

Abstract
This review is focused on the role of Focal Adhesion Kinase (FAK) signaling in cancer stem cells. The recent data demonstrate the important role of FAK in cancer stem cell proliferation, differentiation, motility, and invasion. We showed recently that the transcription factor Nanog binds the FAK promoter and up-regulates FAK expression, and that FAK binds Nanog and phosphorylates it. This review discusses the interaction of FAK, Nanog, Oct-3/4, and Sox-2 signaling pathways that are critical for the regulation of cancer stem cells. The cross-linked signaling of FAK with p53 and Nanog signaling in cancer stem cell and function and targeted therapeutics approaches are discussed.
Keywords: Adhesion, cancer, Focal Adhesion Kinase, inhibitor, metastasis, tumor
CANCER STEM CELLS
Cancer stem cells are cells that can self-renew and differentiate into different lineage-specific cancer cell types which promote tumor formation (Fig. 1) [1]. Cancer stem cells are resistant to chemotherapies and contain specific cell surface markers [2]. The tumors are heterogeneous and contain different types of cells: cancer cells that can’t generate tumors when implanted into the immunocompromised (nude) mice, and cancer stem cells, which can generate tumors [3]. Cancer stem cells were first isolated from AML (acute myeloid leukemia) tumors based on cell surface markers after transplantation of AML cells into immuno deficient (SCID) mice [4]. The leukemia-initiated cells that could produce large numbers of colony forming progenitors were CD34+CD38− [4]. The cancer stem cells were later isolated from different solid tumors: brain [5, 6], breast [7, 8], colon [9], pancreatic [7], lung [8], and prostate [9] tumors. The progression of cancer stem cells to tumors depends on tumor microenvironment or stroma that includes extracellular matrix, endothelial, fibroblasts, and immune cells (macrophages, neutrophils, lymphocytes) (Fig. 1). There are several models of the evolution of normal stem cells into cancer stem cells to drive tumor progression. In one of them, the mutational event happens in transit-amplifying cells, (highly-proliferating cells: progenitor populations) that can either differentiate into tumor cell populations or de-differentiate into mutant or cancer stem cells [3].
Fig. 1. Cancer stem cells andb the microenvironment.

Cancer stem cells are cancer cells that self-renew and differentiate and form tumors. There is a dynamic transition between cancer progenitor cells and cancer stem cells through differentiation and de-differentiation processes. Cancer stem cells are surrounded by cancer stem niche or tumor microenvironment components, such as extracellular matrix (collagen, fibronectin, etc, laminin, etc.) fibroblasts (cancer-associated fibroblasts); endothelial cells, blood vesssels, playing role in angiogenesis and lympangiogenesis; and immune cells, including neutrophils and lymphocytes and macrophages, playing a significant role in inflammation. Tumor microenvironment plays a significant role in cancer stem cell maintenance and transition from epithelial to mesenchymal (EMT) and back (MET) transition
The cancer stem cells can also be generated through epithelial to mesenchymal transition (EMT), when epithelial cells lose the cell-extracellular matrix and cell-cell contacts, and become motile mesenchymal cells [3]. It is known that the primary epithelial cells are able to form distant metastases. The epithelial to mesenchymal transition (EMT) program regulates this metastatic process, where carcinoma cells invade the stroma and translocate to parenchyma cells. The switch back from mesenchymal to epithelial state is mesenchumal epithelial transition (MET), which occurs during carcinoma colonization [10]. The tumor microenvironment induces activation of EMT programs in cancer cells, and EMT regulates the transition of cancer cells into the cancer stem cells [10]. There are also different epigenetic programs that play a role in the transitions between cancer and non cancer stem cells. Thus, it was suggested due to the reversibility of EMT and MET that therapeutic approaches should be developed that target cancer stem cells as well as tumor microenvironment or proposed to develop therapies with a dual effect [10].
IMPORTANT TRANSCRIPTION FACTORS OF STEM CELLS
Many transcription factors play a significant role in the self-renewal of embryonic stem cells and cancer stem cells. Expression of Sox-2, c-Myc, Oct-4, Klf-4 and Lin-28, together with epigenetic changes can reprogram somatic cells into induced pluripotent stem cells (iPSC) [11] (Fig. 2). These transcription factors are often overexpressed in tumors. We will focus below on one of the important transcription factors, Nanog, described in detail below, because we found that it is directly linked to FAK survival signaling [12].
Fig. 2. The transcription factors involved in the transition of cancer stem cells to tumor.
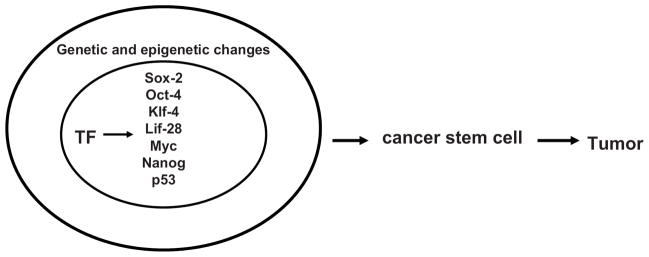
The main transcription factors (shown inside the cancer stem cell nucleus), epigenetic and genetic changes play a significant role in the transition of cancer stem cells to tumor.
NANOG
The name Nanog came from the Irish legend’s name Tir-na-Nog, which translates as “the land of the ever young”. Nanog was discovered by Chambers et al [13] and by Mitsui et al [14] in 2003. The name was given for the ability of Nanog to maintain self-renewal of mouse embryonic cells. Nanog is a homeobox transcription factor that consists of the five domains: N-terminal; homeodomain; C-terminal 1; tryptophan repeat and C-terminal 2 domains, and binds to the (C/G)(G/A)(C/G)C(G/C)ATTAN(G/C) or TAAT(TG)(TG) sequence. The human Nanog 1 gene is localized on chromosome 12; there are several retrogenes with different defects (stop codons, frameshifts, etc) and pseudogenes of Nanog. One of the functional pseudo genes is Nanogp8, which is localized on chromosome 15, and codes for a Nanog 8 protein that is overexpressed in cancer cells and plays a significant role in tumorigenicity [15].
Nanog is required for the maintenance of pluripotency in embryonic stem cells and for germ cell development [16]. Nanog deficiency causes embryonic lethality subsequent to the formation of the inner cell mass E3.5 [16]. For induced pluripotency from human adult dermal fibroblasts only four transcription factors were required Oct3/4;Sox-2;Klf-4 and c-Myc [17]. Nanog was required for establishment of these pre-induced pluripotent cells and thus is required in the final stages of cell reprogramming [16].
NANOG AND P53 IN CANCER STEM CELLS
p53 binds to the Nanog promoter and suppresses Nanog expression after DNA damage [18]. The rapid down-regulation of Nanog correlates with induction of Ser315 p53 phosphorylation, and induction of p53 transcriptional activity [18]. The role of Ser 315 includes the binding of the co repressor mSin3a to the Nanog promoter [18]. The p53-dependent repression of Nanog expression represents one of the mechanisms of maintaining genetic stability in embryonic stem cells by inducing differentiation (Fig. 3).
Fig. 3. The cross-linked signaling between FAK, p53, Mdm-2, and Nanog in cancer stem cells.
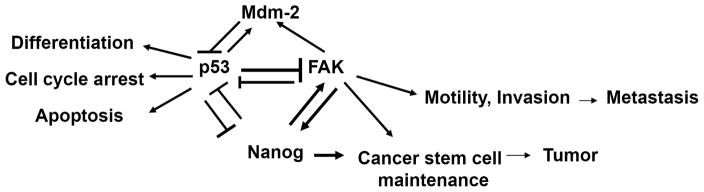
P53 binds FAK promoter and inhibits its promoter activity. FAK sequesters p53 from apoptotic signaling and blocks p53-transcriptional activity. FAK binds Mdm-2 to facilitate p53 ubiquitination and degradation. P53 up-regulates Mdm-2, and represses Nanog. Furthermore, Nanog represses p53 to maintain cancer stem cell pool and block differentiation, cell cycle arrest, apoptosis and other cell growth inhibiting p53 functions to promote tumor growth. Nanog up-regulates FAK, and FAK is able to phosphorylate Nanog and this cross-linked signaling mediates cell motility and invasion of cancer stem cells and play s significant role in tumor metastasis.
p53 has been shown to have a critical role in the reprogramming of pluripotent cells and the self-renewing of stem cells [19]. Disruption and inactivation of p53 pathway induced production of pluripotent stem cells [20]. Kawamura et al showed that decreasing of p53 levels in mouse fibroblasts increased production of the pluripotent (iPS) stem cells by using only two factors, Oct-4 and Sox2 [21]. One of the main players of p53-directed reprogramming was a p53 target, p21 [21]. The p53-deficient cells were genetically unstable, carried numerous DNA damage, short telomeres, and chromosome aberrations [22]. The chimeric mice obtained from p53-deficient iPS cells generated tumors [20]. Thus, p53-p21 and its cross-linked pathways control generation of iPS cells and tumorigenicity.
It was shown that Nanog regulated dedifferentiation of primary mice p53−/− astrocytes into cancer stem-like cells [23]. Another group showed that loss of p53 activated the Hedgehog-Gli pathway that up-regulated Nanog through p53-independent signaling by binding of Gli transcription factors to the Nanog promoter [24]. Nanog was shown to regulate glioma stem cell growth and tumorigenicity [24]. The Hedgehog and p53 pathways are cross-linked and can cross-regulate Nanog expression because p53 directly suppresses Nanog and p53 also suppresses Hedgehog [24]. The authors also propose a model that p53 represses Nanog directly and indirectly through the Hedgehog pathway.
FAK
It is known that Focal Adhesion Kinase plays a significant role in tumor survival [25]. FAK is a 125 kDa tyrosine kinase that contains N-terminal (FERM)-containing, Kinase and C-terminal domains [26]. The Y397-FAK site is the main autophosphorylation site that is phosphorylated once cells attached to the extracellular matrix through the integrin receptors, then Src binds this site and other proteins, such as PI-3-Kinase, Shc, Nck-2, Grb-7 bind, and this turns on activation of ATP-binding K454 site and phosphorylation of Y576/Y577 FAK and other tyrosine sites and causes downstream cytoskeletal and cell morphology changes [26]. FAK was shown to be important for cell adhesion, proliferation, motility, invasion and angiogenesis [27]. Many tumors overexpress FAK mRNA and protein [28]. FAK was used as a target for anti-cancer therapy with several inhibitors developed the targeting ATP-binding site; the Y397 site [29–31], and other FAK functions and activities [32].
FAK AND P53 INTERACTION
The regulatory region or FAK promoter was cloned, and p53 transcription factor was shown to bind FAK promoter and to repress its activity, while the NF-kappa B transcription factor activated its activity [33]. Overexpression of the adenoviral wild type p53 blocked FAK mRNA and protein expression [34]. The overexpression of wild type p53 repressed FAK promoter activity, while overexpression of mutant p53 did not [34]. In addition, mutations of p53 were highly correlated with high FAK expression in a population-based series of breast cancer tumors [34, 35]. In addition, FAK was shown to bind p53 protein and inhibit its transcriptional activity with p53-transcriptional targets: p21, Bax, and Mdm-2 [36]. Another group showed that FAK binds p53 and also that FAK binds Mdm-2 to increase ubiquitination and degradation of p53 [37]. We proposed a model where p53 represses FAK promoter activity and where FAK sequesters p53 from apoptotic signaling to provide survival signaling [38]. Thus, FAK and p53 are involved in regulation of survival/apoptotic signaling in the cancer cells through p53-regulated FAK promoter activity and through FAK-p53 protein binding. Targeting FAK-p53-Mdm-2 proteins is important for developing targeted cancer therapies (Fig. 3).
FAK AND STEM CELLS AND FAK AND NANOG INTERACTION
Targeted deletion of FAK in the mouse epithelial cells significantly decreased mammary tumorigenesis [39]. The ablation of FAK decreased the cancer stem cell pool, demonstrating that FAK maintains the population of the mammary stem cells to promote breast tumorigenesis [39]. In addition, the FAK homologue, Pyk-2, can compensate for the deletion of FAK in the mammary tumor cells, which is important for understanding breast tumorigenesis and developing FAK and Pyk-2 targeted therapies [40].
FAK signaling was important in mesenchymal stem (MSC) motility [41]. Bone marrow or cord blood-derived mesenchymal stem (MSC) cells are able to migrate toward tumors in vitro and in vivo [41]. The stromal cell-derived factor SDF-1 is up-regulated in mesenchymal stem cells and is important for stem cell migration. Stimulation of MSC cells with SDF-1 activated FAK, which was associated with cytoskeletal and actin changes [41]. Thus, the study shows that the FAK pathway is critical for MSC cell motility toward the tumor microenvironment and suggests that targeting tumor and stromal cells can be an effective therapy approach [41].
We demonstrated recently that transcription factor Nanog, which plays a critical role in cancer stem cell functions binds FAK promoter at four binding sites and up-regulates FAK mRNA and protein expression in different cancer cell lines [12]. We showed that colon and breast tumors overexpressed both Nanog and FAK proteins, while normal matched tissues did not. In addition, we showed the interaction of FAK and Nanog proteins and demonstrated that FAK was able to phosphorylate Nanog [12]. Nanog was also phosphorylated by Src, Pyk-2 and PKC. In addition, wild type Nanog increased cancer cell invasion, while Nanog with mutated tyrosines Y35F and Y174F decreased cell invasion [12]. Our data are consistent with the recent data that showed that Oct-3/4 overexpressing glioblastoma cells expressed increased invasion, which was associated with upregulation of FAK and Src expression [42]. Another study showed recently that EphB2 receptor controlled proliferation and migration/invasion of glioblastoma-derived stem-liked neurospheres by interacting with FAK [43]. Thus, the data show an important role of FAK in stem cell proliferation and invasion. FAK function has been shown to be critical for squamous cell carcinomas in TGF-β receptor II-deleted mice [44]. FAK signaling was up-regulated in these mice, which promoted cell survival, proliferation, tumor growth, and carcinogenesis [44]. The data suggested that TGF β receptor II/TGF β and integrin/FAK signaling interacted to control tumor initiation and growth and also the frequency of benign tumor progression to malignant squamous cell carcinomas [44]. The data are consistent with the role of FAK signaling in metastasis, where FAK-integrin beta 1 signaling controlled the initial proliferation of micrometastaic mouse mammary carcinoma cells into the lung parenchyma [45]. The activated FAK-integrin signaling played a significant role in regulating proliferation of metastatic cancer cells under a three-dimensional condition of culture, and inhibition of FAK signaling decreased the extravagation of the cancer cells into lung parenchyma [45]. Thus, FAK plays a significant role in metastasis that includes interactions of tumor cells and tumor microenvironment and host components, such as extracellular matrix, immune cells, blood vessels and organ-specific cells [46]. Thus, the data suggest that therapies that target both tumor and tumor microenvironment should be developed to decrease tumor growth.
FAK, P53 AND NANOG IN CANCER STEM CELLS
In summary, we propose a model of cross-regulation of FAK, p53 and Nanog in cancer stem cells (Fig. 3). FAK can bind Mdm-2 and activate p53 degradation [37]. P53 negatively regulates Nanog and negatively regulates FAK. Thus, targeting complexes of FAK-Mdm-2-p53 with small molecules can up-regulate p53 activity. In addition, simultaneous addition of the drugs targeting these interactions can decrease tumor growth through inhibition of cancer stem cell survival. Up-regulation of p53 will inhibit Nanog and FAK survival signaling pathways to increase cancer stem cell maintenance and cause cell growth inhibition and decrease tumorigenesis. Thus, understanding this cross-linked signaling will provide future anti-cancer therapy approaches.
CONCLUSIONS AND FUTURE PERSPECTIVES
In conclusion, there are many questions that need to be answered, such as understanding down-stream signaling of Nanog-FAK regulation; the other proteins that co-operate with Nanog in FAK up-regulation; the cross-linked signaling of p53-Nanog-FAK regulation; and the role of phosphorylation of Nanog by FAK in tumorigenesis. It is known that p53 can inhibit FAK promoter activity and that p53 can inhibit Nanog expression. At the same time, we showed that Nanog can bind FAK promoter and can up-regulate FAK expression. The question remains to answer: can p53 inhibit Nanog to inhibit FAK expression and thus through this additional indirect mechanism can it regulate stem cell maintenance and survival signaling. There are many questions on posttranslational modifications of Nanog, such as Nanog phosphorylation by different intracellular kinases. There is increased evidence that Nanog is localized not only in the nucleus, but also in the cytoplasm, and Nanog protein has nuclear localization (NLS) and nuclear export (NES) signals. The future studies are required to define the role of Nanog 1 and Nanog 8 in cancer stem biology and tumorigenesis. The nuclear-cytoplasmic shuttling of Nanog, FAK and p53 remains to de studied in detail. It will be interesting to explore the cross-linked signaling of FAK with other cancer stem cell markers, transcription factors and signaling players. The future therapies that target p53, FAK and Nanog pathways can all be developed to regulate cancer stem cell population and decrease tumorigenesis.
Footnotes
CONFLICT OF INTEREST
The author(s) confirm that this article content has no conflict of interest.
References
- 1.Akhtar K, Bussen W, Scott SP. Cancer stem cells - from initiation to elimination, how far have we reached? (Review) Int J Oncol. 2009;34:1491–1503. doi: 10.3892/ijo_00000278. [DOI] [PubMed] [Google Scholar]
- 2.Bhaijee F, Pepper DJ, Pitman KT, Bell D. Cancer stem cells in head and neck squamous cell carcinoma: A review of current knowledge and future applications. Head Neck. 2011 doi: 10.1002/hed.21801. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 3.Scheel C, Weinberg RA. Phenotypic plasticity and epithelial-mesenchymal transitions in cancer and normal stem cells? Int J Cancer. 2011;129:2310–2314. doi: 10.1002/ijc.26311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–648. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 5.Ignatova TN, Kukekov VG, Laywell ED, Suslov ON, Vrionis FD, Steindler DA. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia. 2002;39:193–206. doi: 10.1002/glia.10094. [DOI] [PubMed] [Google Scholar]
- 6.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 7.Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, Bruns CJ, Heeschen C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1:313–323. doi: 10.1016/j.stem.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 8.Tirino V, Camerlingo R, Franco R, Malanga D, La Rocca A, Viglietto G, Rocco G, Pirozzi G. The role of CD133 in the identification and characterisation of tumour-initiating cells in non-small-cell lung cancer. Eur J Cardiothora c Surg. 2009;36:446–453. doi: 10.1016/j.ejcts.2009.03.063. [DOI] [PubMed] [Google Scholar]
- 9.Patrawala L, Calhoun-Davis T, Schneider-Broussard R, Tang DG. Hierarchical organization of prostate cancer cells in xenograft tumors: the CD44+alpha2beta1+ cell population is enriched in tumor-initiating cells. Cancer Res. 2007;67:6796–6805. doi: 10.1158/0008-5472.CAN-07-0490. [DOI] [PubMed] [Google Scholar]
- 10.Scheel C, Weinberg RA. Cancer stem cells and epithelial-mesenchymal transition: Concepts and molecular links. Semin Cancer Biol. 2012 doi: 10.1016/j.semcancer.2012.04.001. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Y, Laterra J. Cancer stem cells: distinct entities or dynamically regulated phenotypes? Cancer Res. 2012;72:576–580. doi: 10.1158/0008-5472.CAN-11-3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ho B, Olson G, Figel S, Gelman I, Cance W, Golubovskaya V. Nanog increases focal adhesion kinase (FAK) promoter activity and expression and directly binds to FAK protein to be phosphorylated. J Biol Chem. 2012 doi: 10.1074/jbc.M111.322883. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chambers I, Colby D, Robertson M, Nichols J, Lee S, Tweedie S, Smith A. Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell. 2003;113:643–655. doi: 10.1016/s0092-8674(03)00392-1. [DOI] [PubMed] [Google Scholar]
- 14.Mitsui K, Tokuzawa Y, Itoh H, Segawa K, Murakami M, Takahashi K, Maruyama M, Maeda M, Yamanaka S. The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell. 2003;113:631–642. doi: 10.1016/s0092-8674(03)00393-3. [DOI] [PubMed] [Google Scholar]
- 15.Jeter CR, Liu B, Liu X, Chen X, Liu C, Calhoun-Davis T, Repass J, Zaehres H, Shen JJ, Tang DG. NANOG promotes cancer stem cell characteristics and prostate cancer resistance to androgen deprivation. Oncogene. 2011;30:3833–3845. doi: 10.1038/onc.2011.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Theunissen TW, Silva JC. Switching on pluripotency: a perspective on the biological requirement of Nanog. Philos Trans R Soc Lond B Biol Sci. 2011;366:2222–2229. doi: 10.1098/rstb.2011.0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 18.Lin T, Chao C, Saito S, Mazur SJ, Murphy ME, Appella E, Xu Y. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat Cell Biol. 2005;7:165–171. doi: 10.1038/ncb1211. [DOI] [PubMed] [Google Scholar]
- 19.Krizhanovsky V, Lowe SW. Stem cells: The promises and perils of p53. Nature. 2009;460:1085–1086. doi: 10.1038/4601085a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hong H, Takahashi K, Ichisaka T, Aoi T, Kanagawa O, Nakagawa M, Okita K, Yamanaka S. Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature. 2009;460:1132–1135. doi: 10.1038/nature08235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kawamura T, Suzuki J, Wang YV, Menendez S, Morera LB, Raya A, Wahl GM, Izpisua Belmonte JC. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature. 2009;460:1140–1144. doi: 10.1038/nature08311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marion RM, Strati K, Li H, Murga M, Blanco R, Ortega S, Fernandez-Capetillo O, Serrano M, Blasco MA. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature. 2009;460:1149–1153. doi: 10.1038/nature08287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.nJH, Kwon S, Jun EK, Kim A, Whang KY, Kim H, Oh S, Yoon BS, You S. Nanog-induced dedifferentiation of p53-deficient mouse astrocytes into brain cancer stem-like cells. Biochem Biophys Res Commun. 2011;412:175–181. doi: 10.1016/j.bbrc.2011.07.070. [DOI] [PubMed] [Google Scholar]
- 24.Po A, Ferretti E, Miele E, De Smaele E, Paganelli A, Canettieri G, Coni S, Di Marcotullio L, Biffoni M, Massimi L, Di Rocco C, Screpanti I, Gulino A. Hedgehog controls neural stem cells through p53-independent regulation of Nanog. EMBO J. 2010;29:2646–2658. doi: 10.1038/emboj.2010.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Golubovskaya VM. Targeting focal adhesion kinase in cancer-part I. Anticancer Agents Med Chem. 2010;10:713. doi: 10.2174/187152010794728693. [DOI] [PubMed] [Google Scholar]
- 26.Hanks SK, Calalb MB, Harper MC, Patel SK. Focal adhesion protein-tyrosine kinase phosphorylated in response to cell attachment to fibronectin. Proc Natl Acad Sci USA. 1992;89:8487–8491. doi: 10.1073/pnas.89.18.8487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Golubovskaya VM, Cance WG. FAK and P53 Protein Interactions. Anticancer Agents Med Chem. 2011;11:617–619. doi: 10.2174/187152011796817619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lark AL, Livasy CA, Calvo B, Caskey L, Moore DT, Yang X, Cance WG. Overexpression of focal adhesion kinase in primary colorectal carcinomas and colorectal liver metastases: immunohistochemistry and real-time PCR analyses. Clin Cancer Res. 2003;9:215–222. [PubMed] [Google Scholar]
- 29.Hochwald SN, Nyberg C, Zheng M, Zheng D, Wood C, Massoll NA, Magis A, Ostrov D, Cance WG, Golubovskaya VM. A novel small molecule inhibitor of FAK decreases growth of human pancreatic cancer. Cell Cycle. 2009;8:2435–2443. doi: 10.4161/cc.8.15.9145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Golubovskaya VM, Nyberg C, Zheng M, Kweh F, Magis A, Ostrov D, Cance WG. A Small Molecule Inhibitor, 1,2,4,5-Benzenetetraamine Tetrahydrochloride, Targeting the Y397 Site of Focal Adhesion Kinase Decreases Tumor Growth. J Med Chem. 2008;51:7405–7416. doi: 10.1021/jm800483v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beierle EA, Ma X, Stewart J, Nyberg C, Trujillo A, Cance WG, Golubovskaya VM. Inhibition of focal adhesion kinase decreases tumor growth in human neuroblastoma. Cell Cycle. 2010;9:1005–1015. doi: 10.4161/cc.9.5.10936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Golubovskaya VM. Focal adhesion kinase as a cancer therapy target. Anticancer Agents Med Chem. 2010;10:735–741. doi: 10.2174/187152010794728648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Golubovskaya V, Kaur A, Cance W. Cloning and characterization of the promoter region of human focal adhesion kinase gene: nuclear factor kappa B and p53 binding sites. Biochim Biophys Acta. 2004;1678:111–125. doi: 10.1016/j.bbaexp.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 34.Golubovskaya VM, Finch R, Kweh F, Massoll NA, Campbell-Thompson M, Wallace MR, Cance WG. p53 regulates FAK expression in human tumor cells. Mol Carcinog. 2008;47:373–382. doi: 10.1002/mc.20395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Golubovskaya VM, Conway-Dorsey K, Edmiston SN, Tse CK, Lark AA, Livasy CA, Moore D, Millikan RC, Cance WG. FAK overexpression and p53 mutations are highly correlated in human breast cancer. Int J Cancer. 2009;125:1735–1738. doi: 10.1002/ijc.24486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Golubovskaya VM, Finch R, Cance WG. Direct Interaction of the N-terminal Domain of Focal Adhesion Kinase with the N-terminal Transactivation Domain of p53. J Biol Chem. 2005;280:25008–25021. doi: 10.1074/jbc.M414172200. [DOI] [PubMed] [Google Scholar]
- 37.Lim ST, Chen XL, Lim Y, Hanson DA, Vo TT, Howerton K, Larocque N, Fisher SJ, Schlaepfer DD, Ilic D. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol Cell. 2008;29:9–22. doi: 10.1016/j.molcel.2007.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cance WG, Golubovskaya VM. Focal adhesion kinase versus p53: apoptosis or survival? Sci Signal. 2008;1:pe22. doi: 10.1126/stke.120pe22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luo M, Fan H, Nagy T, Wei H, Wang C, Liu S, Wicha MS, Guan JL. Mammary epithelial-specific ablation of the focal adhesion kinase suppresses mammary tumorigenesis by affecting mammary cancer stem/progenitor cells. Cancer Res. 2009;69:466–474. doi: 10.1158/0008-5472.CAN-08-3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fan H, Guan JL. Compensatory function of Pyk2 protein in the promotion of focal adhesion kinase (FAK)-null mammary cancer stem cell tumorigenicity and metastatic activity. J Biol Chem. 2011;286:18573–18582. doi: 10.1074/jbc.M110.200717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gao H, Priebe W, Glod J, Banerjee D. Activation of signal transducers and activators of transcription 3 and focal adhesion kinase by stromal cell-derived factor 1 is required for migration of human mesenchymal stem cells in response to tumor cell-conditioned medium. Stem Cells. 2009;27:857–865. doi: 10.1002/stem.23. [DOI] [PubMed] [Google Scholar]
- 42.Kobayashi K, Takahashi H, Inoue A, Harada H, Toshimori S, Kobayashi Y, Goto K, Sugimoto K, Yano H, Ohnishi T, Tanaka J. Oct-3/4 promotes migration and invasion of glioblastoma cells. J Cell Biochem. 2012;113:508–517. doi: 10.1002/jcb.23374. [DOI] [PubMed] [Google Scholar]
- 43.Wang SD, Rath P, Lal B, Richard JP, Li Y, Goodwin CR, Laterra J, Xia S. EphB2 receptor controls proliferation/migration dichotomy of glioblastoma by interacting with focal adhesion kinase. Oncogene. 2012 doi: 10.1038/onc.2012.16. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schober M, Fuchs E. Tumor-initiating stem cells of squamous cell carcinomas and their control by TGF-beta and integrin/focal adhesion kinase (FAK) signaling. Proc Natl Acad Sci U S A. 2011;108:10544–10549. doi: 10.1073/pnas.1107807108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shibue T, Weinberg RA. Integrin beta1-focal adhesion kinase signaling directs the proliferation of metastatic cancer cells disseminated in the lungs. Proc Natl Acad Sci U S A. 2009;106:10290–10295. doi: 10.1073/pnas.0904227106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shibue T, Weinberg RA. Metastatic colonization: settlement, adaptation and propagation of tumor cells in a foreign tissue environment. Semin Cancer Biol. 2011;21:99–106. doi: 10.1016/j.semcancer.2010.12.003. [DOI] [PubMed] [Google Scholar]