

Abstract
There is convincing evidence that nitric oxide (NO) may be a causative factor in the pathogenesis of migraine. We investigated the consequences of NO donors’ administration on meningeal processes related to the development of migraine pain in an animal model of meningeal nociception. The administration in mice of the NO donors nitroglycerin (GTN) and sodium nitroprusside (SNP) produced a delayed meningeal upregulation of interleukin-1ß and inducible NO synthase. A thermal allodynia and hyperalgesia devoid of side effects was produced 1 to 4 h after administration. To clarify the cellular pathways modulated by GTN and SNP, we examined the expression of cellular factors involved in pain modulation, such as protein kinase C (PKC) and its downstream effectors. Western blotting experiments showed an upregulation and increased phosphorylation of PKCγ and PKCε within dura mater after NO donors’ administration. A dramatic PKC-dependent increase of the phosphorylation of cyclic AMP response element binding protein (CREB) and signal transducer and activator of transcription (STAT)-1 was observed, along with an activation of the nuclear factor-κB (NF-κB) pathway, as reflected by a reduction of the inhibitory protein-κ-Bα (IκBα). Furthermore, the PKC blocker, Calphostin C, prevented the GTN and SNP-induced pain hypersensitivity. These results suggest the relevance of the PKC-mediated pathway in the induction of meningeal nociception and might help clarify the etiopathology of migraines. We can suggest PKC as a new target for migraine pain.
Electronic supplementary material
The online version of this article (doi:10.1007/s13311-012-0151-8) contains supplementary material, which is available to authorized users.
Keywords: Migraine, Protein kinase C, CREB, STAT1, NF-κB, NO
Introduction
Despite much research and advanced knowledge in migraine pathophysiology, the mechanisms underlying migraine attacks remain poorly understood. There is convincing evidence that nitric oxide (NO) may be a causative factor in the pathogenesis of migraines [1]. Intravenous infusion or sublingual administration of the NO donor nitroglycerin (GTN) has long been known to induce mild-to-moderate headaches in both healthy subjects and patients suffering from primary headaches [2, 3], but only in migraineurs, this immediate headache is followed by a second headache phase that fulfills the criteria of genuine migraine attacks [1, 2, 4]. NO is a very important molecule in the regulation of cerebral and extracerebral cranial blood flow and arterial diameters, but importantly, it is also involved in nociceptive processing in the central nervous system. In healthy subjects, GTN induces facilitation in the pain processing at the spinal trigeminal nucleus site [5], with a prominent role in the pathophysiology of migraines by modulating pain transmission from intracranial structures to higher centers of the brain. A facilitation of pain processing at the trigeminal level has also been reported during GTN-triggered migraine attacks [6]. In animals, GTN increases neuronal activity in the trigeminal nucleus caudalis and periaqueductal grey matter [7]. The dura mater is among the few intracranial tissues that evoke pain when stimulated, and dural afferent signals can activate the nucleus trigeminalis caudalis. After GTN infusion, the threshold for activation of neurons by stimulation of dural afferents was reduced [8]. Similarly, systemic infusion of the NO donor sodium nitroprusside (SNP) induced an immediate transient and a second delayed increase in activity of neurons in the spinal trigeminal nucleus with meningeal afferent input [9]. GTN also produces a delayed meningeal inflammation mediated by direct actions on the dura mater that does not develop secondary to events within the brain [10]. The migraine is thought to be generated by nociceptive processes within the meninges, followed by activation of trigeminal neurons within the brainstem, but the noxious stimuli initially involved in these nociceptive processes are unknown.
Protein kinase C (PKC) is a family of enzymes located in anatomical regions that regulate pain and their role has been described in acute and chronic pain. At different anatomical levels (peripheral nerve terminal, spinal and supraspinal sites), PKC integrates numerous receptor pathways into final effectors that increase excitatory signaling and decrease inhibitory signaling, thus inducing pain [11]. The activation of PKC has been related to the induction of a painful condition, whereas PKC blockers decreased nociception [11]. Recently, it has been reported that PKC inhibitors reduce NO synthesis from IFN-γ-treated microglia and that specific PKC isoforms are able to regulate inducible NO synthase (iNOS) expression in mouse peritoneal macrophages [12]. It is plausible to suppose that PKC might be activated after the administration of NO donors. Then we investigated the selective involvement of specific PKC isoforms in a painful symptomatology related to a migraine attack in an animal model of meningeal nociception induced by administration of GTN and SNP [9, 13]. To clarify the cellular pathways involved in the induction of meningeal pain hypersensitivity, we focused our attention on PKC downstream effectors. We demonstrated that the activation of a PKC-mediated pathway involving cyclic AMP response element binding protein (CREB) and signal transducer and activator of transcription 1 (STAT1) that appeared to be responsible for meningeal pain hypersensitivity.
Materials and Methods
Animal Treatment
All experiments were carried out in accordance with the European Communities Council Directive of November, 24 1986 (86/609/EEC).
Male Swiss albino mice (20-22 g) from the Morini (San Polo d'Enza, Italy) breeding farm were used. Ten mice were housed per cage (26 × 41 cm). The cages were placed in the experimental room 24 h before the test for acclimatization. The animals were fed a standard laboratory diet and tap water ad libitum, and were kept at 23 ± 1 °C with a 12 h light/dark cycle (i.e., light on at 7 AM). GTN (10 mg/kg; Bioindustria L.I.M., [Novi Ligure, Italy]), dissolved in 10 % ethylene glycol in saline (0.9 % NaCl) and SNP (1 mg/kg, Sigma, [Milan, Italy]), dissolved in saline were administered intraperitoneally, as previously reported [14].
To investigate the role of PKC in the intracellular events modulated by NO donors, the PKC blocker Calphostin C (0.2 μg/mouse i.c.v. [Calbiochem, Milan, Italy]) dissolved in 0.5 % dimethyl sulfoxide, as previously described [15]. Time-course experiments performed in our laboratory showed that the Calphostin C effect peaked 1 to 2 h after intracerebroventricular administration. Animals, therefore, were divided into 2 treatment groups: 1) Calphostin C administered 10 minutes before NO donors injection and protein expression detected 1 or 2 h after GTN/SNP administration; 2) Calphostin C administered 3 h after NO donors, and experiments performed 4 or 6 h after NO donors treatment.
Lipopolysaccharide (LPS) (60 mg/kg i.p., Sigma) was used as positive control of iNOS and interleukin (IL)-1ß expression. After LPS administration, dura mater to conduct Western blotting experiments was removed after 4 and 6 h for iNOS detection, and after 6 h for IL-1ß experiments.
Cold Plate Test
For assessment of cold allodynia, mice were placed on a cold plate that is maintained at a temperature of 4 ± 0.1 ° C. Reaction times were measured with a stopwatch before, and 1, 2, 4, and 6 h after administration of the NO donors. The time between placements of a mouse on the plate and licking or lifting of a hind paw was measured with a digital timer. An arbitrary cut-off time of 60 seconds was adopted and 10 mice per group were used.
Hot Plate Test
Mice were placed inside a stainless steel container, which was thermostatically set at 50.0 ± 0.1 ° C. Reaction times were measured with a stopwatch before, and 1, 2, 4, and 6 h after administration of the NO donors. The endpoint used was the licking of the fore or hind paws. An arbitrary cut-off time of 60 seconds was adopted and 10 mice per group were used.
Motor Coordination
The motor coordination was assessed by using the rota rod test. The apparatus consisted of a base platform and a rotating rod with a diameter of 3 cm and a nonslippery surface. The rod was placed at a height of 15 cm from the base. The rod (30 cm in length) was divided into 5 equal sections by 6 disks. Thus, up to 5 mice were tested simultaneously on the apparatus, with a rod-rotating speed of 16 r.p.m. The integrity of motor coordination was assessed on the basis of the number of falls from the rod in 30 seconds. Those mice scoring <3 and >6 falls in the pre-test were rejected (20 %). The number of falls was measured before (pre-test), and 1, 2, 4, and 6 h after the administration of the NO donors, and 10 mice per group were used.
Locomotor Activity
The locomotor activity was evaluated by using the hole-board test. The apparatus consisted of a 40-cm square plane with 16 flush-mounted cylindrical holes (3-cm diameter) distributed 4 × 4 in an equidistant, grid-like manner. Mice were placed on the center of the board 1 × 1 and were allowed to move around freely for a period of 5 minutes each. Two photo beams, crossing the plane from mid-point to mid-point of opposite sides, thus dividing the plane into 4 equal quadrants, automatically signaled the movement of the animal (counts in 5 minutes) on the surface of the plane (locomotor activity). Miniature photoelectric cells, in each of the 16 holes, recorded (counts in 5 minutes) the exploration of the holes (exploratory activity) by the mice. Experiments were performed 4 h after administration of the NO donors and 10 mice per group were tested.
Western Blot Analysis
Dura mater was collected at 1, 2, 4, and 6 h after GTN or SNP treatment and was homogenized in a buffer containing protease and phosphatase inhibitors. The homogenate was centrifuged at 9,000 x g for 15 minutes at 4 °C; the low speed pellet was discarded and the supernatant was stored at -80 ° C. Protein concentration was quantified using Bradford’s method (protein assay kit [Bio-Rad Laboratories, Milan, Italy]). Membrane homogenates (10-50 μg) were separated on 10 % sodium dodecyl sulphate - PolyAcrylamide Gel (SDS-PAGE), and were then transferred onto nitrocellulose membranes, blocked and probed with specific antibodies against PKCγ phosphorylated on Thr514 (pPKCγ, 1:1000 dilution), c-Fos (1:1000) (Biosource [Camarillo, CA]); PKCγ (1:1000); PKCε (1:800); PKCε phosphorylated on Ser729 (pPKCε, 1:750); iNOS (1:250); IL-1ß (1:1000); Iκ-Bα (1:1000); STAT1 phosphorylated on Tyr701 (pSTAT1, 1:500); ß-actin (1:1000 dilution) (Santa Cruz Biothechnology Inc. [Santa Cruz, CA, USA]); CREB (1:500) or phosphorylated CREB (pCREB) on Ser133 (1:500) (Cell Signalling Technology [Billerica, MA, USA]) followed by peroxidase-conjugated secondary antisera (1:10,000). Blots were developed using enhanced chemiluminescence detection system (Pierce [Milan, Italy]). Optical density measurements were performed by dividing the intensity of the bands by the intensity of the housekeeping protein ß-actin. Measurements in control samples were assigned a relative value of 100 %.
Statistical Analysis
All experimental results are given as the mean ± S.E. mean. The results of Western blotting experiments are the mean of at least 4 independent experiments. Analysis of variance followed by Tukey post hoc test was used for statistical analysis (GraphPad Prism 5.0).
Results
Reduction of the Pain Threshold without Induction of Side Effects by NO Donors
The administration of GTN (10 mg/kg i.p.) and SNP (1 mg/kg i.p.) produced cold allodynia in the cold plate test. The reaction times to the cold stimulus were reduced 1, 2, and 4 h after NO donors’ administration and the pain threshold returned to control values after 6 h (Fig. 1A). After NO donor treatment, a heat hyperalgesia was also observed in the hot plate test with a similar profile and time course to the cold plate test results (Fig. 1B).
Fig. 1.

Nitric oxide (NO) donors decreased the pain threshold without behavioral side effects. Administration of nitroglycerin (GTN) (10 mg/kg i.p.) or sodium nitroprusside (SNP) (1 mg/kg i.p.) induced cold allodynia evaluated in the cold plate test (A), and thermal hyperalgesia, evaluated in the hot plate test (B); 1, 2, and 4 h after injection. GTN and SNP did not alter spontaneous mobility, inspection activity (hole board test) (C), or motor coordination (rota rod test) (D) at any time point. Amphetamine (2 mg/kg i.p.) was used as a positive control, and 10 mice per group were used. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with the control group
The reduction of the pain threshold was not accompanied by the induction of side effects. The spontaneous mobility and exploratory activity of mice treated with GTN and SNP were unmodified in comparison with the control group. In the same experimental conditions, D-amphetamine, used as a positive control, significantly increased both parameters evaluated (Fig. 1C). NO donors did not alter motor coordination of treated animals at any time point (Fig. 1D).
NO Donors Induced IL-1ß, iNOS Expression, and Activation of the Nuclear Factor-κB (NF-κB) Pathway
The meninges are among the few pain-sensitive tissues within the cranium and to evaluate the site of the hyperalgesic action of NO donors, we detected the expression of cellular mediators modulated by NO within dura mater. Interleukin (IL)-1ß, iNOS, and Iκ-Bα were examined by immunoblotting in homogenates of dura mater after administration of GTN (10 mg/kg i.p.) and SNP (1 mg/kg i.p.). NO donors caused a biphasic increase in IL-1ß protein with a rapid rise at 1 to 2 h, followed by a second peak at 6 h. The increase of IL-1ß protein expression produced 6 h after LPS injection (60 mg/kg i.p.) was used as positive control (Fig. 2A).
Fig. 2.

Effect of nitric oxide (NO) donors’ administration on interleukin (IL)-1ß, inducible nitric oxide synthase (iNOS), and Iκ-Bα expression. Protein levels were detected by using an immunoblotting technique. (A) Densitometric measurements of IL-1ß protein expression show a biphasic increase after nitroglycerin (GTN) (10 mg/kg, i.p.; F[4, 18] 19.410) or sodium nitroprusside (SNP) (1 mg/kg i.p.; F[5, 24] 21.740), with a peak protein expression at 1 to 2 h and 6 h. IL-1ß protein expression after lipopolysaccharide (LPS) injection was used as positive control. (B) iNOS protein was not detected in the control animals (C) or in mice receiving GTN or SNP 1 and 2 h after administration within the dura mater. iNOS protein appeared at 4 and 6 h (GTN F[2, 9] 8.125); SNP (F[2, 9] 8.932). LPS was used as control. (C) NO donors activate meningeal NF-κB pathway, as indicated by the reduced levels of Iκ-Bα (GTN F[4, 15] 17.710; SNP 8F[4, 16] 21.670). The columns represent the densitometric quantitation of immunoreactive protein expressed relative to control. Representative immunoblots were reported in each panel. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with the control group
iNOS protein was not constitutively expressed in the dura mater as indicated by the absence of a band in control animals. At 4 and 6 h after GTN (10 mg/kg i.p.) or SNP administration (1 mg/kg i.p.), a 130 kDa band, corresponding to iNOS protein, was detected. iNOS was not expressed at earlier time points (Fig. 2B). LPS was used as positive control. iNOS protein expression was not detected within the brain, in agreement with previous observations [10]. The lack of iNOS expression was observed within the periaqueductal gray matter (PAG) and thalamus, brain areas involved in pain modulation, of GTN- and SNP-treated animals (data not shown).
To investigate transcriptional mechanisms that promote iNOS expression, the activation of NF-κB after NO donor administration was examined. In homogenates of dura mater, we observed a degradation of Iκ-Bα, the protein that constitutively inhibits NF-κB, as indicated by the significant decrease of protein levels, 1 and 2 h after GTN or SNP administration. At later time points (4 and 6 h), the levels of Iκ-Bα increased (Fig. 2C).
Increased Expression and Phosphorylation of PKCγ and PKCε Isoforms by NO Donors
SNP treatment produced a rapid and robust increase of pPKCγ levels within the dura mater that peaked 1 h after treatment. This effect slowly diminished, remaining significant up to 4 h after treatment and returning to control values at 6 h (Fig. 3A).
Fig. 3.

Nitroglycerin (GTN) and sodium nitroprusside (SNP) increased the expression and phosphorylation of protein kinase C (PKC)γ and PKCε isoforms, cyclic AMP response element binding protein (CREB), and signal transducer and activator of transcription 1 (STAT1) within the dura mater. (A) Nitroglycerin (GTN) (10 mg/kg i.p.) and SNP (1 mg/kg i.p.) increased the expression of phosphorylated PKCγ (pPKCγ) at 1, 2, and 4 h after administration. (B) The nitric oxide (NO) donors also increased the expression of pPKCε with a peak at 1 h after administration. (C) An influence on meningeal CREB protein levels was detected as revealed by reduced contents after GTN and SNP treatment. (D) An increased expression of phosphorylated CREB (pCREB) after NO donors’ administration was observed 2 h after SNP treatment. (E) The phosphorylation of STAT1 protein (pSTAT1) was increased 1, 2, and 4 h after SNP (1 mg/kg i.p.) treatment. The columns represent the densitometric quantitation of immunoreactive protein expressed relative to the control. Representative immunoblots are reported on each panel. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with the control group
A robust increase of the phosphorylation of PKCε was produced by both GTN and SNP. Similarly to pPKCγ, the pPKCε levels increased between 1 and 4 h with a peak at 1 h within the dura mater (Fig. 3B). The effect produced by the NO donors disappeared 6 h after administration (Fig. 3B). The intensity of the phosphorylation was comparable to that observed for the PKCγ isoform.
Similar results were obtained after GTN administration. PKCγ and PKCε protein expression were also increased 1, 2, and 4 h after treatment with a similar time course for GTN and SNP.
As potential downstream effectors of PKC, we detected CREB and STAT1. Experiments conducted on dura mater demonstrated that SNP and GTN reduced CREB protein content 2 h after administration, and then the effect disappeared (Fig. 3C).
A robust increase of the pCREB was detected (Fig. 3D). The SNP- and GTN-induced increase of pCREB was significant 1 h after administration, which peaked at 2 h, and then the effect drastically decreased. The pCREB values returned comparable to the control at 4 and 6 h after NO donor administration (Fig. 3D).
A dramatic increase in the phosphorylated form of STAT1 was detected in the dura mater. The pSTAT1 contents were significantly increased 1 h after SNP administration, which peaked at 4 h, and then returned comparable to the control after 6 h. Similar results were obtained after GTN treatment (Fig. 3E).
NO Donors Induce Allodynia and Hyperalgesia through a PKC-Dependent Mechanism
The administration of GTN and SNP produced a cold allodynia (Fig. 4A) and heat hyperalgesia (Fig. 4B) that peaked 2 to 4 h after administration. The intracerebroventricular injection of the PKC blocker Calphostin C (0.2 μg per mouse) in coincidence with the peak of pain hypersensitivity, completely reversed the hypersensitivity to thermal stimuli induced by NO donors, leading to reaction times comparable to the control values. Calphostin C, when administered alone, was devoid of any analgesic activity (Fig. 4A, 4B).
Fig. 4.

Allodynia and hyperalgesia produced by nitric oxide (NO) donors’ administration underlies a protein kinase C (PKC)-mediated pathway of activation. Cold allodynia (A) and heat hyperalgesia (B) induced by nitroglycerin (GTN) (10 mg/kg i.p.) and sodium nitroprusside (SNP) (1 mg/kg i.p.) were reversed by intracerebroventricular injection of the PKC blocker, Calphostin C (calph) (0.2 μg per mouse), and 10 mice per group were used. PKC blockade completely prevented the increased expression of pPKCγ (C) and pPKCε (D) within the dura mater. The protein levels were detected 4 h after SNP administration (C) (calph, 0.2 μg per mouse i.c.v.). The columns represent the densitometric quantitation of immunoreactive protein expressed relative to control. Representative immunoblots are reported in the top of each panel. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with the control group; °P<0.05, °°P < 0.01 and °°°P < 0.001 compared with SNP-treated group. SNP+C = SNP + calphostin C
Calphostin C prevented the increase of pPKCγ (Fig. 4C) and pPKCε (Fig. 4D) induced by SNP within the dura mater, indicating that the doses and administration schedule of Calphostin C in behavioral tests were ideal to block PKC activity.
PKC-Dependent Activation of CREB and STAT1
To investigate the cellular events involved in the PKC-activated pathway, we detected the effects produced on iNOS protein levels. A drastic reduction of iNOS production was detected in the dura mater of mice treated with the PKC blocker, Calphostin C (Fig. 5A). Since iNOS was upregulated 6 h after NO donors’ injection, when PKC phosphorylation was no longer detected, we investigated whether the PKC-mediated regulation of iNOS expression was mediated through the activation of upstream modulators of iNOS. GTN and SNP reduced the Iκ-Bα levels 1-2 h after administration. We, then, investigated if NO donors might induce a PKC-dependent modulation of the NF-κB pathway. Pretreatment with Calphostin C prevented the Iκ-Bα reduction in the dura mater (Fig. 5B).
Fig. 5.
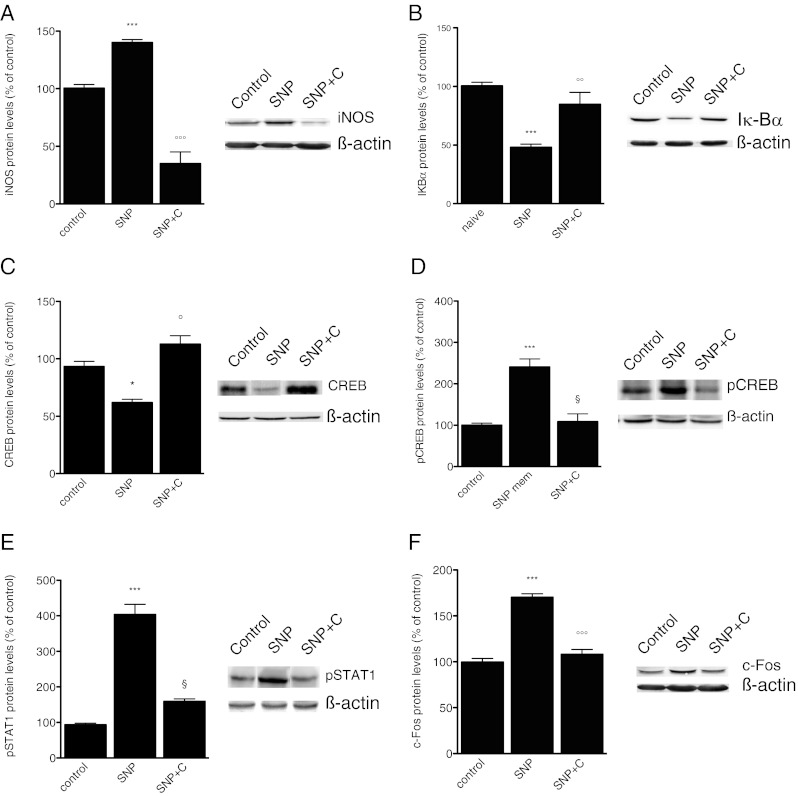
Nitric oxide (NO) donors modulate dural intracellular pathways through a protein kinase C (PKC)-dependent mechanism. Administration of Calphostin C (C) (0.2 μg per mouse i.c.v.) prevented the increased expression of inducible NO synthase (iNOS) (F[2, 12] 5.61) (A), antagonized the reduction of Iκ-Bα levels (F[2, 13] 4.49) (B), prevented the NO donor-induced decrease of cyclic AMP response element binding protein (CREB) (F[2, 12] 5.27) (C), and increase of phosphorylated CRED (pCREB) expression (F[2, 13] 7.22) (D). Calphostin C administration reversed the sodium nitroprusside (SNP)-induced increase of pSTAT1 (F[2, 13] 11.56) (E), and the increased cerebral expression of c-Fos (F[2, 13] 6.11) (F). The columns represent the densitometric quantitation of immunoreactive protein expressed relative to control. Representative immunoblots are reported in the top of each panel. *P < 0.05, ***P < 0.001 compared with the control group. °P < 0.05, °°P < 0.01, and °°°P < 0.001 compared with the SNP-treated group. SNP+C = SNP + calphostin C
CREB and STAT1, respectively, showed a peak activation 2 and 4 h after NO donors’ administration. The modulation of CREB expression was PKC-mediated, since the treatment with Calphostin C, completely reversed the SNP-induced decrease of CREB protein levels (Fig. 5C). The presence of PKC-mediated activation of CREB was demonstrated by the prevention of the pCREB upregulation after Calphostin C administration (Fig. 5D).
A dramatic increase in the phosphorylated form of STAT1 was detected in the dura mater of SNP-treated animals that peaked at 4 h. A PKC-mediated activation of STAT1 was also demonstrated for this transcription factor because the treatment with Calphostin C prevented the phosphorylation of STAT1 (Fig. 5E).
c-Fos contents were measured in several areas within the brainstem and the brain (PAG, trigeminal nucleus caudalis, thalamus) of NO donor-treated mice, and a c-Fos overexpression was observed in all areas. As an example, we reported the c-Fos content from PAG. c-Fos upregulation was inhibited by Calphostin C administration, illustrating that the NO donor-induced neuronal hyperactivation follows a PKC-dependent mechanism (Fig. 5F).
Discussion
The administration of GTN in humans produces intracellular events that lead to a short-lived (minutes) immediate headache followed by a delayed second headache phase only in susceptible individuals (migraineurs). After GTN and SNP administration in mice, we detected a prolonged pain hypersensitivity that appeared 1 h after administration. These results are supported by the observation that intravenous or local application of the NO donor SNP was followed by an increase in spontaneous neuronal activity of trigeminal neurons with meningeal afferent input with a delay of approximately 1 h, indicating that NO produces delayed and long-lasting pro-nociceptive changes in neurons in the trigeminal nucleus [9]. All these results are in good accordance with the clinical “migraine model” in which an infusion of GTN provoked migraine attacks with a similar delay of 1 to several hours [2, 3, 16]. Due to the short half-life of NO donors in the blood plasma, this effect cannot be explained by a direct activation of trigeminal neurons, but rather the NO derived from SNP or GTN is presumed to have triggered a long-term process. We propose the NO-induced PKC-dependent activation of a signaling pathway involving CREB, STAT1, and NF-κB as the mechanisms underlying this process.
Along with pain hypersensitivity, the systemic administration of GTN and SNP produced a delayed meningeal inflammation in mice similar to that observed after NO donors’ infusion [10], as demonstrated by the delayed expression of IL-1ß and iNOS within the dura mater. To elucidate the mechanism responsible for the production of pain hypersensitivity, we first investigated the role of PKC. It is known that PKC represents a second messenger pathway coupled to the induction of c-Fos, the protein product of the immediate early gene c-fos, widely used as marker of neuronal activation and pain [17]. It is also a family of enzymes highly involved in pain modulation [11]. We observed an increased expression and phosphorylation of PKCε and PKCγ, isoforms with a prominent role in pain modulation [11], within meninges, concomitantly with the presence of allodynia and hyperalgesia. Treatment with the PKC inhibitor, Calphostin C, prevented allodynia and hyperalgesia. The same treatment also prevented the increased phosphorylation of PKCε and PKCγ in the dura mater, demonstrating that the Calphostin C activity was related to its ability to block PKC activation, ruling out the presence of any unspecific, PKC-independent effect. Several lines of evidence illustrated that conventional PKC isoenzymes [18, 19], PKCδ [12, 20], PKCη [21], and PKCε [22] are involved in the LPS- and cytokine-induced expression of inflammatory genes, including iNOS, as it emerges from studies conducted on murine microglia/macrophages. Furthermore, Calphostin C dramatically reduced meningeal iNOS expression after GTN and SNP administration. It is plausible to suppose that NO donors might directly activate PKC, and that this NO donor-induced PKC activation contributes to meningeal nociception in this animal model. This hypothesis is further supported by clinical evidence illustrating that tamoxifen, the only agent with documented and appreciable central PKC-inhibitory activity approved for human use [23], has shown promise for treating the migraine, as attested by case reports [24–26] and clinical studies [27, 28]. The positive effects of tamoxifen on the migraine were attributed to its anti-estrogenic activity. Present findings suggest that the tamoxifen-induced anti-migraine activity might be related to its PKC-blocking properties, further supporting the hypothesis that PKC signaling pathway may play an important role in the pathophysiology of the migraine. We can also suggest PKC as an innovative target for migraine pain.
To further elucidate the PKC-mediated pathway after NO donors’ administration, we investigated downstream effectors of PKC, such as NF-κB, STAT1, and CREB, transcription factors also involved in pain modulation. The relevance of NF-κB and STAT pathways in the response to cytokines and inflammation is widely known. The involvement of CREB in pain perception is indicated by the increased CREB phosphorylation in the dorsal horn in the early stages of inflammation [29] and sciatic nerve injury [30].
Mice treated with GTN or SNP showed a consistent activation of NF-κB, CREB, and STAT1 in the dura mater in coincidence with pain hypersensitivity. PKC appears to be an upstream modulator of these transcription factors because the PKC inhibitor, Calphostin C, prevented the IκBα degradation and the CREB and STAT1 hyperphosphorylation, allowing us to hypothesize a role of these transcription factors in the NO donor-induced meningeal pain hypersensitivity. A further correlation between these transcription factors and migraine pain arises from the observation that the activation of the NF-κB pathway is attenuated by parthenolide, the active constituent of feverfew [31] and aspirin [32], anti-inflammatory drugs used for migraine treatment. Recently, it has been observed that triptans, widely used anti-migraine drugs, attenuate capsaicin-induced CREB phosphorylation within the trigeminal nucleus caudalis [33].
The NF-κB and STAT1-dependent signaling pathways are integral to the transcriptional regulation of many inflammatory genes, and these transcriptional factors often cooperatively regulate the transcriptional activation of many genes. It has been demonstrated that IFN-γ-induced STAT1α and TNFα-induced NF-κB synergistically regulate the transcription of the intercellular adhesion molecule-1 and IRF-1 genes [34–36]. Furthermore, the transcriptional coactivator CREB-binding protein cooperates with STAT1 and NF-κB for synergistic transcriptional activation of the gene for CXC ligand 9, an IFNγ-inducible chemokine [37]. We might hypothesize that CREB, STAT1, and NF-κB, acting as downstream effectors of PKC, can cooperatively modulate the sensation of pain to produce a condition of hypersensitivity to noxious stimuli.
Concerning the cell type that might be involved in NO donors’ dural effect, it has been reported that after GTN infusion in rats, a meningeal inflammation characterized by increased IL-6 levels and iNOS expression in dural macrophages was observed [10]. We detected a similar pattern of meningeal inflammation after i.p. administration of NO donors, and we can hypothesize a prominent role of dural macrophages. However, we also observed a PKCγ upregulation. Because PKCγ is a neuronal PKC isoform, we cannot exclude a neuronal involvement.
Although GTN can accumulate and reach toxic levels in adipose tissue and lipid-rich organs, such as the brain, we can exclude that the hypersensitivity observed was related to an altered viability of mice. NO donors did not modify spontaneous mobility, inspection activity, locomotor activity, and were not endowed with visible behavioral side effects at any time point. In regard to the cardiovascular effect, both GTN and SNP systemically administered at the doses used in the present study induced moderate hypotension that lasted 40 and 80 minutes, respectively [38]. No altered cardiovascular parameter was observed 2 and 4 h after treatment [38], and we can suppose that pain hypersensitivity was not subsequent to a hypotensive effect.
Concerning the possible mechanism of action of NO and subsequent steps in the NO-induced cascade, it has been clinically suspected that delayed GTN-induced migraine could be caused by iNOS activation and increased production of NO [1]. However, we observed that the NO donor-induced pain hypersensitivity preceded iNOS expression, suggesting a lack of involvement of iNOS in the induction of the painful symptomatology, typical of this animal model of meningeal nociception. Clinical trials investigating the efficacy of selective iNOS inhibitors for the acute [39] and prophylactic [40] treatment of migraine showed negative results, further supporting the hypothesis that iNOS is not directly involved in the induction of migraine pain. Considering the high iNOS inhibition achieved with these inhibitors, these negative results indicate that iNOS is unlikely to be a suitable target for the anti-migraine therapy.
GTN doses used in animal studies are always higher than those able to cause a migraine attack in migraineurs. Intravenous infusion of GTN can allow the use of a dose that, whereas supramaximal to the dose-producing migraine in humans is much lower than that administered intraperitoneally. After both administration routes, GTN facilitates transmission of afferent fibers to the trigeminal nucleus [8, 41]. However, after GTN infusion, the c-Fos expression was unchanged [8], whereas c-Fos was upregulated after intraperitoneal injection into selected areas of the brain that are primarily involved in the transmission of cephalic pain [7, 42], and hyperalgesia was induced [14]. Based on these data, the murine phenotype we obtained appears to be representative not only for NO-induced pain hypersensitivity, but also for the migraine pain experienced by migraineurs during a migraine attack.
These findings highlight the upregulation and increased phosphorylation of PKCγ and PKCε in an animal model induced by NO donors’ administration, with a time course consistent with migraine attacks. The activation of the PKC-mediated pathways was concomitant with the presence of pain hypersensitivity and appeared to be responsible for the meningeal nociception. We can also suggest PKC as an innovative target for migraine pain.
Electronic Supplementary Material
(PDF 510 kb)
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
References
- 1.Olesen J. The role of nitric oxide (NO) in migraine, tension-type headache and cluster headache. Pharmacol Ther. 2008;120:157–171. doi: 10.1016/j.pharmthera.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 2.Iversen HK, Olesen J, Tfelt-Hansen P. Intravenous nitroglycerin as an experimental model of vascular headache. Basic characteristics. Pain. 1989;38:17–24. doi: 10.1016/0304-3959(89)90067-5. [DOI] [PubMed] [Google Scholar]
- 3.Olesen J, Iversen HK, Thomsen LL. Nitric oxide supersensitivity: a possible molecular mechanism of migraine pain. Neuroreport. 1993;4:1027–1030. doi: 10.1097/00001756-199308000-00008. [DOI] [PubMed] [Google Scholar]
- 4.Christansen I, Thomsen LL, Daugaard D, Ulrich V, Olesen J. Glyceryl trinitrate induces attacks of migraine without aura in sufferers of migraine with aura. Cephalalgia. 1999;19:660–667. doi: 10.1046/j.1468-2982.1999.019007660.x. [DOI] [PubMed] [Google Scholar]
- 5.Di Clemente L, Coppola G, Magis D, et al. Nitroglycerin sensitises in healthy subjects CNS structures involved in migraine pathophysiology: evidence from a study of nociceptive blink reflexes and visual evoked potentials. Pain. 2009;144:156–161. doi: 10.1016/j.pain.2009.04.018. [DOI] [PubMed] [Google Scholar]
- 6.de Tommaso M, Guido M, Libro G, Sciruicchio V, Puca F. Zolmitriptan reverses blink reflex changes induced during the migraine attack in humans. Neurosci Lett. 2000;289:57–60. doi: 10.1016/S0304-3940(00)01255-6. [DOI] [PubMed] [Google Scholar]
- 7.Tassorelli C, Joseph SA. Systemic nitroglycerin induces Fos immunoreactivity in brainstem and forebrain structures of the rat. Brain Res. 1995;682:167–181. doi: 10.1016/0006-8993(95)00348-T. [DOI] [PubMed] [Google Scholar]
- 8.Jones MG, Lever I, Bingham S, Read S, McMahon S, Parson A. Nitric oxide potentiates response of trigeminal neurons to dural or facial stimulation in the rat. Cephalalgia. 2001;21:643–655. doi: 10.1046/j.1468-2982.2001.00213.x. [DOI] [PubMed] [Google Scholar]
- 9.Koulchitsky S, Fischer MJM, De Col R, Schlechtweg PM, Messlinger K. Biphasic response to nitric oxide of spinal trigeminal neurons with meningeal input in rat — possible implications for the pathophysiology of headaches. J Neurophysiol. 2004;92:1320–1328. doi: 10.1152/jn.01210.2003. [DOI] [PubMed] [Google Scholar]
- 10.Reuter U, Bolay H, Jansen-Olesen I, et al. Delayed inflammation in rat meninges: implications for migraine pathophysiology. Brain 2001;124:2490-502. [DOI] [PubMed]
- 11.Velazquez KT, Mohammad H, Sweitzer SM. Protein kinase C in pain: involvement of multiple isoforms. Pharmacol Res. 2007;55:578–589. doi: 10.1016/j.phrs.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhatt KH, Pandey RK, Dahiya Y, Sodhi A. Protein kinase Cδ and protein tyrosine kin ase regulate peptidoglycan-induced nuclear factor-κB activation and inducible nitric oxide synthase expression in mouse peritoneal macrophages in vitro. Mol Immunol. 2010;47:861–870. doi: 10.1016/j.molimm.2009.10.029. [DOI] [PubMed] [Google Scholar]
- 13.Dieterle A, Fisher MJ, Link AS, Neuhuber WL, Messlinger K. Increase in CGRP- and nNOS-immunoreactive neurons in the rat trigeminal ganglion after infusion of an NO donor. J Cephalalagia. 2011;31:31–42. doi: 10.1177/0333102410375725. [DOI] [PubMed] [Google Scholar]
- 14.Tassorelli C, Greco R, Wang D, Sandrini M, Sandrini G, Nappi G. Nitroglycerin induces hyperalgesia in rats – a time course study. Eur J Pharmacol. 2003;464:159–162. doi: 10.1016/S0014-2999(03)01421-3. [DOI] [PubMed] [Google Scholar]
- 15.Galeotti N, Bartolini A, Ghelardini C. The phospholipase C-IP3 pathway is involved in muscarinic antinociception. Neuropsychopharmacology. 2003;28:888–897. doi: 10.1038/sj.npp.1300111. [DOI] [PubMed] [Google Scholar]
- 16.Bank J. Migraine with aura after administration of sublingual nitroglycerin tablets. Headache. 2001;41:84–87. doi: 10.1046/j.1526-4610.2001.111006084.x. [DOI] [PubMed] [Google Scholar]
- 17.Harris JA. Using c-fos as marker of pain. Brain Res Bull. 1998;45:1–8. doi: 10.1016/S0361-9230(97)00277-3. [DOI] [PubMed] [Google Scholar]
- 18.Giroux M, Descoteaux A. Cyclooxygenase-2 expression in macrophages: modulation by protein kinase C-α. J Immunol. 2000;165:3985–3991. doi: 10.4049/jimmunol.165.7.3985. [DOI] [PubMed] [Google Scholar]
- 19.Foey AD, Brennan FM. Conventional protein kinase C and atypical protein kinase Cζ differentially regulate macrophage production of tumour necrosis factor-α and interleukin-10. Immunology. 2004;112:44–53. doi: 10.1111/j.1365-2567.2004.01852.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen CC, Wang JK, Lin SB. Antisense oligonucleotides targeting protein kinase C-α, -ßI, or -δ but not -η inhibit lipopolysaccharide-induced nitric oxide synthase expression in RAW 264.7 macrophages: involvement of a nuclear factor κB-dependent mechanism. J Immunol. 1998;161:6206–6214. [PubMed] [Google Scholar]
- 21.Pham TNQ, Brown BL, Dobson PRM, Richardson VJ. Protein kinase C-eta (PKC-ζ) is required for the development of inducible nitric oxide synthase (iNOS) positive phenotype in human monocytic cells. Nitric Oxide. 2003;9:123–134. doi: 10.1016/j.niox.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 22.Kang J, Yang M, Jou I, Joe E. Identification of protein kinase C isoforms involved in interferon-gamma-induced expression of inducible nitric oxide synthase in murine BV2 microglia. Neurosci Lett. 2001;299:205–208. doi: 10.1016/S0304-3940(01)01515-4. [DOI] [PubMed] [Google Scholar]
- 23.Baltuch GH, Couldwell WT, Villemure JG, Yong VW. Protein kinase C inhibitors suppress cell growth in established and low-passage glioma cell lines: a comparison between staurosporine and tamoxifen. Neurosurgery. 1993;33:495–501. doi: 10.1227/00006123-199309000-00021. [DOI] [PubMed] [Google Scholar]
- 24.Powles TJ. Prevention of migrainous headaches by tamoxifen. Lancet. 1986;2:1344. doi: 10.1016/S0140-6736(86)91483-2. [DOI] [PubMed] [Google Scholar]
- 25.Maggioni F, Palmieri A, Tropea M, Zanchin G. Influence of physiologic modification and of hormonal treatment in a patient with migraine with aura. J Headache Pain. 2008;9:129–131. doi: 10.1007/s10194-008-0018-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smitherman TA, Kolivas ED. Resolution of menstrually related migraine following aggressive treatment for breast cancer. Headache. 2009;50:485–496. doi: 10.1111/j.1526-4610.2009.01594.x. [DOI] [PubMed] [Google Scholar]
- 27.O’Dea JP, Davis EH. Tamoxifen in the treatment of menstrual migraine. Neurology. 1990;40:1470–1471. doi: 10.1212/WNL.40.9.1470. [DOI] [PubMed] [Google Scholar]
- 28.Cuzick J, Forbes JK, Sestak I, et al. Long-term results of tamoxifen prophylaxis for breast cancer – 96-month follow-up of the randomized IBIS-I trial. J Natl Cancer Inst. 2007;99:272–282. doi: 10.1093/jnci/djk049. [DOI] [PubMed] [Google Scholar]
- 29.Ji RR, Rupp F. Phosphorylation of transcription factor CREB in rat spinal cord after formalin-induced hyperalgesia: relationship to c-fos induction. J Neurosci. 1997;17:1776–1785. doi: 10.1523/JNEUROSCI.17-05-01776.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma W, Quirion R. Increased phosphorylation of cyclic AMP response element-binding protein (CREB) in the superficial dorsal horn neurons following partial sciatic nerve ligation. Pain. 2001;93:295–301. doi: 10.1016/S0304-3959(01)00335-9. [DOI] [PubMed] [Google Scholar]
- 31.Reuter U, Chiarugi A, Bolay H, Moskowitz MA. Nuclear factor-kappaB as a molecular target for migraine therapy. Ann Neurol. 2002;51:507–516. doi: 10.1002/ana.10159. [DOI] [PubMed] [Google Scholar]
- 32.Yin MJ, Yamamoto Y, Gaynor RB. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature. 1998;396:77–80. doi: 10.1038/23948. [DOI] [PubMed] [Google Scholar]
- 33.Mitsikostas DD, Knight YE, Lasalandra M, Kavantzas N, Goadsby PJ. Triptans attenuate capsaicin-induced CREB phosphorylation within the trigeminal nucleus caudalis: a mechanism to prevent central sensitization? J Headache Pain. 2011;12:411–417. doi: 10.1007/s10194-011-0352-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jahnke A, Johnson JP. Synergistic activation of intercellular adhesion molecule 1 (ICAM-1) by TNF-alpha and IFN-gamma is mediated by p65/p50 and p65/c-Rel and interferon-responsive factor Stat1 alpha (p91) that can be activated by both IFN-gamma and IFN-alpha. FEBS Lett. 1994;354:220–226. doi: 10.1016/0014-5793(94)01130-3. [DOI] [PubMed] [Google Scholar]
- 35.Ohmori Y, Schreiber RD, Hamilton TA. Synergy between interferon-gamma and tumor necrosis factor-alpha in transcriptional activation is mediated by cooperation between signal transducer and activator of transcription 1 and nuclear factor kappaB. J Biol Chem. 1997;272:14899–14907. doi: 10.1074/jbc.272.23.14899. [DOI] [PubMed] [Google Scholar]
- 36.Pine R. Convergence of TNFalpha and IFNgamma signalling pathways through synergistic induction of IRF-1/ISGF-2 is mediated by a composite GAS/kappaB promoter element. Nucleic Acids Res. 1997;25:4346–4354. doi: 10.1093/nar/25.21.4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hiroi M, Ohmori Y. The transcriptional coactivator CREB-binding protein cooperates with STAT1 and NF-κB for synergistic transcriptional activation of the CXC Ligand 9/monokine induced by Inteferon γ gene. J Biol Chem. 2003;1:651–660. doi: 10.1074/jbc.M204544200. [DOI] [PubMed] [Google Scholar]
- 38.Tassorelli C, Greco R, Cappelletti D, Sandrini G, Nappi G. Comparative analysis of the neuronal activation and cardiovascular effects of nitroglycerin, sodium nitroprusside and L-arginine. Brain Res. 2005;1051:17–24. doi: 10.1016/j.brainres.2005.05.067. [DOI] [PubMed] [Google Scholar]
- 39.Van der Schueren BJ, Lunnon MW, Laurijssens BE, et al. Does the unfavorable pharmacokinetic and pharmacodynamics profile of the iNOS inhibitor GW273629 lead to inefficacy in acute migraine? J Clin Pharmacol. 2009;49:281–290. doi: 10.1177/0091270008329548. [DOI] [PubMed] [Google Scholar]
- 40.Hoivik HO, Laurijssens BE, Harnisch LO, et al. Lack of efficacy of the selective iNOS inhibitor GW274150 in prophylaxis of migraine headache. Cephalalgia. 2010;30:1458–1467. doi: 10.1177/0333102410370875. [DOI] [PubMed] [Google Scholar]
- 41.Tassorelli C, Joseph SA, Buzzi MG, Nappi G. The effects on the central nervous system of nitroglycerin — putative mechanisms and mediators. Prog Neurobiol. 1999;57:607–624. doi: 10.1016/S0301-0082(98)00071-9. [DOI] [PubMed] [Google Scholar]
- 42.Greco R, Tassorelli C, Armentero MC, Sandrini G, Nappi G, Blandini F. Role of central dopaminergic circuitry in pain processing and nitriglycerin-induced hyperalgesia. Brain Res. 2008;1238:215–223. doi: 10.1016/j.brainres.2008.08.022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 510 kb)