

Abstract
Pancreatic cancer is fourth leading cause of cancer-related deaths in the United States of America. In spite of recent advances in the current therapeutic modalities such as surgery, radiation and chemotherapy patients, the average five year survival rate remains still less than 5%. Recently, compounds from natural sources receive ample of attention as anti-cancer agents. Many epidemiological studies published over the past few decades provide a strong correlation between consumption of vegetables, fruits or plant derived products and reduced incidence of cancer. The present review focuses on the potential antitumor effects of various natural products.
Keywords: Benzyl isothiocyanate, Capsaicin, Resveratrol, Green tea, Curcumin
1. Introduction
Pancreatic cancer is the fourth leading cause of cancer related deaths in the United States of America. The average five-year survival rate for pancreatic cancer is less than 5%. Many genetic and environmental risk factors have been associated with pancreatic cancer. Recently, Jones et al. identified 63 core genetic alterations in 12 common pathways of pancreatic tumors [1]. Environmental risk factors include smoking, obesity, diet, alcohol, etc. Pancreatic cancer carcinogenesis is a multi-step process during which oncogenes such as K-ras, RTKs, PI3K/AKT, EGFR and STAT3 are activated and tumor suppressor genes such as p16INK4/Rb, p53 and SMAD4 are deactivated. MicroRNAs (miRNAs) have received ample recent attention as tumor initiators. Ninety five miRNAs were evaluated in pancreatic cancer, eight of which are up-regulated anywhere from 3–2018 fold in pancreatic tumors as compared to normal pancreas [2]. In fact, some of the miRNAs served as potential biomarkers for early detection of pancreatic cancer. For example miR-216 and miR217 are absent or minimally expressed in pancreatic tumors [3].
Pancreatic cancer takes around 10 years from the initial phase to form a tumor and then another five years to transform into metastatic tumor, with patients dying within two years thereafter [4]. Generally, pancreatic cancer is diagnosed at a late stage and responds poorly to current therapeutic modalities, such as chemo- and radiation therapy. Detection of pancreatic cancer at earlier stages would provide greater opportunity to successfully treat this deadly malignancy. Therefore, identification of predictive biomarkers for detecting early stage pancreatic cancer would be a worthwhile task.
Present therapeutic modalities have provided a modest survival advantage over the past few decades. The clinical management of pancreatic cancer depends on the advancement of the disease and the stage at which it is diagnosed. Surgical resection following adjuvant therapy is the standard course of treatment. However, the majority of patients with pancreatic cancer is diagnosed in the later stages and is precluded from surgery, leaving chemotherapy as the only option. Only about 10–15% of patients with advanced pancreatic cancer are eligible for surgical resection of the tumor because of spatial location of pancreas, and 70–80% of resected tumors eventually relapse following surgery. Gemcitabine and 5-flourouracil (5-FU) with or without radiation are the standard chemotherapy options for patients who present with advanced disease. Gemcitabine relieves the majority of pancreatic cancer symptoms as well as having a modest survival advantage over 5-FU [5]. However, gemcitabine and other cytotoxic drugs rarely extend median survival much beyond six months as compared to untreated patients.
In recent years, minimal progress has been made in the systemic treatment of metastatic pancreatic cancer. The limited success of current standard therapies underscores the urgent need to identify new treatment strategies and agents. Many epidemiological studies published over the past few decades provide a strong correlation between consumption of vegetables, fruits or plant-derived products and deceased cancer incidence. The advantages of bioactive dietary agents, or nutraceuticals, are increased cost effectiveness and reduced toxicity. A comprehensive analysis of 200 published articles provides a strong correlation between vegetable and fruit consumption and reduced incidence of pancreatic cancer [6]. The present review focuses on the potential antitumor effects of various natural products.
1.1 Curcumin
Curcumin is a natural compound isolated from the rhizome of Curcuma Longa, commonly called turmeric. Curcumin is commonly used in Indian cuisine as a food spice and coloring agent. It has been used as an anti-inflammatory and anti-oxidant agent in the Indian System of Medicine (Ayurveda) [7; 8]. Numerous pre-clinical studies have shown that curcumin has potential to suppress the growth of various malignancies, including pancreatic cancer. Curcumin is largely known to target a plethora of signaling pathways in pancreatic cancer such as NFkB [9; 10], SP1 [9], STAT3 [11], Notch-1 [12], COX-II [13], ATM/Chk1 [14], WT1 [15], etc., to suppress tumor growth. Chemical structure of curcumin is presented in Fig. 1.
Fig. 1.
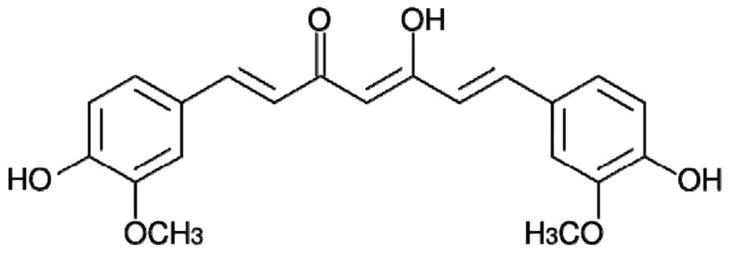
Structure of curcumin
Many studies have addressed NFkB as a prime target of curcumin in various cancer models. Li et al. demonstrated that NFkB and IKK are constitutively active in pancreatic cancer cell lines and inhibition of these molecules by curcumin was associated with growth suppressive activity. Interestingly, NFkB downstream effectors such as COX-2, PGE2and IL-8 were also down-regulated by curcumin treatment. These effector molecules are known to be closely associated with growth and invasiveness of pancreatic cancer [10]. Hidaka et al. showed that curcumin inhibits IL-8 production at a concentration of 10–100 μM through the suppression of NFkB activation, leading to reduced pancreatic cancer growth. However, exogenous addition of recombinant IL-8 did not protect the cells from curcumin-induced death. The authors explained that curcumin treatment enhanced the IL-8 receptors CXCR1 & CXCR2 on the cell surface; however, exogenous addition of IL-8 had no effect on IL-8 receptors. These observations suggest that curcumin inhibits the growth of the pancreatic cancer cells by inhibiting NFkB and IL-8 receptor internalization [16].
Our group has shown that single treatment of BxPC-3 cells with 2.5 μM curcumin for 24 h caused significant G2/M cell cycle arrest and apoptosis [14]. The plasma-achievable concentration of curcumin in humans was estimated to be around 1.8 μM, indicating that the concentration of curcumin used in our studies can be achieved clinically. The G2/M cell cycle arrest by curcumin was associated with DNA damage and the activation (phosphorylation) of ATM and Chk1. Silencing ATM/Chk1 with respective SiRNA blocked the activation of ATM and Chk1 and protected the cells from curcumin mediated G2/M cell cycle arrest and apoptosis in pancreatic cancer cells. Interestingly, normal human pancreatic epithelial (HPDE-6) cells remained unaffected by curcumin treatment [14]. These studies indicate that Chk1 is a potential target of curcumin in pancreatic cancer cells.
Recently, Jutooru et al. have shown that curcumin inhibits the growth of PanC28 and L3.6pl cells in both in vitro and in xenograft models [9]. They demonstrated that curcumin down-regulates the expression of p50 and p65 along with the down-regulation of Specificity Proteins (Sp1, Sp3 and Sp4), which are known to be constitutively active in pancreatic cancer. Both p50 and p65 are Sp-regulated genes, and curcumin-mediated down-regulation of p50 and p65 depends on the modulation of Sp1, Sp3 and Sp4. Hence, it would be interesting to evaluate whether NFkB is a direct target of curcumin or merely the consequence of down-regulation of Sp proteins by curcumin. Furthermore, curcumin potentiated the gemcitabine anti-tumor and anti-angiogenic activity by inhibition of NFkB activation in MIA PaCa-2 cells in an orthotopic model [17].
Curcumin and its analogues were shown to be quite effective against STAT3 signaling in pancreatic cancer [11; 18; 19]. Lin et al. reported that curcumin derivatives FLLL31 and FLLL32 effectively inhibited STAT3 signaling in pancreatic cancer [18]. These derivatives specifically targeted Janus Kinase-2 (JAK-2) and STAT3 to inhibit STAT3 dimerization, thus inhibiting the nuclear localization. Another study by Glienke et al. confirmed the role of curcumin in inhibiting STAT3 phosporylation and down-regulating Survivin/BIRC5 genes. Nonetheless, it was not clear whether inhibition of STAT3 led to down-regulation of Survivin/BIRC5, or whether these are independent targets for curcumin as Survivin promoter contains STAT3 binding site [20]. Another derivative of curcumin, GO-Y030, was found to inhibit STAT3 at much lower doses where curcumin had little or no effect [19].
Accumulating data confirmed that curcumin has profound effect on epigenetic alterations of the cells. Generally, these epigenetic modulators include histone deacetylases (HDACs) and acetyltransferases (HATs), DNA methyltranferases (DNMTs) and MiRNAs. Bora-Tatar et al. investigated the effects of curcumin on HDACs and found that curcumin is more potent than valproic acid sodium butyrate, a well known HDAC inhibitors [21]. Furthermore, curcumin has differential effect on various HDACs. For example, curcumin down regulated the expression of HDACs 1, 3 and 8 but up regulated the expression of HDACs 2 and 4 [22; 23]. Similarly, curcumin also inhibited p300/CREB-binding protein (CBP) HAT activity in vitro and in vivo [24]. The effects of curcumin on DNA methylases are interesting. Molecular docking studies with curcumin and DNMT1 showed that curcumin could covalently block the catalytic thiolate of DNMT1to inhibit DNA methylation [25], but recent study reported that though curcumin inhibits DNMT1 but it has minimal or no pharmacological significance [26].
Micro-RNAs (MiRNAs) are involved in critical biological processes such as development, differentiation, apoptosis and proliferation. MiRNAs exert their effects by imperfect binding with messenger RNA, thus leading to degradation of target mRNA [27]. Altered expressions of miRNAs were reported in various cancers, including pancreatic cancer, thereby functioning as either tumor suppressors or oncogenes [28]. Curcumin and its analogue Difluorocurcumin (CDF) were found to be modulators of pancreatic cancer MiRNAs. Recently, Ali et al. observed that loss of let-7 family and MiRNA-143 expression, along with increased expression of MiRNA-21 in MIA PaCa-2 cells, correlate with Ras GTPase activity. Treatment of MIA PaCa-2 tumor xenografts in vivo with curcumin analogue CDF resulted in the increased expression of let-7i and MiRNA-143 and decreased expression of MiRNA-21, leading to reduced Ras GTPase activity [29]. The same group stated that CDF decreased cell survival, clonogenicity, formation of pancreatospheres, invasive cell migration and CSC function of human pancreatic cancer cells by decreasing EZH2 and increasing the expression of a panel of tumor-suppressive microRNAs, including let-7a, b, c, d, miR-26a, miR-101, miR-146a and miR-200b, c that are typically lost in pancreatic cancer [30]. Interestingly, MiRNA-200 expression is decreased whereas MiRNA-21 expression is up-regulated in gemcitabine-resistant pancreatic cancer cells. Treatment of pancreatic cancer cells with CDF resulted in increased expression of MiRNA-200 and decreased expression of MiRNA-21 in gemcitabine-resistant cells [31]. Hence, CDF shows potential for use along with gemcitabine. Curcumin treatment was reported to up-regulate MiRNA-22 and down-regulate MiRNA-199a. The curcumin-mediated down-regulation of SP1 transcription factor was through up-regulation of MiRNA-22 [32].
Clinical trials with curcumin were promising. Phase I clinical trials have shown that curcumin is relatively safe even at higher doses of 12 g/day in humans. However, curcumin has limited bioavailability because of poor absorption, rapid metabolism and rapid systemic elimination [33]. For example, when curcumin at an oral dose of 2 g/kg was given to rats, a maximum plasma peak level of 1.35 ± 0.25 μg/mL was observed, whereas a similar dose in humans resulted in either undetectable or extremely low serum concentration at 1h [34]. Furthermore, Phase I/II clinical trials on gemcitabine resistant pancreatic cancer patients showed that 8 g oral administration of curcumin resulted in 29–419 ng/mL curcumin in plasma [35]. To increase the bioavailability of curcumin, various approaches were adopted and several synthetic analogues of curcumin were synthesized and novel drug delivery systems were investigated [33]. One such curcumin analogue is difluorinated-curcumin (CDF), which show better bioavailabilty and 10-fold higher concentrations in pancreas as compared to curcumin [13].
Dhillon et al. conducted Phase II clinical trials with curcumin. Twenty-five chemotherapy patients received 8 g/day curcumin. Results showed that two patients demonstrated clinical benefit, whereas one patient had stable disease for 18 months. Peripheral mononuclear cells isolated from the curcumin-treated patients showed reduced activation of NFkB [36]. Another interesting clinical trial on curcumin was performed in gemcitabine-resistant patients. Gemcitabine along with curcumin showed increased median survival time (MST) of 161 days and a one year survival rate of 19% in these patients [35]. Clearly, curcumin can be a potential anticancer therapy; however, further clinical trials are needed to find ways to increase its bioavailability.
1.2 Isothiocyanates
Evidence from epidemiological and case-control studies continues to support the notion that consumption of cruciferous vegetables has substantial chemopreventive activity against various human malignancies, including pancreatic cancer [37; 38]. Cruciferous vegetables are a rich source of glucosinolates, which are converted to isothiocyanates (ITCs) by the enzyme myrosinase and released when plant cells are damaged by cutting or chewing [39]. In addition, intestinal flora plays a major role in releasing ITCs from glucosinolates. Benzyl isothiocyanate (BITC), an agent that is present in cruciferous vegetables such as watercress, cabbage, cauliflower, mustard and horseradish, is widely consumed as part of a routine diet. BITC has been reported to inhibit initiation, growth and metastasis of human cancers in rodents [40; 41; 42; 43; 44; 45; 46; 47]. Previous reports from our laboratory showed that BITC is quite effective in suppressing pancreatic tumor growth by inhibiting various key signaling pathways, such as AKT, STAT3, HDAC, NFkB, etc. [41; 42; 45]. The structure of BITC is shown in Fig. 2.
Fig. 2.
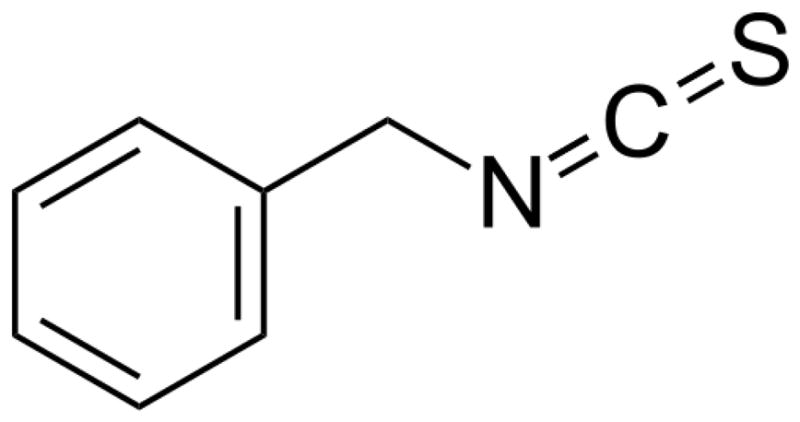
Structure of benzyl isothiocyanate
Signal Transducer Activator Transcription (STAT3) transcription factor is aberrantly activated in a majority of cancers, including pancreatic cancer, and promotes cell survival [48]. More than 50% of breast and lung cancer and around 95% of head and neck cancer clinical samples show hyperactivated STAT3 [49]. Recently, numerous natural and synthetic compounds have been shown to target STAT3 signaling. Results from our laboratory show that BITC induces apoptosis in pancreatic cancer cells in a dose- and time-dependent manner. Mechanistic studies revealed that BITC targets STAT3 signaling to induce apoptosis in pancreatic cancer. The activation (Try-405 & Ser-727) and expression of STAT3 in pancreatic cancer cells lines, such as BxPC-3, PanC-1, Capan-2 and MIA PaCa-2, were suppressed by BITC treatment [45]. Though STAT3 protein levels were down-regulated by BITC, it was not clear whether apoptosis induction was related to STAT3 inhibition. STAT3α overexpression and IL-6 stimulation blocked the effects of BITC and confirmed the role of STAT3 in BITC-induced apoptosis.
Pancreatic tumors can trigger substantial development of new blood vessels through a process called angiogenesis. Neovascularization is a complex process that is mainly triggered by tumor-derived hypoxia inducible factor (HIF-1α) and vascular endothelial growth factor (VEGF) during oxygen deprivation [50]. Recent studies have identified STAT-3 as a direct transcriptional activator of VEGF and HIF-1α under hypoxia [51; 52]. Since BITC substantially down-regulates STAT3 signaling [45], it is logical that BITC would inhibit pancreatic cancer angiogenesis. Boreddy et al. showed that BITC significantly inhibits angiogenesis in both CAM and rat aortic ring assay, indicating that BITC has potential to inhibit tumor angiogenesis [40]. BITC-treated pancreatic cancer cells show reduced secretion of VEGF and MMP-2 under normoxia and hypoxia conditions. Furthermore, BITC was shown to down-regulate various angiogenic factors such as HIF1-α, VEGFR-2, MMP-2, Rho A, Rho C and RAC1,2,3 in a dose-dependent manner in BxPC-3, PanC-1 and HUVEC cells. Interestingly, BITC failed to reduce VEGF, HIF-1α and MMP-2 expression in STAT3 overexpressing BxPC-3 cells [40]. These results clearly indicate that BITC targets STAT3 to suppress pancreatic cancer cell proliferation and angiogenesis.
Phosphatidyl inositol 3 phosphate (PI3K) signaling plays a critical role during embryonic development; however, deregulated PI3K/AKT pathway promotes uncontrolled growth in malignant cells [53]. A recent report has shown that 59% of pancreatic tumors harbor aberrantly activated AKT signaling [54]. Upon activation of PI3K by various stimuli, PDK1 phosphorylates AKT at Ser-308, leading to stabilization of AKT. Complete transcriptional activation of AKT requires another phosphorylation at Ser-475 [55]. PI3K/AKT pathway is negatively regulated by phosphatases such as PTEN, which dephosphorylates PIP3, thus limiting its availability [56]. Results from our laboratory have shown that BITC significantly down-regulates the activation of AKT at both Ser-308 and Ser-475 [41]. Furthermore, BITC also suppresses the phosphorylation of various other key molecules of PI3K/AKT pathway such as PI3K (Tyr-458), PDK1 (Ser-241), mTOR (Ser-2448), etc., indicating that BITC targets PI3K/AKT signaling to induce apoptosis in pancreatic cancer cells. Interestingly, BITC had minimal effect on normal human pancreatic cells, HPDE-6 [41].
Inhibition of AKT phosphorylation by BITC significantly inhibited the phosphorylation of FOXO1 (Ser-256) and FOXO3a, without affecting the protein levels in BxPC-3 and PanC-1 cells. Inhibition of FOXO phosphorylation led to nuclear accumulation of FOXO proteins, resulting in the up-regulation of FOXO transactivated proteins such as Bim, p21 and p27 [41]. BITC also regulated FOXO proteins by acetylation and efficiently decreased the acetylation of FOXO proteins, probably by down-regulating CBP protein levels [41].
NFkB transcription factors play a vital role in the regulation of immunity, inflammation and cell proliferation [57]. Under normal physiological conditions, NFkB is sequestered by IkB in the cytoplasm. Upon cellular stimulation, IkB proteins are phosphorylated at Ser-32/36, releasing NFkB from the complex, which then enters the nucleus and activates kB responsive genes [58]. BITC treatment significantly down-regulated the phosphorylation of NFkB at Ser-276 and Ser-536 in BxPC-3 and Capan-2 cells in a dose- and time-dependent manner [42; 47]. BITC reduced NFkB expression in BxPC-3 cells but not in Capan-2 cells, indicating that BITC acts differentially in different cell lines [42]. Since Capan-2 cells have wild type p53 and BxPC-3 cells have mutated P53, it would be interesting to see whether p53 plays any role in BITC-induced down-regulation of NFkB expression. Mechanistic studies revealed that neither IkB phosphorylation nor expression levels were altered by BITC, whereas IKK expression was dose-dependently down-regulated. Hence, down-regulation of IKK by BITC treatment could be the reason for inhibition of NFkB phosphorylation (Ser-536). Similar to FOXO deacetylation, BITC significantly reduced the acetylation of NFkB in pancreatic cancer cells by down-regulating HADC1 and HDAC3 [42].
Since BITC targets various key survival pathways in pancreatic cancer cells, it would be quite interesting to know the overall mechanism of these actions. As many chemotherapy drugs elicit their anticancer properties by generating reactive oxygen species (ROS), BITC also significantly induced ROS generation in pancreatic cancer cells. Eventually, ROS generation led to DNA damage as demonstrated by increased phosphorylation of H2A.X and G2/M cell cycle arrest through ChK2 phopshorylation (Thr-68) [43; 46; 47]. In addition, ROS generation also led to phosphorylation of various MAP kinases, such as ERK (Thr202/Thy204), JNK (Thr183/Tyr185) and P38 (Thr180/Tyr182) in a dose-dependent manner [43]. Interestingly, ERK, JNK and P38 were phosphorylated in response to BITC treatment but only ERK was involved in BITC-induced cell cycle arrest, whereas all other MAPK were involved in apoptosis induction [43].
BITC (12 μmol/day) was evidently well-tolerated by mice as no symptoms of toxicity such as weight loss and physical inactivity were observed [45]. Tumor growth in BITC-fed mice was substantially reduced as compared to control mice. Tumors appeared to grow more slowly in BITC-fed mice as compared with control mice. For example, six weeks after treatment with 12 μmol BITC, the average tumor volume in control mice was about 1.92 fold higher than that in BITC-treated mice, indicating potential anticancer activity [45]. Interestingly, our results show that mean plasma concentration of BITC in 12 μmol/day treated mice was 6.5±0.1μmol/L (n=10) [41], whereas accumulated BITC concentration in tumors after 46 days was 7.5±0.3 μmol/g (n=10). These results indicate that the therapeutic concentration of BITC could be achieved in vivo by oral feeding. Moreover, mice treated with 12 μmol BITC per day showed reduced (76%) hemoglobin content in matrigel plugs that were implanted in both the flanks of nude mice [40]. Similarly, tumor xenografts excised from treated mice showed about 61% reduced hemoglobin content as compared to control mice [40]. These observations clearly establish that BITC inhibits tumor growth and angiogenesis in vivo.
At this time, it is not clear whether BITC has multiple individual targets or whether it is a tandem effect of upstream molecules. Since STAT3 and NFkB are known to be regulated by AKT, we assume that BITC mainly targets AKT. Further studies are needed to fully understand the mechanism of BITC action.
1.3 Capsaicin
Capsaicin, a homovanillic acid derivative (N-vanillyl-8-methyl-nonenamide), is a principle pungent constituent of all chili pepper plants. The concentration of capsaicin in peppers ranges from 0.1–1% w/w. The structure of capsaicin is shown in Fig 3. Capsaicin has been used to treat pain, inflammation and a variety of diseases, including diabetic neuropathy, rheumatoid arthritis, postmastectomy pain syndrome, cluster headaches and herpes zoster. Although chili pepper is used extensively in South America, Africa and Asia, there is still debate whether consumption of capsaicin is entirely safe as published studies reveal conflicting results [59]. A few studies indicated that capsaicin could potentially induce carcinogenesis in animals. For example, about 60% of rats fed a semisynthetic diet containing 10% chili and Swiss albino mice fed a semisynthetic diet containing 0.03% capsaicin, developed neoplastic changes in liver and cecum, respectively [60; 61]. On the contrary, a majority of the studies, including those from our laboratory, show that capsaicin can suppress cancer growth in vitro and in vivo [62; 63; 64; 65]. The role of capsaicin in human physiology is also controversial. Some studies showed that capsaicin can induce carcinogenesis in gallbladder or gastric cancers [66; 67]. In contrast, bladder biopsies from patients who were fed capsaicin over a five-year period did not show any metaplasia, dysplasia, solid tumor or invasive tumor [68]. Controversial reports on capsaicin clearly suggest a need for controlled epidemiological studies to determine whether capsaicin is cancer preventive or causative [59].
Fig. 3.
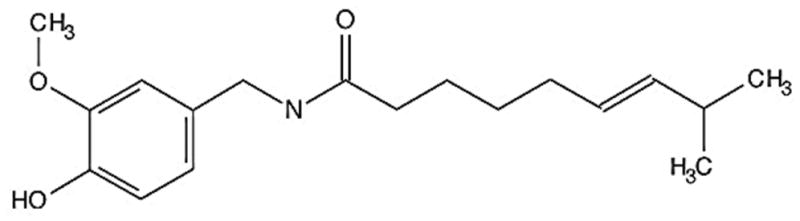
Structure of capsaicin
Studies from our laboratory support the theory that capsaicin can suppress caerulin-induced carcinogenesis in transgenic mice and suppresses pancreatic tumor growth both in vitro and in vivo. Four-week old LSL-KrasG12D/Pdx1-Cre mice developed chromic pancreatitis and PanIN lesions after a single dose of caerulin. However, mice fed a daily diet including 10 or 20 p.p.m of capsaicin for eight weeks significantly reduced the severity of chronic pancreatitis and PanIN lesions [65]. Further analysis of these tumors revealed that capsaicin significantly reduced the phosphorylation of ERK, c-JUN and Hedgehog/GLI1 activation in capsaicin-treated mice as compared to control mice [65]. Hence, capsaicin was quite effective in inhibiting the carcinogenesis of pancreatic cancer. Results from our laboratory further show that capsaicin dose-dependently induces mitochondrial-dependent apoptosis in ASPC-1 and BxPC-3 pancreatic cancer cells [62]. The apoptosis-inducing activity of capsaicin was not mediated through TRPV1 receptor as capsazepine, a specific inhibitor of TRPV1, did not protect the cells from capsaicin-induced death [62]. ROS-mediated JNK activation was found to be involved in capsaicin-induced apoptosis. JNK inhibitor (SP600125) or antioxidants such as N-acetyl cysteine (NAC) significantly attenuated capsaicin-induced cell death in pancreatic cancer cells. Furthermore, oral administration of 5mg/kg capsaicin substantially reduced ASPC-1 xenograft or PanC-1 orthotopically implanted tumor growth in nude mice as compared to control [62; 64].
Interestingly, capsaicin-induced ROS generation was mitochondria-derived. The mechanistic studies revealed that capsaicin inhibited about 2.5–9% and 5–10% complex-I activity and about 8–75% complex-III activity of mitochondrial electron transport chain (ETC) in BxPC-3 and ASPC-1 cells, respectively [63]. These observations were further supported by the fact that BxPC-3 rho cells, which lack intact respiratory chain, were completely resistant to capsaicin-induced ROS generation and apoptosis. Capsaicin failed to cause any ROS generation or induce apoptosis in normal pancreatic epithelial (HPDE-6) cells, indicating the selectivity of capsaicin towards cancer cells [63]. Capsaicin also caused the oxidation of mitochondrial lipid cardiolipin and reduced membrane potential [63]. In addition, capsaicin treatment not only inhibited the enzymatic activities of catalase and SOD but also drastically reduced ATP and glutathione levels in pancreatic cancer cells. Overexpression of catalase or treatment with EUK-134 significantly protected BxPC-3 cells from capsaicin-induced apoptosis, indicating the role of ROS in capsaicin-induced apoptosis [63]. Similarly, tumors from capsaicin-treated mice showed reduced SOD activity and increased GSSG/GSH levels as compared to control mice. In conclusion, capsaicin induces ROS through inhibition of mitochondrial complexes, leading to apoptosis.
Further mechanistic studies revealed that capsaicin targets Trx-ASK1 signaling in pancreatic cancer to induce apoptosis. Previous reports show that apoptosis signal-regulating kinase 1 (ASK1) is activated mostly by oxidative stress. Activated ASK1 further activates both MKK4/MKK7-c-Jun NH2 -terminal kinase (JNK) and MKK3/MKK6-p-38 MAPK signaling cascade [69]. ASK-1 activation is prevented by its endogenous inhibitor thioredoxin (Trx). Oxidation of Trx by oxidative stress or ROS releases ASK-1 from the complex and induces apoptosis. Results from our laboratory show that capsaicin depletes Trx levels and phosphorylates ASK-1 at Thr-845 and increases its kinase activity. This leads to the activation of ASK-1 downstream molecules such as MKK4/7 and JNK in BxPC-3 pancreatic cancer cells [64]. Interestingly, Trx overexpression suppressed apoptosis, whereas ASK-1 overexpression increased capsaicin-induced apoptosis [64]. The role of oxidative stress in capsaicin-induced apoptosis was further substantiated by treatment with β-mercaptoehanol (reducing agent). β-mercaptoehanol blocked capsaicin-mediated activation of ASK-1. On the other hand, Trx inhibitor 1-chloro-2-4-dinitrobenzene increased capsaicin-induced apoptosis in pancreatic cancer cells, indicating the disruption of Trx-ASK-1 complex in capsaicin-induced apoptosis. Similarly, capsaicin-treated tumors show reduced Trx levels and increased ASK-1 phosphorylation as compared to control tumors [64].
Taken together, capsaicin targets mitochondrial electron transport chain to generate ROS, and generated ROS activates JNK and disrupts Trx-ASK-1 interaction to induce mitochondrial apoptosis in pancreatic cancer.
1.4 Green tea
Green tea produced from Camellia sinensis var. sinensis is one of the most widely consumed beverages in the world [70]. Green tea chemical composition is quite complex, but the significant therapeutic properties could be attributed to its flavonoid (polyphenol) components. The main flavonoids present in green tea include catechins, such as (−)-epigallocatechin-3-gallate (EGCG), that represents approximately 59% of the total catechins: (−)-epigallocatechin (EGC) (19% approximately); (−)-epicatechin-3-gallate (ECG) (13.6% approximately); and (−)-epicatechin (EC) (6.4% approximately) [71]. Chemical structures of EGCG is presented in Fig. 4. Out of these catechins, EGCG received ample attention as an anticancer drug. Plasma bioavailability of green tea polyphenols appears to be quite variable. For example, although EGCG concentration is five times more abundant than EGC in green tea, plasma levels of EGC were higher as compared to EGCG in rats given 0.6% green tea phenols (GTP) in their drinking water over a period of 28 days. However, when a similar dose was given to mice, EGCG levels were much higher in plasma as compared to EGC, indicating that bioavailability of catechins may vary with species [72]. A recent pharmacokinetics study of catechins in humans showed that oral administration of 20 mg tea solids/kg resulted in average plasma peak concentrations of 223, 124 and 78 ng/mL for EGC, EC and EGCG, respectively, indicating that therapeutic concentrations of catechins could be achievable in humans [73].
Fig. 4.
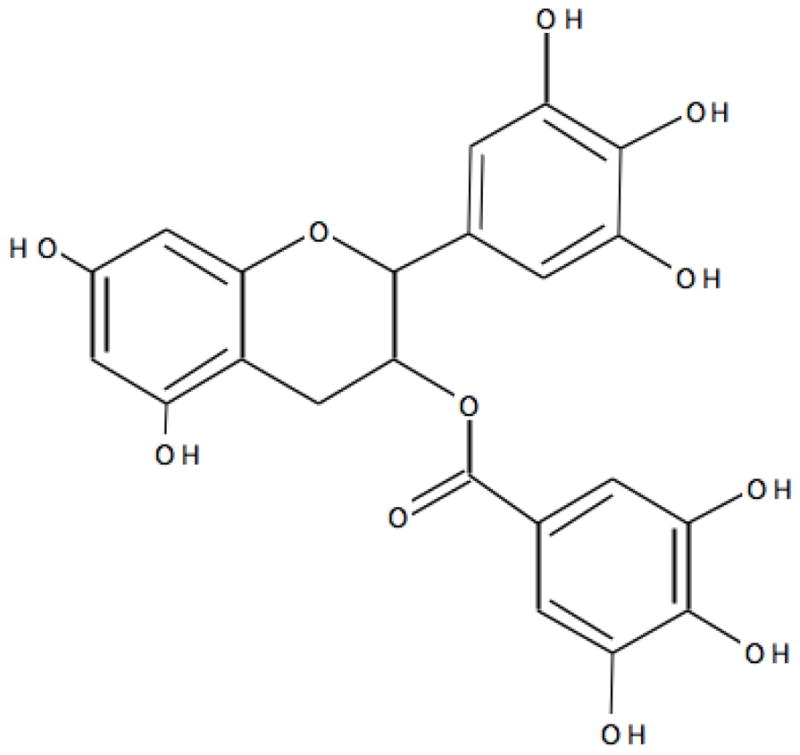
Structure of (−)-epigallocatechin-3-gallate (EGCG)
Green tea catechins are known to inhibit the growth of various cancers by targeting multiple signaling pathways [74]; however few of these targets such as HSP90, FAK and STAT3 were evaluated in pancreatic cancer. Recently, Zhang et al. used green tea extract (GTE) to suppress pancreatic tumor growth [75]. GTE altered 32 protein expressions in HPAF-II cells, which were involved in drug resistance, motility and metabolism. Particularly, GTE altered the expression of heat-shock proteins such as Hsp-90, Hsp-75 and Hsp-27. In addition, GTE inhibited the phosphorylation of AKT and p53, leading to apoptosis and suppression of pancreatic tumor growth [75]. On the other hand, EGCG decreased the association of p23 and Hsp-70 with Hsp-90, while having little effect on ATP binding to HSp-90 [76]. Proteolytic fingerprinting revealed that EGCG directly binds to C-terminal domain of the Hsp-90, leading to inhibition of co-chaperone association and thereby inducing the degradation of Hsp-90 and apoptosis [76]. Mechanistic studies further revealed that ROS was involved in EGCG-induced cell death, as EGCG dose-dependently induced ROS generation in pancreatic cancer cells. Eventually, ROS activated JNK and cell cycle arrest, leading to apoptosis. These results were corroborated by studies with pharmacological inhibitors, where JNK inhibitor and NAC completely rescued the cells from EGCG-induced effects [77; 78]. In addition, EGCG significantly inhibited the pluripotency maintenance factors such as Nanog, c-Myc and Oct-4 of pancreatic cancer stem cells (CSC), thus inhibiting the self-renewal potency of pancreatic CSCs [79]. Interestingly, EGCG also inhibited sonic hedgehog signaling (smoothened, patched, Gli1 and Gli2) and EMT activators such as Snail, Slug, ZEB1 and TCF/LEF [79]. Furthermore, green tea catechins were shown to be effective in potentiating antitumor effects of chemodrugs such as pterostilbene, celecoxib, TRAIL, thymoquine and gemcitabine [80; 81; 82; 83; 84].
Pancreatic cancer entails angiogenesis for early metastasis to various organs such as liver and lymph nodes. Green tea polyphenols were quite effective in suppressing angiogenesis and metastasis of pancreatic cancer. Shankar et al. showed that EGCG inhibits viability, tube formation and migration of HUVECs. Similarly, ASPC-1 xenografts treated with EGCG showed reduced expression of VEGF, CD31, MMPs and ERK phosphorylation, demonstrating the potential anti-angiogenic and anti-metastasis activity of EGCG [85]. Recently, Vu et al. proved that BxPC-3 and ASPC-1 cells treated with EGCG showed reduced phosphorylation of focal adhesion kinase (FAK), insulin like growth factor (IGF-1R) and mammalian target of rapamycin (mTOR), which play a critical role in pancreatic cancer invasion and metastasis. Interestingly, EGCG treatment did not induce cleavage of PARP, indicating that EGCG induces apoptosis independent of caspase 3 in pancreatic cancer cells in contrast to other studies [86]. In another study, EGCG-mediated pancreatic cancer growth suppression and anti-metastatic potentials were mediated by STAT3 inhibition, and IL-6 pre-treatment protected the cells from EGCG-induced cell death and VEGF secretion [84; 87]. Moreover, EGCG treatment significantly protected pancreas of hamster from N-Nitrosobis (2-oxopropyl)amine (BOP)-induced carcinogenesis [88; 89; 90].
Interestingly, Thakur et al. reported that green tea polyphenols increased p53 transcriptional activity and p53 acetylation at Lys373 and Lys382 residues by inhibiting class 1 HDACs, resulting in the accumulation of cells in G0/G1 phase [91]. EGCG has proven to be a potent inhibitor of DNMT1 by forming hydrogen bonds with different residues in the catalytic pocket of DNMT [92; 93]. Furthermore, Tsang and Kwok performed microarray analysis on curcumin-treated hepatocytes and found that EGCG modified the expression of 61 miRNAs [94]. Taken together, green tea catechins appear to be promising anticancer agents for pancreatic cancer. Nevertheless, further clinical studies are required.
1.5 Resveratrol
Resveratrol, trans-3,5,4′-trihydroxy-trans-stilbene, is a naturally occurring polyphenol synthesized by a variety of plant species in response to injury, ultraviolet (UV) irradiation and fungal attack [95]. Resveratrol has been detected in more than 70 plant species, including red grapes, peanuts, berries and pines [96]. Red grape skin is particularly rich in resveratrol, which contains about 50 to 100 μg of resveratrol per gram wet weight [97]. Evidence from epidemiological studies indicates an inverse relationship between red wine consumption and the risk of cardiovascular disease, the so-called “French Paradox” [98]. A chemical structure of resveratrol is presented in Fig. 5. In Asian folk medicine, dried roots of Poligonum cuspidatum, a rich source of trans-resveratrol, were used for treating inflammation or hyperlipidemia [99]. Extensive data on human cell culture and mouse models prove that resveratrol has beneficial effects on various biological processes, including the prevention of cardiovascular diseases and cancer. The anti-carcinogenic effects of resveratrol appear to be closely associated with its antioxidant activity. It has been shown to inhibit cyclooxygenase, hydroperoxidase, protein kinase C, Bcl-2 phosphorylation, AKT, focal adhesion kinase, NFκB, matrix metalloprotease-9 and cell cycle regulators [97]. However, the protective effects of resveratrol are hampered by poor bioavailability (less than 1%) due to extensive metabolism in the intestine and liver. For example, 25 mg oral dose of resveratrol resulted in undetectable levels of unmetabolized-resveratrol in plasma after 0.5–2 h [100]. Efforts have been made to increase the bioavailbility and antitumor activity of resveratrol by synthesizing its analogues [101; 102; 103]. Resveratrol is shown to target various signaling pathways in pancreatic cancer such as hedgehog, FOXO, leukotriene A4 hydrolase, macrophage inhibitory cytokine-1, Src and STAT3.
Fig. 5.
Structure of resveratrol
Resveratrol induces apoptosis in pancreatic cancer cells through mitochondrial-dependent pathway. The authors Sun et al. suggested that resveratrol generates moderate ROS which are greatly increased when combined with ionizing radiation [104]. Interestingly, resveratrol exhibited lower toxicity to normal pancreatic cells [105; 106]. Resveratrol induced apoptosis in INS-1E insulinoma cells by inhibiting AKT signaling [107]. Furthermore, resveratrol-treated PanC-1 and ASPC-1 pancreatic cancer cells showed growth inhibition and cell cycle arrest in G0/G1 phase [108; 109; 110]. Roy et al. reported that resveratrol-induced cell cycle arrest was associated with the up-regulation of key cell cycle molecules such as p21/CIP1, p27/KIP1 and inhibition of Cyclin D1 expression in MIA PaCa-2, PanC-1 and ASPC-1 pancreatic cancer cells [111]. Resveratrol-induced growth inhibition and internucleosomal DNA fragmentation-induced apoptosis in Capan-2 and PanC-28 were enhanced at low pH conditions, indicating resveratrol probably has potential to target tumor microenvironment [106].
Resveratrol specifically binds to sulfonylurea receptors (SUR), a regulatory sub unit of ATP-sensitive K+ channels [112]. Moreover, resveratrol has higher binding affinity towards SUR than glibenclamide; hence resveratrol displaces the glibenclamide from SUR. In addition, resveratrol induced SUR1-specific apoptosis in embryonic human kidney 293 cells expressing SUR1. Interestingly, resveratrol failed to induce SUR-specific apoptosis in SUR1 knockout mice and cells expressing SUR2A or SUR2B or the mutant SUR1 isoform, indicating the specificity of the resveratrol towards SUR-1 [112]. Interestingly, resveratrol induces apoptosis in pancreatic cancer cells by targeting hedgehog pathway. Low concentrations of resveratrol decreased the expression of hedgehog pathway members such as Gli1, Ptc1, CCND1 and Blc2 [108]. Roy et al. demonstrated that resveratrol targets FOXO phosphorylation through the inhibition of PI3K/AKT and MEK/ERK signaling in pancreatic cancer cells [111]. Decreased phosphorylation of FOXO proteins led to nuclear accumulation of FOXO proteins, up-regulation of pro-apoptoic protein Bim, and eventually apoptosis. Furthermore, silencing FOXO proteins abrogated resveratrol-induced cell cycle arrest and apoptosis in pancreatic cancer cells [113]. Similarly, mice bearing orthotopically implanted PanC-1 xenografts and treated with resveratrol showed significant inhibition of tumor growth, which was associated with reduced phosphorylation of ERK, PI3K, AKT, FOXO1 and FOXO3a and induction of FOXO-transactive genes [111].
Based on structural similarities of resveratrol, Oi et al. hypothesized that resveratrol could bind to leukotriene A4 hydrolase (LTA4H). As predicted, resveratrol was found to bind to LTA4H and suppressed the proliferation and anchorage-independent growth of pancreatic cancer [114]. In addition, inhibitory effects of resveratrol were reduced in LTA4H silenced cells. Resveratrol inhibited tumor formation in a xenograft mouse model of human pancreatic cancer by inhibiting LTA4H activity [114]. Oligonucleotide microarray analysis of resveratrol treated S2-013 pancreatic cancer cells revealed that resveratrol specifically up-regulates MIC1 gene. Silencing of MIC1 gene in CD18 cells resulted in significant protection against resveratrol-induced growth inhibition [115]. Pancreatic cancer cells harboring constitutively active STAT3 oncogene are specific targets for resveratrol. Investigations from Kotha et al. revealed that resveratrol inhibits Src tyrosine kinase activity, thereby inhibiting the constitutive activation of STAT3 in v-Src-transformed fibroblasts, MDA-MB and pancreatic cancer cells [116]. Interestingly, resveratrol induced irreversible cell cycle arrest in STAT3-activated cells, but absence of aberrant STAT3 activity showed reversible cell cycle arrest and minimal loss of cell viability. Furthermore, resveratrol inhibited the growth and development of PanIN lesions in Kras G12D mice model. Similarly, resveratrol inhibited the pluripotency maintaining factors such as Nanog, Sox-2, c-Myc and Oct-4 and EMT inducers [117].
Resveratrol is also a potential candidate for the epigenetic modulation. Multiple studies have shown that resveratrol can induce the expression of SIRT1 and down regulate the expression of p300 [118; 119]. However, resveratrol had weak effect on DNA methylation in breast cancer cells [120]. Furthermore, resveratrol decreased the levels of the miR-155 by up-regulating miR-663, a microRNA targeting JunB and JunD [121].
Resveratrol sensitizes pancreatic cancer cells to various chemotherapy drugs, including gemcitabine, which has potential significance for cancer treatment. The chemosensitization of tumor cells by resveratrol appears to be mediated through its ability to modulate multiple cell-signaling molecules, such as drug transporters, NFkB and STAT3 [122; 123; 124].
2. Conclusion
Continuing support from epidemiological studies encourages the use of natural products as anticancer agents. However, many of the epidemiologic studies had severe limitations, such as potential misclassification of subjects by exposure, poor control of extenuating factors and possible recall bias. And while some isolated substances may not exhibit potential anti-cancer properties, they can be used as through chemical modification to synthesize better and more effective chemotherapy drugs. Therefore, better controlled epidemiological studies are needed to firmly establish and define the role of nutraceuticals in cancer prevention.
Acknowledgments
This work was supported in part by R01 grants CA106953 and CA129038 (to Sanjay K. Srivastava) awarded by the National Cancer Institute, NIH.
Footnotes
Conflict of interest statement
None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, Hong SM, Fu B, Lin MT, Calhoun ES, Kamiyama M, Walter K, Nikolskaya T, Nikolsky Y, Hartigan J, Smith DR, Hidalgo M, Leach SD, Klein AP, Jaffee EM, Goggins M, Maitra A, Iacobuzio-Donahue C, Eshleman JR, Kern SE, Hruban RH, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang Y, Li M, Wang H, Fisher WE, Lin PH, Yao Q, Chen C. Profiling of 95 microRNAs in pancreatic cancer cell lines and surgical specimens by real-time PCR analysis. World Journal of Surgery. 2009;33:698–709. doi: 10.1007/s00268-008-9833-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Szafranska AE, Davison TS, John J, Cannon T, Sipos B, Maghnouj A, Labourier E, Hahn SA. MicroRNA expression alterations are linked to tumorigenesis and non-neoplastic processes in pancreatic ductal adenocarcinoma. Oncogene. 2007;26:4442–4452. doi: 10.1038/sj.onc.1210228. [DOI] [PubMed] [Google Scholar]
- 4.Costello E, Neoptolemos JP. Pancreatic cancer in 2010: new insights for early intervention and detection. Nature Reviews Gastroenterology & Hepatology. 2011;8:71–73. doi: 10.1038/nrgastro.2010.214. [DOI] [PubMed] [Google Scholar]
- 5.Shore S, Raraty MG, Ghaneh P, Neoptolemos JP. Review article: chemotherapy for pancreatic cancer. Alimentary Pharmacology & Therapeutics. 2003;18:1049–1069. doi: 10.1111/j.1365-2036.2003.01781.x. [DOI] [PubMed] [Google Scholar]
- 6.Block G, Patterson B, Subar A. Fruit, vegetables, and cancer prevention: a review of the epidemiological evidence. Nutrition and Cancer. 1992;18:1–29. doi: 10.1080/01635589209514201. [DOI] [PubMed] [Google Scholar]
- 7.Aggarwal BB, Sundaram C, Malani N, Ichikawa H. Curcumin: the Indian solid gold. Advances in Experimental Medicine and Biology. 2007;595:1–75. doi: 10.1007/978-0-387-46401-5_1. [DOI] [PubMed] [Google Scholar]
- 8.Strimpakos AS, Sharma RA. Curcumin: preventive and therapeutic properties in laboratory studies and clinical trials. Antioxidants & Redox Signaling. 2008;10:511–545. doi: 10.1089/ars.2007.1769. [DOI] [PubMed] [Google Scholar]
- 9.Jutooru I, Chadalapaka G, Lei P, Safe S. Inhibition of NFkappaB and pancreatic cancer cell and tumor growth by curcumin is dependent on specificity protein down-regulation. The Journal of Biological Chemistry. 2010;285:25332–25344. doi: 10.1074/jbc.M109.095240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li L, Aggarwal BB, Shishodia S, Abbruzzese J, Kurzrock R. Nuclear factor-kappaB and IkappaB kinase are constitutively active in human pancreatic cells, and their down-regulation by curcumin (diferuloylmethane) is associated with the suppression of proliferation and the induction of apoptosis. Cancer. 2004;101:2351–2362. doi: 10.1002/cncr.20605. [DOI] [PubMed] [Google Scholar]
- 11.Glienke W, Maute L, Wicht J, Bergmann L. Curcumin inhibits constitutive STAT3 phosphorylation in human pancreatic cancer cell lines and downregulation of survivin/BIRC5 gene expression. Cancer Investigation. 2010;28:166–171. doi: 10.3109/07357900903287006. [DOI] [PubMed] [Google Scholar]
- 12.Wang Z, Zhang Y, Banerjee S, Li Y, Sarkar FH. Notch-1 down-regulation by curcumin is associated with the inhibition of cell growth and the induction of apoptosis in pancreatic cancer cells. Cancer. 2006;106:2503–2513. doi: 10.1002/cncr.21904. [DOI] [PubMed] [Google Scholar]
- 13.Padhye S, Banerjee S, Chavan D, Pandye S, Swamy KV, Ali S, Li J, Dou QP, Sarkar FH. Fluorocurcumins as cyclooxygenase-2 inhibitor: molecular docking, pharmacokinetics and tissue distribution in mice. Pharmaceutical Research. 2009;26:2438–2445. doi: 10.1007/s11095-009-9955-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sahu RP, Batra S, Srivastava SK. Activation of ATM/Chk1 by curcumin causes cell cycle arrest and apoptosis in human pancreatic cancer cells. British Journal of Cancer. 2009;100:1425–1433. doi: 10.1038/sj.bjc.6605039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glienke W, Maute L, Wicht J, Bergmann L. Wilms’ tumour gene 1 (WT1) as a target in curcumin treatment of pancreatic cancer cells. European Journal of Cancer. 2009;45:874–880. doi: 10.1016/j.ejca.2008.12.030. [DOI] [PubMed] [Google Scholar]
- 16.Hidaka H, Ishiko T, Furuhashi T, Kamohara H, Suzuki S, Miyazaki M, Ikeda O, Mita S, Setoguchi T, Ogawa M. Curcumin inhibits interleukin 8 production and enhances interleukin 8 receptor expression on the cell surface:impact on human pancreatic carcinoma cell growth by autocrine regulation. Cancer. 2002;95:1206–1214. doi: 10.1002/cncr.10812. [DOI] [PubMed] [Google Scholar]
- 17.Kunnumakkara AB, Guha S, Krishnan S, Diagaradjane P, Gelovani J, Aggarwal BB. Curcumin potentiates antitumor activity of gemcitabine in an orthotopic model of pancreatic cancer through suppression of proliferation, angiogenesis, and inhibition of nuclear factor-kappaB-regulated gene products. Cancer Research. 2007;67:3853–3861. doi: 10.1158/0008-5472.CAN-06-4257. [DOI] [PubMed] [Google Scholar]
- 18.Lin L, Hutzen B, Zuo M, Ball S, Deangelis S, Foust E, Pandit B, Ihnat MA, Shenoy SS, Kulp S, Li PK, Li C, Fuchs J, Lin J. Novel STAT3 phosphorylation inhibitors exhibit potent growth-suppressive activity in pancreatic and breast cancer cells. Cancer Research. 2010;70:2445–2454. doi: 10.1158/0008-5472.CAN-09-2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hutzen B, Friedman L, Sobo M, Lin L, Cen L, De Angelis S, Yamakoshi H, Shibata H, Iwabuchi Y, Lin J. Curcumin analogue GO-Y030 inhibits STAT3 activity and cell growth in breast and pancreatic carcinomas. International Journal of Oncology. 2009;35:867–872. doi: 10.3892/ijo_00000401. [DOI] [PubMed] [Google Scholar]
- 20.Gu L, Chiang KY, Zhu N, Findley HW, Zhou M. Contribution of STAT3 to the activation of survivin by GM-CSF in CD34+ cell lines. Experimental Hematology. 2007;35:957–966. doi: 10.1016/j.exphem.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 21.Bora-Tatar G, Dayangac-Erden D, Demir AS, Dalkara S, Yelekci K, Erdem-Yurter H. Molecular modifications on carboxylic acid derivatives as potent histone deacetylase inhibitors: Activity and docking studies. Bioorganic Medicinal Chemistry. 2009;17:5219–5228. doi: 10.1016/j.bmc.2009.05.042. [DOI] [PubMed] [Google Scholar]
- 22.Liu HL, Chen Y, Cui GH, Zhou JF. Curcumin, a potent anti-tumor reagent, is a novel histone deacetylase inhibitor regulating B-NHL cell line Raji proliferation. Acta Pharmacologica Sinica. 2005;26:603–609. doi: 10.1111/j.1745-7254.2005.00081.x. [DOI] [PubMed] [Google Scholar]
- 23.Meja KK, Rajendrasozhan S, Adenuga D, Biswas SK, Sundar IK, Spooner G, Marwick JA, Chakravarty P, Fletcher D, Whittaker P, Megson IL, Kirkham PA, Rahman I. Curcumin restores corticosteroid function in monocytes exposed to oxidants by maintaining HDAC2. American Journal of Respiratory Cell and Molecular Biology. 2008;39:312–323. doi: 10.1165/rcmb.2008-0012OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Balasubramanyam K, Varier RA, Altaf M, Swaminathan V, Siddappa NB, Ranga U, Kundu TK. Curcumin, a novel p300/CREB-binding protein-specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase-dependent chromatin transcription. The Journal of Biological Chemistry. 2004;279:51163–51171. doi: 10.1074/jbc.M409024200. [DOI] [PubMed] [Google Scholar]
- 25.Liu Z, Xie Z, Jones W, Pavlovicz RE, Liu S, Yu J, Li PK, Lin J, Fuchs JR, Marcucci G, Li C, Chan KK. Curcumin is a potent DNA hypomethylation agent. Bioorganic and Medicinal Chemistry Letters. 2009;19:706–709. doi: 10.1016/j.bmcl.2008.12.041. [DOI] [PubMed] [Google Scholar]
- 26.Medina-Franco JL, Lopez-Vallejo F, Kuck D, Lyko F. Natural products as DNA methyltransferase inhibitors: a computer-aided discovery approach. Molecular Diversity. 2011;15:293–304. doi: 10.1007/s11030-010-9262-5. [DOI] [PubMed] [Google Scholar]
- 27.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nature Reviews Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 28.Rachagani S, Kumar S, Batra SK. MicroRNA in pancreatic cancer: pathological, diagnostic and therapeutic implications. Cancer Letters. 2010;292:8–16. doi: 10.1016/j.canlet.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ali S, Ahmad A, Aboukameel A, Bao B, Padhye S, Philip PA, Sarkar FH. Increased Ras GTPase activity is regulated by miRNAs that can be attenuated by CDF treatment in pancreatic cancer cells. Cancer Letters. 2012;319:173–181. doi: 10.1016/j.canlet.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30.Bao B, Ali S, Banerjee S, Wang Z, Logna F, Azmi AS, Kong D, Ahmad A, Li Y, Padhye S, Sarkar FH. Curcumin analogue CDF inhibits pancreatic tumor growth by switching on suppressor microRNAs and attenuating EZH2 expression. Cancer Research. 2012;72:335–345. doi: 10.1158/0008-5472.CAN-11-2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ali S, Ahmad A, Banerjee S, Padhye S, Dominiak K, Schaffert JM, Wang Z, Philip PA, Sarkar FH. Gemcitabine sensitivity can be induced in pancreatic cancer cells through modulation of miR-200 and miR-21 expression by curcumin or its analogue CDF. Cancer Research. 2010;70:3606–3617. doi: 10.1158/0008-5472.CAN-09-4598. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Sun M, Estrov Z, Ji Y, Coombes KR, Harris DH, Kurzrock R. Curcumin (diferuloylmethane) alters the expression profiles of microRNAs in human pancreatic cancer cells. Molecular Cancer Therapeutics. 2008;7:464–473. doi: 10.1158/1535-7163.MCT-07-2272. [DOI] [PubMed] [Google Scholar]
- 33.Anand P, Kunnumakkara AB, Newman RA, Aggarwal BB. Bioavailability of curcumin: problems and promises. Molecular Pharmaceutics. 2007;4:807–818. doi: 10.1021/mp700113r. [DOI] [PubMed] [Google Scholar]
- 34.Shoba G, Joy D, Joseph T, Majeed M, Rajendran R, Srinivas PS. Influence of piperine on the pharmacokinetics of curcumin in animals and human volunteers. Planta Medica. 1998;64:353–356. doi: 10.1055/s-2006-957450. [DOI] [PubMed] [Google Scholar]
- 35.Kanai M, Yoshimura K, Asada M, Imaizumi A, Suzuki C, Matsumoto S, Nishimura T, Mori Y, Masui T, Kawaguchi Y, Yanagihara K, Yazumi S, Chiba T, Guha S, Aggarwal BB. A phase I/II study of gemcitabine-based chemotherapy plus curcumin for patients with gemcitabine-resistant pancreatic cancer. Cancer Chemotherapy and Pharmacology. 2011;68:157–164. doi: 10.1007/s00280-010-1470-2. [DOI] [PubMed] [Google Scholar]
- 36.Dhillon N, Aggarwal BB, Newman RA, Wolff RA, Kunnumakkara AB, Abbruzzese JL, Ng CS, Badmaev V, Kurzrock R. Phase II trial of curcumin in patients with advanced pancreatic cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2008;14:4491–4499. doi: 10.1158/1078-0432.CCR-08-0024. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Y, Talalay P, Cho CG, Posner GH. A major inducer of anticarcinogenic protective enzymes from broccoli: isolation and elucidation of structure. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:2399–2403. doi: 10.1073/pnas.89.6.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stoner GD, Morse MA. Isothiocyanates and plant polyphenols as inhibitors of lung and esophageal cancer. Cancer Letters. 1997;114:113–119. doi: 10.1016/s0304-3835(97)04639-9. [DOI] [PubMed] [Google Scholar]
- 39.Shapiro TA, Fahey JW, Wade KL, Stephenson KK, Talalay P. Chemoprotective glucosinolates and isothiocyanates of broccoli sprouts: metabolism and excretion in humans. Cancer epidemiology, biomarkers & prevention. 2001;10:501–508. [PubMed] [Google Scholar]
- 40.Boreddy SR, Sahu RP, Srivastava SK. Benzyl isothiocyanate suppresses pancreatic tumor angiogenesis and invasion by inhibiting HIF-alpha/VEGF/Rho-GTPases: pivotal role of STAT-3. PloS One. 2011;6:e25799. doi: 10.1371/journal.pone.0025799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boreddy SR, Pramanik KC, Srivastava SK. Pancreatic tumor suppression by benzyl isothiocyanate is associated with inhibition of PI3K/AKT/FOXO pathway. Clinical Cancer Research. 2011;17:1784–1795. doi: 10.1158/1078-0432.CCR-10-1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Batra S, Sahu RP, Kandala PK, Srivastava SK. Benzyl isothiocyanate-mediated inhibition of histone deacetylase leads to NF-kappaB turnoff in human pancreatic carcinoma cells. Molecular Cancer Therapeutics. 2010;9:1596–1608. doi: 10.1158/1535-7163.MCT-09-1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sahu RP, Zhang R, Batra S, Shi Y, Srivastava SK. Benzyl isothiocyanate-mediated generation of reactive oxygen species causes cell cycle arrest and induces apoptosis via activation of MAPK in human pancreatic cancer cells. Carcinogenesis. 2009;30:1744–1753. doi: 10.1093/carcin/bgp157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sahu RP, Epperly MW, Srivastava SK. Benzyl isothiocyanate sensitizes human pancreatic cancer cells to radiation therapy. Frontiers in Bioscience (Elite Ed) 2009;1:568–576. doi: 10.2741/e55. [DOI] [PubMed] [Google Scholar]
- 45.Sahu RP, Srivastava SK. The role of STAT-3 in the induction of apoptosis in pancreatic cancer cells by benzyl isothiocyanate. Journal of the National Cancer Institute. 2009;101:176–193. doi: 10.1093/jnci/djn470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang R, Loganathan S, Humphreys I, Srivastava SK. Benzyl isothiocyanate-induced DNA damage causes G2/M cell cycle arrest and apoptosis in human pancreatic cancer cells. The Journal of Nutrition. 2006;136:2728–2734. doi: 10.1093/jn/136.11.2728. [DOI] [PubMed] [Google Scholar]
- 47.Srivastava SK, Singh SV. Cell cycle arrest, apoptosis induction and inhibition of nuclear factor kappa B activation in anti-proliferative activity of benzyl isothiocyanate against human pancreatic cancer cells. Carcinogenesis. 2004;25:1701–1709. doi: 10.1093/carcin/bgh179. [DOI] [PubMed] [Google Scholar]
- 48.Wei D, Le X, Zheng L, Wang L, Frey JA, Gao AC, Peng Z, Huang S, Xiong HQ, Abbruzzese JL, Xie K. Stat3 activation regulates the expression of vascular endothelial growth factor and human pancreatic cancer angiogenesis and metastasis. Oncogene. 2003;22:319–329. doi: 10.1038/sj.onc.1206122. [DOI] [PubMed] [Google Scholar]
- 49.Darnell JE. Validating Stat3 in cancer therapy. Nature medicine. 2005;11:595–596. doi: 10.1038/nm0605-595. [DOI] [PubMed] [Google Scholar]
- 50.Semenza GL. HIF-1 and mechanisms of hypoxia sensing. Current Opinion in Cell Biology. 2001;13:167–171. doi: 10.1016/s0955-0674(00)00194-0. [DOI] [PubMed] [Google Scholar]
- 51.Xu Q, Briggs J, Park S, Niu G, Kortylewski M, Zhang S, Gritsko T, Turkson J, Kay H, Semenza GL, Cheng JQ, Jove R, Yu H. Targeting Stat3 blocks both HIF-1 and VEGF expression induced by multiple oncogenic growth signaling pathways. Oncogene. 2005;24:5552–5560. doi: 10.1038/sj.onc.1208719. [DOI] [PubMed] [Google Scholar]
- 52.Jung JE, Lee HG, Cho IH, Chung DH, Yoon SH, Yang YM, Lee JW, Choi S, Park JW, Ye SK, Chung MH. STAT3 is a potential modulator of HIF-1-mediated VEGF expression in human renal carcinoma cells. FASEB Journal. 2005;19:1296–1298. doi: 10.1096/fj.04-3099fje. [DOI] [PubMed] [Google Scholar]
- 53.Finkielsztein A, Kelly GM. Altering PI3K-Akt signalling in zebrafish embryos affects PTEN phosphorylation and gastrulation. Biology of the Cell. 2009;101:661–678. doi: 10.1042/BC20090034. 664 p following 678. [DOI] [PubMed] [Google Scholar]
- 54.Schlieman MG, Fahy BN, Ramsamooj R, Beckett L, Bold RJ. Incidence, mechanism and prognostic value of activated AKT in pancreas cancer. British Journal of Cancer. 2003;89:2110–2115. doi: 10.1038/sj.bjc.6601396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 56.Osaki M, Oshimura M, Ito H. PI3K-Akt pathway: its functions and alterations in human cancer. Apoptosis. 2004;9:667–676. doi: 10.1023/B:APPT.0000045801.15585.dd. [DOI] [PubMed] [Google Scholar]
- 57.Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annual Review of Immunology. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 58.Sun Z, Andersson R. NF-kappaB activation and inhibition: a review. Shock. 2002;18:99–106. doi: 10.1097/00024382-200208000-00001. [DOI] [PubMed] [Google Scholar]
- 59.Bode AM, Dong Z. The two faces of capsaicin. Cancer Research. 2011;71:2809–2814. doi: 10.1158/0008-5472.CAN-10-3756. [DOI] [PubMed] [Google Scholar]
- 60.Hoch-Ligeti C. Production of liver tumours by dietary means; effect of feeding chilies [Capsicum frutescens and annuum (Linn.)] to rats. Acta - Unio Internationalis Contra Cancrum. 1951;7:606–611. [PubMed] [Google Scholar]
- 61.Toth B, Gannett P. Carcinogenicity of lifelong administration of capsaicin of hot pepper in mice. In Vivo. 1992;6:59–63. [PubMed] [Google Scholar]
- 62.Zhang R, Humphreys I, Sahu RP, Shi Y, Srivastava SK. In vitro and in vivo induction of apoptosis by capsaicin in pancreatic cancer cells is mediated through ROS generation and mitochondrial death pathway. Apoptosis. 2008;13:1465–1478. doi: 10.1007/s10495-008-0278-6. [DOI] [PubMed] [Google Scholar]
- 63.Pramanik KC, Boreddy SR, Srivastava SK. Role of mitochondrial electron transport chain complexes in capsaicin mediated oxidative stress leading to apoptosis in pancreatic cancer cells. PloS One. 2011;6:e20151. doi: 10.1371/journal.pone.0020151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pramanik KC, Srivastava SK. Apoptosis Signal-Regulating Kinase 1-Thioredoxin Complex Dissociation by Capsaicin Causes Pancreatic Tumor Growth Suppression by Inducing Apoptosis. Antioxidants & Redox Signaling. 2012;17:1417–1432. doi: 10.1089/ars.2011.4369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bai H, Li H, Zhang W, Matkowskyj KA, Liao J, Srivastava SK, Yang GY. Inhibition of chronic pancreatitis and pancreatic intraepithelial neoplasia (PanIN) by capsaicin in LSL-KrasG12D/Pdx1-Cre mice. Carcinogenesis. 2011;32:1689–1696. doi: 10.1093/carcin/bgr191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Serra I, Yamamoto M, Calvo A, Cavada G, Baez S, Endoh K, Watanabe H, Tajima K. Association of chili pepper consumption, low socioeconomic status and longstanding gallstones with gallbladder cancer in a Chilean population. International Journal of Cancer. 2002;102:407–411. doi: 10.1002/ijc.10716. [DOI] [PubMed] [Google Scholar]
- 67.Lopez-Carrillo L, Hernandez Avila M, Dubrow R. Chili pepper consumption and gastric cancer in Mexico: a case-control study. American journal of epidemiology. 1994;139:263–271. doi: 10.1093/oxfordjournals.aje.a116993. [DOI] [PubMed] [Google Scholar]
- 68.Dasgupta P, Chandiramani V, Parkinson MC, Beckett A, Fowler CJ. Treating the human bladder with capsaicin: is it safe? European Urology. 1998;33:28–31. doi: 10.1159/000019531. [DOI] [PubMed] [Google Scholar]
- 69.Nishitoh H, Saitoh M, Mochida Y, Takeda K, Nakano H, Rothe M, Miyazono K, Ichijo H. ASK1 is essential for JNK/SAPK activation by TRAF2. Molecular Cell. 1998;2:389–395. doi: 10.1016/s1097-2765(00)80283-x. [DOI] [PubMed] [Google Scholar]
- 70.Cabrera C, Artacho R, Gimenez R. Beneficial effects of green tea--a review. Journal of the American College of Nutrition. 2006;25:79–99. doi: 10.1080/07315724.2006.10719518. [DOI] [PubMed] [Google Scholar]
- 71.McKay DL, Blumberg JB. The role of tea in human health: an update. Journal of the American College of Nutrition. 2002;21:1–13. doi: 10.1080/07315724.2002.10719187. [DOI] [PubMed] [Google Scholar]
- 72.Okushio K, Matsumoto N, Kohri T, Suzuki M, Nanjo F, Hara Y. Absorption of tea catechins into rat portal vein. Biological & Pharmaceutical Bulletin. 1996;19:326–329. doi: 10.1248/bpb.19.326. [DOI] [PubMed] [Google Scholar]
- 73.Lee MJ, Maliakal P, Chen L, Meng X, Bondoc FY, Prabhu S, Lambert G, Mohr S, Yang CS. Pharmacokinetics of tea catechins after ingestion of green tea and (−)-epigallocatechin-3-gallate by humans: formation of different metabolites and individual variability. Cancer Epidemiology, Biomarkers & Prevention. 2002;11:1025–1032. [PubMed] [Google Scholar]
- 74.Yang CS, Wang X. Green tea and cancer prevention. Nutrition and Cancer. 2010;62:931–937. doi: 10.1080/01635581.2010.509536. [DOI] [PubMed] [Google Scholar]
- 75.Zhang L, Pang E, Loo RR, Rao J, Go VL, Loo JA, Lu QY. Concomitant inhibition of HSP90, its mitochondrial localized homologue TRAP1 and HSP27 by green tea in pancreatic cancer HPAF-II cells. Proteomics. 2011;11:4638–4647. doi: 10.1002/pmic.201100242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li Y, Zhang T, Jiang Y, Lee HF, Schwartz SJ, Sun D. (−)-Epigallocatechin-3-gallate inhibits Hsp90 function by impairing Hsp90 association with cochaperones in pancreatic cancer cell line Mia Paca-2. Molecular Pharmaceutics. 2009;6:1152–1159. doi: 10.1021/mp900037p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Qanungo S, Das M, Haldar S, Basu A. Epigallocatechin-3-gallate induces mitochondrial membrane depolarization and caspase-dependent apoptosis in pancreatic cancer cells. Carcinogenesis. 2005;26:958–967. doi: 10.1093/carcin/bgi040. [DOI] [PubMed] [Google Scholar]
- 78.Shankar S, Suthakar G, Srivastava RK. Epigallocatechin-3-gallate inhibits cell cycle and induces apoptosis in pancreatic cancer. Frontiers in Bioscience. 2007;12:5039–5051. doi: 10.2741/2446. [DOI] [PubMed] [Google Scholar]
- 79.Tang SN, Fu J, Nall D, Rodova M, Shankar S, Srivastava RK. Inhibition of sonic hedgehog pathway and pluripotency maintaining factors regulate human pancreatic cancer stem cell characteristics. International Journal of Cancer. 2012;131:30–40. doi: 10.1002/ijc.26323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kostin SF, McDonald DE, McFadden DW. Inhibitory effects of (−)-epigallocatechin-3-gallate and pterostilbene on pancreatic cancer growth in vitro. Journal of Surgical Research. 2012;177:255–262. doi: 10.1016/j.jss.2012.04.023. [DOI] [PubMed] [Google Scholar]
- 81.Hardtner C, Multhoff G, Falk W, Radons J. (−)-Epigallocatechin-3-gallate, a green tea-derived catechin, synergizes with celecoxib to inhibit IL-1-induced tumorigenic mediators by human pancreatic adenocarcinoma cells Colo357. European Journal of Pharmacology. 2012;684:36–43. doi: 10.1016/j.ejphar.2012.03.039. [DOI] [PubMed] [Google Scholar]
- 82.Basu A, Haldar S. Combinatorial effect of epigallocatechin-3-gallate and TRAIL on pancreatic cancer cell death. International Journal of Oncology. 2009;34:281–286. [PubMed] [Google Scholar]
- 83.Tan M, Norwood A, May M, Tucci M, Benghuzzi H. Effects of (−)epigallocatechin gallate and thymoquinone on proliferation of a PANC-1 cell line in culture. Biomedical Sciences Instrumentation. 2006;42:363–371. [PubMed] [Google Scholar]
- 84.Tang SN, Fu J, Shankar S, Srivastava RK. EGCG enhances the therapeutic potential of gemcitabine and CP690550 by inhibiting STAT3 signaling pathway in human pancreatic cancer. PloS One. 2012;7:e31067. doi: 10.1371/journal.pone.0031067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shankar S, Ganapathy S, Hingorani SR, Srivastava RK. EGCG inhibits growth, invasion, angiogenesis and metastasis of pancreatic cancer. Frontiers in Bioscience. 2008;13:440–452. doi: 10.2741/2691. [DOI] [PubMed] [Google Scholar]
- 86.Vu HA, Beppu Y, Chi HT, Sasaki K, Yamamoto H, Xinh PT, Tanii T, Hara Y, Watanabe T, Sato Y, Ohdomari I. Green tea epigallocatechin gallate exhibits anticancer effect in human pancreatic carcinoma cells via the inhibition of both focal adhesion kinase and insulin-like growth factor-I receptor. Journal of Biomedical Biotechnology. 2010;2010:290516. doi: 10.1155/2010/290516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhu BH, Chen HY, Zhan WH, Wang CY, Cai SR, Wang Z, Zhang CH, He YL. (−)-Epigallocatechin-3-gallate inhibits VEGF expression induced by IL-6 via Stat3 in gastric cancer. World Journal of Gastroenterology. 2011;17:2315–2325. doi: 10.3748/wjg.v17.i18.2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Takabayashi F, Tahara S, Kaneko T, Harada N. Effect of green tea catechins on oxidative DNA damage of hamster pancreas and liver induced by N-Nitrosobis(2-oxopropyl)amine and/or oxidized soybean oil. Biofactors. 2004;21:335–337. doi: 10.1002/biof.552210165. [DOI] [PubMed] [Google Scholar]
- 89.Majima T, Tsutsumi M, Nishino H, Tsunoda T, Konishi Y. Inhibitory effects of beta-carotene, palm carotene, and green tea polyphenols on pancreatic carcinogenesis initiated by N-nitorsobis(2-oxopropyl)amine in Syrian golden hamsters. Pancreas. 1998;16:13–18. doi: 10.1097/00006676-199801000-00003. [DOI] [PubMed] [Google Scholar]
- 90.Hiura A, Tsutsumi M, Satake K. Inhibitory effect of green tea extract on the process of pancreatic carcinogenesis induced by N-nitrosobis-(2-oxypropyl)amine (BOP) and on tumor promotion after transplantation of N-nitrosobis-(2-hydroxypropyl)amine (BHP)-induced pancreatic cancer in Syrian hamsters. Pancreas. 1997;15:272–277. doi: 10.1097/00006676-199710000-00009. [DOI] [PubMed] [Google Scholar]
- 91.Thakur VS, Gupta K, Gupta S. Green tea polyphenols increase p53 transcriptional activity and acetylation by suppressing class I histone deacetylases. International Journal of Oncology. 2012;41:353–361. doi: 10.3892/ijo.2012.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fang MZ, Wang Y, Ai N, Hou Z, Sun Y, Lu H, Welsh W, Yang CS. Tea polyphenol (−)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Research. 2003;63:7563–7570. [PubMed] [Google Scholar]
- 93.Pandey M, Shukla S, Gupta S. Promoter demethylation and chromatin remodeling by green tea polyphenols leads to re-expression of GSTP1 in human prostate cancer cells. International Journal of Cancer. 2010;126:2520–2533. doi: 10.1002/ijc.24988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tsang WP, Kwok TT. Epigallocatechin gallate up-regulation of miR-16 and induction of apoptosis in human cancer cells. Journal of Nutritional Biochemistry. 2010;21:140–146. doi: 10.1016/j.jnutbio.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 95.Penumathsa SV, Maulik N. Resveratrol: a promising agent in promoting cardioprotection against coronary heart disease. Canadian Journal of Physiology and Pharmacology. 2009;87:275–286. doi: 10.1139/Y09-013. [DOI] [PubMed] [Google Scholar]
- 96.Baliga MS, Meleth S, Katiyar SK. Growth inhibitory and antimetastatic effect of green tea polyphenols on metastasis-specific mouse mammary carcinoma 4T1 cells in vitro and in vivo systems. Clinical Cancer Research. 2005;11:1918–1927. doi: 10.1158/1078-0432.CCR-04-1976. [DOI] [PubMed] [Google Scholar]
- 97.Athar M, Back JH, Tang X, Kim KH, Kopelovich L, Bickers DR, Kim AL. Resveratrol: a review of preclinical studies for human cancer prevention. Toxicology and Applied Pharmacology. 2007;224:274–283. doi: 10.1016/j.taap.2006.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kundu JK, Surh YJ. Molecular basis of chemoprevention by resveratrol: NF-kappaB and AP-1 as potential targets. Mutation Research. 2004;555:65–80. doi: 10.1016/j.mrfmmm.2004.05.019. [DOI] [PubMed] [Google Scholar]
- 99.Wu JM, Hsieh TC. Resveratrol: a cardioprotective substance. Annals of the New York Academy of Sciences. 2011;1215:16–21. doi: 10.1111/j.1749-6632.2010.05854.x. [DOI] [PubMed] [Google Scholar]
- 100.Goldberg DM, Yan J, Soleas GJ. Absorption of three wine-related polyphenols in three different matrices by healthy subjects. Clinical Biochemistry. 2003;36:79–87. doi: 10.1016/s0009-9120(02)00397-1. [DOI] [PubMed] [Google Scholar]
- 101.Mannal PW, Alosi JA, Schneider JG, McDonald DE, McFadden DW. Pterostilbene inhibits pancreatic cancer in vitro. Journal of Gastrointestinal Surgery. 2010;14:873–879. doi: 10.1007/s11605-010-1164-4. [DOI] [PubMed] [Google Scholar]
- 102.Hong YB, Kang HJ, Kim HJ, Rosen EM, Dakshanamurthy S, Rondanin R, Baruchello R, Grisolia G, Daniele S, Bae I. Inhibition of cell proliferation by a resveratrol analog in human pancreatic and breast cancer cells. Experimental & Molecular Medicine. 2009;41:151–160. doi: 10.3858/emm.2009.41.3.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bernhaus A, Ozsvar-Kozma M, Saiko P, Jaschke M, Lackner A, Grusch M, Horvath Z, Madlener S, Krupitza G, Handler N, Erker T, Jaeger W, Fritzer-Szekeres M, Szekeres T. Antitumor effects of KITC, a new resveratrol derivative, in AsPC-1 and BxPC-3 human pancreatic carcinoma cells. Investigational New Drugs. 2009;27:393–401. doi: 10.1007/s10637-008-9183-7. [DOI] [PubMed] [Google Scholar]
- 104.Sun W, Wang W, Kim J, Keng P, Yang S, Zhang H, Liu C, Okunieff P, Zhang L. Anti-cancer effect of resveratrol is associated with induction of apoptosis via a mitochondrial pathway alignment. Advances in Experimental Medicine and Biology. 2008;614:179–186. doi: 10.1007/978-0-387-74911-2_21. [DOI] [PubMed] [Google Scholar]
- 105.Zhou JH, Cheng HY, Yu ZQ, He DW, Pan Z, Yang DT. Resveratrol induces apoptosis in pancreatic cancer cells. Chinese Medical Journal. 2011;124:1695–1699. [PubMed] [Google Scholar]
- 106.Shamim U, Hanif S, Albanyan A, Beck FW, Bao B, Wang Z, Banerjee S, Sarkar FH, Mohammad RM, Hadi SM, Azmi AS. Resveratrol-induced apoptosis is enhanced in low pH environments associated with cancer. Journal of Cellular Physiology. 2012;227:1493–1500. doi: 10.1002/jcp.22865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bortolotti C, Kunit T, Moder A, Hufnagl C, Schmidt S, Hartl A, Langelueddecke C, Furst J, Geibel JP, Ritter M, Jakab M. The phytostilbene resveratrol induces apoptosis in INS-1E rat insulinoma cells. Cellular Physiology and Biochemistry. 2009;23:245–254. doi: 10.1159/000218171. [DOI] [PubMed] [Google Scholar]
- 108.Mo W, Xu X, Xu L, Wang F, Ke A, Wang X, Guo C. Resveratrol inhibits proliferation and induces apoptosis through the hedgehog signaling pathway in pancreatic cancer cell. Pancreatology. 2011;11:601–609. doi: 10.1159/000333542. [DOI] [PubMed] [Google Scholar]
- 109.Ding XZ, Adrian TE. Resveratrol inhibits proliferation and induces apoptosis in human pancreatic cancer cells. Pancreas. 2002;25:e71–76. doi: 10.1097/00006676-200211000-00024. [DOI] [PubMed] [Google Scholar]
- 110.Cui J, Sun R, Yu Y, Gou S, Zhao G, Wang C. Antiproliferative effect of resveratrol in pancreatic cancer cells. Phytotherapy Research. 2010;24:1637–1644. doi: 10.1002/ptr.3157. [DOI] [PubMed] [Google Scholar]
- 111.Roy SK, Chen Q, Fu J, Shankar S, Srivastava RK. Resveratrol inhibits growth of orthotopic pancreatic tumors through activation of FOXO transcription factors. PloS One. 2011;6:e25166. doi: 10.1371/journal.pone.0025166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hambrock A, de Oliveira Franz CB, Hiller S, Grenz A, Ackermann S, Schulze DU, Drews G, Osswald H. Resveratrol binds to the sulfonylurea receptor (SUR) and induces apoptosis in a SUR subtype-specific manner. The Journal of Biological Chemistry. 2007;282:3347–3356. doi: 10.1074/jbc.M608216200. [DOI] [PubMed] [Google Scholar]
- 113.Chen Q, Ganapathy S, Singh KP, Shankar S, Srivastava RK. Resveratrol induces growth arrest and apoptosis through activation of FOXO transcription factors in prostate cancer cells. PloS One. 2010;5:e15288. doi: 10.1371/journal.pone.0015288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Oi N, Jeong CH, Nadas J, Cho YY, Pugliese A, Bode AM, Dong Z. Resveratrol, a red wine polyphenol, suppresses pancreatic cancer by inhibiting leukotriene A(4)hydrolase. Cancer Research. 2010;70:9755–9764. doi: 10.1158/0008-5472.CAN-10-2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Golkar L, Ding XZ, Ujiki MB, Salabat MR, Kelly DL, Scholtens D, Fought AJ, Bentrem DJ, Talamonti MS, Bell RH, Adrian TE. Resveratrol inhibits pancreatic cancer cell proliferation through transcriptional induction of macrophage inhibitory cytokine-1. Journal of Surgical Researh. 2007;138:163–169. doi: 10.1016/j.jss.2006.05.037. [DOI] [PubMed] [Google Scholar]
- 116.Kotha A, Sekharam M, Cilenti L, Siddiquee K, Khaled A, Zervos AS, Carter B, Turkson J, Jove R. Resveratrol inhibits Src and Stat3 signaling and induces the apoptosis of malignant cells containing activated Stat3 protein. Molecular Cancer Therapeutics. 2006;5:621–629. doi: 10.1158/1535-7163.MCT-05-0268. [DOI] [PubMed] [Google Scholar]
- 117.Shankar S, Nall D, Tang SN, Meeker D, Passarini J, Sharma J, Srivastava RK. Resveratrol inhibits pancreatic cancer stem cell characteristics in human and KrasG12D transgenic mice by inhibiting pluripotency maintaining factors and epithelial-mesenchymal transition. PloS One. 2011;6:e16530. doi: 10.1371/journal.pone.0016530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, Scherer B, Sinclair DA. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- 119.Gracia-Sancho J, Villarreal G, Jr, Zhang Y, Garcia-Cardena G. Activation of SIRT1 by resveratrol induces KLF2 expression conferring an endothelial vasoprotective phenotype. Cardiovasc Research. 2010;85:514–519. doi: 10.1093/cvr/cvp337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Paluszczak J, Krajka-Kuzniak V, Baer-Dubowska W. The effect of dietary polyphenols on the epigenetic regulation of gene expression in MCF7 breast cancer cells. Toxicology Letters. 2010;192:119–125. doi: 10.1016/j.toxlet.2009.10.010. [DOI] [PubMed] [Google Scholar]
- 121.Tili E, Michaille JJ, Adair B, Alder H, Limagne E, Taccioli C, Ferracin M, Delmas D, Latruffe N, Croce CM. Resveratrol decreases the levels of miR-155 by upregulating miR-663, a microRNA targeting JunB and JunD. Carcinogenesis. 2010;31:1561–1566. doi: 10.1093/carcin/bgq143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gupta SC, Kannappan R, Reuter S, Kim JH, Aggarwal BB. Chemosensitization of tumors by resveratrol. Annals of the New York Academy of Sciences. 2011;1215:150–160. doi: 10.1111/j.1749-6632.2010.05852.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Harikumar KB, Kunnumakkara AB, Sethi G, Diagaradjane P, Anand P, Pandey MK, Gelovani J, Krishnan S, Guha S, Aggarwal BB. Resveratrol, a multitargeted agent, can enhance antitumor activity of gemcitabine in vitro and in orthotopic mouse model of human pancreatic cancer. International Journal of Cancer. 2010;127:257–268. doi: 10.1002/ijc.25041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Fulda S, Debatin KM. Sensitization for tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by the chemopreventive agent resveratrol. Cancer Research. 2004;64:337–346. doi: 10.1158/0008-5472.can-03-1656. [DOI] [PubMed] [Google Scholar]