

Significance
Reduced oxygen supply—hypoxia—is a near-universal feature of solid tumors that can alter how tumors respond to therapies. We investigated the transition from normoxia to hypoxia in model brain cancer systems, using single-cell proteomics and data analysis tools based on physicochemical concepts. This approach permits the simplification of otherwise complex biology. We find a hypoxia-induced switch within a mammalian target of rapamycin (mTOR) signaling network. At the switching point, mTOR is predicted, and then shown by experiment, to be unresponsive to inhibition. These results may help explain the undistinguished performance of mTOR inhibitors in certain clinical trials.
Keywords: cancer biology, microfluidics, single-cell proteomics, brain cancer
Abstract
Hypoxia is a near-universal feature of cancer, promoting glycolysis, cellular proliferation, and angiogenesis. The molecular mechanisms of hypoxic signaling have been intensively studied, but the impact of changes in oxygen partial pressure (pO2) on the state of signaling networks is less clear. In a glioblastoma multiforme (GBM) cancer cell model, we examined the response of signaling networks to targeted pathway inhibition between 21% and 1% pO2. We used a microchip technology that facilitates quantification of a panel of functional proteins from statistical numbers of single cells. We find that near 1.5% pO2, the signaling network associated with mammalian target of rapamycin (mTOR) complex 1 (mTORC1)—a critical component of hypoxic signaling and a compelling cancer drug target—is deregulated in a manner such that it will be unresponsive to mTOR kinase inhibitors near 1.5% pO2, but will respond at higher or lower pO2 values. These predictions were validated through experiments on bulk GBM cell line cultures and on neurosphere cultures of a human-origin GBM xenograft tumor. We attempt to understand this behavior through the use of a quantitative version of Le Chatelier’s principle, as well as through a steady-state kinetic model of protein interactions, both of which indicate that hypoxia can influence mTORC1 signaling as a switch. The Le Chatelier approach also indicates that this switch may be thought of as a type of phase transition. Our analysis indicates that certain biologically complex cell behaviors may be understood using fundamental, thermodynamics-motivated principles.
In most solid organ cancers, increased interstitial pressure, vascular constriction, abnormal leaky blood vessels, and edema result in a hypoxic microenvironment, particularly in the center of the tumor (1–5). Hypoxia, in part by stabilizing the hypoxia-inducible transcription factor (HIF), can increase the biological aggressiveness of tumors, promoting glycolysis, cellular proliferation, and angiogenesis; it can also make tumors less responsive to many therapies (6–9).
Signaling through mammalian target of rapamycin (mTOR) is often a critical component of the hypoxic response (10–13). Amplification and activating mutations of receptor tyrosine kinases; mutation of phosphoinositide 3-kinase (PI3K) and its regulatory subunits; and loss of the phosphatase and tensin homolog (PTEN) tumor suppressor protein can lead to elevated growth factor-independent activation of mTOR signaling (10, 14). The hypoxic microenvironment indirectly regulates mTOR, in part by regulating intracellular ATP levels (15), to promote tumor cell growth and proliferation. Such regulation can occur via activation of hypoxia-inducible factor-1α (HIF-1α)-dependent glycolysis, and by stimulating angiogenesis (16). Most models of mTOR signaling in cancer assume a continuous relationship among the level of growth factor receptor pathway signaling, and/or ATP and nutrient levels, and the degree of mTORC1 activation. However, most signaling cascades actually behave as excitable devices with built-in excitability thresholds, enabling them to integrate diverse temporal and spatial inputs to produce specific signaling responses (17). It is not known how physical perturbations such as altering oxygen partial pressure (pO2) can influence the excitability of signaling networks, and whether such effects yield continuous or discrete transitions. This question is important because if mTOR signaling becomes uninhibitable at levels of hypoxia that are frequently reached within the center of a tumor, a potentially targetable mechanism of drug resistance can be identified.
We set out to study how varying pO2 from 21% (ambient) to 1% (hypoxia) influences mTOR complex 1 (mTORC1) and HIF-1α signaling within model glioblastoma multiforme (GBM) cancer cells that exhibit persistent mTORC1 activation (18, 19). We used the single-cell barcode chip (SCBC) (20, 21) to investigate U87 EGFRvIII cells [GBM cells that stably express the epidermal growth factor receptor-activating mutation (EGFRvIII)]. The SCBC is an integrated microfluidics platform (22) designed for the quantification of a panel of functional proteins from statistical numbers of single cells (21). The panel, which was designed to capture key aspects of both HIF-1α and mTORC1 signaling (9, 12), included three secreted proteins [VEGF, IL-6, and matrix metalloprotease-1 (MMP1)], one cytoplasmic protein (HIF-1α), and three cytoplasmic phosphoproteins [phospho(p)-mTOR, p-ERK1, and p-P70 ribosomal protein S6 kinase (p-P70S6K)]. Advantages of these proteins are the availability of high-quality antibody pairs for our assays, and the fact that they are produced by single cells at a level that allows us to accurately convert single-cell fluorescence signal into copy numbers detected per cell.
An SCBC cell data set, which is comprised of a statistical number of single-cell assays, yields three types of independent observables. The first observables are averaged levels of each assayed protein from single cells; the second are the protein fluctuations, which are histograms of the observation frequency vs. the measured protein levels; and the third are the correlations between the various assayed proteins. The latter two observables are unique to single-cell multiplex proteomics assays, and all three observation types are used to understand how changes in pO2 influence mTORC1 and HIF-1α signaling. We provide the interpretation in three stages of increasing level of detail, where the last stage is a theory with predictive capabilities. We first discuss a mean-field qualitative model that provides a context for discussing how the average effect of other proteins influences the fluctuations of a specific protein in question. The experimentally measured fluctuations, when interpreted within this model, point toward a pO2-dependent deregulation of mTORC1 signaling, and imply that mTORC1 signaling will be difficult to inhibit near 1.5% pO2. This picture is shown to be correct through the use of the ATP-competitive mTOR inhibitor PP242 (23) on the GBM cell lines as well as a neurosphere culture model grown from a human-derived GBM xenograft tumor that also expresses the EGFRvIII mutation. We then attempt to understand the pO2-dependent deregulation of mTORC1 in two more detailed ways. We first present a steady-state kinetic model to capture the relationships among O2, p-mTOR, HIF-1α, and PP242. The kinetic approach indicates that there is a switch in mTORC1 signaling near 1.5% pO2, and that there is a value of pO2 near 1.5% for which mTOR is uninhibitable. Finally, we discuss a quantitative version of the Le Chatelier's principle that relies on the single-cell proteomics assays as input (24), and, unlike the mean field model, allows for the explicit treatment of protein–protein correlations. The theory is validated by using it to predict the effect of changes of pO2 on the mean numbers of the assayed proteins. This prediction fails between 2% and 1.5% pO2, which implies that changing pO2 through this range is a strong perturbation to the cells. The theory then shows that the deregulation of mTORC1 signaling is associated with a phase transition in the signaling network. The implication is that, near 1.5% pO2, the network switches from one set of protein–protein interactions to another. At the switching point, the network is unstable, and the coordinated signaling between mTOR and its effector proteins is lost.
Results and Discussion
Single-Cell Proteomic Assays Use the SCBC Platform.
The SCBC platform (Fig. 1A; SI Appendix, Fig. S1) contains 240 1.7-nL-volume microchambers. Each microchamber has an upper assaying compartment that contains a nine-element DNA barcode. A second compartment, separated by a valve, serves as a lysis buffer reservoir. Eight elements of the barcode are converted to a miniature antibody array for assaying a panel of proteins by loading a DNA–antibody conjugate mixture (Materials and Methods), and one element provides an alignment marker (Fig. 1B). Cells are loaded from an upstream inlet into the SCBC and distributed randomly among the microchambers. Controlling the cell loading density enables about half of the microchambers to contain a single cell, whereas others may be empty or contain two or more cells. After cell loading and counting, the microchip is incubated in a controlled O2 environment for 7 h (Fig. 1A), followed by an on-chip cell lysis (Fig. 1B; SI Appendix, Fig. S1E). An O2 sensor (0.1% accuracy) measured both the level and the equilibration rate of the pO2. Secreted proteins are captured during incubation, and intracellular proteins are captured following lysis. A detection antibody mixture and the fluorescent probes are loaded afterward to complete the on-chip immunoassay, which is read with a GenePix array scanner (see Fig. 1B for detailed execution scheme). The incubation time was chosen to ensure cell viability at all pO2 explored (SI Appendix, Fig. S1B), and to enable capture of sufficient numbers of secreted proteins. Additional experimental details, plus assay cross-reactivity and calibration, are provided (Materials and Methods; SI Appendix, Fig. S2 and Table S2).
Fig. 1.

SCBC platform, experimental flowchart, and representative data. This SCBC design permits incubation of the cells within controlled pO2 environments, followed by multiplexed and quantitative assays of functional (secreted, membrane, and/or cytoplasmic) proteins from quantized cell populations. (A) Drawing of the custom-built hypoxia setup with real-time pO2 monitoring. The photograph is of an SCBC with the microchambers (red) and control valve layers (green) delineated with food dyes. (Lower Right) Side view of a single-cell microchamber with a representative readout image from the SCBC device. Each barcode fluorescent stripe corresponds to a specific protein assay. Signals from three microchambers with different cell numbers, indicated as 5, 1, and 3, are shown. (B) SCBC assay steps. DNA barcodes are converted into antibody barcodes using a mixture of DNA–antibody conjugates. Cells are then loaded and isolated into the upper chamber and incubated at a desired pO2, during which time secreted proteins are captured on designated barcode stripes. The chip is then cooled to near 0 °C, and the valve connecting the lysis buffer chamber is opened, leading to cell lysis within 15 min. The intracellular proteins are released and captured onto designated barcode stripes. (C) Scatter plots of assayed protein levels measured from U87 EGFRvIII single cells at 21% (blue dots) and 1% (red dots) pO2. The averaged fluorescence intensity with SEM is overlaid for each protein. Statistical uniqueness is evaluated by two-tailed Student t test assuming unequal variance (NS, not significant; *P < 0.05; **P < 0.005; ***P < 0.0005). (D) Western blotting results for several of the cytoplasmic proteins from U87 EGFRvIII cells assayed at 21% and 1% pO2. (E) Scatter plots of the assayed levels of p-P70S6K and p-mTOR at 21% pO2 for individual microchambers containing one, two, or three cells, indicating the statistical uniqueness of data sets representing different quantized cell populations.
For the U87 EGFRvIII cell line, we collected single-cell data at 21%, 3%, 2%, 1.5%, and 1% pO2. At each condition, we assayed ∼100 single cells, 60 zero-cell chambers, and 50 two-cell chambers. After background subtraction, scatter plots of the single-cell proteomic data (Fig. 1C) can be compared against bulk cell population protein assays using Western blotting or sandwich ELISA (Fig. 1D; SI Appendix, Fig. S2A). The statistical uniqueness of one-cell data was established via comparison against two-cell data (Fig. 1E). For each protein measured, the fluorescence intensity is converted into copy numbers detected using calibration data (SI Appendix, Fig. S2C) that relied on standard proteins. For a given protein, a histogram of copy number vs. frequency of observation reflects the fluctuations of that protein.
Protein Fluctuations Reveal a Deregulation in mTORC1 Signaling near 1.5% pO2.
Fig. 2 A and B show the single-cell fluctuations for the four cytoplasmic proteins at different pO2 values. HIF-1α has a unique profile compared with the phosphoproteins related to mTORC1 signaling (including mTOR and its effectors: P70S6K and ERK1) (12). As pO2 decreases, the HIF-1α fluctuations evolve from a narrow and peaked histogram into a widely dispersed profile, with the average shifting to higher copy numbers. By contrast, the three phosphoprotein fluctuations exhibit broad widths at 21%, 3%, 2%, and 1% pO2, but are sharply peaked at 1.5% pO2 (Fig. 2B), which has implications for a signaling network transition.
Fig. 2.

Measured single-cell fluctuations for four cytoplasmic proteins as a function of pO2, and a simulation of fluctuations for a hypothetical protein. (A) Single-cell fluctuation profiles for HIF-1α at various pO2. (B) Single-cell fluctuations for p-mTOR, p-P70S6K, and p-ERK1 at various pO2. Note that these fluctuations exhibit a sharpening at 1.5% pO2. (C) Single-cell fluctuation profiles from a Monte Carlo simulation that assumes a hypothetical protein participates in varying numbers of functional processes. Note the comparison of this simulation to the measured fluctuations of HIF-1α.
Protein fluctuations can be highly informative toward understanding protein functional activity. A widely dispersed fluctuation can indicate a highly active protein that is involved in multiple functional processes. A narrow, sharp fluctuation, by contrast, represents a protein with limited interactions. To illustrate this point, we carried out Monte Carlo simulations to generate histograms for a hypothetical functional protein at several degrees of activity. The protein was assumed to participate in up to four independent functional processes. Each process required a range of protein copy numbers and had an associated probability that it was active in any given single cell (see Materials and Methods for detail). The simulated histograms (Fig. 2C; SI Appendix, Table S3) reveal that the fluctuations are increasingly dispersed as the number of potentially active functional processes increases. This plot effectively emulates the fluctuations of HIF-1α as pO2 is lowered (Fig. 2A). The implication is that HIF-1α is increasingly activated as the cells transition from normoxia to hypoxia. This conclusion may be drawn by simply inspecting the fluctuation profiles of HIF-1α, but it is also in strong agreement with the literature (8, 9).
By analogy with the above discussion of HIF-1α, we hypothesized that the phosphoproteins associated with mTORC1 signaling (Fig. 2B) become isolated from cell signaling processes at ∼1.5% pO2. Such isolation has implications because mTORC1 is considered an important drug target in GBM (and other) tumors. The decoupling of mTORC1 from its effector proteins within this hypoxic window could account for a level of resistance to mTOR kinase inhibitors. We tested this prediction by assaying for the effects of the mTOR inhibitor PP242 (23) on the phosphorylation levels of mTOR, P70S6K, and ERK1, as a function of pO2, on bulk U87 EGFRvIII cell cultures, because those cells were the ones analyzed using the SCBC platform. We also tested our prediction on a tumor model by similarly analyzing neurosphere cultures derived from the human origin GBM39 xenograft (25). This model also carried the EGFRvIII mutation. GBM neurospheres can provide realistic tumor models relative to cell lines (26), and have even been shown to exhibit stem-like behaviors under hypoxic stress (27). Inhibition of mTOR by PP242 leads to down-regulation of the phosphorylation of both mTOR and P70S6K, and increased phosphorylation of ERK1, due to the activation of a negative feedback loop downstream of mTORC1 that targets the PI3K pathway (28). The protein assays used here were multiplexed sandwich ELISA immunoassays from statistical numbers of cells based upon a published technique (29). The cells were assayed in the presence of a 3-μM solution of PP242, or a DMSO control, under varying pO2. As shown in Fig. 3, the results clearly support the prediction. We found that mTORC1 signaling is inhibited by PP242 for both U87 EGFRvIII cells and for the GBM39 neurosphere cultures at 21%, 3%, and 1% pO2, but is much less inhibited between 2% and 1.5% pO2.
Fig. 3.

The influence of the mTOR inhibitor PP242 on the assayed protein levels for GBM cell lines and xenograft neurosphere tumor models, as a function of pO2. (A and B) Bar graphs showing the changes in protein copy number, as measured from bulk-cell lysate of the U87 EGFRvIII cells and the GBM39 tumor model. Protein level changes are normalized by the number listed below the corresponding protein name. (Insets) Fluorescence images of the developed assays of the highly expressed mTOR effector, p-P70S6K. (C) Plot of protein concentrations at various pO2 joined with spline fit for control and PP242-treated U87 EGFRvIII cells (Upper) and GBM39 neurospheres (Lower). Note that the drug treated and untreated levels coincide for p-mTOR, p-P70S6K, and p-ERK1 for both model systems near 1.5–2% pO2. Error bars represent SDs of the measurements.
We now look toward achieving a better mechanistic understanding of the behavior of mTOR signaling near 1.5% pO2 via a steady-state kinetic model.
Steady-State Kinetic Model Identifies a Switch in mTORC1 Signaling near 1.5% pO2.
As master regulators of hypoxic GBM cells, HIF-1α and mTORC1 act in an integrated way (12). Our data suggest that their interplay is critical for the signaling network transition. PP242, as an ATP competitive inhibitor, can directly inhibit mTORC1 activity. mTORC1 activity will also be inhibited by HIF-1α–dependent transcriptional regulation, which can occur through REDD1 (regulated in development and DNA damage responses 1) or BINP3 (BCL2/adenovirus E1B 19-kDa protein-interacting protein 3), when exposed to hypoxia (30–32). Furthermore, our measurements (Fig. 3), and other reports (33), indicate that the HIF-1α expression level can be suppressed by addition of PP242 under hypoxia. Thus, because HIF-1α can repress mTORC1, suppression of HIF-1α could potentially promote mTORC1 activity. This effect may compete against PP242 direct inhibition of mTORC1 during the course of hypoxia, thus providing a potential mechanistic explanation of the undruggability of mTORC1 signaling between 1.5% and 2% pO2; this is summarized by the network hypothesis illustrated in Fig. 4A.
Fig. 4.

The network hypothesis and accompanying steady-state kinetic model describing relationships among HIF-1α, p-mTOR, PP242, and pO2 in U87 EGFRvIII cells reveal a switch in mTOR regulation below 1.5% pO2. Bracketed protein names indicate the concentration of that protein in pg/mL. (A) The network drawing indicates (net) effective activating (arrow) and inhibiting (bar) interactions. The functional forms of those interactions represent the fitted or predicted parameters, using steady-state kinetic relationships. (B) The levels of HIF-1α fit well to a steady-state kinetic model predicting a hyperbolic increase in HIF-1α with decreasing pO2. (C) [p-mTOR] exhibits an inverse linear relationship with [HIF-1α]. (D) The change in HIF-1α levels upon addition of a 3-µM solution of PP242 exhibits a quadratic dependence upon [HIF-1α]. (E) The fitted parameters from the model are used to calculate [p-mTOR] in terms of pO2 in the presence and absence of PP242, and compared against experiments (the points connected by lighter lines). The calculation predicts a pO2 level where the solid red and blue lines cross, or where PP242 does not inhibit p-mTOR. However, the model also predicts PP242 activates p-mTOR at pO2 levels above this crossing point, which is clearly not observed. This disagreement implies that different regulators of mTOR are important in the regime of moderate-to-severe hypoxia.
This Fig. 4A network is a greatly simplified version of what is known from the literature, but we are able to work with it here because, as a steady-state kinetic model, it only requires that the flux into and out of a particular protein channel equal a constant value, for a given set of physical conditions. Thus, we are accounting for the net influence of the network components on each other, but not necessarily the direct influence. The network of Fig. 4A has the nuance that the indicated protein–protein and protein–molecule interactions are not necessarily linear relationships. We combined steady-state chemical kinetic analysis with the fitting of data from calibrated microwell-based sandwich ELISAs on proteins collected from lysed U87 EGFRvIII cells (Fig. 3 A and C; SI Appendix, Table S5). The details of our approach are in SI Appendix, Method III. In Fig. 4 B–D, we present the relationships between HIF-1α and pO2, and p-mTOR and HIF-1α, and the influence of PP242 on HIF-1α.
With these relationships in hand, we can calculate the dependence of the p-mTOR level on pO2 using, as input, only the measured pO2 values, the presence or absence of PP242, and the fitted parameters (Fig. 4E; SI Appendix, Method III). This result is of interest in three ways. First, the kinetic model accurately captures the p-mTOR levels in the absence of PP242 inhibition, for all values of pO2; second, it predicts a pO2 level for which p-mTOR is not influenced by PP242. For the parameters fitted here, this level is near 1.25% pO2, but can be shifted to slightly higher pO2 levels by altering some of the fitting parameters, while keeping them within their statistical margins of error. Third, for any of the fitted parameters, the kinetic model also predicts PP242 inhibition of mTOR at pO2 levels above the crossing point, and PP242 activation of mTOR below the crossing point, which is clearly not observed experimentally. The implication is that new regulators of mTOR, not included in the model of Fig. 4A, would have to be invoked to account for the observed behavior at very low pO2 levels.
The influence of hypoxia and PP242 on the GBM39 model exhibits many similarities to that observed for U87 EGFRvIII cells, but only certain aspects of the kinetic model translate to that system. For example, HIF-1α exhibits a clear hyperbolic dependence on decreasing pO2 in both models, but the other relationships are not as clear for GBM39. This finding is not surprising, given that the GBM39 protein assays are sampling a neurosphere model of a tumor, which is comprised of a heterogeneous mixture of cellular phenotypes.
The kinetic model gives some mechanistic insight into the switch in mTORC1, partly through its failure to predict the influence of PP242 below 1.5% pO2. This failure presumably arises because certain protein–protein interactions are neglected in this range. Those interactions are implicit in the protein fluctuations. Thus, we turn to the quantitative Le Chatelier’s principle, because it explicitly recognizes individual protein–protein correlations, and the predictive nature of this theory may help shed light on the uninhibitability of mTORC1 between 1.5% and 2% pO2.
Application of a Quantitative Le Chatelier's Principle to the Single-Cell Data Identifies a Phase Transition in mTORC1 Signaling Between 2% and 1.5% pO2.
For the Le Chatelier approach, the goal is to understand whether a change in pO2 constitutes a strong or a weak perturbation to the U87 EGFRvIII cells. We previously reported on the development and validation of this approach (24). In that earlier work, we used the theory to predict how the levels of a panel of secreted proteins from a human macrophage cell line, stimulated with lipopolysaccharide to emulate gram-negative bacteria, would respond to the addition of neutralizing antibodies. The theory requires single-cell data as input, and can predict how the levels of certain proteins will respond to a weak perturbation. A strong perturbation is implied when the theoretical prediction and the experimental measurement are in strong disagreement.
For the theory, we first use the measured data to compute the mean number 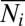 of molecules for each protein i per cell, and the mean of the joint numbers of proteins i and j,
of molecules for each protein i per cell, and the mean of the joint numbers of proteins i and j, 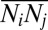 .
.
Thereby we compute the covariance matrix 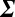 , which is a symmetric P × P matrix, where P is the size of the protein panel assayed, and the matrix elements Σij represent the covariance between proteins i and j (SI Appendix, Table S6). Given the protein–protein covariance matrix
, which is a symmetric P × P matrix, where P is the size of the protein panel assayed, and the matrix elements Σij represent the covariance between proteins i and j (SI Appendix, Table S6). Given the protein–protein covariance matrix 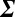 , we write the quantitative Le Chatelier's principle as the matrix equation
, we write the quantitative Le Chatelier's principle as the matrix equation 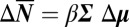 , where
, where 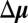 is a column vector whose P components give the change in the chemical potentials of the P proteins due to the change in external conditions.
is a column vector whose P components give the change in the chemical potentials of the P proteins due to the change in external conditions. 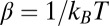 , where T is the temperature and kB is Boltzmann’s constant (theoretic details can be found in ref. 24). This matrix equation relates the change
, where T is the temperature and kB is Boltzmann’s constant (theoretic details can be found in ref. 24). This matrix equation relates the change 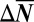 in the mean number of molecules of each protein to external perturbations, such as O2 pressure changes, or addition of a drug. Applying this approach to the single-cell data, we found that the state of the signaling network at 3% pO2 was only weakly perturbed from that at 21% pO2 (Fig. 5A). The change between 3% and 2% pO2 was a stronger perturbation (we correctly predict the signs of the changes in protein levels, but the predicted levels for proteins IL-6 and MMP1 deviate significantly from experiment). We could not predict the measured changes between 2% and 1.5% pO2. We could, however, describe the changes between 1.5% and 1% pO2. We do not show a prediction for VEGF at low pO2 because, in this range, VEGF appears decoupled from the other proteins (SI Appendix, Fig. S3).
in the mean number of molecules of each protein to external perturbations, such as O2 pressure changes, or addition of a drug. Applying this approach to the single-cell data, we found that the state of the signaling network at 3% pO2 was only weakly perturbed from that at 21% pO2 (Fig. 5A). The change between 3% and 2% pO2 was a stronger perturbation (we correctly predict the signs of the changes in protein levels, but the predicted levels for proteins IL-6 and MMP1 deviate significantly from experiment). We could not predict the measured changes between 2% and 1.5% pO2. We could, however, describe the changes between 1.5% and 1% pO2. We do not show a prediction for VEGF at low pO2 because, in this range, VEGF appears decoupled from the other proteins (SI Appendix, Fig. S3).
Fig. 5.

The use of a quantitative Le Chatelier principle reveals an oxygen partial pressure-dependent phase transition in the mTORC1 signaling network within model GBM cells. (A) Measured and predicted changes for the panel of assayed proteins, as pO2 is changed between specified levels. The agreement between experiment and prediction for 21–3% and 1.5–1% implies that these pO2 changes constitute only a weak perturbation on the signaling network. The change from 3% to 2% pO2 represents a somewhat stronger perturbation, whereas for the range 2–1.5% pO2, a strong perturbation is indicated by the qualitative disagreement between prediction and experiment. (B) The coordination of mTOR-associated signaling modes, as a function of pO2, is reflected in an analysis of the relevant eigenvalues (mode strength) and their composition of the protein–protein covariance matrix (mode composition). The coordination of mTOR with its effectors, p-ERK and p-P70S6K, dominates the composition of the three lowest-amplitude eigenvectors, which exhibit singular behavior between 2% and 1.5% pO2. Experimentally determined points are connected by solid lines; dashed lines imply that the amplitudes of the three eigenvectors will reach a (shallow) minimum (loss of mTOR signaling coordination), which is indicative of a phase transition. Each column of the pie charts represents the compositions of the three lowest-amplitude eigenvectors at the corresponding pO2; they reflect a shift in the coordination of mTOR signaling across the phase transition. Note the importance of HIF-1α in these eigenvectors at pO2 ≥ 2%, and the importance of p-ERK below 2%.
Based on these observations, we hypothesized that the states corresponding to ∼2–21% pO2 represented one phase of the signaling network, whereas those between 1% and 1.5% pO2 represented a second phase, with a phase transition occurring in between. We tested this hypothesis by analyzing the protein–protein covariance matrix to view the coordination of mTORC1 signaling as pO2 was varied. This approach goes beyond measuring specific protein–protein pairwise interactions, because it accounts for all of the proteins that are simultaneously assayed from each single cell. For the analysis, the eigenvalues (Fig. 5B) of the covariance matrix describe the strength of the coordinated protein–protein interaction modes, and the eigenvectors (Fig. 5C) describe the composition of those modes. Such an analysis draws from the Gibbs phase rule (34, 35), which states that, at a phase transition, a degree of freedom is lost for each coexisting phase. Consider the water liquid/solid phase transition. Away from the transition, temperature can be readily varied by warming or cooling, but at the transition when ice and water coexist, it is not possible to change the temperature without altering the pressure.
The nature of the hypoxia-induced transition is that, at the phase transition, the signaling network undergoes a switch in connectivity during which the functional phosphoproteins related to mTORC1 signaling are isolated and inactivated, and this is reflected in how the fluctuations of Fig. 2B sharpen at 1.5% pO2, but more rigorously in Fig. 5B. Above 2% pO2, these eigenvectors capture 75–95% of the covariance, and hence signaling network coordination, among the proteins HIF-1α, p-P70S6K, and p-mTOR; below 1.5% pO2, they capture 80–100% of the covariance among p-P70S6K, p-mTOR, and p-ERK1. The amplitudes of these eigenvectors are strongly influenced by pO2, and they each point to a minimum between 1.5% and 2% pO2 (Fig. 5B). Because the cell is a finite system, the minimum will likely not be sharp. This eigenvalue singularity indicates a loss of degrees of freedom (or the loss of mTORC1 signaling coordination) and thereby points to the existence of a phase transition associated with mTORC1 signaling between 1.5% and 2% pO2. Recall the quantitative Le Chatelier's principle 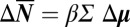 , where the vector
, where the vector 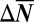 of change in protein numbers has P components. The matrix equation tells us that we can identify P linearly independent ways in which an external perturbation can influence the response of the proteins within the network. If the matrix
of change in protein numbers has P components. The matrix equation tells us that we can identify P linearly independent ways in which an external perturbation can influence the response of the proteins within the network. If the matrix 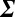 is singular (i.e., it has one or more zero eigenvalues), there are fewer independently allowable variations. This is the loss of degrees of freedom. This analysis leads to the surprising prediction that mTORC1 signaling will be intrinsically uncontrollable in the U87 EGFRvIII cells between 1.5% and 2% pO2, but may be influenced at higher or lower pO2 values. The proof follows from the near-zero eigenvalues of the covariance matrix; the associated eigenvectors are those localized on the phosphoproteins associated with mTORC1 signaling. Near the transition, even large changes in the chemical potentials of p-mTOR and its effector proteins p-ERK and p-P70S6K result in very small changes in their mean numbers.
is singular (i.e., it has one or more zero eigenvalues), there are fewer independently allowable variations. This is the loss of degrees of freedom. This analysis leads to the surprising prediction that mTORC1 signaling will be intrinsically uncontrollable in the U87 EGFRvIII cells between 1.5% and 2% pO2, but may be influenced at higher or lower pO2 values. The proof follows from the near-zero eigenvalues of the covariance matrix; the associated eigenvectors are those localized on the phosphoproteins associated with mTORC1 signaling. Near the transition, even large changes in the chemical potentials of p-mTOR and its effector proteins p-ERK and p-P70S6K result in very small changes in their mean numbers.
The hypoxia-induced phase transition is a multidimensional transition that behaves in a complementary manner to a regular transition of the inverse relation 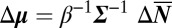 . The latter implies that near a phase coexistence where
. The latter implies that near a phase coexistence where  has a low eigenvalue, large changes of the number of molecules (extensive variables) will barely influence the chemical potential (the conjugated intensive variables); this bears an analogy to the liquid/solid transition of water where finite changes of the internal energy (the extensive variable) via the addition of heat do not alter the temperature (the conjugated intensive variable). Given that intensive and extensive variables come in conjugate pairs and are interchangeable through Legendre transforms (35), both transition manners can be appreciated.
has a low eigenvalue, large changes of the number of molecules (extensive variables) will barely influence the chemical potential (the conjugated intensive variables); this bears an analogy to the liquid/solid transition of water where finite changes of the internal energy (the extensive variable) via the addition of heat do not alter the temperature (the conjugated intensive variable). Given that intensive and extensive variables come in conjugate pairs and are interchangeable through Legendre transforms (35), both transition manners can be appreciated.
Conclusion
We found that in model GBM cell lines and in a mouse GBM xenograft neurosphere model, the change in mTOR signaling from normoxia to hypoxia involves a discontinuous transition between two phases—i.e., changing pO2 induces a switch in mTORC1 signaling. These results point to a fundamentally different approach toward understanding and predicting certain cellular behaviors, and may also provide a clue toward understanding the clinical failure of mTOR inhibitors on GBM tumors (36, 37). Our measurements were guided by the existing biological literature, but our concern was not with capturing the detailed biomolecular interactions within the cells, but rather on understanding how the state of the signaling network is influenced by physical (pO2) or molecular (therapeutic) perturbations. The approach is driven by recently developed experimental tools for quantitating the levels of a panel of functional proteins from single cells, and the theory is grounded in well-established physicochemical principles.
Materials and Methods
Cell Lines and Reagents.
U87 EGFRvIII cells were constructed as previously described (38) and routinely maintained in DMEM (American Type Culture Collection) containing 10% FBS in humidified atmosphere of 5% CO2 and 95% air at 37 °C (all percentage concentrations are vol/vol unless otherwise specified). GBM39 human glioblastoma cells were generated as previously described (25) and maintained in NeuroCult-XF Proliferation Medium (STEMCELL Technologies, Inc.) containing 20 ng/mL EGF (Sigma) and FGF (Sigma) and 1 µg/mL heparin (Sigma) in humidified atmosphere of 5% CO2 and 95% air at 37 °C. The DNA and antibody reagents are listed in the SI Appendix, Table S1. The DNA–antibody conjugates were synthesized as described in SI Appendix, Method I and validated with standard proteins by DNA spot microarray before use (29).
Protein Assays from Bulk Cell Culture.
The validation of the DNA–antibody conjugates involved separate calibrations for each of the different immunoassays (SI Appendix, Fig. S2C), as well as quantitating the cross-reactivity between those immunoassays (SI Appendix, Fig. S2B). All bulk protein assays in this study started with spotted DNA microarrays that were obtained from the Institute for Systems Biology (Seattle, WA). The spotted arrays and the flow patterned barcode arrays used the same DNA oligomer pairs (SI Appendix, Table S1) for each detected protein. The description of the microwell-based multiplexed immunoassays from statistical numbers of cells followed our previously published protocols (29).
mTOR Kinase Inhibition Assay.
U87 EGFRvIII cells were cultured in DMEM with 1% FBS at a density of 150,000 cells/mL and at O2 levels controlled to be 21%, 3%, 2%, 1.5%, or 1% for 7 h, with or without addition of 3 µM of the mTOR kinase inhibitor 2-(4-amino-1-isopropyl-1H-pyrazolo[3,4-d]pyrimidin-3-yl)-1H-indol-5-ol (PP242; Sigma-Aldrich). GBM39 neurosphere cells were dissociated with TripLE (Invitrogen) to form a single-cell suspension, and then cultured in laminin (Sigma) precoated dishes with NeuroCult-XF Proliferation Medium at a density of 150,000 cells/mL and at various conditions identical to U87 EGFRvIII cells above. Following incubation, the treated cells were then washed with cold PBS to remove residual media. A mixture of cell lysis buffer (Cell Signaling) containing 20 mM Tris⋅HCl, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, and 1 µg/mL leupeptin; Complete Protease Inhibitor (Roche); and Phosphatase Inhibitor Mixture 2 (Sigma) was added, and the mixture was stored on ice for 10 min. The cell extract was then collected and spun at 14,000 × g at 4 °C for 10 min. The resulting supernatant was recentrifuged to remove remaining cell debris. The cell lysate and media were then added into corresponding wells for profiling secreted and intracellular proteins. This protocol followed closely a previously published optimized protocol (21).
SCBC Fabrication and Operation.
DNA barcode arrays are flow patterned using molded polydimethylsiloxane (PDMS) microfluidics templates. This procedure has been previously described (39), and the quality of the flow-patterned barcodes is reflected in the data of SI Appendix, Fig. S1C.
For SCBC fabrication, the PDMS microfluidic chip for the single-cell assay was fabricated by two-layer soft lithography (SI Appendix, Fig. S1 A and B). The fabrication of these chips has been previously described (21), and with a few specific differences, as described here. The channel surface of the as-fabricated PDMS chip was coated with collagen type 1 (BD Biosciences; 0.1 mg/mL in deionized water) before thermally bonded to the DNA barcode slide to form the working device. The collagen coating promoted cell adherence during the on-chip cell culture.
Protocols of single-cell proteomic assays that were used for profiling secreted, intracellular, and membrane proteins from single cells are derived from previously published work, but with the several modifications described in detail in SI Appendix, Method II.
Protein Calibration and Error Analysis for Bulk and SCBC Assays.
Calibration curves for bulk protein measurement.
The assay for generating calibration curves was performed under conditions identical to the mTOR kinase inhibition assay described previously, except that standard proteins were used instead of cell lysate or medium. A mixture of standard proteins was serially diluted in 1× PBS and added into different wells. Fluorescence signals were collected and plotted vs. protein concentrations (SI Appendix, Fig. S2C and Table S2).
Calibration curves for SCBC measurement.
These calibrations were performed within an SCBC and under exact the same condition as the single-cell proteomic assay described above, except that standard proteins were used, rather than cells. A mixture of standard proteins from the SCBC assayed panel was serially diluted in 1× PBS and flowed into the SCBC microchannels. Fluorescence signals were collected to generate the calibration curves (SI Appendix, Fig. S2C). Because the volume of the microchambers is known, these calibration curves enable a transformation from the fluorescence intensity to number of molecules for each protein assayed, under the caveat that the standard proteins may not be exactly the same as their counterparts from the GBM cells.
Experimental error in SCBC assays.
The error analysis of proteomic measurement via SCBC was intensively discussed in our previous work (21, 24). Briefly, the experimental error comes from barcode-structured protein assays. We previously demonstrated that location of a cell within a microchamber does not contribute significantly to measurement error (21, 24). The width of a histogram that plots the frequency at which a particular (binned) protein level is observed vs. that level (which represents the single cell fluctuations) is dominated by the biology (the cell-to-cell heterogeneity) rather than the experiment. The experimental measurement error is generally less than 10% for this fashion of measurement.
Monte Carlo Simulation of Protein Fluctuation Profile.
The simulation is designed to capture the protein fluctuation profile for different degrees of protein activity, and is programmed by R (www.r-project.org). The protein is assumed to be able to participate in up to four independent functional processes. Each process has a required range of protein concentration (protein copy number for a given volume) represented by a Gaussian distribution and a fraction active, which is the likelihood of the protein carrying out this process. If the protein does not carry out any process, its concentration is set as an inactive baseline. The more active the functional protein is, the more processes it will participate in (up to four in this simulation).
The hypothesized processes and their required concentrations and fraction active values are listed in SI Appendix, Table S3.
In the Fig. 2C, one active process means that protein is confined to be involved only in process 1 or doing nothing; two active processes represents that the protein can access both process 1 and process 2 and so on. Finally, four active processes indicate the protein is able to participate into all four parallel functional processes listed above. The Gaussian distributions for representing the required range of protein copy number have been set to have a fixed coefficient of variation as 0.15. A total of 200 single-cell events are generated for each case, and the corresponding histogram is plotted in Fig. 2C. The averaged required protein copy number and fraction active value for each process are arbitrarily chosen for calculation convenience, and can be altered freely without affecting the final conclusion of the simulation.
Supplementary Material
Acknowledgments
This work was funded by National Cancer Institute Grant 5U54 CA119347 (to J.R.H.), National Institute of Neurological Disorders and Stroke Grant R01 NS73831 (to P.S.M.), the Ben and Catherine Ivy Foundation, the Jean Perkins Foundation, and the Grand Duchy of Luxembourg. F.R. is a Director of Research with Fonds National de Recherche Scientifique, Belgium.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1303060110/-/DCSupplemental.
References
- 1.Provenzano PP, et al. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21(3):418–429. doi: 10.1016/j.ccr.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goel S, et al. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev. 2011;91(3):1071–1121. doi: 10.1152/physrev.00038.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bertout JA, Patel SA, Simon MC. The impact of O2 availability on human cancer. Nat Rev Cancer. 2008;8(12):967–975. doi: 10.1038/nrc2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown JM, Wilson WR. Exploiting tumour hypoxia in cancer treatment. Nat Rev Cancer. 2004;4(6):437–447. doi: 10.1038/nrc1367. [DOI] [PubMed] [Google Scholar]
- 5.Collingridge DR, Piepmeier JM, Rockwell S, Knisely JP. Polarographic measurements of oxygen tension in human glioma and surrounding peritumoural brain tissue. Radiother Oncol. 1999;53(2):127–131. doi: 10.1016/s0167-8140(99)00121-8. [DOI] [PubMed] [Google Scholar]
- 6.Harris AL. Hypoxia—a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2(1):38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- 7.Tannock IF. Conventional cancer therapy: Promise broken or promise delayed? Lancet. 1998;351(Suppl. 2):9–16. doi: 10.1016/s0140-6736(98)90327-0. [DOI] [PubMed] [Google Scholar]
- 8.Semenza GL. Oxygen sensing, homeostasis, and disease. N Engl J Med. 2011;365(6):537–547. doi: 10.1056/NEJMra1011165. [DOI] [PubMed] [Google Scholar]
- 9.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3(10):721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 10.Guertin DA, Sabatini DM. The pharmacology of mTOR inhibition. Sci Signal. 2009;2(67):pe24. doi: 10.1126/scisignal.267pe24. [DOI] [PubMed] [Google Scholar]
- 11.Menon S, et al. Chronic activation of mTOR complex 1 is sufficient to cause hepatocellular carcinoma in mice. Sci Signal. 2012;5(217):ra24. doi: 10.1126/scisignal.2002739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer. 2008;8(11):851–864. doi: 10.1038/nrc2501. [DOI] [PubMed] [Google Scholar]
- 13.Fan QW, et al. A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell. 2006;9(5):341–349. doi: 10.1016/j.ccr.2006.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akhavan D, Cloughesy TF, Mischel PS. mTOR signaling in glioblastoma: Lessons learned from bench to bedside. Neuro Oncol. 2010;12(8):882–889. doi: 10.1093/neuonc/noq052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Houghton PJ, et al. Initial testing (stage 1) of the mTOR kinase inhibitor AZD8055 by the pediatric preclinical testing program. Pediatr Blood Cancer. 2012;58(2):191–199. doi: 10.1002/pbc.22935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Majumder PK, et al. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat Med. 2004;10(6):594–601. doi: 10.1038/nm1052. [DOI] [PubMed] [Google Scholar]
- 17.Kholodenko B, Yaffe MB, Kolch W. Computational approaches for analyzing information flow in biological networks. Sci Signal. 2012;5(220):re1. doi: 10.1126/scisignal.2002961. [DOI] [PubMed] [Google Scholar]
- 18.Wang MY, et al. Mammalian target of rapamycin inhibition promotes response to epidermal growth factor receptor kinase inhibitors in PTEN-deficient and PTEN-intact glioblastoma cells. Cancer Res. 2006;66(16):7864–7869. doi: 10.1158/0008-5472.CAN-04-4392. [DOI] [PubMed] [Google Scholar]
- 19.Tanaka K, et al. Oncogenic EGFR signaling activates an mTORC2-NF-κB pathway that promotes chemotherapy resistance. Cancer Discov. 2011;1(6):524–538. doi: 10.1158/2159-8290.CD-11-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma C, et al. A clinical microchip for evaluation of single immune cells reveals high functional heterogeneity in phenotypically similar T cells. Nat Med. 2011;17(6):738–743. doi: 10.1038/nm.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shi Q, et al. Single-cell proteomic chip for profiling intracellular signaling pathways in single tumor cells. Proc Natl Acad Sci USA. 2012;109(2):419–424. doi: 10.1073/pnas.1110865109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Quake SR, Scherer A. From micro- to nanofabrication with soft materials. Science. 2000;290(5496):1536–1540. doi: 10.1126/science.290.5496.1536. [DOI] [PubMed] [Google Scholar]
- 23.Apsel B, et al. Targeted polypharmacology: Discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nat Chem Biol. 2008;4(11):691–699. doi: 10.1038/nchembio.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shin YS, et al. Protein signaling networks from single cell fluctuations and information theory profiling. Biophys J. 2011;100(10):2378–2386. doi: 10.1016/j.bpj.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sarkaria JN, et al. Use of an orthotopic xenograft model for assessing the effect of epidermal growth factor receptor amplification on glioblastoma radiation response. Clin Cancer Res. 2006;12(7 Pt 1):2264–2271. doi: 10.1158/1078-0432.CCR-05-2510. [DOI] [PubMed] [Google Scholar]
- 26.Joo KM, et al. Patient-specific orthotopic glioblastoma xenograft models recapitulate the histopathology and biology of human glioblastomas in situ. Cell Rep. 2013;3(1):260–273. doi: 10.1016/j.celrep.2012.12.013. [DOI] [PubMed] [Google Scholar]
- 27.Bar EE, Lin A, Mahairaki V, Matsui W, Eberhart CG. Hypoxia increases the expression of stem-cell markers and promotes clonogenicity in glioblastoma neurospheres. Am J Pathol. 2010;177(3):1491–1502. doi: 10.2353/ajpath.2010.091021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carracedo A, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118(9):3065–3074. doi: 10.1172/JCI34739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bailey RC, Kwong GA, Radu CG, Witte ON, Heath JR. DNA-encoded antibody libraries: A unified platform for multiplexed cell sorting and detection of genes and proteins. J Am Chem Soc. 2007;129(7):1959–1967. doi: 10.1021/ja065930i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DeYoung MP, Horak P, Sofer A, Sgroi D, Ellisen LW. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev. 2008;22(2):239–251. doi: 10.1101/gad.1617608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Horak P, et al. Negative feedback control of HIF-1 through REDD1-regulated ROS suppresses tumorigenesis. Proc Natl Acad Sci USA. 2010;107(10):4675–4680. doi: 10.1073/pnas.0907705107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Y, et al. Bnip3 mediates the hypoxia-induced inhibition on mammalian target of rapamycin by interacting with Rheb. J Biol Chem. 2007;282(49):35803–35813. doi: 10.1074/jbc.M705231200. [DOI] [PubMed] [Google Scholar]
- 33.Chen H, et al. mTOR activates hypoxia-inducible factor-1α and inhibits neuronal apoptosis in the developing rat brain during the early phase after hypoxia-ischemia. Neurosci Lett. 2012;507(2):118–123. doi: 10.1016/j.neulet.2011.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gibbs JW. On the equilibrium of heterogeneous substances. Trans Conn Acad Arts and Sci. 1874;3:343–524. [Google Scholar]
- 35.Alberty RA. Legendre transform in chemical thermodynamics. Chem Rev. 1994;94:1457–1482. [Google Scholar]
- 36.Cloughesy TF, et al. Antitumor activity of rapamycin in a phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med. 2008;5(1):e8. doi: 10.1371/journal.pmed.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chang SM, et al. North American Brain Tumor Consortium and the National Cancer Institute Phase II study of CCI-779 in patients with recurrent glioblastoma multiforme. Invest New Drugs. 2005;23(4):357–361. doi: 10.1007/s10637-005-1444-0. [DOI] [PubMed] [Google Scholar]
- 38.Mellinghoff IK, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005;353(19):2012–2024. doi: 10.1056/NEJMoa051918. [DOI] [PubMed] [Google Scholar]
- 39.Shin YS, et al. Chemistries for patterning robust DNA microbarcodes enable multiplex assays of cytoplasm proteins from single cancer cells. ChemPhysChem. 2010;11(14):3063–3069. doi: 10.1002/cphc.201000528. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.