

Abstract
In this study, we demonstrated that knockdown of Programmed cell death 4 (Pdcd4), a novel tumor suppressor, decreased the expressions of epithelial-specific proteins and increased the expressions of mesenchymal-specific proteins in vitro and in vivo, suggesting that knockdown of Pdcd4 results in epithelial to mesenchymal transition (EMT). Knockdown of Pdcd4 increased the rate of wound closure and migration capacity in wound-healing assays and Boyden chamber migration assays, respectively, indicating that Pdcd4 knockdown promotes cell migration. Pdcd4 knockdown also altered the adhesion capacity of GEO cells to extracellular matrix including laminin, collagen IV, and fibronectin. To test whether knockdown of Pdcd4 promotes metastasis in vivo, parental, control, and Pdcd4 knockdown cells were injected into the cecal wall (orthotopic implantation) of nude mice. Tumors are formed on cecum in all injected mice. However, only mice injected with Pdcd4 knockdown cells developed hepatic and local lymph node metastases. Immunohistochemical staining analyses showed that c-Myc and Snail/Slug expressions were up-regulated in the tumors from injection with Pdcd4 knockdown cells. These results implicated that promotion of metastasis by Pdcd4 knockdown was contributed by up-regulation of c-Myc and Snail/Slug in nude mice. Taken together, our data demonstrated that knockdown of Pdcd4 leads to EMT, alternation of adhesion, and promotion of migration and metastasis.
Keywords: Pdcd4, EMT, migration, metastasis, orthotopic implantation, colon cancer
1. Introduction
Metastasis, the spread of tumor cells from a primary tumor site to a secondary site is the major cause of death in cancer patients. Detachment of tumor cells from primary tumor and invasion into surrounding normal tissues are important steps for tumor metastasis. During these steps, tumor cells require epithelial to mesenchymal transition (EMT) to lose cell-cell adhesion and gain motility.1 EMT was first recognized as a conserved process during embryonic development. During EMT, cells acquire a fibroblastoid phenotype, lose epithelial cell polarity, down-regulate epithelial-specific proteins and induce various mesenchymal-specific proteins, and increase migration through extracellular matrix.2 Loss of E-cadherin expression, an epithelial-specific protein, is a major indication of EMT, which is usually concomitant with the increase of mesenchymal N-cadherin expression, a mesenchymal-specific protein. This cadherin switch leads to loss of the affinity with epithelial neighbors and gain of the affinity for mesenchymal cells, resulting in increased migration and invasion.3 E-cadherin can be inactivated or silenced by various mechanisms including mutations and gene down-regulation through promoter hypermethylation and histone deacetylation. In addition, the zinc-finger transcription factors, Snail and Slug, have been reported to repress E-cadherin expression and increase tumor invasion in various malignancies.4, 5 Snail and Slug inhibit E-cadherin expression by binding to the proximal E-box of the E-cadherin promoter.4 Over-expression of Snail or Slug in epithelial cells has been shown to induce EMT and enhance invasion capacity.5, 6
Programmed cell death 4 (Pdcd4), a novel tumor suppressor, is frequently down-regulated in several cancerous tissues compared to adjacent normal tissues including colon cancer tissue.7 Pdcd4 was first identified and its cDNA was cloned when cells were treated with apoptosis inducers.8 Over-expression of Pdcd4 has been shown to inhibit cell proliferation in the neuroendocrine Bon-1 cells,9 whereas knockdown of Pdcd4 promoted cell proliferation.10 Ectopic expression of pdcd4 cDNA in mouse JB6 cells inhibits 12-O-tetradecanoylphorbol-13-acetate (TPA)-induced transformation and tumor phenotype.11, 12 Conversely, down-regulation of Pdcd4 expression by pdcd4 antisense resulted in an increase in TPA-induced transformation.13 In consistence with these observations, transgenic mice that over-expressing pdcd4 cDNA in the skin showed significant reduction in 7,12-dimethylbenz(a)anthracene (DMBA)/TPA-induced skin papilloma formation and carcinoma incidence.14 Knockout of Pdcd4 in mice led to an increase in DMBA/TPA-induced papilloma.15 These findings suggest that Pdcd4 is able to inhibit the early stage of carcinogenesis.
In addition to inhibit tumor promotion, Pdcd4 has also been demonstrated to suppress tumor invasion. Ectopic expression of pdcd4 cDNA suppressed invasion and intravasation in colon tumor RKO cell16, 17 and prostaglandin E2-induced invasion in breast tumor MCF7 cells,18 and ovarian tumor OVCA433 and SKOV3 cells.19 Knockdown of Pdcd4 expression promoted invasion in colon HT29 and GEO cells20, 21 as well as breast cancer MCF7 and D47T cells.22 Pdcd4 knockdown in colon tumor HT29 and GEO cells led to a fibroblast-like morphological change and promoted invasion.20 Concurrently, Pdcd4 knockdown resulted in down-regulation of E-cadherin expression, translocation of β-catenin into nucleus, and activation of β-catenin dependent transcription.20 Down-regulation of E-cadherin by Pdcd4 knockdown in colon HT29 cells was contributed, at least in part, by elevation of Snail expression since knockdown of Snail expression in the Pdcd4 knockdown cells reversed the E-cadherin expression.21 The expression of c-Myc, the downstream targets of β-catenin dependent transcription, was found to be up-regulated by Pdcd4 knockdown and knockdown of c-Myc inhibited invasion.21 Recently, we found that c-Myc stimulated MAP4K1 expression leading to activation of AP-1 dependent transcription in the Pdcd4 knockdown cells.23 Since AP-1 dependent transcription regulates several events required for cell invasion,24 these findings suggest that c-Myc contributes to invasion induced by Pdcd4 knockdown. Although knockdown of Pdcd4 has been demonstrated to down-regulate E-cadherin expression and promote invasion in cell culture systems, it is unclear whether Pdcd4 knockdown causes EMT and promotes metastasis in vivo.
In this study, we demonstrated that Pdcd4 knockdown led to EMT, enhancement of cell migration, alternation of cell-matrix adhesion, and promotion of metastasis in nude mice. We also demonstrated that c-Myc and Snail/Slug expression were up-regulated in the primary tumors derived from injection of Pdcd4 knockdown cells, revealing a mechanism that knockdown of Pdcd4 promotes metastasis in vivo.
2. Materials and Methods
2.1. Cell lines and culture conditions
GEO cells were kindly provided by Dr. Douglas Boyd (MD Anderson Cancer Center) and HT29 cells were purchased from American Type Culture Collection (ATCC, Manassas, VA). GEO-shLacZ, GEO-shPdcd4, HT29-shLacZ, and HT29-shPdcd4 cells were generated as described previously.20 All cells were grown in McCoy’s medium containing 10% FBS, 2 mM L-glutamine, and 100U/ml penicillin-streptomycin and incubated at 37°C with 5% CO2 in a humidified incubator.
2.2. Western blot analysis
Aliquots containing 20 or 40 μg of protein were separated on a SDS-PAGE, and transferred to nitrocellulose membranes as described previously.11 Subsequently, the membrane was incubated with antibodies against α-catenin (1:200 dilution, Santa Cruz Biotechnology, Santa Cruz), γ-catenin (1:200 dilution, Santa Cruz Biotechnology), N-cadherin (1:500 dilution, BD Biosciences, San Jose, CA), Fibronectin (1:500 dilution, BD Biosciences), or GAPDH (1:2000 dilution, Santa Cruz Biotechnology) followed by horseradish peroxidase-linked secondary antibody. The target protein was visualized by chemiluminescence (Denville Scientific, Metuchen, NJ).
2.3. Migration and wound-healing assays
For Boyden chamber migration assay, cells were incubated in McCoy medium containing 0.1% FBS for 24 h prior to migration assays. Cells (1 × 105 cells) were suspended in McCoy medium containing 0.1% BSA (McCoy-BSA) and seeded onto Transwell (8-μm pore size, Corning, NY). EGF was diluted into McCoy-BSA at 20 ng/ml and added to the lower well. Chambers were incubated at 37°C for 24 h. The un-migrated cells on the upper surface of the filter were removed with cotton swabs. The filter then was fixed and stained with 0.1% (w/v) crystal violet in ethanol. The number of cells that traversed the filter into the lower compartment was determined by counting 7 random areas under Olympus CKX 41 microscope. For wound-healing assays, 5 × 104 cells were plated in each well in a 24-well plate. Once the cells reached 100% confluence, a “wound” was created using a sterile 200 μl pipette tip. The wounded monolayer was then washed 3 times with complete medium to remove cell debris. Photographs of the wounded area were taken at the time 0 and 48 h. The width of the cell-free gap was measured by Olympus CKX41 microscope using Qcapture Pro software (QImaging, Surrey, BC, Canada). The percent of wound closure was determined by using the following formula: [Δ length/length (0 hour)] × 100%.
2.4. Cell adhesion assays
The adhesion assays were performed as follows. GEO-shLacZ or GEO-shPdcd4 cells were prepared in serum-free McCoy’s medium, and 1 × 104 cells per well were seeded on 24-well plates coated with either Matrigel, collagen I, collagen IV, laminin, or fibronectin (1 μg/well). Cells seeded on fibronectin were incubated at 37°C for 30 min while other cells were incubated for 15 min. After incubation, plates were washed twice with phosphate-buffered saline (PBS) to remove un-attached cells and stained with 0.1% (w/v) crystal violet in ethanol at room temperature for 20 min. After wash with distilled water three times, the stained cells were dissolved with 0.5% (v/v) Triton X-100. The optical density was measured at 595 nm and the cell numbers were estimated by using cell-number standard curve.
2.5. Orthotopic implantation of tumor cells
Five to six weeks old female Athymic nude (Hsd:Athymic Nude-Foxn1nu) and SCID (ICRTaac:TCR-Prkdc<scid>) mice weighting 18 to 22 g were purchased from Harlan (Indianapolis, IN) and Toconic (Hudson, NY), respectively. Mice were maintained in the University of Kentucky facility under specific pathogen-free condition. Animal care procedures and experimental protocols were approved by the Institutional Animal Care and Use Committee based on guidelines from the NIH. All mice were fed with commercial diet, given water ad libitum, and subjected to a 12 h-light/12 h-dark cycle. The mice were anesthetized intraperitoneally with ketamine:xylazine cocktail (ketamine, 90mg/kg; xylazine, 10mg/kg). A small abdominal incision was made, and the cecum was exteriorized. Cells (2 × 106 cells) in 100 μl of PBS were injected into the cecal wall from the serosal side using a 30-gauge needle on a 1-ml syringe. A visible bulla between the submucosal and subserosal tissues was formed. The cecum was returned to the abdominal cavity, and the peritoneum and skin were then closed with 4-0 Vicryl suture and metal clips, respectively. At 8-week post-injection, the lung, liver, lymph node, and cecum samples were collected.
2.6. Immunohistochemical (IHC) analyses
The tissue samples were fixed with 4% neutral buffered paraformaldehyde, embedded in paraffin, and sectioned into 4-μm slices. Tissue sections were de-waxed by heating at 60°C, washed in xylene, and re-hydrated through a graded series of ethanol and water. After antigen retrieval, inactivation of endogenous peroxidase with 3% H2O2, and block of tissues with normal goat serum, the tissues were incubated with antibodies against Ki-67 (predilute, LifeTechnologies, Grand Island, NY), α-catenin (1:250 dilution, Abcam, Cambridge, MA), β-catenin (1:100 dilution, Cell Signaling), N-cadherin (1:300 dilution, Abcam), E-cadherin (1:400 dilution, Cell Signal Technology, Danvers, MA), c-Myc (1:100 dilution, Abcam), Snail/Slug (1:150 dilution, Abcam), and Pdcd4 (1:1500 dilution)20 followed by biotin-linked secondary antibody using Histostain-Plus kit (LifeTechnologies). The tissue sections were visualized using 3,3′-diaminobenzidine (DAB) and counterstained with haematoxylin.
2.7. Statistical analysis
Differences in the migration, cell adhesion, and proliferation index between control (GEO-shLacZ) and Pdcd4 knockdown (GEO-shPdcd4) cells were analyzed using one-way ANOVA (http://faculty.vassar.edu/lowry/anova1u.html). Differences in the presence of tumor nodules in cecum, lymph node, liver, and lung between groups were compared using Fisher’s exact test (http://faculty.vassar.edu/lowry/tab2x2.html). Differences were considered statistically significant at the P < 0.05 level. The statistical trend of liver metastasis with injecting HT29, HT29-shLacZ, and HT29-shPdcd4 cells was defined as P ≤ 0.1.
3. Results
3.1. Knockdown of Pdcd4 leads to EMT
We demonstrated that knockdown of Pdcd4 in colon HT29 and GEO cells resulted in a fibroblast-like transition and down-regulation of E-cadherin.20 This finding suggests that knockdown of Pdcd4 may led to EMT. The expression switch from epithelial to mesenchymal marker genes is a hallmark of EMT. To analyze these changes induced by Pdcd4 knockdown, the expression of epithelial and mesenchymal marker proteins in control (GEO-shLacZ and HT29-shLacZ) and Pdcd4 knockdown (GEO-shPdcd4 and HT29-shPdcd4) cells were examined. The control and Pdcd4 knockdown cells were generated by stable expression of lacZ shRNA and pdcd4 shRNA, respectively.20 Knockdown of Pdcd4 decreased the expression of epithelial marker proteins (α-catenin and γ-catenin) and concomitantly increased the protein level of mesenchymal marker protein (N-cadherin) in both GEO and HT29 cells (Figure 1A). Interestingly, knockdown of Pdcd4 increased fibronectin expression in GEO cells but it was undetectable in the HT29 cells. To analyze whether the Pdcd4 knockdown cells maintained the mesenchymal marker expression in vivo, the GEO-shLacZ and GEO-shPdcd4 cells were injected into the cecum of nude mice. After 8 weeks, the tumors on cecum were collected, fixed with paraformaldehyde, sectioned, and stained with antibodies recognizing N-cadherin, β-catenin, E-cadherin, or α-catenin. The GEO-shLacZ derived tumors showed weak to moderate N-cadherin and β-catenin staining (mesenchymal markers, Figure 1B and D) but moderate to strong E-cadherin and α-catenin expressions (epithelial markers, Figure 1F and H). In contrast, GEO-shPdcd4 derived tumors showed strong N-cadherin and β-catenin expressions (Figure 1C and E), while E-cadherin and α-catenin staining was not detectable (Figure 1G and I). The β-catenin expression in GEO-shPdcd4 derived tumors was high in the nucleus (Figure 1E). In contrast, β-catenin was weakly expressed in the cytoplasm and nucleus in GEO-shLacZ derived tumors (Figure 1D). These findings suggested that the β-catenin dependent transcription was activated in the GEO-shPdcd4 derived tumors. Interestingly, knockdown of Pdcd4 in cultured GEO cells showed a decrease in β-catenin expression21 whereas the β-catenin expression was increased in the tumors derived from Pdcd4 knockdown GEO cells (GEO-shPdcd4). These Western blotting and IHC staining results indicate that knockdown of Pdcd4 leads to the decrease in expression of epithelial proteins but increase in expression of mesenchymal proteins.
Fig. 1.


Knockdown of Pdcd4 down-regulates epithelial proteins and up-regulates mesenchymal proteins. (A) Expression of epithelial proteins: α-catenin and γ-catenin, mesenchymal proteins: N-cadherin and fibronectin, and GAPDH were examined in control (GEO-shLacZ and HT29-shLacZ) and Pdcd4 knockdown (GEO-shPdcd4 and HT29-shPdcd4) cells using Western blotting analysis. (B–I) GEO-shLacZ and GEO-shPdcd4 cells were orthotopically injected into mice as described in Materials and Methods. After 8 weeks, the tumor nodules in the cecum were collected, fixed, and embedded. Paraffin sections from GEO-shLacZ (B, D, F, H) and GEO-shPdcd4 (C, E, G, I) derived tumors were stained using IHC with antibodies against N-cadherin (B, C), β-catenin (D, E), E-cadherin (F, G), and α-catenin (H, I). Scale bar: 50 μm.
3.2. Knockdown of Pdcd4 promotes cell migration
We have demonstrated that knockdown of Pdcd4 promoted cell invasion.20 Although promotion of cell invasion usually increases migration ability, some studies did show that increase in cell invasion has a little effect on cell motility.25 Thus, it is necessary to find out whether knockdown of Pdcd4 promotes cell migration. The cell motility of Pdcd4 knockdown (GEO-shPdcd4) cells and control (GEO-shLacZ) cells were measured with wound-healing assays and Boyden chamber migration assays. The rate of wound closure was used to measure cell mobility in wound healing assays. In a confluent monolayer cells, the ability of cells (adjacent to the scratch) to heal the “wound” was evaluated under a microscope. GEO-shPdcd4 cells completed wound closure 100% within 48 h, but the control cells (GEO-shLacZ) only showed approximately 25% wound closure, indicating that the rate of wound closure was approximately 4-fold faster in GEO-shPdcd4 cells than that in GEO-shLacZ cells (Figure 2A and B). In addition to wound healing assays, we also performed Boyden chamber migration assays to measure cell mobility. Control or Pdcd4 knockdown cells were serum starved for 24 h and placed in the upper well of the transwell. The attractant (EGF, 20 ng/ml) was placed in the lower well. After 24 h, the unmigrated cells were removed and cells that had migrated to the lower face of the membrane were stained with 0.1% (w/v) crystal violet. As shown in Figure 2C, the migration ability of GEO-shPdcd4 cells was approximately 9-fold higher than that of GEO-shLacZ cells. These results suggested that knockdown of Pdcd4 promoted cell migration in GEO cells.
Fig. 2.

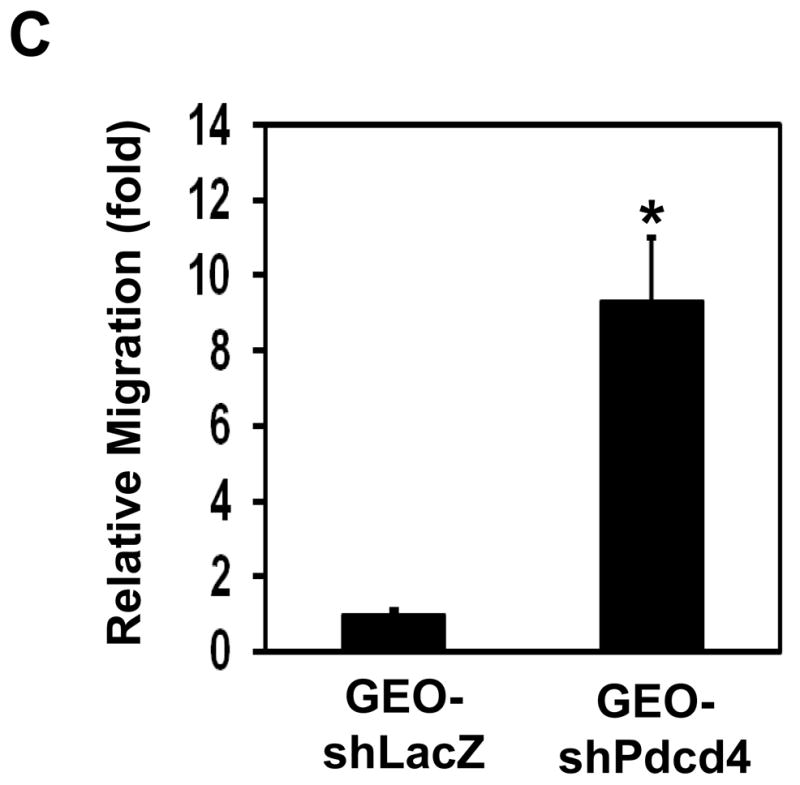
Knockdown of Pdcd4 promotes cell migration. (A) Wound-healing assays. GEO-shLacZ and GEO-shPdcd4 cells were seeded onto a 24-well plate. At 100% confluence, cell monolayer was scratched with a plastic tip. At 0 h and 48 h after the scratch, the distance of wounded monolayer was measured under a microscope equipped with a digital camera. The representative images are shown. (B) The percentage of wound closure. Each value is expressed as the mean ± standard deviation (SD) of three independent experiments. The asterisk indicates significant difference compared with GEO-shLacZ cells as determined by one-way ANOVA (P<0.01). (C) Boyden chamber migration assays. The number of cells that has migrated to the lower side of membrane surface was determined by counting 7 areas. The migration capacity of GEO-shLacZ cells is designed as 1. Two independent experiments were performed with triplicates for each sample. The representative data are shown and expressed as the mean ± SD. The asterisk indicates a significant difference compared with GEO-shLacZ cells as determined by one-way ANOVA (P<0.05).
3.3. Cell adhesion to extracellular matrices is altered by Pdcd4 knockdown
Cell-matrix adhesions are essential for cell migration, tissue organization, and differentiation. For tumor invasion, tumor cells must be able to form new cell-matrix and cell-cell attachments, and break the existing ones. Since knockdown of Pdcd4 enhances migration (Figure 2) and Matrigel invasion,20 it is of interest to examine whether knockdown of Pdcd4 alters cell-matrix interaction. GEO-shLacZ and GEO-shPdcd4 cells were seeded on 24-well plates coated with various EMC, Matrigel, collagen I, collagen IV, laminin, or fibronectin. After incubation, each well was washed with PBS to remove the unattached cells. The number of cells attached to the well was determined. As shown in Figure 3, attachment to collagen IV or laminin-coated plates was significantly decreased in GEO-shPdcd4 cells compared with GEO-shLacZ cells. In contrast, the adhesive capacity to fibronectin is about 2-fold higher in GEO-shPdcd4 cells than that in GEO-shLacZ cells, revealing a functional link between fibronectin-mediated adhesion and Pdcd4 knockdown-induced EMT. These results demonstrate that knockdown of Pdcd4 alters cell-matrix adhesion.
Fig. 3.

Pdcd4 knockdown alters the adhesive capacity to ECM components. The cells attached to the ECM component were determined as described in Materials and Methods. The number of adhered GEO-shLacZ cells is designated as 100%. Two independent experiments were performed with 5 replicates for each sample. The representative data are shown and expressed as the mean ± SD. The asterisk indicates a significant difference compared with GEO-shLacZ cells as determined by one-way ANOVA (P<0.05).
3.4. Knockdown of Pdcd4 promotes metastasis and tumor growth in mice
Previously, we have demonstrated that knockdown of Pdcd4 promotes Matrigel invasion in vitro.20 To provide in vivo evidence that knockdown of pdcd4 promotes metastasis, cells (GEO, GEO-shLacZ, or GEO-shPdcd4 cells) were injected into cecal wall of nude mice using orthotopic implantation method. The advantage of orthotopic implantation is that the cells are injected into a similar organ environment in which the colon cancer stages, dissemination patterns, and aggressiveness closely replicates all relevant metastatic sites observed in humans.26 Eight weeks post-injection, samples of lung, liver, lymph node, and cecum were collected and the tumor nodules of each sample were determined. Tumors were formed in the cecum in the mice injected with GEO (six out of six), GEO-shLacZ (ten out of ten), or GEO-shPdcd4 (eight out of eight) cells (Table 1). Local lymph node metastasis (Table 1) and hepatic metastasis (Figure 4A and Table 1) occurred in all mice injected with GEO-shPdcd4 cells (eight out of eight). In contrast, no mice injected with GEO (parental) or GEO-shLacZ (control) cells showed hepatic or local lymph node metastasis (Table 1). In addition, none of the three injected cell types caused pulmonary metastasis (Table 1). We also injected HT29, HT29-shLacZ, and HT29-shPdcd4 cells into cecal wall of SCID mice. Four out of five mice injected with HT29-shPdcd4 showed liver metastasis. On the contrary, zero out of four mice injected with HT29 and one out of seven mice injected with HT29-shLacZ cells had liver metastasis (supplementary Table S1). These results indicate that Pdcd4 knockdown in colon cells promotes their metastatic capacity in mice.
Table 1.
Tumorigenicity and metastatic ability of GEO, GEO-shLacZ, and GEO-shPdcd4 cells in nude mice.
| Cell line | Tumorigenicity | Regional lymph node metastasis | Liver metastasis | Lung metastasis |
|---|---|---|---|---|
| GEO | 6/6 | 0/6 | 0/6 | 0/6 |
| GEO-shLacZ | 10/10 | 0/10 | 0/10 | 0/10 |
| GEO-shPdcd4 | 8/8 | 8/8a,b | 8/8a,b | 0/8 |
Values are number of positive mice/number of injected mice
P = 0.00033 as compared with GEO cells
P = 0.00002 as compared with GEO-shLacZ cells
Fig. 4.


Knockdown of Pdcd4 promotes metastasis and proliferation in nude mice. GEO-shLacZ and GEO-shPdcd4 cells were orthotopically injected into mice as described in Materials and Methods. After 8 weeks, the tumor nodules in the cecum and liver were collected, fixed, and embedded. (A) The representative tumor nodules in cecum and liver are shown. Arrows indicate the tumor nodules. (B) Paraffin sections from GEO-shLacZ and GEO-shPdcd4 derived tumors were stained with Ki-67 antibody. The representative images are shown. The brown color indicates the positive staining of Ki-67 in the nuclei. (C) The proliferation index. The proliferation index was calculated as number of Ki-67 positive staining cells per 500 cells counted. Two independent tumors were chosen with counting 3 areas each. The representative data are shown and expressed as the mean ± SD. The asterisk indicates significant difference compared with GEO-shLacZ cells as determined by one-way ANOVA (p<0.0005).
Interestingly, the size of GEO-shPdcd4 derived tumor in the cecal wall is much bigger than GEO-shLacZ derived tumor (control) (Figure 4A). This observation suggests that knockdown of Pdcd4 may increase cell proliferation in vivo. To confirm this, the tissues of both GEO-shLacZ and GEO-shPdcd4 derived tumors were stained with Ki-76 antibody, a proliferation marker. The GEO-shPdcd4 derived tumor tissues showed strong positive staining of Ki-67 in the nuclei while the GEO-shLacZ derived tumors showed weak nuclear staining (Figure 4B). The proliferation index of GEO-shPdcd4 derived tumors was 45% whereas the proliferation index of GEO-shLacZ derived tumors were 13% (Figure 4C). The proliferation index was calculated as number of cells with positive Ki-67 staining per 500 cells counted × 100%. These results suggest that knockdown of Pdcd4 promotes cell proliferation in vivo, which is in agreement with our previous findings that knockdown of Pdcd4 increases cell proliferation in the HT29 cells.10
3.5. Histological observations
Histological examination of primary and metastatic tumors demonstrated a moderately differentiated adenocarcinoma in the cecum when GEO-shLacZ cells were injected (Figure 5A). In contrast, the GEO-shPdcd4 cell derived tumors formed a poorly differentiated adenocarcinoma in the cecum (Figure 5B). Several large tumors from GEO-shPdcd4 cells injection were found to have central necrosis. As in the primary cecal tumors from the GEO-shPdcd4 injection, the metastatic tumors in the liver were poorly differentiated (Figure 5C).
Fig. 5.

Histological and IHC analyses of GEO-shLacZ and GEO-shPdcd4 derived tumors. (AC) Histological analyses of tumor samples. Paraffin sections from GEO-shLacZ derived colon tumor (A), GEO-shPdcd4 derived colon tumor (B), and GEO-shPdcd4 derived metastatic liver tumor (C) were stained by haematoxylin and eosin. The representative images are shown. T: tumor tissue, N: normal tissue, Nec: Necrosis. Scale bar: 100 μm. (D–I) IHC analyses of tumor samples. Paraffin sections from GEO-shLacZ (D, F, H) and GEO-shPdcd4 (E, G, I) derived tumor were stained by IHC with antibodies against Pdcd4 (D, E), c-Myc (F, G), and Snail/Slug (H, I). Representative images are shown. Scale bar: 50 μm.
3.6. Knockdown of Pdcd4 up-regulates c-Myc and Snail/Slug expression in mice
In the previous studies, we have demonstrated that knockdown of Pdcd4 up-regulated c-Myc and Snail expression in the cultured cells.21 c-Myc is a transcription factor that activates genes involved in cell growth and represses genes involved in anti-proliferation and anti-metastasis.27 Snail is a transcription repressor, which inhibits E-cadherin expression. Over-expression of Snail in epithelial cells promotes cell invasion.5 To investigate whether Pdcd4 knockdown up-regulates c-Myc and Snail expression in vivo, IHC staining of tissue slides from GEO-shLacZ and GEO-shPdcd4 derived tumors using antibodies against Pdcd4, c-Myc, or Snail/Slug (antibody recognizes both Snail and Slug) were performed. As expected, GEO-shLacZ derived tumors showed a strong, cytoplasmic and nuclear Pdcd4 staining (Figure 5D) while GEO-shPdcd4 derived tumors had no or very weak staining of Pdcd4 (Figure 5E). The GEO-shLacZ derived tumors were weakly stained for c-Myc (Figure 5F) whereas GEO-shPdcd4 derived tumors were moderately to strongly stained for c-Myc in the nuclei (Figure 5G). The GEO-shLacZ derived tumors only showed a weak Snail/Slug expression (Figure 5H) but the Snail/Slug staining was strong in the nuclei in the GEO-shPdcd4 derived tumors (Figure I). These results indicate that Pdcd4 knockdown up-regulates c-Myc and Snail/Slug expression in vivo.
4. Discussion
In this study, we demonstrated that knockdown of Pdcd4 decreased expression of epithelial proteins (α-catenin and γ-catenin) and increased expression of mesenchymal proteins (N-cadherin and fibronectin) in cultured cells (Figure 1A). In addition, the GEO-shPdcd4 derived tumors showed a lower E-cadherin and α-catenin expressions and a higher N-cadherin and β-catenin expressions comparing to GEO-shLacZ derived tumors. In combination with our previous in vitro findings,20 these results suggest that knockdown of Pdcd4 leads to EMT. This conclusion is further supported by the findings that knockdown of Pdcd4 promotes migration (Figure 2), decreases collagen IV and laminin adhesion, and increases fibronectin adhesion (Figure 3) because EMT usually increases cell motility and alters cell-matrix adhesion.2 Since fibronectin mainly interacts with integrins,28 the finding of increased adhesion with fibronectin by Pdcd4 knockdown (Figure 3) suggests that integrins might be up-regulated in the Pdcd4 knockdown cells. Integrins are heterodimeric transmembrane proteins consisting of α and β subunits. There are 18 known α subunits and 8 β subunits that combine into 25 different integrins. Which integrin components and how integrins are up-regulated by Pdcd4 knockdown remain unclear.
The finding that the GEO-shPdcd4 derived tumors display a lower level of E-cadherin than GEO-shLacZ derived tumors (Figure 1) suggests a weak cell-cell interaction in the Pdcd4 knockdown cells in vivo. E-cadherin is a transmembrane glycoprotein on the cell surface, which plays a major role in cell-cell interaction in epithelial cells. The extracellular domain of E-cadherin mediates hemophilic, calcium-dependent interactions required for adhesion, whereas the intracellular domain interacts with the actin cytoskeleton through α-catenin, p120, and β-catenin.29 Loss of E-cadherin expression is frequently found during tumor progression in most epithelial cancers. Immunohistochemical staining of human cancer tissues showed that the level of E-cadherin expression often is inversely correlated with the tumor grade and stage.30, 31 Over-expression of E-cadherin results in reversion from invasive to benign tumor phenotype in cultured tumor cells.32 Reduced E-cadherin expression in colon tumor cells correlates with the increased invasive capacity.33 Using Rip1Tag2 mice of pancreatic β-cell carcinogenesis, Perl et al. demonstrated that the loss of E-cadherin mediated cell-cell adhesion is causally involved in the progression from adenoma to carcinoma.34 Thus, E-cadherin functions as an invasion suppressor.
How does knockdown of Pdcd4 promote invasion and/or metastasis? One possible mechanism is through activation of β-catenin dependent transcription. Given that E-cadherin is the binding partner of β-catenin, a decrease in E-cadherin expression results in an increase of free β-catenin in cytoplasm. The free β-catenin is then rapidly phosphorylated by glycogen synthase kinase 3β (GSK3β) in the adenomatous polyposis coli (APC)-axin-GSK3β-casein kinase I complex, and subsequently degraded by the proteasome pathway. If the APC is mutated or GSK3β activity is blocked by Wnt signaling, β-catenin will not be degraded and will translocate into nucleus leading to the activaton of β-catenin dependent transcription.35 Several target genes of β-catenin dependent transcription have been found to involve in the tumor invasion and metastasis.36 Hence, down-regulation of E-cadherin is believed to promote invasion or metastasis through activation of β-catenin dependent transcription. Our IHC staining data showed that GEO-shPdcd4 derived tumors displayed very weak expression of E-cadherin and strong accumulation of β-catenin in the nucleus (Figure 1), suggesting that promotion of metastasis by Pdcd4 knockdown is through activation of β-catenin dependent transcription. In addition, c-Myc, a downstream target of β-catenin dependent transcription, showed a stronger staining in the GEO-shPdcd4 derived tumors than in GEO-shLacZ derived tumors (Figure 5), indicating that c-Myc expression was up-regulated in the GEO-shPdcd4 derived tumors. Recently, c-Myc has been demonstrated to activate JNK signaling pathway and AP-1 dependent transcription in Pdcd4 knockdown cells.23 Since AP-1 regulates several required events for tumor migration and invasion,24 activation of AP-1 dependent transcription by c-Myc suggests that c-Myc regulates invasion/metastasis in the Pdcd4 knockdown cells. This argument is further supported by the finding that knockdown of c-Myc in the GEO-shPdcd4 cells inhibits invasive capacity.21 These findings reveal that Pdcd4 knockdown promotes metastasis, at least in part, through up-regulation of c-Myc expression in vivo.
Previously, we demonstrated that knockdown of Snail in GEO-shPdcd4 cells activates E-cadherin promoter activity.21 In addition to the in vitro data, here, we found that Snail/Slug expression is inversely correlated with E-cadherin expression in the GEO-shPdcd4 derived tumors (Figure 1 and 5), suggesting that Snail regulates E-cadherin expression in vivo. Snail is a key transcription repressor for regulating E-cadherin expression. Over-expression of Snail in epithelial cells promotes invasion.5 Conversely, knockdown of Snail up-regulates E-cadherin expression and inhibits invasion.37 Recent studies by Evdokimova et al. showed that the 5′untranslated region (5′UTR) of snail mRNA forms a stable secondary structure and translation of snail mRNA is regulated by Y-box binding protein 1 (YB-1), suggesting that protein translational control contributes to the regulation of Snail expression.38 Pdcd4 is a translation inhibitor that preferentially inhibits translation of mRNA with secondary structure at 5′UTR.39 Therefore, Pdcd4 may directly inhibit translation of Snail. Alternatively, Pdcd4 may directly interact with helix-loop-helix domain of Twist1 to inhibit YB-1 expression resulting in suppressing Snail expression.40 In addition, knockdown of Pdcd4 has been recently demonstrated to activate AKT kinase activity and increase the phosphorylation of 4E binding protein (4EBP) and translation initiation factor (eIF) 4B, two AKT downstream targets.10 Hyperphosphorylation of 4EBP dissociates the cap binding protein eIF4E and results in the activation of cap-dependent translation.41 Increased phosphorylation of eIF4B has been shown to enhance translation and neoplastic transformation.42 Thus, it is possible that Pdcd4 regulates Snail translation through activation of eIF4E and eIF4B. Whether Pdcd4 regulates Snail translation needs further investigation.
Taken together, the results in this study demonstrate that knockdown of Pdcd4 promotes migration and metastasis. In addition, over-expression of Pdcd4 inhibits tumor cell invasion.16–18 Thus, Pdcd4 might function as a suppressor of tumor invasion and metastasis. Agents for elevating Pdcd4 expression might be a promising therapeutic strategy in cancer treatment.
Supplementary Material
Acknowledgments
We greatly thank Dr. Daret St. Clair and Mrs. Heather Russell-Simmons for reading this manuscript and Ms. Yan Zhang for her technical assistance. This study was supported by a National Institute of Health grant (RO1CA129015).
Footnotes
Conflict of interest Statement
There is no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2(6):442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 2.Grunert S, Jechlinger M, Beug H. Diverse cellular and molecular mechanisms contribute to epithelial plasticity and metastasis. Nat Rev Mol Cell Biol. 2003;4(8):657–665. doi: 10.1038/nrm1175. [DOI] [PubMed] [Google Scholar]
- 3.Hulit J, Suyama K, Chung S, Keren R, Agiostratidou G, Shan W, et al. N-cadherin signaling potentiates mammary tumor metastasis via enhanced extracellular signal-regulated kinase activation. Cancer Res. 2007;67(7):3106–3116. doi: 10.1158/0008-5472.CAN-06-3401. [DOI] [PubMed] [Google Scholar]
- 4.Batlle E, Sancho E, Franci C, Dominguez D, Monfar M, Baulida J, et al. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2000;2(2):84–89. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- 5.Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, et al. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2(2):76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- 6.Bolos V, Peinado H, Perez-Moreno MA, Fraga MF, Esteller M, Cano A. The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: a comparison with Snail and E47 repressors. J Cell Sci. 2003;116(Pt 3):499–511. doi: 10.1242/jcs.00224. [DOI] [PubMed] [Google Scholar]
- 7.Mudduluru G, Medved F, Grobholz R, Jost C, Gruber A, Leupold JH, et al. Loss of programmed cell death 4 expression marks adenoma-carcinoma transition, correlates inversely with phosphorylated protein kinase B, and is an independent prognostic factor in resected colorectal cancer. Cancer. 2007;110(8):1697–1707. doi: 10.1002/cncr.22983. [DOI] [PubMed] [Google Scholar]
- 8.Shibahara K, Asano M, Ishida Y, Aoki T, Koike T, Honjo T. Isolation of a novel mouse gene MA-3 that is induced upon programmed cell death. Gene. 1995;166(2):297–301. doi: 10.1016/0378-1119(95)00607-9. [DOI] [PubMed] [Google Scholar]
- 9.Goke R, Barth P, Schmidt A, Samans B, Lankat-Buttgereit B. Programmed cell death protein 4 suppresses CDK1/cdc2 via induction of p21(Waf1/Cip1) Am J Physiol Cell Physiol. 2004;287(6):C1541–1546. doi: 10.1152/ajpcell.00025.2004. [DOI] [PubMed] [Google Scholar]
- 10.Guo X, Li W, Wang Q, Yang HS. AKT Activation by Pdcd4 Knockdown Up-Regulates Cyclin D1 Expression and Promotes Cell Proliferation. Genes Cancer. 2011;2(8):818–828. doi: 10.1177/1947601911431082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang HS, Jansen AP, Nair R, Shibahara K, Verma AK, Cmarik JL, et al. A novel transformation suppressor, Pdcd4, inhibits AP-1 transactivation but not NF-kappaB or ODC transactivation. Oncogene. 2001;20(6):669–676. doi: 10.1038/sj.onc.1204137. [DOI] [PubMed] [Google Scholar]
- 12.Yang HS, Knies JL, Stark C, Colburn NH. Pdcd4 suppresses tumor phenotype in JB6 cells by inhibiting AP-1 transactivation. Oncogene. 2003;22(24):3712–3720. doi: 10.1038/sj.onc.1206433. [DOI] [PubMed] [Google Scholar]
- 13.Cmarik JL, Min H, Hegamyer G, Zhan S, Kulesz-Martin M, Yoshinaga H, et al. Differentially expressed protein Pdcd4 inhibits tumor promoter-induced neoplastic transformation. Proc Natl Acad Sci U S A. 1999;96(24):14037–14042. doi: 10.1073/pnas.96.24.14037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jansen AP, Camalier CE, Colburn NH. Epidermal expression of the translation inhibitor programmed cell death 4 suppresses tumorigenesis. Cancer Res. 2005;65(14):6034–6041. doi: 10.1158/0008-5472.CAN-04-2119. [DOI] [PubMed] [Google Scholar]
- 15.Schmid T, Jansen AP, Baker AR, Hegamyer G, Hagan JP, Colburn NH. Translation inhibitor Pdcd4 is targeted for degradation during tumor promotion. Cancer Res. 2008;68(5):1254–1260. doi: 10.1158/0008-5472.CAN-07-1719. [DOI] [PubMed] [Google Scholar]
- 16.Leupold JH, Yang HS, Colburn NH, Asangani I, Post S, Allgayer H. Tumor suppressor Pdcd4 inhibits invasion/intravasation and regulates urokinase receptor (u-PAR) gene expression via Sp-transcription factors. Oncogene. 2007;26(31):4550–4562. doi: 10.1038/sj.onc.1210234. [DOI] [PubMed] [Google Scholar]
- 17.Yang HS, Matthews CP, Clair T, Wang Q, Baker AR, Li CC, et al. Tumorigenesis suppressor Pdcd4 down-regulates mitogen-activated protein kinase kinase kinase kinase 1 expression to suppress colon carcinoma cell invasion. Mol Cell Biol. 2006;26(4):1297–1306. doi: 10.1128/MCB.26.4.1297-1306.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nieves-Alicea R, Colburn NH, Simeone AM, Tari AM. Programmed cell death 4 inhibits breast cancer cell invasion by increasing tissue inhibitor of metalloproteinases-2 expression. Breast Cancer Res Treat. 2009;114(2):203–209. doi: 10.1007/s10549-008-9993-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wei N, Liu SS, Chan KK, Ngan HY. Tumour suppressive function and modulation of programmed cell death 4 (PDCD4) in ovarian cancer. PLoS One. 2012;7(1):e30311. doi: 10.1371/journal.pone.0030311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Q, Sun Z, Yang HS. Downregulation of tumor suppressor Pdcd4 promotes invasion and activates both beta-catenin/Tcf and AP-1-dependent transcription in colon carcinoma cells. Oncogene. 2008;27(11):1527–1535. doi: 10.1038/sj.onc.1210793. [DOI] [PubMed] [Google Scholar]
- 21.Wang Q, Sun ZX, Allgayer H, Yang HS. Downregulation of E-cadherin is an essential event in activating beta-catenin/Tcf-dependent transcription and expression of its target genes in Pdcd4 knockdown cells. Oncogene. 2010;29(1):128–138. doi: 10.1038/onc.2009.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Santhanam AN, Baker AR, Hegamyer G, Kirschmann DA, Colburn NH. Pdcd4 repression of lysyl oxidase inhibits hypoxia-induced breast cancer cell invasion. Oncogene. 2010;29(27):3921–3932. doi: 10.1038/onc.2010.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Q, Zhang Y, Yang HS. Pdcd4 knockdown up-regulates MAP4K1 expression and activation of AP-1 dependent transcription through c-Myc. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research (0) doi: 10.1016/j.bbamcr.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ozanne BW, McGarry L, Spence HJ, Johnston I, Winnie J, Meagher L, et al. Transcriptional regulation of cell invasion: AP-1 regulation of a multigenic invasion programme. Eur J Cancer. 2000;36(13 Spec):1640–1648. doi: 10.1016/s0959-8049(00)00175-1. [DOI] [PubMed] [Google Scholar]
- 25.Jiang Y, Liu XQ, Rajput A, Geng L, Ongchin M, Zeng Q, et al. Phosphatase PRL-3 is a direct regulatory target of TGFbeta in colon cancer metastasis. Cancer Res. 2011;71(1):234–244. doi: 10.1158/0008-5472.CAN-10-1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cespedes MV, Espina C, Garcia-Cabezas MA, Trias M, Boluda A, Gomez del Pulgar MT, et al. Orthotopic microinjection of human colon cancer cells in nude mice induces tumor foci in all clinically relevant metastatic sites. Am J Pathol. 2007;170(3):1077–1085. doi: 10.2353/ajpath.2007.060773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O’Connell BC, Cheung AF, Simkevich CP, Tam W, Ren X, Mateyak MK, et al. A large scale genetic analysis of c-Myc-regulated gene expression patterns. J Biol Chem. 2003;278(14):12563–12573. doi: 10.1074/jbc.M210462200. [DOI] [PubMed] [Google Scholar]
- 28.Akiyama SK, Olden K, Yamada KM. Fibronectin and integrins in invasion and metastasis. Cancer Metastasis Rev. 1995;14(3):173–189. doi: 10.1007/BF00690290. [DOI] [PubMed] [Google Scholar]
- 29.Kemler R. Classical cadherins. Semin Cell Biol. 1992;3(3):149–155. doi: 10.1016/s1043-4682(10)80011-x. [DOI] [PubMed] [Google Scholar]
- 30.Madhavan M, Srinivas P, Abraham E, Ahmed I, Mathew A, Vijayalekshmi NR, et al. Cadherins as predictive markers of nodal metastasis in breast cancer. Mod Pathol. 2001;14(5):423–427. doi: 10.1038/modpathol.3880329. [DOI] [PubMed] [Google Scholar]
- 31.Dorudi S, Sheffield JP, Poulsom R, Northover JM, Hart IR. E-cadherin expression in colorectal cancer. An immunocytochemical and in situ hybridization study. Am J Pathol. 1993;142(4):981–986. [PMC free article] [PubMed] [Google Scholar]
- 32.Vleminckx K, Vakaet L, Jr, Mareel M, Fiers W, van Roy F. Genetic manipulation of E-cadherin expression by epithelial tumor cells reveals an invasion suppressor role. Cell. 1991;66(1):107–119. doi: 10.1016/0092-8674(91)90143-m. [DOI] [PubMed] [Google Scholar]
- 33.Kinsella AR, Lepts GC, Hill CL, Jones M. Reduced E-cadherin expression correlates with increased invasiveness in colorectal carcinoma cell lines. Clin Exp Metastasis. 1994;12(4):335–342. doi: 10.1007/BF01753841. [DOI] [PubMed] [Google Scholar]
- 34.Perl AK, Wilgenbus P, Dahl U, Semb H, Christofori G. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature. 1998;392(6672):190–193. doi: 10.1038/32433. [DOI] [PubMed] [Google Scholar]
- 35.Gregorieff A, Clevers H. Wnt signaling in the intestinal epithelium: from endoderm to cancer. Genes Dev. 2005;19(8):877–890. doi: 10.1101/gad.1295405. [DOI] [PubMed] [Google Scholar]
- 36.Fuchs SY, Ougolkov AV, Spiegelman VS, Minamoto T. Oncogenic beta-catenin signaling networks in colorectal cancer. Cell Cycle. 2005;4(11):1522–1539. doi: 10.4161/cc.4.11.2129. [DOI] [PubMed] [Google Scholar]
- 37.Olmeda D, Jorda M, Peinado H, Fabra A, Cano A. Snail silencing effectively suppresses tumour growth and invasiveness. Oncogene. 2007;26(13):1862–1874. doi: 10.1038/sj.onc.1209997. [DOI] [PubMed] [Google Scholar]
- 38.Evdokimova V, Tognon C, Ng T, Ruzanov P, Melnyk N, Fink D, et al. Translational activation of snail1 and other developmentally regulated transcription factors by YB-1 promotes an epithelial-mesenchymal transition. Cancer Cell. 2009;15(5):402–415. doi: 10.1016/j.ccr.2009.03.017. [DOI] [PubMed] [Google Scholar]
- 39.Yang HS, Cho MH, Zakowicz H, Hegamyer G, Sonenberg N, Colburn NH. A novel function of the MA-3 domains in transformation and translation suppressor Pdcd4 is essential for its binding to eukaryotic translation initiation factor 4A. Mol Cell Biol. 2004;24(9):3894–3906. doi: 10.1128/MCB.24.9.3894-3906.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shiota M, Izumi H, Tanimoto A, Takahashi M, Miyamoto N, Kashiwagi E, et al. Programmed cell death protein 4 down-regulates Y-box binding protein-1 expression via a direct interaction with Twist1 to suppress cancer cell growth. Cancer Res. 2009;69(7):3148–3156. doi: 10.1158/0008-5472.CAN-08-2334. [DOI] [PubMed] [Google Scholar]
- 41.Gingras AC, Raught B, Sonenberg N. mTOR signaling to translation. Curr Top Microbiol Immunol. 2004;279:169–197. doi: 10.1007/978-3-642-18930-2_11. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Y, Wang Q, Guo X, Miller R, Guo Y, Yang HS. Activation and up-regulation of translation initiation factor 4B contribute to arsenic-induced transformation. Mol Carcinog. 2011;50(7):528–538. doi: 10.1002/mc.20733. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.