessed using an unpaired, two-tailed Student t test. Where the difference between more than two sets of data was analyzed, one-way ANOVA was used with Bonferroni post hoc test. Where ionophore was used, the statistical analysis of the response to ionophore was conducted separately to physiological agonists. P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
The studies were supported by the Wellcome Trust (V.B.O. and V.J.H.), European Union Marie Curie Actions FP7 (to S.R.C. and C.P.T.), the British Heart Foundation (V.B.O. and P.W.C.), and National Institutes of Health Grant R01 HL34303 (to R.C.M.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1222419110/-/DCSupplemental.
References
- 1.Daleke DL. Phospholipid flippases. J Biol Chem. 2007;282(2):821–825. doi: 10.1074/jbc.R600035200. [DOI] [PubMed] [Google Scholar]
- 2.Ravichandran KS. Find-me and eat-me signals in apoptotic cell clearance: Progress and conundrums. J Exp Med. 2010;207(9):1807–1817. doi: 10.1084/jem.20101157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heemskerk JW, Bevers EM, Lindhout T. Platelet activation and blood coagulation. Thromb Haemost. 2002;88(2):186–193. [PubMed] [Google Scholar]
- 4.Zwaal RF, Comfurius P, Bevers EM. Surface exposure of phosphatidylserine in pathological cells. Cell Mol Life Sci. 2005;62(9):971–988. doi: 10.1007/s00018-005-4527-3. [DOI] [PubMed] [Google Scholar]
- 5.Daleke DL. Regulation of transbilayer plasma membrane phospholipid asymmetry. J Lipid Res. 2003;44(2):233–242. doi: 10.1194/jlr.R200019-JLR200. [DOI] [PubMed] [Google Scholar]
- 6.Andree HA, et al. Binding of vascular anticoagulant alpha (VAC alpha) to planar phospholipid bilayers. J Biol Chem. 1990;265(9):4923–4928. [PubMed] [Google Scholar]
- 7.Koopman G, et al. Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood. 1994;84(5):1415–1420. [PubMed] [Google Scholar]
- 8.Meers P, Mealy T. Phospholipid determinants for annexin V binding sites and the role of tryptophan 187. Biochemistry. 1994;33(19):5829–5837. doi: 10.1021/bi00185a022. [DOI] [PubMed] [Google Scholar]
- 9.Schlaepfer DD, Mehlman T, Burgess WH, Haigler HT. Structural and functional characterization of endonexin II, a calcium- and phospholipid-binding protein. Proc Natl Acad Sci USA. 1987;84(17):6078–6082. doi: 10.1073/pnas.84.17.6078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koster JJ, Boustead CM, Middleton CA, Walker JH. The sub-cellular localization of annexin V in cultured chick-embryo fibroblasts. Biochem J. 1993;291(Pt 2):595–600. doi: 10.1042/bj2910595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Junker M, Creutz CE. Ca(2+)-dependent binding of endonexin (annexin IV) to membranes: Analysis of the effects of membrane lipid composition and development of a predictive model for the binding interaction. Biochemistry. 1994;33(30):8930–8940. doi: 10.1021/bi00196a010. [DOI] [PubMed] [Google Scholar]
- 12.Edwards HC, Crumpton MJ. Ca(2+)-dependent phospholipid and arachidonic acid binding by the placental annexins VI and IV. Eur J Biochem. 1991;198(1):121–129. doi: 10.1111/j.1432-1033.1991.tb15994.x. [DOI] [PubMed] [Google Scholar]
- 13.Boustead CM, Brown R, Walker JH. Isolation, characterization and localization of annexin V from chicken liver. Biochem J. 1993;291(Pt 2):601–608. doi: 10.1042/bj2910601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi J, Gilbert GE. Lactadherin inhibits enzyme complexes of blood coagulation by competing for phospholipid-binding sites. Blood. 2003;101(7):2628–2636. doi: 10.1182/blood-2002-07-1951. [DOI] [PubMed] [Google Scholar]
- 15.Otzen DE, Blans K, Wang H, Gilbert GE, Rasmussen JT. Lactadherin binds to phosphatidylserine-containing vesicles in a two-step mechanism sensitive to vesicle size and composition. Biochim Biophys Acta. 2012;1818(4):1019–1027. doi: 10.1016/j.bbamem.2011.08.032. [DOI] [PubMed] [Google Scholar]
- 16.Gao C, et al. Procoagulant activity of erythrocytes and platelets through phosphatidylserine exposure and microparticles release in patients with nephrotic syndrome. Thromb Haemost. 2012;107(4):681–689. doi: 10.1160/TH11-09-0673. [DOI] [PubMed] [Google Scholar]
- 17.Hurley WL, Finkelstein E. Identification of leukocyte surface proteins. Methods Enzymol. 1990;184:429–433. doi: 10.1016/0076-6879(90)84303-x. [DOI] [PubMed] [Google Scholar]
- 18.Perrotta PL, Perrotta CL, Snyder EL. Apoptotic activity in stored human platelets. Transfusion. 2003;43(4):526–535. doi: 10.1046/j.1537-2995.2003.00349.x. [DOI] [PubMed] [Google Scholar]
- 19.Albanyan AM, Murphy MF, Rasmussen JT, Heegaard CW, Harrison P. Measurement of phosphatidylserine exposure during storage of platelet concentrates using the novel probe lactadherin: A comparison study with annexin V. Transfusion. 2009;49(1):99–107. doi: 10.1111/j.1537-2995.2008.01933.x. [DOI] [PubMed] [Google Scholar]
- 20.Albanyan AM, Harrison P, Murphy MF. Markers of platelet activation and apoptosis during storage of apheresis- and buffy coat-derived platelet concentrates for 7 days. Transfusion. 2009;49(1):108–117. doi: 10.1111/j.1537-2995.2008.01942.x. [DOI] [PubMed] [Google Scholar]
- 21.Suzuki J, Umeda M, Sims PJ, Nagata S. Calcium-dependent phospholipid scrambling by TMEM16F. Nature. 2010;468(7325):834–838. doi: 10.1038/nature09583. [DOI] [PubMed] [Google Scholar]
- 22.Castoldi E, Collins PW, Williamson PL, Bevers EM. Compound heterozygosity for 2 novel TMEM16F mutations in a patient with Scott syndrome. Blood. 2011;117(16):4399–4400. doi: 10.1182/blood-2011-01-332502. [DOI] [PubMed] [Google Scholar]
- 23.Schoenwaelder SM, et al. Two distinct pathways regulate platelet phosphatidylserine exposure and procoagulant function. Blood. 2009;114(3):663–666. doi: 10.1182/blood-2009-01-200345. [DOI] [PubMed] [Google Scholar]
- 24.Mills KI, et al. Inhibition of mitochondrial function in HL60 cells is associated with an increased apoptosis and expression of CD14. Biochem Biophys Res Commun. 1999;263(2):294–300. doi: 10.1006/bbrc.1999.1356. [DOI] [PubMed] [Google Scholar]
- 25.Aronis A, Madar Z, Tirosh O. Mechanism underlying oxidative stress-mediated lipotoxicity: exposure of J774.2 macrophages to triacylglycerols facilitates mitochondrial reactive oxygen species production and cellular necrosis. Free Radic Biol Med. 2005;38(9):1221–1230. doi: 10.1016/j.freeradbiomed.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 26.Thomas CP, et al. Phospholipid-esterified eicosanoids are generated in agonist-activated human platelets and enhance tissue factor-dependent thrombin generation. J Biol Chem. 2010;285(10):6891–6903. doi: 10.1074/jbc.M109.078428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tavoosi N, et al. Molecular determinants of phospholipid synergy in blood clotting. J Biol Chem. 2011;286(26):23247–23253. doi: 10.1074/jbc.M111.251769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dougherty RM, Galli C, Ferro-Luzzi A, Iacono JM. Lipid and phospholipid fatty acid composition of plasma, red blood cells, and platelets and how they are affected by dietary lipids: A study of normal subjects from Italy, Finland, and the USA. Am J Clin Nutr. 1987;45(2):443–455. doi: 10.1093/ajcn/45.2.443. [DOI] [PubMed] [Google Scholar]
- 29.Vance JE. Phosphatidylserine and phosphatidylethanolamine in mammalian cells: Two metabolically related aminophospholipids. J Lipid Res. 2008;49(7):1377–1387. doi: 10.1194/jlr.R700020-JLR200. [DOI] [PubMed] [Google Scholar]
- 30.Ramstrom S, O’Neill S, Dunne E, Kenny D. Annexin V binding to platelets is agonist, time and temperature dependent. Platelets. 2010;21(4):289–296. doi: 10.3109/09537101003660564. [DOI] [PubMed] [Google Scholar]
- 31.Freyssinet JM, Toti F. Formation of procoagulant microparticles and properties. Thromb Res. 2010;125(Suppl 1):S46–S48. doi: 10.1016/j.thromres.2010.01.036. [DOI] [PubMed] [Google Scholar]
- 32.Bevers EM, Williamson PL. Phospholipid scramblase: An update. FEBS Lett. 2010;584(13):2724–2730. doi: 10.1016/j.febslet.2010.03.020. [DOI] [PubMed] [Google Scholar]
- 33.Lentz BR. Exposure of platelet membrane phosphatidylserine regulates blood coagulation. Prog Lipid Res. 2003;42(5):423–438. doi: 10.1016/s0163-7827(03)00025-0. [DOI] [PubMed] [Google Scholar]
- 34.Falls LA, Furie B, Furie BC. Role of phosphatidylethanolamine in assembly and function of the factor IXa-factor VIIIa complex on membrane surfaces. Biochemistry. 2000;39(43):13216–13222. doi: 10.1021/bi0009789. [DOI] [PubMed] [Google Scholar]
- 35.Morrissey JH, Tajkhorshid E, Rienstra CM. Nanoscale studies of protein-membrane interactions in blood clotting. J Thromb Haemost. 2011;9(Suppl 1):162–167. doi: 10.1111/j.1538-7836.2011.04300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harker LA, et al. Interruption of vascular thrombus formation and vascular lesion formation by dietary n-3 fatty acids in fish oil in nonhuman primates. Circulation. 1993;87(3):1017–1029. doi: 10.1161/01.cir.87.3.1017. [DOI] [PubMed] [Google Scholar]
- 37.Lagarde M, Chen P, Véricel E, Guichardant M. Fatty acid-derived lipid mediators and blood platelet aggregation. Prostaglandins Leukot Essent Fatty Acids. 2010;82(4-6):227–230. doi: 10.1016/j.plefa.2010.02.017. [DOI] [PubMed] [Google Scholar]
- 38.O’Donnell VB, et al. Catalytic consumption of nitric oxide by prostaglandin H synthase-1 regulates platelet function. J Biol Chem. 2000;275(49):38239–38244. doi: 10.1074/jbc.M001802200. [DOI] [PubMed] [Google Scholar]
- 39.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37(8):911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 40.Hemker HC, et al. Calibrated automated thrombin generation measurement in clotting plasma. Pathophysiol Haemost Thromb. 2003;33(1):4–15. doi: 10.1159/000071636. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.