

Summary
Ligation of P2X7 receptors with a “danger signal”, extracellular ATP (eATP), has recently been shown to result in production of intracellular reactive-oxygen-species (ROS) in macrophages. We show that primary gingival epithelial cells (GECs) produce sustained, robust cellular ROS upon stimulation by eATP. The induction of ROS was mediated by P2X7 receptor signaling coupled with NADPH-oxidase activation, as determined by pharmacological inhibition and RNA-interference. Furthermore, Porphyromonas gingivalis, an oral opportunistic pathogen, up-regulated the antioxidant glutathione response, modulated eATP-induced cytosolic and mitochondrial ROS generated the through P2X7/NADPH-oxidase interactome, and subsequently blocked oxidative-stress in GECs via temporal secretion of a P. gingivalis effector, nucleoside-diphosphate-kinase (Ndk). An ndk-deficient P. gingivalis mutant lacked the ability to inhibit ROS production and persist intracellularly following eATP stimulation. Treatment with recombinant Ndk significantly diminished eATP-evoked ROS production. P. gingivalis infection elicited a strong, time-dependent increase in anti-oxidative mitochondrial UCP2 levels, whereas ndk-deficient mutant did not cause any change. The results reveal a novel signaling cascade that is tightly coupled with eATP signaling and ROS regulation. Ndk by P. gingivalis counteracts these antimicrobial signaling activities by secreting Ndk, thus contributing to successful persistence of the pathogen.
Keywords: Porphyromonas gingivalis, opportunistic bacteria, persistence, reactive-oxygen-species, purinergic signaling, NADPH-oxidase, epithelial cells
Introduction
Infected, stressed, or dying cells release small molecules called “danger signals” that specifically interact with “danger signal receptors” on neighboring cells to alter the immune response and control colonization by pathogens. Among these host-derived molecules, ATP, contributes to bacterial clearance by stimulating the secretion of cytokines, recruitment of immune cells, and sensing of pathogens (Coutinho-Silva et al., 2012). Recent studies suggest that extracellular ATP (eATP) released from immune cells can directly function as a sensor against intracellular pathogens upon ligation of the purinergic receptor, P2X7 (Miller et al., 2011). The P2X7 receptor has recently emerged as an important multi-level mediator in controlling intracellular infections by regulating the innate immune response, apoptosis, intracellular trafficking, and generation of reactive oxygen species (ROS) (Chaves et al., 2009, Wiley et al., 2011, Franchi et al., 2007, Spooner et al., 2011). Although ROS were previously known mainly as toxic agents, a growing body of evidence also indicates that intracellular ROS levels play a major role as secondary signaling molecules, and tightly control a number of diverse biological functions critical for cellular proliferation, production of cytokines, senescence, and cell death and survival, and the immune response, all of which appear to be closely associated with cellular/mitochondrial oxidative-stress (Spooner et al., 2011, D’Autreaux et al., 2007, Bryan et al., 2012, Boots et al., 2009).
Besides the observation that eATP can stimulate pathogen elimination via the P2X7 receptor, P2X7 can also promote microbial existence in host tissues (Franchi et al., 2007, Lammas et al., 1997, Lees et al., 2010, Coutinho-Silva et al., 2009). Consistent with this view, the ROS-mediated signaling for immune function can be exploited by “opportunistic pathogens” to modulate host responses in order to enhance survival and persistence in their target host cells.
Within the gingival compartment, gingival epithelial cells (GECs) are among the first host cells targeted by invading microbes, and these epithelial cells function as a critical component of the immune response since they have the ability to recognize, respond to, and eliminate pathogens without major impairment to the host (Yilmaz, 2008a). Porphyromonas gingivalis, a successful host-adapted anaerobe capable of intracellular survival, replication, and intercellular spreading within the gingival epithelium, can withstand the challenge of host-mediated immune responses working to destroy pathogens and interrupt their colonization (Yilmaz, 2008a, Avila et al., 2009, Hajishengallis et al., 2011, Henry et al., 2012, Darveau et al., 1998). The infection by P. gingivalis can render the GECs resistant to mitochondrion-dependent cell death by inhibiting mitochondrial permeability changes, caspase-9 activation, and cytochrome c release, as well as inactivating mitochondrial pro-apoptotic Bad via Akt/phosphoinositide 3-kinase (PI3K) signaling (Yao et al., 2010, Yilmaz et al., 2004, Mao et al., 2007, Boisvert et al., 2010). We previously demonstrated that primary GECs express functional P2X7 receptors and are highly sensitive to stimulation by the “danger signal”, eATP. Conversely, in order to protect its host cell. P. gingivalis can diminish eATP/P2X7 receptor-mediated apoptosis of primary GECs (Yilmaz et al., 2008b). Secretion of a nucleoside-diphosphate-kinase (Ndk) homolog by the pathogen has been proposed to consume eATP, thereby inhibiting P2X7 ligation (Spooner et al., 2012a, Yilmaz et al., 2008b). Thus, P. gingivalis can successfully evade host-mediated defense systems through its secreted effectors, converting epithelial cells into a prime reservoir for the organism’s survival and further propagation in the oral cavity (Choi et al., 2011, Spooner et al., 2012, Hasegawa et al., 2008, Tribble et al., 2006, Hajishengallis, 2009, Yilmaz, 2008a).
P. gingivalis displays qualities of a self-limiting, host-balanced pathogen; however, key determinants of the organism’s ability to adapt to the epithelium are not fully understood. Moreover, the mechanisms of ROS induction by the stress-induced eATP on epithelial cells - the preferred target cells for many pathogens - still remain to be characterized. Similarly, the molecular machinery that mediates eATP-mediated ROS production and the functional effects of ROS on the intracellular life of P. gingivalis in GECs have never been investigated.
In this study, we demonstrate for the first time that gingival epithelial cells can generate significant amount of cellular ROS in response to eATP treatment, and this is dependent on P2X7 signaling coupled with membrane-bound NADPH-oxidase and the mitochondrial respiratory chain. Moreover, P. gingivalis infection modulates eATP- P2X7 evoked cytosolic and mitochondrially-generated ROS production and activates an anti-oxidative-stress response in primary GECs. However, infection by the ndk-deficient mutant lacks the ability to protect GECs against eATP-mediated oxidative-stress and to survive intracellulary in ATP-treated infected GECs. Pre-treatment of the infected GECs with N-Acetyl Cysteine (NAC), a potent inhibitor of ROS, largely restored the intracellular survival of the ndk-deficient strain. Temporal secretion of the Ndk protein by P. gingivalis appears to serve as a central effector in directing these specific cellular actions.
Results
Extracellular ATP(eATP) induces ROS through P2X7 receptor activation in primary GECs
We previously showed that primary GECs, which represent a physiologically relevant epithelial model for oral infection, express functional P2X7 receptors and that GEC treatment with eATP induces a high level of cell death (Yilmaz et al., 2008b). Therefore, we initially examined the potential effect of eATP on GEC oxidation by measuring cellular ROS production under the same conditions as previously used to induce GEC apoptosis, i.e. after stimulation with 3 mM ATP during 24 h period. As shown in Fig. 1A, cellular ROS was quantified by cytofluorimetry using CM-H2DCFDA (DCF), a non-fluorescent dye emitting green fluorescent after cellular oxidation. The results revealed that ATP strongly enhanced the cellular ROS levels in GECs when compared to the untreated control cells (Fig. 1A).
Fig. 1. ATP induces cellular ROS through P2X7 receptor in GECs.


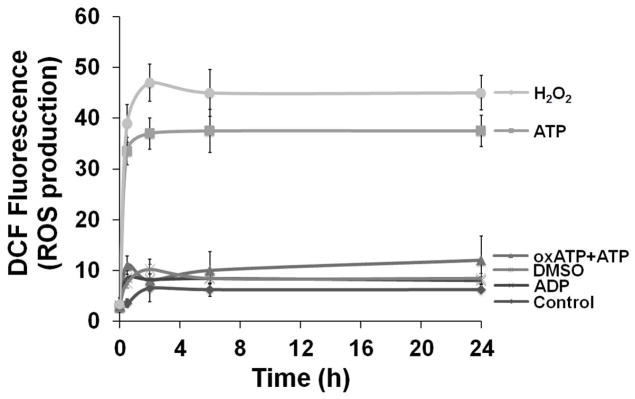
A. Generation of ROS in primary GECs after treatment with ATP measured using CM-H2DCFDA (DCF, a dye oxidized to green fluorescent by cellular H2O2) staining by cytofluorimetry at 24 h. High fluorescence population (right-shift of the green fluorescence distribution peak) indicates strong production of ROS upon ATP treatment at 24 h.
B. Kinetics of cellular ROS in living cells upon P2X7 agonist (3 mM ATP), antagonist (5 mM oxATP), and non-specific nucleotide (3 mM ADP) treatments were examined using DCF by fluorescence microscopy imaging combined with DIC during 24 h. The DIC verified the total number of cells in the samples.
C. The mean fluorescence intensities were determined by NIH-Image J analysis for 30 min, 2, 6, and 24 h time periods. The fluorescence intensity was normalized to the level of fluorescence recorded prior to stimulation with ATP. 100 μM H2O2 is used as positive control. 10 mM DMSO (diluent for DCF dye) was used to determine background control levels. The results are representative of at least 3 separate experiments performed in duplicate.
We next measured ATP-elicited ROS generation in live DCF-labeled GECs as a function of time using epifluorescence microscopy combined with differential interference contrast (DIC) microscopy (Fig. 1B and C). H2O2-treated samples were used as positive control, and untreated and DMSO-treated cells were used as controls for determining the background values for the DCF fluorescence intensity analysis. In the same experiments, the potential role of the P2X7 receptor in eATP-mediated ROS formation was determined using an irreversible inhibitor of the P2X7 receptor, oxidized-ATP (oxATP), and the non-specific nucleotide ADP as controls (Fig 1B and C). The results illustrate that ATP induced a rapid and large increase in the levels of ROS, which were similar to the ROS production in H2O2-treated cells. Pre-treatment of the GECs with oxATP abrogated the amount of ROS in ATP-treated cells, whereas ADP treatment did not induce any ROS (Fig. 1C). The specific agonist of P2X7, 3′-O-(4-benzoyl) benzoyl-ATP (BzATP), stimulated comparable levels of ROS (not shown), suggesting that ATP-induced ROS largely originates from P2X7 ligation.
P2X7 depletion abolishes eATP-dependent ROS production
We further verified the involvement of P2X7 receptor in the ATP-evoked cellular ROS generation by depleting P2X7 using shRNA interference. A stable HIGK (human immortalized gingival keratinocytes) cell line derived from a primary GEC source by transformation with HPV E6/E7 was utilized for the receptor depletion experiments (Oda et al., 1996). As displayed in Fig. 2, there was a significant effect of P2X7 depletion on ATP-evoked cytosolic ROS production, further indicating that the P2X7 receptor is responsible for most of the ATP-induced ROS responses in epithelial cells.
Fig. 2. Depletion of P2X7 receptor diminishes ATP-induced ROS production.

A target-specific P2X7 antibody was used to confirm the inhibition of P2X7 expression by Western-blotting. A non-target antibody (β-tubulin) was used as endogenous control. Generation of ROS by ATP following 30 min, 2 and 3 h post-incubations was significantly inhibited in stable P2X7 knockdown cells (HIGKs). ROS levels were determined using quantitative DCF staining by fluorescence microscopy and DIC analysis as described in experimental procedures. (**) denotes statistical significance (P = 0.001 t-test)
P2X7-dependent ROS production from NADPH oxidase synergizes with mitochondrial ROS
In macrophages and other professional phagocytes, a major source of ROS is due to activation of the NADPH-oxidase enzyme complex, consisting of multiple subunits of phospho-proteins localized on the plasma membrane and the cytosol (Bedard et al., 2007). Although ROS production has traditionally been associated with professional phagocytes, recent studies have demonstrated that epithelial cells also have a functionally active NADPH-oxidase complex for ROS production and oxidative-stress responses (Cha et al., 2010). In order to examine the role of this enzyme in ATP-mediated ROS generation in primary GECs, we pre-incubated cells with diphenyleneiodonium chloride (DPI), an inhibitor of NADPH-oxidase, for 30 min before treatment with ATP, and analyzed the kinetics of ROS production using DCF staining by epifluorescence and DIC microscopy. The analysis showed that DPI substantially blocks ATP-mediated early ROS production, implying that NADPH-oxidase is at least partially responsible for the generation of ROS via P2X7 ligation (Fig. 3A and B).
Fig. 3. ATP-P2X7 activated ROS is associated with NADPH-oxidase and mitochondrion.




A. 10 μM DPI, an inhibitor of NADPH-oxidase, inhibited the ATP-induced cellular ROS levels similar to the control (untreated) in primary GECs as revealed by DCF staining.
B. The quantitative NIH-Image J analysis of DCF (green) fluorescence intensities determined cellular ROS levels in samples.
C. The pre-labeling of the cells with MitoSOX Red (red fluorescent), highly selective dye for O2− in the mitochondria, demonstrated that ATP induces strong mROS visualized by fluorescence and DIC microscopy. Results displayed that mROS can be inhibited by DPI and oxATP indicating that mROS generation associated with NADPH-oxidase and P2X7 coupling.
D. The levels of mROS were determined by NIH-Image J analysis of MitoSOX Red fluorescence in samples. Data represents at least three independent experiments.
It has been shown that NADPH-oxidase-induced cytosolic ROS can synergistically promote mitochondrial ROS (mROS), which in turn can induce oxidative stress (Ahmed et al., 2012, Wosniak et al., 2009). We examined mROS production following treatment with ATP using the mitochondrial superoxide indicator (MitoSOX Red) (Fig. 3C and B). The dye MitoSOX Red detects superoxide in the mitochondria of living cells, emitting red fluorescence when it becomes oxidized. Our results show ATP also induces steady high levels of mROS, as compared with H202 treatment. Interestingly, NADPH-oxidase can synergistically enhance ATP-stimulated ROS production by stimulating ROS release from mitochondria, as detected by the marked inhibition of mROS by DPI treatment (Fig 3. and B). The P2X7 inhibitor, oxATP, also inhibits eATP-induced mROS, implying that eATP has its effect via surface-bound P2X7. The partial effect of the oxATP (an inhibitor of P2X7) on mROS generation substantiates the possibility of NADPH-oxidase and P2X7 receptor cross-talk.
P. gingivalis infection activates an anti-oxidative cell response against eATP-induced ROS
The main antioxidant system utilized by eukaryotic cells to combat oxidative-stress is the production of glutathione, which can reduce the effects of cellular ROS (Fernandez-Checa et al., 1998, Jefferies et al., 2003). Since P. gingivalis has the ability to interfere with eATP/P2X7 signaling and there is growing evidence for persistent microbes modulating host cell ROS production and oxidative-stress for maintaining long-term persistence (Spooner et al., 2011, Coutinho-Silva et al., 2012), we next evaluated whether P. gingivalis infection affects eATP-induced oxidative-stress by measuring intracellular glutathione levels in the ATP pre-treated and P. gingivalis-infected GECs (Fig. 4A). The total glutathione (oxidized “GSSG” over reduced “GSH”) was quantified using a specific chemiluminescence assay. ATP-treated cells alone exhibited a steady and high GSSG/GSH ratio over time that was similar to the H202 treated samples, suggesting a response to excessive ROS accumulation and oxidative-stress. However, P. gingivalis-infected cells with and without ATP pre-treatment showed lower GSSG/GSH ratios compared with control levels, especially at 6 h, 24 h post-infection, indicating that the infection can inhibit eATP-induced oxidative stress by increasing the intracellular GSH “antioxidant” defense system in primary GECs (Fig. 4A).
Fig. 4. P. gingivalis activates an antioxidant, GSH, response against ATP-mediated ROS.


A. The reduced glutathione (GSH) and oxidized glutathione (GSSG) levels of primary GECs were measured using chemiluminescence detection. P. gingivalis-infected cells w±/o 3 mM ATP pre-treatment (Wt P.g and ATP+Wt P.g respectively) showed significantly decreased GSH/GSSG ratios compare to uninfected ATP-treated cells or H2O2-treated cells at 6, 24 h.
B. Fluorescent ThiolTracker Violet (blue fluorescent) was also used to quantify intracellular reduced GSH levels at 24 h. NIH-Image J analysis performed to calculate % GSH levels. All samples were normalized against untreated (control) sample. (*) and (**) denote statistical significance (P = 0.01 and P = 0.001 t-test respectively).
Further examination of intracellular GSH levels in living primary GEC cultures was accomplished using ThiolTracker-Violet Stain and fluorescence microscopy (Fig. 4B) (Mandavilli et al., 2010). Cells pretreated for 24 h with ATP displayed substantially depleted GSH levels, reflective of an oxidative-stress response, whereas cells infected for 24 h with P. gingivalis had GSH levels similar to untreated cells and ADP-stimulated cells, as detected by intense ThiolTracker Violet staining. Furthermore, ATP-induced depletion of GSH in GECs was markedly abrogated by the infection (Fig. 4B).
Ndk of P. gingivalis confers resistance against eATP-P2X7 induced cellular ROS
Observation of the infection-mediated induction of GSH in response to ATP-dependent oxidative stress prompted us to study the effect of infection on cellular ROS generation (Fig. 5A and B). Since we previously showed that P. gingivalis can interfere with P2X7 activity by production of the Ndk protein, we also infected GECs with ndk-deficient mutant and the complemented-strain (Yilmaz et al., 2008b) to test whether the P. gingivalis-mediated antioxidant response against ATP signaling may be associated with Ndk activity. Infection of the GECs provoked high levels of ROS, which was transient (Fig. 5A), with ROS levels declining at 3 h post-infection (not shown). At 6 and 24 h post-infection, the amount of ROS in infected cells was similar to the levels in untreated control cells (Fig. 5B). The levels of ROS produced from the cells treated with ATP prior to infection were also significantly reduced over the course of infection, implying that P. gingivalis infection can block ATP-mediated ROS production in a time-dependent manner. The ndk-deficient mutant did not block the ATP-mediated ROS, while GECs infected with the complemented-strain and incubated with ATP displayed comparable levels of ROS induced by the wild strain, strongly suggesting a critical role for Ndk in this activity (Fig. 5A and B).
Fig. 5. P. gingivalis Ndk modulates ATP-P2X7 mediated cellular ROS.
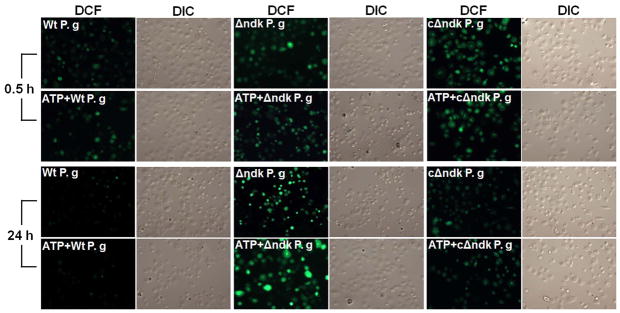

A. Primary GECs pre-treated with 3 mM ATP and infected with P. gingivalis, or Δndk (mutant strain) or cΔndk strain (complemented-strain), were studied for cytosolic ROS production at 30 min, 2, 6 and 24 h time periods using DCF staining by fluorescence and DIC microscopy. The representative images of 30 min and 24 h samples are displayed.
B. The mean fluorescence intensities were measured by NIH-Image J analysis. Data represents at least three independent experiments. (**) denotes statistical significance (P = 0.001 t-test).
Inhibition of P2X7-dependent mitochondrial ROS by modulation of UCP2
Because P. gingivalis can modulate and inhibit mitochondrial activity in GECs, we next investigated whether the infection can control mROS and potentially utilize the Ndk enzyme for this effect. Thus, we first analyzed the kinetics of mROS production following ATP treatment of GECs infected with P. gingivalis or the ndk-deficient mutant, and then studied the expression of a transcriptionally regulated mitochondrial anti-oxidative gene, specifically the gene encoding the uncoupling protein, UCP2 (Fig 6A, B, C).
Fig. 6. P2X7 receptor dependent mROS and UCP2 is inhibited by infection.

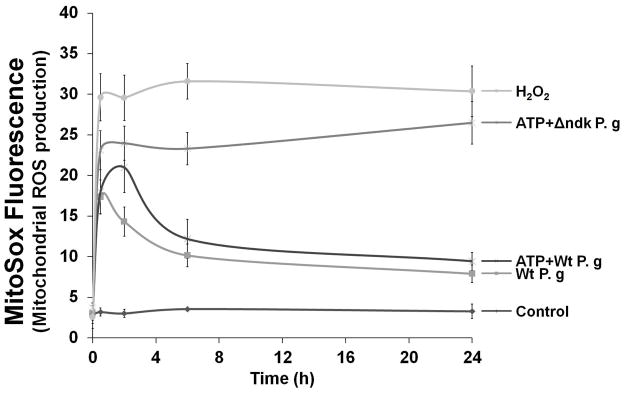

A. Primary GECs pre-treated with 3 mM ATP and infected with P. gingivalis, or Δndk (mutant strain), were studied for mitochondrial superoxide production at 30 min, 2, 6, and 24 h time periods using MitoSOX Red staining by fluorescence and DIC microscopy. The representative images of 30 min and 24 h samples are displayed.
B. The mean fluorescence intensities were measured by NIH-Image J analysis. Data represents at least three independent experiments.
C. The primary GECs pre-treated with 3 mM ATP and infected with P. gingivalis, or Δndk strain for 24 h, were analyzed for mRNA levels of UCP2 by quantitative PCR using iQ SYBR Green. (**) denotes statistical significance (P = 0.001 t-test). Data are representative of at least two experiments in duplicate.
The results showed that P. gingivalis triggered a rapid but transient increase in mROS in the initial hours of infection, which was also partially inhibited by DPI treatment (not shown). The infection by the wild-strain inhibited ATP-induced mROS (maximally at 6 h post-infection) and the ndk-deficient mutant lacked entirely the ability to block ATP-mediated mROS (Fig. 6A and B). In the earlier time points of P. gingivalis infection, UCP2 mRNA levels were up-regulated, suggesting that the infection activates this anti-oxidative mechanism (not shown). ATP treatment initially did not induce any changes in UCP2 gene transcription. However, the treatment caused a 3-fold increase at 24 h post-treatment (Fig. 6C). However, infection by the wild-strain decreased ATP-induced UCP2 up-regulation to basal levels at 24 h post-infection, whereas the ndk-deficient mutant did not have any effect (Fig. 6C).
Secretion of Ndk from P. gingivalis during infection contributes to persistence by inhibiting eATP-mediated cellular ROS
Our previous analysis of culture supernatants and pellets from the wild-strain and ndk-deficient P. gingivalis strains showed that the ATPase activities were significantly lower in the mutant-strain compared with the wild-strain, suggesting that the P. gingivalis Ndk homolog was actively secreted and consumed extracellular ATP (Yilmaz et al., 2008b). Since infection with the ndk-deficient mutant lacked the ability to block the high ROS production mediated by eATP, we examined the ATPase activities of the supernatants collected from primary GECs infected with the wild-strain or mutant-strain (Fig. 7A). ATP hydrolysis from the supernatant of cells infected with the wild-strain after normalization against control (supernatant from uninfected cells) reached a maximum at 6 h (~0.1 μM/sec) and continued at a similar level for 24 h post-infection. Supernatants from cells infected with the mutant strain displayed a similar activity as the uninfected controls (Fig. 7A). Thus, the observed hydrolysis rate in the supernatant from cells infected with wild-type P. gingivalis was similar to the ATPase activity reported for MgsA (a homolog of the human AAA+ family proteins) in E. coli, which was 0.06 μM/sec (Page et al., 2011).
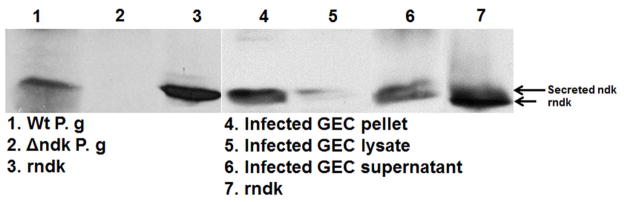
Fig. 7. Ndk secretion of P. gingivalis during GEC infection contributes to intracellular survival.
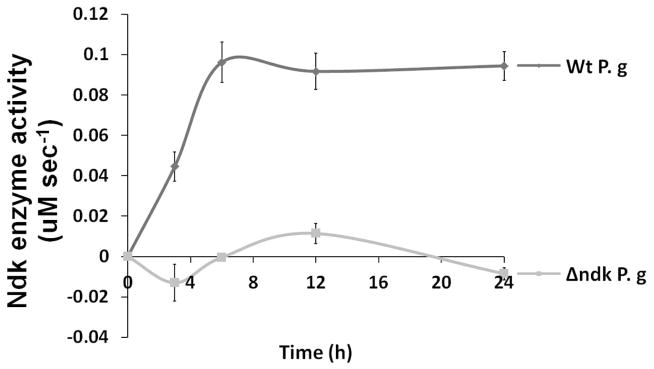

A. ATP hydrolysis of primary GEC culture supernatants collected from cells infected with P. gingivalis or Δndk strain during 24 h determined using colorimetric ATPase assay as described in experimental procedures. The ATPase activity measured by calculating amount of enzyme that catalyzes the reaction of 1 μM ATPsubstrate per minute.
B. The western-blot analyses of the TSB media with P. gingivalis or Δndk strain and rNdk (lanes 1, 2, 3 respectively), as well as the ammonium sulfate concentrated culture supernatants from primary GEC infected with P. gingivalis for 12 h (lane 4), the infected GEC lysate (lane 5), and pellet of the infected GEC (lane 6), rNdk (lane 7) were performed using anti-Ndk specific polyclonal antibodies.
C. Antibiotic Protection Assay was performed to evaluate the effect of ATP on bacterial killing in primary GECs. The GECs, pre-treated w±/o 50 mM NAC (an inhibitor of ROS) or 5U/ml Apyrase (a nucleotide-degrading enzyme), infected with P. gingivalis or Δndk strain, were incubated w±/o 3 mM ATP during 24 h. The colony forming unit for the wild type strain without any treatments was set as 100% intracellular survival. The results were then expressed as percentages. (*) and (**) denote statistical significance (P = 0.01 and P = 0.001 t-test respectively). Data are representative of at least two experiments in duplicate.
Secretion of Ndk protein from infected cells was next confirmed by examining the supernatants from infected cells, using specific polyclonal antibodies to the recombinant Ndk (rNdk) and Western-blot analysis. The 24 h bacterial culture supernatants and rNdk were used as controls (Fig. 7B, lanes 1, 2 and 3 respectively). The results demonstrated that Ndk can be present intracellularly (detected in the insoluble fraction) and secreted extracellularly during infection of primary GECs (Fig. 7B). The analyses indicated that the earliest detection of ~15 kDa Ndk in the bacterial growth culture media was 24 h, whereas 6 h (not shown) and 12 h infected GEC culture supernatants contained the Ndk (Fig. 7B, lanes 4 and 6). The densitometry analysis by NIH-Image J revealed a ~30% increase in the levels of Ndk detected in the GEC culture supernatants at 12 h post-infection, compared to 6 h post-infection (not shown). A protein sequence analysis was performed to validate the identities of the detected bands in the ~15 kDa range using mass spectrometry (Qstar Elite, Applied Biosystems). Scaffold Proteome Software (Searle, 2010) analysis further validated the bands with higher than 95% probability as hypothetical protein PG1018, which corresponds to P. gingivalis Ndk (Yilmaz et al., 2008b).
Lactate dehydrogenase (LDH) release assays were simultaneously performed in the infected GEC supernatants to assess the status of host cell plasma membrane integrity and host cell viability (not shown). The LDH results confirmed that the infected GECs were intact throughout P. gingivalis infection, as reported earlier (Yilmaz et al., 2004, Mao et al., 2007, Yilmaz et al., 2008b, Kuboniwa et al., 2008, Yao et al., 2010), while the ATP and staurosporine-treated cells showed significantly elevated levels of LDH in the cell supernatants (not shown).
Since previous studies reported that P2X7-mediated ROS generation plays a role in eliminating intracellular infections in professional phagocytes (Coutinho-Silva et al., 2012), we evaluated the viability of intracellular P. gingivalis and the ndk-deficient strain following ATP treatment of infected GECs using the antibiotic protection assay (Fig. 7D). The results showed that the ndk-deficient mutant, which is already less capable of intracellular survival than the wild type strain (Yilmaz et al., 2008b), was greatly affected by ATP treatment of infected cells, as only ~40% of the mutant bacteria could be recovered (Fig. 7D). To directly examine the involvement of ROS-mediated killing of intracellular bacteria, the infected and ATP-treated GECs were pre-incubated with 50 mM NAC, a potent inhibitor of ROS. NAC significantly enhanced the survival of ATP-treated intracellular bacteria, since ~70% of the ndk-deficient bacteria were rescued in ATP-treated host cells that had been incubated with NAC (Fig. 7D). In addition, the incubation of infected and ATP-treated GECs with an ATP-hydrolyzing enzyme, apyrase (5 μg/ml), rescued substantially the intracellular ndk-deficient bacteria, further demonstrating the role of Ndk in inhibiting P2X7-dependent ROS-mediated bacterial killing (Fig. 7D).
eATP-mediated cellular ROS production is decreased by rNdk
Our previous study demonstrated that treatment with purified rNdk inhibits ATP-mediated plasma membrane permeabilization in a dose-dependent manner, demonstrating the ability of Ndk to interfere with P2X7 ligation (Yilmaz et al., 2008b). We next aimed to determine whether rNdk can block ATP-induced ROS formation in primary GECs. The host cells were pre-incubated with rNdk and then treated with ATP (Fig. 8A). ATP-induced ROS was substantially reduced in a time-dependent manner in uninfected GECs.
Fig. 8. rNdk blocks ATP-mediated cellular ROS and decreases extracellular ATP release.


A. Primary GECs were pre-treated with 5 μg/ml rNdk and incubated with 3mM ATP for 30 min, 3 and 6 h. rFimA (5 μg/ml) protein from P. gingivalis (Yilmaz et al., 2002) was used as negative control (not shown). The cells were measured for global ROS production by DCF staining as described in experimental procedures. (*) denotes statistical significance (P = 0.01 t-test). Data are representative of at least two experiments in duplicate.
B. HIGK cells and pannexin-1 deficient HIGK cells (via RNAi) were infected with P. gingivalis or Δndk strain and studied for extracellular ATP release during 24 h period. Extracellular ATP levels were measured by a luciferase-based assay. Light emission was measured as relative light units (rlu) and absorbance from replicate samples of uninfected cell supernatants was subtracted from each sample. Data are representative of at least two experiments in duplicate.
Similarly, when we studied ATP release cells infected with the ndk-deficient strain versus cells infected with the wild type P. gingivalis, significantly more ATP was measured in the supernatant from primary GECs or the HIGK cells infected with the ndk-deficient mutant than cells infected with wild type P. gingivalis. The release of ATP from P. gingivalis-infected cells was transient (peaking to maximum at 30 min) and rapidly declined at 3 h, decreasing to control levels at 6 h post-infection (Fig. 8B). These results were consistent with the time periods for the ROS inhibition observed during the infection. However, ATP release from the ndk-deficient strain infected cells was constant, slowly declining only after at 24 h post-infection.
The potential source of the eATP in the oral cavity was next investigated. Since pannexin-1 was previously shown to mediate ATP release from macrophages and other cells (Sridharan et al., 2010, Xia et al., 2012, Sandilos et al., 2012, Chekeni et al., 2010), we evaluated whether pannexin-1 may be a pathway for ATP release from GECs during P. gingivalis infection. Thus, depletion of pannexin-1 from HIGK cells through RNAi abolished the release of ATP from cells infected with both the ndk-deficient and the wild type strain of P. gingivalis (Fig. 8B). These results strongly support the concept that P2X7/pannexin-1 channels, which are critical for inflammasome activation and IL-1β secretion, are also modulated by the intracellular infection (Yilmaz et al., 2010).
Discussion
The human oral mucosa maintains a barrier and signaling function in the presence of a diverse microbial consortium, which involves highly sophisticated relationships between the epithelium and its specialized microbial colonizers. While these interactions often exist in commensal harmony within the oral cavity, harmful outcomes are frequently associated with “potential pathogens”, which have evolved a variety of complex strategies allowing safe encounters with the immune defense system and adaptation to host metabolic functions for successful co-existence with their host (Avila et al., 2009, Yilmaz, 2008a, McKenzie et al., 2012). P. gingivalis, a key member of the subgingival microbiota and a major contributor to severe forms of periodontal diseases, colonizes the gingival epithelium in the absence of overt disease (Colombo et al., 2007, Rudney et al., 2001, Jenkinson et al., 2005). P. gingivalis can replicate in GECs, and exhibit cell-to-cell intracellular spreading, without hampering host-cell mitosis (Yilmaz et al., 2006, Kuboniwa et al., 2008). The infection impacts multi-apoptotic pathways including mitochondrion-dependent and P2X7 signaling networks (Yao et al., 2010, Yilmaz et al., 2008a)
Generation of ROS can serve as a key intracellular signaling mediator for a variety of cellular functions but can also be used by host cells as a mechanism to combat intracellular pathogens (Spooner et al., 2011, Said-Sadier et al., 2010, Abdul-Sater et al., 2010). However, sustained generation of ROS can lead to induction of oxidative stress, which in turn may trigger cell death, an inflammatory response, and perturbed tissue balance (Pourova et al., 2010). Recent studies on the cell biology of chronic infections revealed new roles for host metabolic pathways such as activation of cytosolic ROS and oxidative-stress pathways in modulating immune response and persistent infections in specialized immune cells (Cruz et al., 2007a, Forman et al., 2010, Hasan et al., 2006, Yang et al., 2007).
P2X7 signaling has emerged as a key mediator for regulation of cellular ROS and oxidative stress in professional phagocytes following ligation of eATP (Mancino et al., 2001, Chen et al., 2006, Ferrari et al., 2006). eATP is extensively documented as a “danger” molecule released from infected and stressed cells, and it can trigger a range of specific signaling events critical for inflammasome activation, cell death, membrane traffic/organelle function, and control of infections upon ligation with P2X7. Nevertheless, there are currently no reported studies on the potential role of eATP as a mediator of ROS responses in gingival epithelial cells and the associated signaling networks involved in ROS formation and signaling. Our study demonstrates for the first time that eATP induces elevated levels of ROS in the epithelial cells, which are the result of coordinated P2X7 receptor-dependent NADPH-oxidase activation and subsequent mitochondrial-derived ROS.
Furthermore, we show that P. gingivalis infection modulates global ROS derived from plasma membrane NADPH-oxidase and mitochondria in GECs and inhibits P2X7-dependent NADPH oxidase-mediated oxidative stress. The ndk-deficient mutant can not block the eATP-mediated ROS production, indicating the importance of Ndk in this process. GSH, a major antioxidant that maintains cellular redox status, appears to play a role in the P. gingivalis-mediated anti-oxidative stress response during infection of GECs. P. gingivalis also modulates mRNA levels of mitochondrial UCP2, which acts as a regulator of mitochondria derived superoxide and protector against oxidative stress (Chu et al., 2009, Deng et al., 2012, Emre et al., 2010). ATP evoked up-regulation of UCP2 was similar to the effect of H202 treatment, suggesting that ATP may have its effects through sustained production of ROS. The prompt but transient activation of UCP2 by P. gingivalis infection suggests that the infection may protect cells from increased mROS through the rapid induction of anti-oxidative genes. P. gingivalis infection can suppress ATP-mediated oxidative-stress at longer times by reducing the UCP2 levels close to normal, while the ndk-deficient mutant completely lacks this ability. These results are consistent with our previous finding that P. gingivalis suppresses ATP-induced cell death (Yilmaz et al., 2008a, Yilmaz, 2008b).
We previously reported that P. gingivalis produces a homolog of Ndk, which inhibits eATP-dependent apoptosis of GECs (Spooner et al., 2012). Ndk has recently emerged as a multi-functional effector used by opportunistic pathogens such as Mycobacterium tuberculosis and Pseudomonas aeruginosa to establish successful chronic infections (Sun et al., 2010, Zaborina et al., 1999, Chakrabarty, 1998, Kamath et al., 2000) Our study demonstrates that a microbial Ndk is up-regulated during intracellular infection and detected in the cytosol, and can be successfully secreted outside of the infected host cells. Examination of the infected GEC supernatants also revealed that the secreted Ndk is functionally active and contains ATPase activity that is at least comparable to the activity of established ATPases (Page et al., 2011). Our results also reveal a protective role of the Ndk against ATP-mediated killing of intracellular bacteria, as shown to the higher sensitivity of the ndk-deficient strain to killing, compared to the wild type P. gingivalis strain. This phenotype is consistent with the previous observation that cells infected with the mutant strain are more sensitive to ATP-induced cell death than the wild-type strain (Yilmaz et al., 2008b). Moreover, the finding of NAC pre-incubation significantly rescued the ndk-deficient strain from ATP-induced bacterial killing suggests that Ndk is centrally involved in protection of the pathogen against ROS-mediated bacterial removal, thus contributing to persistence of infection.
Intriguingly, different species of Ndk are found localized in sub-cellular compartments, especially mitochondria, a major source of cellular ATP (Tsuiki et al., 1999). Exact mechanisms of the Ndk need to be further characterized. Based on the increasing view of Ndk as a pleiotropic molecule (Spooner et al., 2012), it is tempting to suggest that P. gingivalis Ndk may function beyond depleting extracellular ATP before ligation of P2X7 and plays at least a dual role in governing the aforementioned actions important for effectual colonization.
Overall, this initial study addresses an emerging aspect of P2X7-dependent activation of specific ROS signaling cascades in epithelial cells. It also reveals that the NADPH-oxidase/mitochondrion interactome can be a target for microorganisms seeking to alter ROS generation in order to enhance bacterial survival and persistence. Future studies on the oxidative stress response of infected cells may lead to design of novel therapies aimed at interfering with or reducing the severity of infections caused by these opportunistic pathogens.
Experimental Procedures
Bacteria and Cell Culture
P. gingivalis ATCC 33277 was cultured anaerobically for 24 h at 37°C in trypticase soy broth (TSB) supplemented with yeast extract (1 mg ml-1), haemin (5 μg ml-1) and menadione (1 μg ml-1). The nucleoside diphosphate kinase (ndk)-deficient mutant and the complementation strain were created as described previously (Yilmaz et al., 2008b) and cultured anaerobically for 24 h at 37 °C in the same TSB media. Erythromycin (10μg/ml) was added to the media for culturing the mutant strain. The media of the complemented strain was supplemented with erythromycin (10 μg/ml) and tetracycline (3μg/ml). All bacteria were grown for 24 h, harvested by centrifugation at 6000 g and 4°C for 10 min, washed twice, and resuspended in Dulbecco’s Phosphate-buffered saline (Sigma) pH 7.3 before they were incubated with host cells. The number of bacteria was determined using a Klett-Summerson photometer.
Primary GECs were obtained after oral surgery from healthy gingival tissue as previously described (Yilmaz et al., 2004). Cells were cultured as monolayers in serum-free keratinocyte growth medium (KGM) (Lonza) at 37°C in 5% CO2. GECs were used for experimentation at ~75–80% confluence and cultured for 24 h or 48 h before infection with bacterial cells or exposure to other test reagents in KGM.
Measurement of ROS Production
Cells were incubated with 3 mM ATP or ADP, 2 mM BzATP, 100 μM H2O2 and further incubated with 5 mM oxATP or 10 μM diphenyleneiodoniumchloride (DPI), and/or infected with P. gingivalis strains at a multiplicity of infection (m.o.i.) of 100 in 6-well microscopic slide chamber system (Warner instruments) for 30 min, 2, 6, and 24 h time periods. All the ATP treatments were performed for 1 h prior to infections, as described previously (Yilmaz et al., 2008b).
(Cells were pre-labeled with fluorescent probe 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA) (Invitrogen) which was solubilized at 10 mM in dimethyl sulfoxide (DMSO) (Sigma) and then diluted to 10 μM in Hanks balanced salt solution (HBSS). The cells were washed twice with HBSS containing MgCl2 and CaCl2 (Gibco, Invitrogen), incubated with 10 μM CM-H2DCFDA in this buffer for 30 min at 37°C, and then washed once with fresh medium. This dye reacts with intracellular thiol-chloromethyl groups upon oxidation yielding a fluorescent adduct that is trapped inside the cells without leakage (regardless of whether the cells are damaged or have compromised membranes). MitoSOX Red (Invitrogen) was utilized in parallel assays for mROS measurements. MitoSOX Red was prepared similarly as CM-H2DCFDA and added to a final concentration of 10 μM. Cells were pre-labeled with MitoSOX Red for a period of 30 min at 37°C, washed twice with HBSS and the media was replaced with fresh medium.
The fluorescence intensities of CM-H2DCFDA (green) or MitoSOX Red, (red) inside living cells were analyzed using an epifluorescence (DM IRE2 HC inverted scope, Leica Microsystems GmbH) microscopy equipped with DIC. The single exposure images were collected sequentially in fluorescence and DIC modes by a cooled CCD camera (Qimaging). The DIC verified the total number of cells in the samples. The fluorescent intensities were quantified by NIH-Image J analysis as previously described (Yilmaz et al., 2010). Untreated and uninfected cells were used as control to set up the background values, and 100 μM H2O2 treated samples were used as positive control. Results are representative images of 400 cells studied per sample from at least three individual experiments performed in duplicate. We also employed cytofluorimetry for the 24 h samples to further evaluate the live cell global ROS measurements as we described previously (Cruz et al., 2007). The simultaneous Trypan blue dye exclusion staining was performed to confirm the host cell viability. Additional light microscopy (phase-contrast) analysis was employed to further verify cell numbers and examine cell morphology and size before and after treatments.
Generation of P2X7 and Pannexin-1 Knockdown Epithelial Cells
HIGK (human immortalized gingival keratinocytes) cell line (Oda et al., 1996) stably expressing shRNA against P2X7 (TRCN0000045095 and TRCN0000045097), and Pannexin-1 (TRCN0000156046 and TRCN0000155348) were generated by transducing the cells with lentiviral particles purchased from Sigma. Transduction was performed following the manufacturer’s instructions. Non-target shRNA control cells were also generated using an irrelevant sequence (SHC002V, Sigma). Briefly, cells were plated at ~70% confluence 24 h prior to transduction and then the corresponding lentiviral transduction particles were added at m.o.i. of 3 overnight. Fresh media were added the next day, and stable infected cells were selected by addition of media containing 5 μg/ml puromycin (Sigma).
Western-blot analysis was performed to verify the knockdown. The samples were subjected to 10% SDS-PAGE gel electrophoresis. Proteins were blotted onto nitrocellulose membrane, blocked in 5% dried milk solution diluted in Tris-buffered saline (TBS) containing 0.01% Tween 20, incubated with anti-P2X7 antibody at a dilution of 1:1000 (Aviva systems biology), and treated with HRP conjugated secondary antibody at 1:2000 (Cell Signaling). The blot was then stripped and probed with anti-β-tubulin antibody 1:1000 and HRP conjugated secondary antibody (Cell signaling) at 1:2000 used as control. Results were visualized by an ECL detection kit (Amersham Biosciences).
Measurement of Intracellular Glutathione
The ratio of reduced glutathione (GSH) and oxidized glutathione (GSSG) of the cells was determined using a luminescence-based GSH/GSSG-Glo Assay kit (Promega). This assay is built on the conversion of a luciferin derivative into luciferin, catalyzed by glutathione S-transferase in the presence of glutathione. Luminescence signal of each sample was measured with a multi-well plate reader and GSSG/GSH ratio was calculated in accordance with the procedure provided. Luminescence for each sample homogenate was converted to GSH concentration based on a standard curve created by serial dilution of a 5 mM GSH standard in the homogenization buffer per the manufacturer’s instructions. The experiments were repeated at least two times in duplicate.
ThiolTracker Violet stain (Invitrogen) was utilized to quantify the intracellular GSH in living cells, since GSH represents the majority of intracellular free thiols in the cell. Briefly, primary GECs were treated with w±/o 3 mM ATP and/or infected with P. gingivalis strains in 6-well microscopic slide chamber system (Warner instruments). After designated incubation period, the cells were washed and incubated in Dulbecco’s PBS containing 10 μM ThiolTracker Violet for 30 min at 37°C. Then the cells were viewed using an epifluorescence microscopy (Zeiss Axio imager A1) and images of cells were taken by a cooled CCD camera (Qimaging).
Analysis of Mitochondrial Uncoupling Protein 2 (UCP2)Gene Transcription
Transcription of ucp2 gene was measured by quantitative real time PCR, using primers specific for human ucp2 gene (5′CCGGGGCCTCTGGAAAG 3′ forward, 5′CCCAAGCGGAGAAAGGA 3′ reverse) and a CFX96 Real-Time PCR Detection System (BioRad). Human gapdh gene (5′GAAATCCCATCACCATCTTCCAGG 3′ forward, 5′GAGCCCCAGCCTTCTCCATG 3′ reverse) was used as an internal control. Briefly, RNA was prepared and 1μg per sample was reverse-transcribed from uninfected, infected, and infected and 5 mM ATP treated primary GECs. cDNA were constructed using Superscript III First Strand cDNA Synthesis Kit (Invitrogen). Quantitative PCR was conducted at 95°C for 3 min, followed by 45 cycles at 95°C for 30 sec, and at 60°C for 30 sec. Gene expression analysis was performed using the CFX Manager Software (BioRad). The measured expression of GADPH was used as a normalization factor for the target gene. For each condition, non-treated and non-infected GECs were assigned as control. The control quantity, which is set to 1, was used by the software to further normalize the relative quantities for the ucp2 gene.
ATP Hydrolysis Assay
The ATPase activity of the supernatants collected from primary GECs infected with P. gingivalis strains following 3, or 6, 12, and 24 h post-infection was measured by monitoring the release of free γ-phosphate (Pi) from ATP in 96-well plates using a high-sensitivity colorimetric ATPase Assay kit (Innova Biosciences) as per manufacturer’s recommendations. 100 μl assay samples were incubated with 0.5 mM ATP for 30 min at room temperature, reaction was terminated, and absorbance at 630 nm was measured. Inorganic phosphate (Pi)standards provided by the company were used to calculate a standard curve of the enzymatic activity. The results were determined by calculating the amount of enzyme that catalyzes the reaction of 1 μM ATP substrate per minute.
Ndk Analysis by Western-blotting
The supernatants collected from the TSB grown P. gingivalis strains’ cultures or culture supernatants of the GECs infected with P. gingivalis strains for the indicated time points, were collected and assayed by a colorimetric BCA protein (Pierce) to determine the protein concentrations. Equal amount of protein containing samples were then precipitated with ammonium sulfate, (NH4)2SO4, (Sigma)using the powder form of (NH4)2SO4by drop-wise to the protein solution method combined with ultrafiltration technique. The samples were subjected to 15% SDS-PAGE gel electrophoresis. Proteins were blotted onto nitrocellulose membrane, blocked in 5% dried milk solution diluted in TBS with 0.01% Tween 20, incubated with purified anti-rNdk polyclonal antibodies at a dilution of 1:500 (GenScript), and treated with HRP conjugated secondary antibody at 1:1000 (Cell Signaling). 1 μg recombinant Ndk (rNdk) used as control (Yilmaz et al, 2008b). Results were visualized by an ECL detection kit (Amersham Biosciences). Densitometric scanning was quantitated using NIH-Image J analysis. The experiments were repeated at least 3 separate times.
Protein sequence analysis was performed using a LC/MS/MS (QSTAR Elite Applied Biosystems). In short, the samples were run on 15% SDS-PAGE gel, followed by Coomassie brilliant blue staining. Protein bands corresponding to the Western-blot were excised and used for protein sequencing analysis. Scaffold 3 (Proteome Software Inc.) was used to validate MS/MS based peptide/protein identification. Protein probabilities were assigned by the Protein Prophet Algorithm.
LDH Viability Assay
A colorimetric Lactate Dehydrogenase (LDH) release assay (Abcam) was simultaneously performed in the GEC culture supernatants infected with P. gingivalis strains to evaluate the loss of cellular integrity as per manufacturer’s recommendations. The cell culture supernatants collected from the GECs incubated with 3 mM ATP or 1 μM staurosporine (Sigma) or uninfected/untreated GECs were used as control. Dilute LDH sample (1:10) provided by the company was used as assay positive control. 20 μl of each sample was added to the substrate solution and the absorbance at 450 nm was measured using a 96-well plate reader.
Antibiotic Protection Assay
Intracellular viability of P. gingivalis strains in GECs was determined by the antibiotic protection assay described previously (Choi et al., 2011). In brief, bacteria in KGM were incubated with GECs in 6-well plates for 24 h at 37°C. 3 mM ATP was added at 3 h post-infection and incubated for another 21 h to complete 24 h incubation. Cells were pre-treated with a broad spectrum ROS inhibitor, NAC (50 mM), 1 h prior to infection or with Apyrase (5U/ml), 2 h prior to infection and the subsequent ATP treatment at 3 h post-infection. After washing with PBS, remaining external bacteria were killed with metronidazole (200 μg ml-1) and gentamicin (300 μg ml-1) and further incubated for 24 h. GECs were washed and lysed with sterile distilled water, and intracellular bacteria were enumerated by culture on blood agar supplemented with haemin and menadione.
Extracellular ATP Release Assay
Extracellular ATP levels were measured by a luciferase-based assay (ATP Bioluminescence Assay kit, Roche Diagnostics) according to the manufacturer’s recommendations. Light emission was measured as relative light units (rlu) and absorbance from replicate samples of uninfected cell supernatants was subtracted from each sample.
Statistical Analysis
Data are presented as mean ± standard deviation (SD). Statistical significance was analyzed by using, where appropriate, a two-tailed Student’s t-test. P values less or equal than 0.01 were considered statistically significant.
Acknowledgments
This study was supported by NIDCR grants R01DE016593 (to O.Y) and R01DE019444 (to D.M.O). We also would like to thank to Dr. Najwane Said-Sadier (UC Merced) for preparation of the P2X7 and Pannexin-1 knockdown cells.
References
- Abdul-Sater AA, Said-Sadier N, Lam VM, Singh B, Pettengill MA, Soares F, et al. Enhancement of reactive oxygen species production and chlamydial infection by the mitochondrial Nod-like family member NLRX1. The Journal of biological chemistry. 2010;285:41637–41645. doi: 10.1074/jbc.M110.137885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed KA, Sawa T, Ihara H, Kasamatsu S, Yoshitake J, Rahaman MM, et al. Regulation by mitochondrial superoxide and NADPH oxidase of cellular formation of nitrated cyclic GMP: potential implications for ROS signalling. The Biochemical journal. 2012;441:719–730. doi: 10.1042/BJ20111130. [DOI] [PubMed] [Google Scholar]
- Avila M, Ojcius DM, Yilmaz O. The oral microbiota: living with a permanent guest. DNA Cell Biol. 2009;28:405–411. doi: 10.1089/dna.2009.0874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- Boisvert H, Duncan MJ. Translocation of Porphyromonas gingivalis gingipain adhesin peptide A44 to host mitochondria prevents apoptosis. Infection and immunity. 2010;78:3616–3624. doi: 10.1128/IAI.00187-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boots AW, Hristova M, Kasahara DI, Haenen GR, Bast A, van der Vliet A. ATP-mediated activation of the NADPH oxidase DUOX1 mediates airway epithelial responses to bacterial stimuli. The Journal of biological chemistry. 2009;284:17858–17867. doi: 10.1074/jbc.M809761200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan N, Ahswin H, Smart N, Bayon Y, Wohlert S, Hunt JA. Reactive oxygen species (ROS) - a family of fate deciding molecules pivotal in constructive inflammation and wound healing. Eur Cell Mater. 2012;24:249–265. doi: 10.22203/ecm.v024a18. [DOI] [PubMed] [Google Scholar]
- Cha B, Lim JW, Kim KH, Kim H. HSP90beta interacts with Rac1 to activate NADPH oxidase in Helicobacter pylori-infected gastric epithelial cells. The international journal of biochemistry & cell biology. 2010;42:1455–1461. doi: 10.1016/j.biocel.2010.04.015. [DOI] [PubMed] [Google Scholar]
- Chakrabarty AM. Nucleoside diphosphate kinase: role in bacterial growth, virulence, cell signalling and polysaccharide synthesis. Molecular microbiology. 1998;28:875–882. doi: 10.1046/j.1365-2958.1998.00846.x. [DOI] [PubMed] [Google Scholar]
- Chaves SP, Torres-Santos EC, Marques C, Figliuolo VR, Persechini PM, Coutinho-Silva R, Rossi-Bergmann B. Modulation of P2X(7) purinergic receptor in macrophages by Leishmania amazonensis and its role in parasite elimination. Microbes Infect. 2009;11:842–849. doi: 10.1016/j.micinf.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, et al. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature. 2010;467:863–867. doi: 10.1038/nature09413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Brosnan CF. Regulation of immune response by P2X7 receptor. Critical reviews in immunology. 2006;26:499–513. doi: 10.1615/critrevimmunol.v26.i6.30. [DOI] [PubMed] [Google Scholar]
- Choi CH, DeGuzman JV, Lamont RJ, Yilmaz O. Genetic transformation of an obligate anaerobe, P. gingivalis for FMN-green fluorescent protein expression in studying host-microbe interaction. PLoS One. 2011;6:e18499. doi: 10.1371/journal.pone.0018499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu AC, Ho PW, Kwok KH, Ho JW, Chan KH, Liu HF, et al. Mitochondrial UCP4 attenuates MPP+ - and dopamine-induced oxidative stress, mitochondrial depolarization, and ATP deficiency in neurons and is interlinked with UCP2 expression. Free radical biology & medicine. 2009;46:810–820. doi: 10.1016/j.freeradbiomed.2008.12.015. [DOI] [PubMed] [Google Scholar]
- Colombo AV, da Silva CM, Haffajee A, Colombo AP. Identification of intracellular oral species within human crevicular epithelial cells from subjects with chronic periodontitis by fluorescence in situ hybridization. Journal of periodontal research. 2007;42:236–243. doi: 10.1111/j.1600-0765.2006.00938.x. [DOI] [PubMed] [Google Scholar]
- Coutinho-Silva R, Correa G, Sater AA, Ojcius DM. The P2X(7) receptor and intracellular pathogens: a continuing struggle. Purinergic Signal. 2009;5:197–204. doi: 10.1007/s11302-009-9130-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutinho-Silva R, Ojcius DM. Role of extracellular nucleotides in the immune response against intracellular bacteria and protozoan parasites. Microbes Infect. 2012 doi: 10.1016/j.micinf.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz CM, Rinna A, Forman HJ, Ventura AL, Persechini PM, Ojcius DM. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J Biol Chem. 2007;282:2871–2879. doi: 10.1074/jbc.M608083200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Autreaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007;8:813–824. doi: 10.1038/nrm2256. [DOI] [PubMed] [Google Scholar]
- Darveau RP, Belton CM, Reife RA, Lamont RJ. Local chemokine paralysis, a novel pathogenic mechanism for Porphyromonas gingivalis. Infection and immunity. 1998;66:1660–1665. doi: 10.1128/iai.66.4.1660-1665.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng S, Yang Y, Han Y, Li X, Wang X, Zhang Z, Wang Y. UCP2 inhibits ROS-mediated apoptosis in A549 under hypoxic conditions. PLoS One. 2012;7:e30714. doi: 10.1371/journal.pone.0030714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emre Y, Nubel T. Uncoupling protein UCP2: when mitochondrial activity meets immunity. FEBS letters. 2010;584:1437–1442. doi: 10.1016/j.febslet.2010.03.014. [DOI] [PubMed] [Google Scholar]
- Fernandez-Checa JC, Garcia-Ruiz C, Colell A, Morales A, Mari M, Miranda M, Ardite E. Oxidative stress: role of mitochondria and protection by glutathione. Biofactors. 1998;8:7–11. doi: 10.1002/biof.5520080102. [DOI] [PubMed] [Google Scholar]
- Ferrari D, Pizzirani C, Adinolfi E, Lemoli RM, Curti A, Idzko M, et al. The P2X7 receptor: a key player in IL-1 processing and release. Journal of immunology. 2006;176:3877–3883. doi: 10.4049/jimmunol.176.7.3877. [DOI] [PubMed] [Google Scholar]
- Forman HJ, Maiorino M, Ursini F. Signaling functions of reactive oxygen species. Biochemistry. 2010;49:835–842. doi: 10.1021/bi9020378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchi L, Kanneganti TD, Dubyak GR, Nunez G. Differential requirement of P2X7 receptor and intracellular K+ for caspase-1 activation induced by intracellular and extracellular bacteria. J Biol Chem. 2007;282:18810–18818. doi: 10.1074/jbc.M610762200. [DOI] [PubMed] [Google Scholar]
- Hajishengallis G. Porphyromonas gingivalis-host interactions: open war or intelligent guerilla tactics? Microbes Infect. 2009;11:637–645. doi: 10.1016/j.micinf.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, et al. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe. 2011;10:497–506. doi: 10.1016/j.chom.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan N, Yusuf N, Toossi Z, Islam N. Suppression of Mycobacterium tuberculosis induced reactive oxygen species (ROS) and TNF-alpha mRNA expression in human monocytes by allicin. FEBS letters. 2006;580:2517–2522. doi: 10.1016/j.febslet.2006.03.071. [DOI] [PubMed] [Google Scholar]
- Hasegawa Y, Tribble GD, Baker HV, Mans JJ, Handfield M, Lamont RJ. Role of Porphyromonas gingivalis SerB in gingival epithelial cell cytoskeletal remodeling and cytokine production. Infect Immun. 2008;76:2420–2427. doi: 10.1128/IAI.00156-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry LG, McKenzie RM, Robles A, Fletcher HM. Oxidative stress resistance in Porphyromonas gingivalis. Future microbiology. 2012;7:497–512. doi: 10.2217/fmb.12.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefferies H, Coster J, Khalil A, Bot J, McCauley RD, Hall JC. Glutathione. ANZ J Surg. 2003;73:517–522. doi: 10.1046/j.1445-1433.2003.02682.x. [DOI] [PubMed] [Google Scholar]
- Jenkinson HF, Lamont RJ. Oral microbial communities in sickness and in health. Trends in microbiology. 2005;13:589–595. doi: 10.1016/j.tim.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Kamath S, Chen ML, Chakrabarty AM. Secretion of nucleoside diphosphate kinase by mucoid Pseudomonas aeruginosa 8821: involvement of a carboxy-terminal motif in secretion. Journal of bacteriology. 2000;182:3826–3831. doi: 10.1128/jb.182.13.3826-3831.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuboniwa M, Hasegawa Y, Mao S, Shizukuishi S, Amano A, Lamont RJ, Yilmaz O. P. gingivalis accelerates gingival epithelial cell progression through the cell cycle. Microbes and infection/Institut Pasteur. 2008;10:122–128. doi: 10.1016/j.micinf.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammas DA, Stober C, Harvey CJ, Kendrick N, Panchalingam S, Kumararatne DS. ATP-induced killing of mycobacteria by human macrophages is mediated by purinergic P2Z(P2X7) receptors. Immunity. 1997;7:433–444. doi: 10.1016/s1074-7613(00)80364-7. [DOI] [PubMed] [Google Scholar]
- Lees MP, Fuller SJ, McLeod R, Boulter NR, Miller CM, Zakrzewski AM, et al. P2X7 receptor-mediated killing of an intracellular parasite, Toxoplasma gondii, by human and murine macrophages. J Immunol. 2010;184:7040–7046. doi: 10.4049/jimmunol.1000012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancino G, Placido R, Di Virgilio F. P2X7 receptors and apoptosis in tuberculosis infection. Journal of biological regulators and homeostatic agents. 2001;15:286–293. [PubMed] [Google Scholar]
- Mandavilli BS, Janes MS. Detection of intracellular glutathione using ThiolTracker violet stain and fluorescence microscopy. Curr Protoc Cytom. 2010;Chapter 9(Unit 9 35) doi: 10.1002/0471142956.cy0935s53. [DOI] [PubMed] [Google Scholar]
- Mao S, Park Y, Hasegawa Y, Tribble GD, James CE, Handfield M, et al. Intrinsic apoptotic pathways of gingival epithelial cells modulated by Porphyromonas gingivalis. Cell Microbiol. 2007;9:1997–2007. doi: 10.1111/j.1462-5822.2007.00931.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie RM, Johnson NA, Aruni W, Dou Y, Masinde G, Fletcher HM. Differential response of Porphyromonas gingivalis to varying levels and duration of hydrogen peroxide-induced oxidative stress. Microbiology. 2012;158:2465–2479. doi: 10.1099/mic.0.056416-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CM, Boulter NR, Fuller SJ, Zakrzewski AM, Lees MP, Saunders BM, et al. The role of the P2X(7) receptor in infectious diseases. PLoS Pathog. 2011;7:e1002212. doi: 10.1371/journal.ppat.1002212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda D, Bigler L, Lee P, Blanton R. HPV immortalization of human oral epithelial cells: a model for carcinogenesis. Exp Cell Res. 1996;226:164–169. doi: 10.1006/excr.1996.0215. [DOI] [PubMed] [Google Scholar]
- Page AN, George NP, Marceau AH, Cox MM, Keck JL. Structure and biochemical activities of Escherichia coli MgsA. The Journal of biological chemistry. 2011;286:12075–12085. doi: 10.1074/jbc.M110.210187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pourova J, Kottova M, Voprsalova M, Pour M. Reactive oxygen and nitrogen species in normal physiological processes. Acta physiologica. 2010;198:15–35. doi: 10.1111/j.1748-1716.2009.02039.x. [DOI] [PubMed] [Google Scholar]
- Rudney JD, Chen R, Sedgewick GJ. Intracellular Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis in buccal epithelial cells collected from human subjects. Infection and immunity. 2001;69:2700–2707. doi: 10.1128/IAI.69.4.2700-2707.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Said-Sadier N, Padilla E, Langsley G, Ojcius DM. Aspergillus fumigatus stimulates the NLRP3 inflammasome through a pathway requiring ROS production and the Syk tyrosine kinase. PLoS One. 2010;5:e10008. doi: 10.1371/journal.pone.0010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandilos JK, Chiu YH, Chekeni FB, Armstrong AJ, Walk SF, Ravichandran KS, Bayliss DA. Pannexin 1, an ATP release channel, is activated by caspase cleavage of its pore-associated C-terminal autoinhibitory region. The Journal of biological chemistry. 2012;287:11303–11311. doi: 10.1074/jbc.M111.323378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Searle BC. Scaffold: a bioinformatic tool for validating MS/MS-based proteomic studies. Proteomics. 2010;10:1265–1269. doi: 10.1002/pmic.200900437. [DOI] [PubMed] [Google Scholar]
- Spooner R, Yilmaz O. The role of reactive-oxygen-species in microbial persistence and inflammation. Int J Mol Sci. 2011;12:334–352. doi: 10.3390/ijms12010334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spooner R, Yilmaz O. Nucleoside-diphosphate-kinase: a pleiotropic effector in microbial colonization under interdisciplinary characterization. Microbes Infect. 2012;14:228–237. doi: 10.1016/j.micinf.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridharan M, Adderley SP, Bowles EA, Egan TM, Stephenson AH, Ellsworth ML, Sprague RS. Pannexin 1 is the conduit for low oxygen tension-induced ATP release from human erythrocytes. American journal of physiology Heart and circulatory physiology. 2010;299:H1146–1152. doi: 10.1152/ajpheart.00301.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Wang X, Lau A, Liao TY, Bucci C, Hmama Z. Mycobacterial nucleoside diphosphate kinase blocks phagosome maturation in murine RAW 264.7 macrophages. PLoS One. 2010;5:e8769. doi: 10.1371/journal.pone.0008769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tribble GD, Mao S, James CE, Lamont RJ. A Porphyromonas gingivalis haloacid dehalogenase family phosphatase interacts with human phosphoproteins and is important for invasion. Proc Natl Acad Sci U S A. 2006;103:11027–11032. doi: 10.1073/pnas.0509813103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuiki H, Nitta M, Furuya A, Hanai N, Fujiwara T, Inagaki M, et al. A novel human nucleoside diphosphate (NDP) kinase, Nm23-H6, localizes in mitochondria and affects cytokinesis. Journal of cellular biochemistry. 1999;76:254–269. doi: 10.1002/(sici)1097-4644(20000201)76:2<254::aid-jcb9>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Wiley JS, Sluyter R, Gu BJ, Stokes L, Fuller SJ. The human P2X7 receptor and its role in innate immunity. Tissue Antigens. 2011;78:321–332. doi: 10.1111/j.1399-0039.2011.01780.x. [DOI] [PubMed] [Google Scholar]
- Wosniak J, Jr, Santos CX, Kowaltowski AJ, Laurindo FR. Cross-talk between mitochondria and NADPH oxidase: effects of mild mitochondrial dysfunction on angiotensin II-mediated increase in Nox isoform expression and activity in vascular smooth muscle cells. Antioxidants & redox signaling. 2009;11:1265–1278. doi: 10.1089/ars.2009.2392. [DOI] [PubMed] [Google Scholar]
- Xia J, Lim JC, Lu W, Beckel JM, Macarak EJ, Laties AM, Mitchell CH. Neurons respond directly to mechanical deformation with pannexin-mediated ATP release and autostimulation of P2X7 receptors. The Journal of physiology. 2012;590:2285–2304. doi: 10.1113/jphysiol.2012.227983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CS, Lee HM, Lee JY, Kim JA, Lee SJ, Shin DM, et al. Reactive oxygen species and p47phox activation are essential for the Mycobacterium tuberculosis-induced pro-inflammatory response in murine microglia. Journal of neuroinflammation. 2007;4:27. doi: 10.1186/1742-2094-4-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao L, Jermanus C, Barbetta B, Choi C, Verbeke P, Ojcius DM, Yilmaz O. Porphyromonas gingivalis infection sequesters pro-apoptotic Bad through Akt in primary gingival epithelial cells. Molecular oral microbiology. 2010;25:89–101. doi: 10.1111/j.2041-1014.2010.00569.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz O. The chronicles of Porphyromonas gingivalis: the microbium, the human oral epithelium and their interplay. Microbiology. 2008a;154:2897–2903. doi: 10.1099/mic.0.2008/021220-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz O, Yao L, Maeda K, Rose TM, Lewis EL, Duman M, et al. ATP scavenging by the intracellular pathogen Porphyromonas gingivalis inhibits P2X7-mediated host-cell apoptosis. Cellular microbiology. 2008b;10:863–875. doi: 10.1111/j.1462-5822.2007.01089.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz O, Jungas T, Verbeke P, Ojcius DM. Activation of the phosphatidylinositol 3-kinase/Akt pathway contributes to survival of primary epithelial cells infected with the periodontal pathogen Porphyromonas gingivalis. Infect Immun. 2004;72:3743–3751. doi: 10.1128/IAI.72.7.3743-3751.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz O, Sater AA, Yao L, Koutouzis T, Pettengill M, Ojcius DM. ATP-dependent activation of an inflammasome in primary gingival epithelial cells infected by Porphyromonas gingivalis. Cellular microbiology. 2010;12:188–198. doi: 10.1111/j.1462-5822.2009.01390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz O, Verbeke P, Lamont RJ, Ojcius DM. Intercellular spreading of Porphyromonas gingivalis infection in primary gingival epithelial cells. Infection and immunity. 2006;74:703–710. doi: 10.1128/IAI.74.1.703-710.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz O, Watanabe K, Lamont RJ. Involvement of integrins in fimbriae-mediated binding and invasion by Porphyromonas gingivalis. Cellular microbiology. 2002;4:305–314. doi: 10.1046/j.1462-5822.2002.00192.x. [DOI] [PubMed] [Google Scholar]
- Zaborina O, Li X, Cheng G, Kapatral V, Chakrabarty AM. Secretion of ATP-utilizing enzymes, nucleoside diphosphate kinase and ATPase, by Mycobacterium bovis BCG: sequestration of ATP from macrophage P2Z receptors? Molecular microbiology. 1999;31:1333–1343. doi: 10.1046/j.1365-2958.1999.01240.x. [DOI] [PubMed] [Google Scholar]