

Abstract
Systemic lupus erythematosus (SLE) is characterized by imbalance redox state and increased apoptosis. The activation, proliferation and cell death of lymphocytes are dependent on intracellular levels of glutathione and controlled production of reactive oxygen species (ROS). Changes in the intracellular redox environment of cells, through oxygen-derived free radical production known as oxidative stress, have been reported to be critical for cellular immune dysfunction, activation of apoptotic enzymes and apoptosis. The shift in the cellular GSH-to-GSSG redox balance in favor of the oxidized species, GSSG, constitutes an important signal that can decide the fate of the abnormal apoptosis in the disease. The current review will focus on four main areas: (1) general description of oxidative stress markers in SLE, (2) alteration of redox state and complication of disease (3) role of redox mechanisms in the initiation and execution phases of apoptosis, and (4) intracellular glutathione and its checkpoints with lymphocyte apoptosis represent novel targets for pharmacological intervention in SLE.
Keywords: Reactive oxygen species, redox state, apoptosis, glutathione, systemic lupus erythematosus (SLE)
1. Introduction
Systemic lupus erythematosus is a multifactorial autoimmune disease characterized by the presence of autoantibodies, especially against nuclear components. The assortments of autoantibodies produced are broad and as a consequence the manifestations of the disease are diverse [1]. Although it is believed that the etiology of SLE is multifactorial, including genetic, hormonal and environmental triggers, the molecular mechanisms underlying this systemic autoimmune response remain largely unknown. A key issue in the pathogenesis of lupus is how intracellular antigens become exposed and targeted by the immune system [2]. In this regard, excessive production of ROS, altered redox state [3] and a defect in regulation of apoptosis [4] are considered as imperative factors involved in the production, expansion of antibody flares and various clinical features in SLE. The depletion of intracellular glutathione is an indicator for ROS formation and may be involved in dysregulation of apoptosis in disease [5]. The oxidative damage mediated by ROS resulting in the defect in control of apoptosis or programmed cell death and delayed clearance of apoptotic cells may prolong interaction between ROS and apoptotic cell macromolecules generating neoepitopes that subsequently broad spectrum of autoantibody formation leading to the tissue damage in SLE [6,7]. An increase in MDA-modified proteins, anti-SOD and anti-catalase antibodies in the sera of SLE patients support a critical role for oxidative stress in disease development. The positive relationships between oxidative stress markers and apoptosis reinforce the contribution of oxidative stress in the perturbation of apoptosis in SLE [8,9]. This chapter the role of redox state in apoptosis perturbation, exploring how the imbalance in redox status not only can directly responsible for tissue damage, but also promote development through the imbalance of immunological tolerance and the generation of neo-antigenic determinants from one side and of autoantibodies from the other.
2. Oxidative stress and its biomarkers for SLE
2.1. Reactive oxygen species (ROS)
A diverse number of stimuli have been shown to induce apoptosis, many of which are also known to compromise the fine balance between intracellular oxidants and their defense systems. Under aerobic condition, the participation of oxygen in redox reactions is unavoidable and a variety of highly reactive chemical entities are produced. These are commonly referred to as ROS. ROS are short-lived molecules produced by normal cellular metabolism and aid in a multitude of physiological and pathological processes. ROS and reactive nitrogen species (RNS) are well recognized for playing a dual role as both deleterious and beneficial species, since they can be either harmful or beneficial to living systems [10–12]. Beneficial effects of ROS occur at low/moderate concentrations and are associated with cellular responses to noxia, for example in defense against infectious agents and in cellular signaling. Another example of ROS at low/moderate concentrations is the induction of a mitogenic response. The harmful effect of free radicals causing potential biological damage is termed oxidative stress and nitrosative stress [13,14].This occurs when there is an overproduction of ROS/RNS or a deficiency of enzymatic and non-enzymatic antioxidants. Reactive intermediates are produced by reactions involving enzymes such as nicotinamide adenine dinucleotide phosphate (NADPH) [15], nitric oxide synthase [16], or by nonenzymatic reactions through the mitochondrial electron transport chain [17], and reduced transition metals [18]. ROS include, superoxide anion radical (O2−·), peroxy radical (ROO−), hydrogen peroxide (H2O2), singlet oxygen (1O2), perhydroxyl radical (HO2·) and extremely reactive ·OH, Table 1.
TABLE 1.
List of important reactive oxygen intermediates and their sources and modes of action
| Source | Modes of action | |
|---|---|---|
| Super oxide (SO) | NADPH oxidase | Oxidative stress |
| Xanthine oxidase | Redox signaling | |
| Complex I/complex III (mitochondria) | ||
| 5-lipoxygenase | ||
| Cyclooxygenase | ||
| Uncoupled nitric oxide synthase | ||
| Hydrogen peroxide (H2O2) | Peroxisomes | Redox signaling |
| Super oxide dismutase | Oxidative stress | |
| Hydroxyl radical OH | Fenton reaction | Oxidative stress |
| Haber-Weiss reaction | ||
| Hypochlorous acid (HOCL) | Myeloperoxidase (MPO) | Chlorination |
| Oxidative stress |
2.2. Reactive Nitrogen species (NOS)
ROS can also interact with nitric oxide (NO), the product of NO synthases, whose expression is usually accompanied by inflammatory lesion. These results in the conversion of NO to various reactive nitrogen species which include nitrosonium cation (NO+), nitroxyl anion (NO−) and peroxynitrite (ONOO·) (Table 2).
TABLE 2.
List of important reactive nitrogen intermediates and their sources and modes of action
| Source | Modes of action/effects | |
|---|---|---|
| Nitric oxide (NO) | Nitric oxide synthase | Nitrosylation of metal centers |
| Redox signaling | ||
| Peroxynitrite (ONOO−) | Reaction between NO and SO | Nitration |
| Oxidation | ||
| Oxidative stress | ||
| Nitrogen dioxide radical (NO2·) | Reaction between NO and O2 | Nitration |
| Oxidation | ||
| Oxidative stress | ||
| Dinitrogen trioxide (N2O3) | Reaction between NO and NO2 | Nitrosation |
| Nitrosative stress |
2.3. Sources of Oxygen Radicals and their scavengers
The source of oxygen radicals can be endogenous as well as exogenous. as well as endogenous sources include non-enzymatic reactions through the mitochondrial electron transport chain during oxidative metabolism to generate ATP [19]. It has been estimated that 3–5% of total electron flux results in the formation of ROS, which in a typical human (even at rest) corresponds to the production of approximately 2 kg of O2−· per annum. Additional sources of O2−· generation come from nicotinamide adenine dinucleotide phosphate oxidase (NADPH) [15], nitric oxide synthase in peroxisomes [20], neutrophils (oxidative burst) [21], lysosome and microsomes [22,23]. Hydroxyl radical is an extremely reactive oxidant, which are generated as a result of interaction of H2O2 with transition metal ions such as Fe2+ or Cu2+ (Fenton reaction) and O2 (Haber-Weiss reaction) as well as from peroxynitrite which is rapidly formed via the reaction between NO· and O2−· Hydrogen peroxide is formed through dismutation of O2−· catalyzed by the enzyme superoxide dismutase, and is also produced by several other oxidase enzymes (e.g. amino acid oxidases) (Figure 1). Likewise, activated phagocytes are capable of generating O2−· H2O2 and NO· species (as well as hypochlorous acid, HOCl), all of which may contribute to tissue injury during inflammation. In vivo, peroxynitrite is formed in macrophages, endothelial cells, platelets, leukocytes, neurons, etc by the reaction between O2−· and NO· [24,25]. Tissue inflammation and chronic infection lead to the overproduction of O2−· and NO·, which rapidly combine to yield peroxynitrite: O2−· + NO·→ ONO2−·. In addition, ROS may amplify the inflammation process by gene involved in the inflammatory response, particularly via activation of the nuclear transcription factor NF-kβ, which in turn up regulates pro-inflammatory cytokines and leukocyte adhesion molecules. Exogenous sources include radiation, UV light, x-rays, gamma rays, chemicals that react to form peroxides, ozone and singlet oxygen, chemicals that promote superoxide formation, quinones, nitroaromatics, bipyrimidiulium herbicides, chemicals that are metabolized to radicals e.g., polyhalogenated alkanes, phenols, aminophenols etc. [26–28].
Figure 1.

Endogenous sources of reactive oxygen species. Major endogenous sources of ROS production include a variety of metallo-enzymes (such as NAD(P)H oxidase and nitric oxide synthase), mitochondrial complexes I and III, transitional metals (Fe2+ and Cu2+).
The formation of superoxide anion radical leads to a cascade of other ROS. Superoxide dismutate catalyzes the conversion of superoxide anion to H2O2 and oxygen. Without SOD this reaction is spontaneous and rapid, but the SOD-catalyzed reaction is four orders of magnitude faster. Clearly, O2−· is more toxic than H2O2 and its rapid removal is critical. H2O2 is reduced by three general mechanisms (Figure 2). First, it is the substrate for two enzymes, catalase and glutathione peroxidase, which catalyze the conversion of H2O2 to H2O + O2; this presumably is functions as a detoxification mechanism. Second, H2O2 is converted by myeloperoxidase (MPO) in neutrophils to hypochlorous acid (HOCl). This appears to be a mechanism for a physiological toxic agent, since HOCl is a strong oxidant that acts as a bactericidal agent in phagocytic cells. Reaction of HOCl with H2O2 yields singlet oxygen (1O2) and water. The biological significance of singlet oxygen is unclear. Third, H2O2 is converted in a spontaneous reaction catalyzed by Fe2+ (Fenton reaction) to the highly reactive ·OH. The hydroxyl radical reacts promptly with any biological molecule like lipid, protein and DNA and cause severe consequence in the pathogenesis of SLE.
Figure 2.
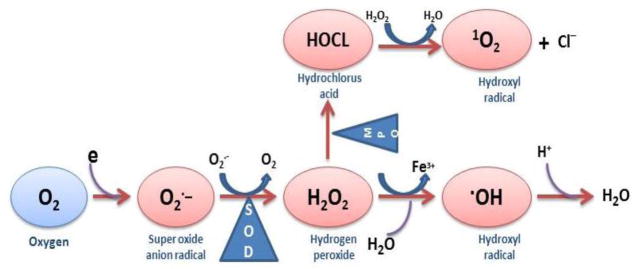
Formation of superoxide anion radical leads to a cascade of other ROS.
2.4. Validated Biomarkers of Oxidative stress
Oxidation and the production of free radicals are an integral part of human metabolism, and oxidative stress is related to many diseases, including autoimmune disease like SLE. The use of biomarkers for oxidative stress may provide further evidence of a causal relationship between oxidative damage to macromolecules (DNA, lipids, and proteins). Biomarkers may yield information relevant to disease outcome on three progressive levels: (i) as measurable endpoints of damage to proteins/amino acids, oxidised lipids, oxidised DNA bases, (ii) as functional markers of, for example, blood flow, platelet aggregation, or cognitive function, and (iii) as endpoints related to specific disease. A valid biomarker should be:
a major product of oxidative modification that may be implicated directly in the development of disease. The reactive oxygen species (or the oxidative damage it causes) should always be demonstrable at the site of injury.
a stable product, not susceptible to artifactual induction or loss during storage.
representative of the balance between oxidative damage generation and clearance of ROS (i.e. the steady state, but also possibly applicable to the measurement of cumulative oxidative damage).
determined by an assay that is specific, sensitive, reproducible and robust.
free of confounding factors from dietary intake.
accessible in a target tissue or a valid surrogate tissue such as a leukocyte and
measurable within the limits of detection of a reliable analytical procedure.
A wide variety of functional assays, both in vivo and ex vivo, include various measures of total reactive oxygen species (ROS measured in blood components; erythrocytes, lymphocytes, neutrophils), lipid oxidation (thiobarbituric acid-reactive substances, exhaled pentane/ethane, low-density lipoprotein resistance to oxidation, isoprostanes), protein oxidation (protein carbonyls) and DNA oxidation (oxidized DNA bases such as 8-OHdG, autoantibodies to oxidized DNA, modified comet assay) Table-3. Besides these markers of oxidative stress, the intracellular levels of glutathione are considered as a reliable and novel marker for measuring oxidative stress due to its close association with immune regulation in autoimmune disease.
TABLE 3.
List of Oxidative stress Biomarkers and their methods of detection
| Markers of Oxidative Stress | Markers/probes | Detection Methods |
|---|---|---|
| Reactive Oxygen Species | 2′,7′dichlorofluorescein (DCF) | Spectroflurometry, Flow cytometry |
| Nitric oxide | 2,3-naphthotriazole (NAT) | Spectroflurometry |
| Lipid peroxidation | Malonaldehyde TBARS | TBARS-spectrophotometric assay |
| HPLC-based TBARS assay | ||
| GC-MS | ||
| F2-isoprostanes (8-iso-PGF2) | Immunoassays (ELISA, Blotting) | |
| 4-Hydroxynonenal (4-HNE) | GC-MS, LC-MS | |
| Proteins carbonyl Groups | carbonyl derivatives of proline, lysine, arginine and threonine residues | DNPH spectrophotometric assay |
| Immunoassays (ELISA, Blotting) | ||
| One- and two-dimensional electrophoresis | ||
| Mass Spectroscopy | ||
| DNA Damage | 8-hydroxy-2′-deoxyguanosine | Immunoassays (ELISA, Blotting) |
| HPLC-ECD | ||
| LC-MS, GC-MS | ||
| DNA strand breaks | Comet assay | |
| Flow cytometry | ||
| Total anti-oxidant capacity | 2,2′-azinobis(3-ethylbenzthiazoline-6-sulphonic acid) | Spectrophotometry |
| Chemiluminescence assay | ||
| Anti-oxidant molecule/enzymes | Spectrophotometry | |
| Glutathione | 5, 5′ dithio-bis 2-nitrobenzoic acid (DTNB) | Chemiluminescence assay |
| Mercury orange | HPLC | |
| Flow cytometry | ||
| Superoxide dismutase | Nitroblue tetrazolium (NBT) | Spectrophotometry |
| Flow cytometry | ||
| Anti-SOD antibody | ||
| Catalase | H202 | Spectrophotometry |
| Anti-catalase antibody | Flow cytometry | |
| Glutathione Peroxidase | NADPH | Spectrophotometry |
| Glutathione Reductase | Spectrophotometry | |
| Non-enzymatic antioxidants | Spectrophotometry | |
| Vitamin C, Vitamin E, Vitamin C and Vitamin A (carotenoids) | Chemiluminescence assay | |
| Proteins like Albumin, Transferrin, Cerolopasmin. Pyruvate. Ubiquinol, Bilirubin | HPLC |
3. Alteration of redox state and its effect on SLE pathogenic mechanism
3.1. Oxidative stress and SLE
Oxidative stress is believed to play a major role in the initiation and progression of autoimmune disease by excessive free radical formation. An increase in ROS production or a decrease in ROS-scavenging capacity due to exogenous stimuli or endogenous metabolic alterations can disrupt redox homeostasis, lead to an overall increase intracellular ROS levels, or oxidative stress [29,30]. Among the ROS, ·OH is the most potent damaging radical, and can react with all biological macromolecules (lipids, proteins, nucleic acids and carbohydrates). It can lead to the formation of DNA-protein cross-links, single- and double-strand breaks, base damage, lipid peroxidation and protein fragmentation [31,32] (Figure 3). This oxygen species may penetrate cellular membranes and react with nuclear DNA [7]. Murine models of SLE demonstrate abnormally high levels of ·NO compared with normal mice, whereas systemic blockade of ·NO production reduces disease activity. Elevated serum nitrate levels correlate with indices of disease activity and, along with serum titers of anti-(ds DNA) antibodies, serve as indicators of SLE [33,34]. Excessive oxidative stress is thought to play an important role in the pathogenesis of autoimmune diseases by enhancing inflammation, inducing apoptotic cell death and breaking down immunological tolerance [35,36]. Free radical production and altered redox status can modulate expression of a variety of immune and inflammatory molecules [37] leading to inflammatory processes, exacerbating inflammation and affecting tissue damage [38]. ROS generation also provides oxidant for thiol oxidation or peroxynitrite formation which can be a basis for antibody modification [39]. Convincing evidence for the association of oxidative/nitrosative stress and SLE diseases has been shown by increased levels of validated biomarkers of oxidative stress in the disease. Increased levels of 8-oxodG, a marker of oxidative DNA damage in the immune complex derived DNA, have been found in lymphocytes and serum from SLE patients, reinforcing ROS in disease etiology [40,41]. The level of protein oxidation markers correlating with severity of disease in SLE patients further supports the role of protein oxidation in SLE [42]. Elevated levels of F2 isoprostanes (prostaglandin-like substances derived from lipid peroxidation) in serum and urine from SLE patients have been reported [43]. It has been reported that ·OH, could lead to neoantigens like ·OH damaged human serum albumin (HSA), which in turn could initiate autoimmunity in SLE. These reports support the role of oxidative stress in the pathogenesis of SLE [44,45].
Figure 3.
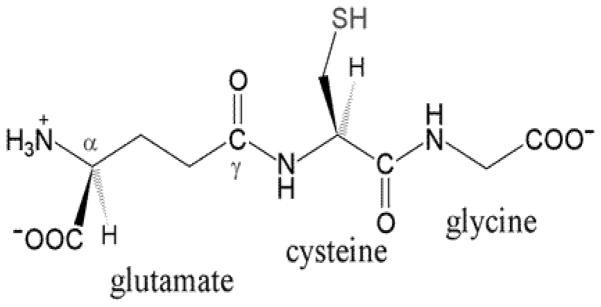
Glutathione (L-γ-glutamyl-L-cysteinyl-glycine) is a linear tripeptide formed from the aminoacids glycine, cysteine, and glutamate.
The primary target of ROS is lipids in the cell membrane and lipid peroxidation (LPO) impairs cell structure and function [7]. An increase in malondialdehyde (MDA), a product of lipid peroxidation, has been reported in serum/plasma/erythrocyte [8,46–49] as well as in lymphocytes [50] in patients with SLE. The increased level of lipid peroxidation was positively correlated with severity of the disease and organ damage especially in nephritis patients [50–53]. All cell types, including lymphocytes and other immune cells, have a complex machinery of antioxidant enzymes (superoxide dismutase, catalase, glutathione peroxidase, glutathione reductase, thioredoxin etc.) and antioxidant molecule (reduced glutathione, vitamins) for regulating oxidant reactions in the cells prevent free radical mediated cytotoxicity [54]. The circulating human erythrocytes are able to scavenge O2·− and H2O2 by SOD, CAT, and GPx-dependent mechanisms may be important in regulating such reactions. The first line of defense against ROS is provided by SOD which catalyzes dismutation of O2·− into H2O2. The H2O2 is then transformed into H2O and O2 by catalase. GPx is a selenoprotein that reduces lipidic or nonlipidic hydroperoxides as well as H2O2 utilizing glutathione [55].
Antioxidant enzymes such as SOD, CAT and GPx have been studied in serum and erythrocytes as well as in lymphocyte in patients with SLE. Decreased activity of SOD has been reported in the serum/plasma/erythrocyte from SLE patients [49,56]. The decreased activity of SOD leads to excessive accumulation of O2·− that would otherwise have been enzymatically converted to H2O2. Increased O2·− levels have potential to initiate the lipid peroxidation chain reaction in patients with SLE. Increased as well as decreased activities of CAT from erythrocytes of SLE patients have been reported by various groups [46,48,49,57]. The activity of glutathione peroxidase is controversial in SLE patients however, and most showed decreased activity of GPx in SLE patients [46,52,57,58].
Adequate concentrations of glutathione are required for a variety of functions, including protection of the cell from oxidative damage quenching of oxidant species, lymphocyte activation, natural killer cell activation and lymphocyte-mediated cytotoxicity [59,60]. The depletion of intracellular glutathione has been associated with many autoimmune inflammatory diseases including SLE [61]. A decrease in the level of intracellular GSH showed a correlation with the severity of disease especially with nephritis patients [48,49]. Decreased intracellular GSH may be ascribed to ROS-induced GSH oxidation or GSH export from cells [62]. In addition to the important role of GSH in the maintenance of redox state in the cell, GSH oxidation is a major contributor to lymphocyte apoptosis mediated by oxidants [63].
3.2. Genetic link of oxidative stress in SLE
Associations between oxidative stress and SLE have generally been investigated by measuring DNA damage, free radicals and anti-oxidants. Both increased ROS levels and deceased intracellular glutathione have been previously reported in conjugation with disease flare. However, greater insight into the relationship between oxidative stress and disease susceptibility may be gained by studying functional polymorphisms in genes that control levels of cellular oxidative damage. Candidate pathways include anti-oxidant and DNA repair mechanisms which are modulated by individual genetic variation. A number of polymorphisms in the genes coding for superoxide dismutase, catalase and glutathione peroxidase (SOD2, CAT and GPX1, respectively) have been identified and associated with SLE risk, however these association have been reported as ethical dependent. The Nrf2-Keap1 pathway is important in protecting against oxidative stress and inflammation. A significant association between the NRF2-653 G/A polymorphism and the development of nephritis among Mexican, childhood-onset female SLE patients [64]. The CAT polymorphism (−330CC genotype) showed a significant association with thrombocytopenia, renal manifestations, as well as production of anti-snRNP and anti-Scl-70 antibodies in SLE patients [65]. A member of S-transferase superfamily, GSTM1 has been reported to be associated with anti-RO antibodies suggesting that the dysfunction of this gene may contribute to the anti-Ro autoantibody response or to photosensitivity [66,67]. However, another group has reported no association of GSTM1 polymorphism with anti-Ro autoantibodies, or clinical features in American, and Mexican-American SLE patients [68]. These reports showed that the polymorphism association of oxidative stress genes ethical dependent and further studies should be directed at evaluating their association.
4. Redox mechanisms and their effect in initiation and execution of apoptosis
4.1. Antioxidant defense system
The effect of ROS is limited by the presence of various regulatory systems that maintain redox homeostasis. A relatively large number of compounds have been shown to possess some measurement of antioxidant activities. They maintain a balance between the production and metabolism of ROS and protect the cell from oxidative damage [29,69]. The antioxidant enzymes include SOD, CAT and glutathione related enzymes; GPx, GR and GST. The non-enzymatic scavengers are vitamins E, C and A and thiol containing compounds such as glutathione [70].
4.1.1. Glutathione
Reduced glutathione (L-γ-glutamyl-L-cysteinylglycine) is the most prevalent cellular thiol and the most abundant low molecular weight peptide present in all cells [71] (Figure 4). The role of GSH as a reductant is extremely important in the highly oxidizing environment of the erythrocyte. GSH levels in human tissues normally range from 0.1 to 10 mM, most concentrated in the liver (upto 10 mM), spleen, kidney, lens, erythrocytes and leucocytes [72]. In healthy cells and tissues, more than 90% of the total glutathione pool is in the reduced form (GSH) and less than 10% exists in the oxidized form (GSSG). Glutathione is required for many critical cellular processes and plays a particularly important role in the maintenance and regulation of the thiol-redox status of the cell [73]. The GSH/GSSG ratio is a useful measurement for determining oxidative stress [74] and changes in this ratio appear to correlate with cell proliferation [75], differentiation [76] and apoptosis [77]. This led to attention to the role of thiol status in the onset and progression of autoimmune and inflammatory diseases, including rheumatoid arthritis [78] and, SLE [61] as well as the effectiveness of thiol repletion therapies in the treatment of these diseases. Cellular GSH levels affect T helper cell maturation [79], T cell proliferation [80], as well as susceptibility to ROS secreted by inflammatory cells. Additionally, many correlations exist between immune system dysfunction and alterations in GSH levels in the cells. It is reported that GSH depletion in antigen presenting cells inhibits Th1-related cytokine production like IFN-γ and IL-12 and supports the Th2- mediated humoral immune response [80]. Furthermore, when antigen presenting cells have high intracellular GSH levels they secrete cytokines that favor the development of Th1 cells [81]. In addition, it is reported that specific cytokines can alter GSH levels in antigen presenting cells [81]. Exposure to IFN-γ, a Th1 cytokine, resulted in increased GSH levels, whereas exposure to IL-4, a Th2 cytokine, resulted in decreased intracellular GSH. Because GSH has a significant impact on the immune system’s ability to activate the appropriate Th response, altering its levels may have significant implications in Th1/Th2-related diseases like SLE.
Figure 4.

Oxidative damage of lipid, protein and DNA by reactive oxygen species. The ROS generated in cells may cause damage to macromolecules including DNA, lipid, and protein.
4.1.2. Superoxide dismutase (SOD)
Superoxide dismutase is a metalloprotein, considered to be the first line of defense against free radicals. It catalyzes the dismutation of superoxide radical into oxygen and hydrogen peroxide [82]. The superoxide radical, if not scavenged effectively, may directly inactivate several enzymes like CAT and GPx which are needed to eliminate hydrogen peroxide from intracellular medium [83]. Three forms of this enzyme found in human: SOD1 located in the cytoplasm, SOD2 in the mitochondria and SOD3 is extracellular [84]. SOD1 is a dimer [82], while SOD2 and SOD3 are tetrameric [85,86]. SOD1 and SOD3 contain copper and zinc, while SOD2 has manganese in its reactive center. Several groups reported decreased activity of SOD and auto-antibody against SOD in SLE patients [46,48,50]. It has been reported that antibodies to SOD are potentially responsible for increased oxidative damage in SLE patients.
4.1.3. Catalase
Catalase is found in peroxisomes (80%) and cytosol (20%) of all aerobic cells. It is responsible for dealing with the large amounts of hydrogen peroxide generated within the cells. Catalase converts hydrogen peroxide to water and oxygen without the production of free radicals [87,88]. The concentration of CAT is highest in the liver, kidney and erythrocyte and low in connective tissues [89]. In these tissues, it is mainly particle bound (mitochondria and peroxisomes), where it exists in soluble state in erythrocytes. Catalase does not show significant activity under physiological conditions, due to its relatively low affinity for hydrogen peroxide but becomes an important enzyme when the concentration of H2O2 is raised e.g., by exposing cells to drugs and chemicals that increase intracellular H2O2 generation [18]. In mice, CAT activity has been found to be related to the biosynthesis of proteins involved in inflammation [90]. The CAT polymorphism (−330CC genotype) showed a significant association with thrombocytopenia, renal manifestations, as well as production of anti-snRNP and anti-Scl-70 antibodies in SLE patients [65]. It has been showed that elevated levels of auto-antibodies against catalase are associated with oxidative stress in patients with SLE [8].
4.1.4. Glutathione peroxidase
Glutathione peroxidase is a tetrameric protein (85 KDa), which has four atoms of selenium bound as selenocysteine moieties that confer catalytic activity [91]. It has a lower Km value for H2O2 than CAT and considered more important when low amounts of H2O2 are generated. It plays an important role in the defense mechanism against oxidative damage in erthrocytes by catalyzing the reduction of H2O2 and variety of lipid hydro-peroxides [92] using GSH as the reducing substrate. In SLE patients, decreased activity of GPx [46] leads to a change in redox ratio in favor of oxidized glutathione. Glutathione, a strong natural antioxidant molecule not only controls oxidative stress of the cells but is also involved in regulation of apoptosis pathway [62] and cytokine network [79] in SLE.
5. Interplay between intracellular glutathione and apoptosis: novel targets for pharmacological intervention in SLE
5.1. Apoptosis and SLE
Apoptosis is a form of actively induced programmed cell death, with the characteristic features of chromatin condensation, DNA fragmentation and apoptotic body formation. Apoptotic bodies composed of numerous nucleolus bodies and organelles are normally removed by phagocytes as soon as they are formed. Failure to remove apoptotic bodies leads to the release of autoantigens that may cause autoimmunity [93]. Progressive studies on SLE demonstrated that lymphocytes apoptosis might play an important role in the pathogenesis of disease. During the process of apoptosis, release of excessive quantity of intact nucleosomes has been suggested to be a source of nuclear antigens that drive an immune response, inducing anti-DNA and anti-histone antibody production [93,94]. If the apoptotic cells are not phagocytosed immediately, they undergo post-translational modification altering antigenicity that may provide a source of nuclear antigens to drive the autoantibody response in SLE [4]. Tolerance of self-antigens requires the deletion of autoreactive T- and B-cells by apoptosis. Therefore defects in inducing apoptosis could lead to the persistence of autoreactive T- or B-cells [95]. Thus, defective apoptosis leading to prolonged survival of pathogenic lymphocytes could be one cause of SLE [96]. This hypothesis is supported by observations in murine lupus models of MRL/lpr mice are characterized by the presence of lpr gene, associated with defective Fas (CD95) receptor on the surface of lymphocytes. The interaction of Fas and Fas ligand (FasL) transduces an active signal for cellular apoptosis [97]. Defective Fas mediated apoptosis in MRL/lpr mice results in massive lymphoproliferation and development of a severe lupus-like disease with immune glomerulonephritis [98]. Gld/gld mice, characterized by a mutation in the FasL gene leads to a non-functional FasL molecule, also develop lymphoproliferation, hypergammaglobulinaemia and immunoglobulin deposits in the kidneys [99]. The lpr/gld gene mutations were not found in patients with lupus [100]. Despite this lack of relationship between mutations, or deficiency, in apoptotic genes and SLE, there is agreement that apoptosis plays an important role. This is based on observations of excessive apoptosis of T and B lymphocytes and on defective clearance of apoptotic cells in patients with SLE [4,101]. Furthermore, Fas mutations have been found in children presenting early signs of autoimmunity including cutaneous vasculitis, glomerulitis, thrombocytopenia, anemia and neutropenia with non-malignant lymphoproliferative syndrome [102,103]. In human SLE, high levels of soluble Fas (sFas) have been measured, and in vitro and in vivo studies in mice have shown that soluble Fas impairs apoptosis induced by Fas ligand [104]. Thus it appears that Fas-mediated apoptosis is defective in human SLE, leads to loss of control of autoreactive B cells by CD4+ T cells and autoantibody production. There are convincing literatures supporting the role of T lymphocyte perturbation in SLE patients. Increased levels of apoptotic total T lymphocytes and its subsets; CD4 and CD8 lymphocyte have been reported by various groups in SLE patients [50,61,105,106]. Increased T-lymphocyte apoptosis in lupus was reported to correlate with disease activity in SLE patients [107]. The study from SLE patients showed an increase expression of Fas/Fas ligand and caspase-3 activity in T cells with a consequent increase in T lymphocyte apoptosis [50,108]. Further study showed up-regulation of apoptosis-related protein Bcl-2 and Fas in T and B lymphocytes of patients with juvenile-onset SLE [109]. It proposed that sFas may be a common feature in diseases involving Fas-mediated organ damage and that it might modulate both autoimmune response and FasL-mediated tissue damage in SLE. A recent study on SLE patients showed increased levels of Fas/FasL in SLE patients related to depletion of intracellular glutathione.
Taken together, apoptosis of lymphocytes may be defective in patients with SLE and Fas/FasL-mediated signaling pathways could be crucial in the process. Altered lymphocyte apoptosis in patients with SLE could contribute to an overload of nucleosome in circulation that could initiate an autoimmune response that might break tolerance, resulting in the autoimmune phenomena.
5.2. Interaction between glutathione and apoptosis
A decrease in cellular GSH concentration has long been reported to be an early event in the apoptotic cascade induced by death receptor activation [5], mitochondrial apoptotic signaling [110], and oxidative stress [62,111]. Convincing evidence showed that GSH depletion during apoptosis is an indicator for ROS formation and oxidative stress and may be tied to pathogenesis in many autoimmune diseases including SLE [50]. Changes in the intracellular thiol-disulfide (GSH/GSSG) balance are considered major determinants in the redox status/signaling of the cell [112]. GSH constitutes the major intracellular antioxidant defense against RS and oxidative stress. GSH has been shown to scavenge a wide variety of RS, including superoxide anion (O2·−), hydroxyl radical (·OH), singlet oxygen (1O2), protein−, and DNA radicals, by donating electrons and becoming oxidized to glutathiyl radical (GS·). Generation of disulfide bonds between two GSH leads to further formation of GSSG. GSH also catalytically detoxifies cells from peroxides such as hydroperoxides (H2O2), peroxynitrite (OONO−), and lipid peroxides (LOO·) by the action of GSH peroxidases (GPX) and peroxiredoxins (PXR). Accumulation of GSSG on oxidative stress has been observed to be toxic to the cell. GSSG has been shown to directly induce apoptosis by the activation SAPK/MAPK pathway [5,113]. There are several lines of evidence linking intracellular ROS levels and induction of apoptosis in numerous cells lines. It has been shown that hydrogen peroxide induce apoptosis in the blastocyst and neutrophil and this is prevented by catalase [114]. Further, ROS generated by sodium arsenite in eosinophil induce apoptosis. Peroxynitrite has been shown to cause DNA fragmentation and apoptosis first identified in several cell types such as thymocytes, HL-60 cells, cultured cortical neurons, and PC12 cells. Peroxynitrite induces apoptosis in a number of cell types in culture, including pheochromocytoma derived PC12 cells, cortical neurons, HL-60 cells and rat thymocytes [22]. However, a few reports have shown contradictory roles including inhibitory and stimulatory effects of ROS on apoptosis [115].
GSH depletion in response to oxidants has been widely reported, and linked to cell death [116]. GSH is essential for cell survival as demonstrated by observations that glutamate cysteine ligase (GCL) knockout mice die from massive apoptotic cell death [117], and that the knockdown of GCL in distinct cell types induces time-dependent apoptosis [118,119]. GSH levels have been shown to influence caspase activity, transcription factor activation, Bcl-2 expression and function, ceramide production, thiol-redox signaling, and phosphatidylserine externalization. A remarkable feature of cells undergoing apoptosis is that they rapidly and selectively release a large fraction of their intracellular GSH into the extracellular space [120,121]. GSH peroxidase (GPX) has been shown to protect against apoptosis induced by Fas activation [122]. Replenishing GSH pools by NAC is known to protect against apoptosis. The apoptosis-inducing effects can be blocked by glutathione and N-acetylcysteine [115]. Glutathione depletion has been reported to involve in extrinsic/death receptor as well as intrinsic pathway of apoptosis.
Induction of apoptosis via the extrinsic pathway is triggered by the activation of the death receptors Fas (CD95/Apo-1), TNF-related apoptosis-inducing ligand (TRAIL) receptors 1 and 2 (DR4/DR5), and TNF receptor 1 (TNFR1) by their respective ligands FasL, TRAIL, and TNF-a. Activation of death receptors leads to formation of the death-inducing signaling complex, which includes the Fas-associated death domain (FADD), initiator caspase 8 or 10, and the cellular FADD-like interleukin-1 beta-converting enzyme (FLICE)-inhibitory protein (FLIP) leading to the activation of initiator caspases. Activation of NF-kβ antagonizes programmed cell death induced by TNFR1, and GSH depletion has been shown to down-regulate TNF-induced NF-kβ activation and sensitize to apoptotic cell death [123]. GSH depletion is necessary for the formation of the apoptosome [124] and also triggers cell death by modulation of the permeability transition pore of the mitochondria and the activation of executioner caspases [125,126]. In addition, GSH depletion activates the intrinsic apoptotic pathway initiator Bax and Cyt C release [127]. Released Cyt C requires cytosolic GSH levels to be depleted for its pro-apoptotic action [128]. Depletion of intracellular GSH also overcomes Bcl-2-mediated resistance to apoptosis [129]. The antiapoptotic role of Bcl-2 has been linked to GSH content by several studies, where it was reported that Bcl-2 regulates GSH content and distribution in different cellular compartments [130,131]. A recent study suggests that Bcl-2 regulates mitochondrial GSH content by a direct interaction of the BH3 groove with GSH [132], while the antiapoptotic effect of Bcl-xl has also been attributed to the regulation of GSH homeostasis by preventing GSH loss [133] Figure 5.
Figure 5.

Overview on the mechanisms of glutathione induced cell death. Apoptosis includes cellular shrinking, chromatin condensation and margination at the nuclear periphery with the eventual formation of membrane-bound apoptotic bodies that contain organelles, cytosol and nuclear fragments and are phagocytosed without triggering inflammatory processes. Depletion of GSH might be a prerequisite for the generation of ROS. GSH depletion and the concomitant decrease in GSH/GSSG ratio might increase the availability of GSSG to exert its redox signaling and modulate both intrinsic as well as extrinsic pathway of apoptosis. GSH depletion triggers the permeability transition pore of the mitochondria, the pro-apoptotic function of released Cyt C, the formation of the apoptosome, and the activation of executioner caspases and lead to intrinsic apoptosis pathway. Furthermore, GSH depletion might trigger aggregation of death receptors, activation of Fas/FasL and caspase-3.
In SLE patients, depletion of glutathione has been associated with various immune abnormalities including deregulation of apoptosis, abnormal cytokine and chemokine production and various clinical features. There are several lines of evidence correlating the depletion of intracellular glutathione with generation of ROS/RNS and progression of apoptosis in SLE patients [50,61,78]. It has been reported that glutathione levels were diminished in RBC and total lymphocyte as well as lymphocyte subsets in SLE patients [48,50,57,78]. Depletion of glutathione is correlated with severity of the disease and allied with oxidative stress and apoptosis [50,78]. The diminished levels of glutathione in the RBC and lymphocytes positively associated with increased levels of oxidative stress makers such as ROS, lipid peroxidation in SLE patients [50]. A negative association of the levels of GSH levels with apoptosis of T lymphocytes, CD4+, CD8+ T lymphocyte sub-sets and intracellular activated caspase-3 may support the role of reduced glutathione in the alteration of T lymphocyte apoptosis in the disease state [49,100]. These results suggest that glutathione played a role in depletion of CD4+ T lymphocyte in SLE patients. The role of glutathione as a therapeutic molecule to replenish depleted glutathione has been related to reduction in autoantibody. It has been show diminished GSH/GSSG ratios in the kidneys of 8-month-old versus 4-month-old (NZB x NZW) F1 mice, and treatment with N-acetylcysteine (NAC), a precursor of GSH and stimulator of its de novo biosynthesis, prevented decline of GSH/GSSG ratios, reduced autoantibody production and development of glomerulonephritis and prolonged survival of (NZB x NZW) F1 mice [134]. Intracellular glutathione has been shown to be involved in regulating several immune mechanisms in human body. While GSH scavenges •OH, 1O2, and NO directly, it catalytically detoxifies hydrogen peroxides (H2O2), OONO−, and lipid peroxides by activation of glutathione peroxidases. Perricone and his group have shown that modulation of intracellular glutathione can inhibit complement-mediated damage in autoimmune diseases [135]. Because glutathione is the major intracellular antioxidant defense within a cell, it is proposed that its depletion might be a prerequisite for modulating the apoptotic machinery in autoimmune disease like SLE (Figure 6). Inhibition of GSH depletion by either high extracellular GSH or NAC may prevent increased ROS formation and control abnormal apoptosis as well as several other abnormal immune responses, cytokine as well as chemokine production in SLE patients.
Figure 6.

A general schematic showing the interaction of oxidative stress and apoptosis in SLE. Excessive oxidative stress is thought to have an important role in the pathogenesis of autoimmune diseases by enhancing inflammation, inducing apoptotic cell death and breaking down the immunological tolerance. The ROS generation also provides oxidant for lipid membrane damage, protein oxidation and DNA damage. The decrease in clearance of apoptosis provides interaction of intracellular molecules with ROS, which can be a basis for autoantibody production in SLE. The depletion of glutathione favours this cascade.
5.3. Antioxidants: N-acetylcysteine and cysteamine
Antioxidants might be a beneficial adjunctive therapy in the treatment of SLE. ROS, the superoxide anion, hydroxyl radicals, and hydrogen peroxide are generated during immune responses associated with neutrophil and macrophage activity [136]. ROS directly damage endothelium, leading to vascular permeability and edema. Evidently, glutathione has been involved in regulating several immune mechanisms in human body. While GSH scavenges ·OH, 1O2, and NO directly, it catalytically detoxifies H2O2, OONO−, and lipid peroxides by the action of glutathione peroxidases. Because glutathione is the major intracellular antioxidant defense within a cell, it is proposed that its depletion might be a prerequisite for modulating the apoptotic machinery in autoimmune disease like SLE. Recent studies have shown that restoration of intracellular glutathione by supplementing with precursor like N-acetylcysteine and cysteamine had produced beneficial effect. In a study testing the immunomodulatory effects of the nonenzymatic antioxidants N-acetylcysteine and cysteamine, significant benefit was observed in glomerulonephritis and mortality in the NZB/W F1 murine model of SLE [134]. N-acetylcysteine suppressed autoantibody formation and prolonged survival of the mice [75]. Cysteamine inhibited the development of renal insufficiency and improved survival significantly. Prospective clinical studies are currently ongoing to assess whether NAC treatment can reduce GSH depletion, correct T-cell-signaling defects and provide clinical benefit to patients with SLE (www.clinicaltrials.gov IND 101320). Another pilot study by Li et al. showed that NAC safely improves lupus disease activity by blocking mTOR in T lymphocytes in SLE patients [137].
6. Conclusion
Recent studies showed a strong association between oxidative stress and apoptosis in animal models as well as in SLE patients. Moreover the depletion of glutathione is closely related to perturbation of apoptosis in SLE patients. However, more mechanistic studies in vitro and in vivo are required for addressing how the interplay between glutathione and apoptosis may lead to a break intolerance and aggressiveness of lupus disease activity. N-acetyl cysteine has been the first phase of clinical trial for the therapeutic management of SLE patients. Further studies should be directed to evaluate a role of the glutathione precursor; NAC in oxidative stress, apoptosis and cultivating severity of disease, which has shown as a promising therapy in SLE.
Acknowledgments
This study was made possible by funding from National Institutes of Health R01AR060366 & R21AI094377.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Amital H, Shoenfeld Y. Autoimmunity and autoimmune diseases such as systemic lupus erythematosus. In: Lahita Robert G, editor. Systemic Lupus Erythematosus. 4. Amsterdam, The Netherlands: Elsevier Publication; 2004. pp. 3–27. [Google Scholar]
- 2.Graham KL, Utz PJ. Sources of autoantigens in systemic lupus erythematosus. Curr Opin Rheumatol. 2005;17:513–517. doi: 10.1097/01.bor.0000171215.87993.6b. [DOI] [PubMed] [Google Scholar]
- 3.Kurien BT, Scofield RH. Free radical mediated peroxidative damage in systemic lupus erythematosus. Life Sci. 2003;73:1655–1666. doi: 10.1016/s0024-3205(03)00475-2. [DOI] [PubMed] [Google Scholar]
- 4.Munoz LE, van Bavel C, Franz S, Berden J, Herrmann M, van der Vlag J. Apoptosis in the pathogenesis of systemic lupus erythematosus. Lupus. 2008;17:371–375. doi: 10.1177/0961203308089990. [DOI] [PubMed] [Google Scholar]
- 5.Franco R, Cidlowski JA. Apoptosis and glutathione: beyond an antioxidant. Cell Death Differ. 2009;16:1303–1314. doi: 10.1038/cdd.2009.107. [DOI] [PubMed] [Google Scholar]
- 6.Griffiths HR. Is the generation of neo-antigenic determinants by free radicals central to the development of autoimmune rheumatoid disease? Autoimmun Rev. 2008;7:544–549. doi: 10.1016/j.autrev.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 7.Ahsan H, Ali A, Ali R. Oxygen free radicals and systemic autoimmunity. Clin Exp Immunol. 2003;131:398–404. doi: 10.1046/j.1365-2249.2003.02104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mansour RB, Lassoued S, Gargouri B, El Gaid A, Attia H, Fakhfakh F. Increased levels of autoantibodies against catalase and superoxide dismutase associated with oxidative stress in patients with rheumatoid arthritis and systemic lupus erythematosus. Scand J Rheumatol. 2008;37:103–108. doi: 10.1080/03009740701772465. [DOI] [PubMed] [Google Scholar]
- 9.Ben Mansour R, Lassoued S, Elgaied A, Haddouk S, Marzouk S, Bahloul Z, Masmoudi H, Attia H, Aifa MS, Fakhfakh F. Enhanced reactivity to malondialdehyde-modified proteins by systemic lupus erythematosus autoantibodies. Scand J Rheumatol. 39:247–253. doi: 10.3109/03009740903362511. [DOI] [PubMed] [Google Scholar]
- 10.Valko M, Izakovic M, Mazur M, Rhodes CJ, Telser J. Role of oxygen radicals in DNA damage and cancer incidence. Mol Cell Biochem. 2004;266:37–56. doi: 10.1023/b:mcbi.0000049134.69131.89. [DOI] [PubMed] [Google Scholar]
- 11.Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 12.Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160:1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 13.Kovacic P, Jacintho JD. Mechanisms of carcinogenesis: focus on oxidative stress and electron transfer. Curr Med Chem. 2001;8:773–796. doi: 10.2174/0929867013373084. [DOI] [PubMed] [Google Scholar]
- 14.Ridnour LA, Isenberg JS, Espey MG, Thomas DD, Roberts DD, Wink DA. Nitric oxide regulates angiogenesis through a functional switch involving thrombospondin-1. Proc Natl Acad Sci U S A. 2005;102:13147–13152. doi: 10.1073/pnas.0502979102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vignais PV. The superoxide-generating NADPH oxidase: structural aspects and activation mechanism. Cell Mol Life Sci. 2002;59:1428–1459. doi: 10.1007/s00018-002-8520-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wei CC, Wang ZQ, Durra D, Hemann C, Hille R, Garcin ED, Getzoff ED, Stuehr DJ. The three nitric-oxide synthases differ in their kinetics of tetrahydrobiopterin radical formation, heme-dioxy reduction, and arginine hydroxylation. J Biol Chem. 2005;280:8929–8935. doi: 10.1074/jbc.M409737200. [DOI] [PubMed] [Google Scholar]
- 17.Turrens JF, Boveris A. Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem J. 1980;191:421–427. doi: 10.1042/bj1910421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev. 1979;59:527–605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- 19.Suski JM, Lebiedzinska M, Bonora M, Pinton P, Duszynski J, Wieckowski MR. Relation between mitochondrial membrane potential and ROS formation. Methods Mol Biol. 2012;810:183–205. doi: 10.1007/978-1-61779-382-0_12. [DOI] [PubMed] [Google Scholar]
- 20.Zahrt TC, Deretic V. Reactive nitrogen and oxygen intermediates and bacterial defenses: unusual adaptations in Mycobacterium tuberculosis. Antioxid Redox Signal. 2002;4:141–159. doi: 10.1089/152308602753625924. [DOI] [PubMed] [Google Scholar]
- 21.Nagy G, Koncz A, Telarico T, Fernandez D, Ersek B, Buzas E, Perl A. Central role of nitric oxide in the pathogenesis of rheumatoid arthritis and systemic lupus erythematosus. Arthritis Res Ther. 2010;12:210. doi: 10.1186/ar3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahmad R, Rasheed Z, Ahsan H. Biochemical and cellular toxicology of peroxynitrite: implications in cell death and autoimmune phenomenon. Immunopharmacol Immunotoxicol. 2009;31:388–396. doi: 10.1080/08923970802709197. [DOI] [PubMed] [Google Scholar]
- 23.Murphy MP, Packer MA, Scarlett JL, Martin SW. Peroxynitrite: a biologically significant oxidant. Gen Pharmacol. 1998;31:179–186. doi: 10.1016/s0306-3623(97)00418-7. [DOI] [PubMed] [Google Scholar]
- 24.Calcerrada P, Peluffo G, Radi R. Nitric oxide-derived oxidants with a focus on peroxynitrite: molecular targets, cellular responses and therapeutic implications. Curr Pharm Des. 2011;17:3905–3932. doi: 10.2174/138161211798357719. [DOI] [PubMed] [Google Scholar]
- 25.Radi R, Peluffo G, Alvarez MN, Naviliat M, Cayota A. Unraveling peroxynitrite formation in biological systems. Free Radic Biol Med. 2001;30:463–488. doi: 10.1016/s0891-5849(00)00373-7. [DOI] [PubMed] [Google Scholar]
- 26.Mena S, Ortega A, Estrela JM. Oxidative stress in environmental-induced carcinogenesis. Mutat Res. 2009;674:36–44. doi: 10.1016/j.mrgentox.2008.09.017. [DOI] [PubMed] [Google Scholar]
- 27.Ortona E, Margutti P, Matarrese P, Franconi F, Malorni W. Redox state, cell death and autoimmune diseases: a gender perspective. Autoimmun Rev. 2008;7:579–584. doi: 10.1016/j.autrev.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 28.Shadyro OI, Yurkova IL, Kisel MA. Radiation-induced peroxidation and fragmentation of lipids in a model membrane. Int J Radiat Biol. 2002;78:211–217. doi: 10.1080/09553000110104065. [DOI] [PubMed] [Google Scholar]
- 29.Halliwell B. Reactive oxygen species in living systems: source, biochemistry, and role in human disease. Am J Med. 1991;91:14S–22S. doi: 10.1016/0002-9343(91)90279-7. [DOI] [PubMed] [Google Scholar]
- 30.Wiseman H, Halliwell B. Damage to DNA by reactive oxygen and nitrogen species: role in inflammatory disease and progression to cancer. Biochem J. 1996;313 (Pt 1):17–29. doi: 10.1042/bj3130017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stohs SJ, Bagchi D. Oxidative mechanisms in the toxicity of metal ions. Free Radic Biol Med. 1995;18:321–336. doi: 10.1016/0891-5849(94)00159-h. [DOI] [PubMed] [Google Scholar]
- 32.Lloyd RV, Hanna PM, Mason RP. The origin of the hydroxyl radical oxygen in the Fenton reaction. Free Radic Biol Med. 1997;22:885–888. doi: 10.1016/s0891-5849(96)00432-7. [DOI] [PubMed] [Google Scholar]
- 33.Wanchu A, Khullar M, Deodhar SD, Bambery P, Sud A. Nitric oxide synthesis is increased in patients with systemic lupus erythematosus. Rheumatol Int. 1998;18:41–43. doi: 10.1007/s002960050055. [DOI] [PubMed] [Google Scholar]
- 34.Gilkeson G, Cannon C, Oates J, Reilly C, Goldman D, Petri M. Correlation of serum measures of nitric oxide production with lupus disease activity. J Rheumatol. 1999;26:318–324. [PubMed] [Google Scholar]
- 35.Kurien BT, Scofield RH. Autoimmunity and oxidatively modified autoantigens. Autoimmun Rev. 2008;7:567–573. doi: 10.1016/j.autrev.2008.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kumagai S, Jikimoto T, Saegusa J. Pathological roles of oxidative stress in autoimmune diseases. Rinsho Byori. 2003;51:126–132. [PubMed] [Google Scholar]
- 37.Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270:296–299. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- 38.Tsai KJ, Hung IJ, Chow CK, Stern A, Chao SS, Chiu DT. Impaired production of nitric oxide, superoxide, and hydrogen peroxide in glucose 6-phosphate-dehydrogenase-deficient granulocytes. FEBS Lett. 1998;436:411–414. doi: 10.1016/s0014-5793(98)01174-0. [DOI] [PubMed] [Google Scholar]
- 39.Crane FL, Low H. Reactive oxygen species generation at the plasma membrane for antibody control. Autoimmun Rev. 2008;7:518–522. doi: 10.1016/j.autrev.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 40.Lunec J, Herbert K, Blount S, Griffiths HR, Emery P. 8-Hydroxydeoxyguanosine. A marker of oxidative DNA damage in systemic lupus erythematosus. FEBS Lett. 1994;348:131–138. doi: 10.1016/0014-5793(94)00583-4. [DOI] [PubMed] [Google Scholar]
- 41.Evans MD, Cooke MS, Akil M, Samanta A, Lunec J. Aberrant processing of oxidative DNA damage in systemic lupus erythematosus. Biochem Biophys Res Commun. 2000;273:894–898. doi: 10.1006/bbrc.2000.3078. [DOI] [PubMed] [Google Scholar]
- 42.Morgan PE, Sturgess AD, Davies MJ. Evidence for chronically elevated serum protein oxidation in systemic lupus erythematosus patients. Free Radic Res. 2009;43:117–127. doi: 10.1080/10715760802623896. [DOI] [PubMed] [Google Scholar]
- 43.Abou-Raya A, el-Hallous D, Fayed H. 8-Isoprostaglandin F2 alpha: a potential index of lipid peroxidation in systemic lupus erythematosus. Clin Invest Med. 2004;27:306–311. [PubMed] [Google Scholar]
- 44.Avalos I, Chung CP, Oeser A, Milne GL, Morrow JD, Gebretsadik T, Shintani A, Yu C, Stein CM. Oxidative stress in systemic lupus erythematosus: relationship to disease activity and symptoms. Lupus. 2007;16:195–200. doi: 10.1177/0961203306075802. [DOI] [PubMed] [Google Scholar]
- 45.Sheikh Z, Ahmad R, Sheikh N, Ali R. Enhanced recognition of reactive oxygen species damaged human serum albumin by circulating systemic lupus erythematosus autoantibodies. Autoimmunity. 2007;40:512–520. doi: 10.1080/08916930701574331. [DOI] [PubMed] [Google Scholar]
- 46.Turgay M, Durak I, Erten S, Ertugrul E, Devrim E, Avci A, Turgay F. Oxidative stress and antioxidant parameters in a Turkish group of patients with active and inactive systemic lupus erythematosus. APLAR Journal of Rheumatology. 2007;10:101–106. [Google Scholar]
- 47.Kurien BT, Scofield RH. Lipid peroxidation in systemic lupus erythematosus. Indian J Exp Biol. 2006;44:349–356. [PubMed] [Google Scholar]
- 48.Shah D, Kiran R, Wanchu A, Bhatnagar A. Oxidative stress in systemic lupus erythematosus: relationship to Th1 cytokine and disease activity. Immunol Lett. 2010;129:7–12. doi: 10.1016/j.imlet.2010.01.005. [DOI] [PubMed] [Google Scholar]
- 49.Turi S, Nemeth I, Torkos A, Saghy L, Varga I, Matkovics B, Nagy J. Oxidative stress and antioxidant defense mechanism in glomerular diseases. Free Radic Biol Med. 1997;22:161–168. doi: 10.1016/s0891-5849(96)00284-5. [DOI] [PubMed] [Google Scholar]
- 50.Shah D, Aggarwal A, Bhatnagar A, Kiran R, Wanchu A. Association between T lymphocyte sub-sets apoptosis and peripheral blood mononuclear cells oxidative stress in systemic lupus erythematosus. Free Radic Res. 2011;45:559–567. doi: 10.3109/10715762.2011.555765. [DOI] [PubMed] [Google Scholar]
- 51.Taysi S, Gul M, Sari RA, Akcay F, Bakan N. Serum oxidant/antioxidant status of patients with systemic lupus erythematosus. Clin Chem Lab Med. 2002;40:684–688. doi: 10.1515/CCLM.2002.117. [DOI] [PubMed] [Google Scholar]
- 52.Tewthanom K, Janwityanuchit S, Totemchockchyakarn K, Panomvana D. Correlation of lipid peroxidation and glutathione levels with severity of systemic lupus erythematosus: a pilot study from single center. J Pharm Pharm Sci. 2008;11:30–34. doi: 10.18433/j3c885. [DOI] [PubMed] [Google Scholar]
- 53.Hassan SZ, Gheita TA, Kenawy SA, Fahim AT, El-Sorougy IM, Abdou MS. Oxidative stress in systemic lupus erythematosus and rheumatoid arthritis patients: relationship to disease manifestations and activity. Int J Rheum Dis. 2011;14:325–331. doi: 10.1111/j.1756-185X.2011.01630.x. [DOI] [PubMed] [Google Scholar]
- 54.Young IS, Woodside JV. Antioxidants in health and disease. J Clin Pathol. 2001;54:176–186. doi: 10.1136/jcp.54.3.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Michiels C, Raes M, Toussaint O, Remacle J. Importance of Se-glutathione peroxidase, catalase, and Cu/Zn-SOD for cell survival against oxidative stress. Free Radic Biol Med. 1994;17:235–248. doi: 10.1016/0891-5849(94)90079-5. [DOI] [PubMed] [Google Scholar]
- 56.Shah D, Kiran R, Wanchu A, Bhatnagar A. Relationship between T lymphocyte subsets and cortisol in systemic lupus erythematosus. Kathmandu Univ Med J (KUMJ) 2009;7:213–219. doi: 10.3126/kumj.v7i3.2726. [DOI] [PubMed] [Google Scholar]
- 57.Shah D, Wanchu A, Bhatnagar A. Interaction between oxidative stress and chemokines: possible pathogenic role in systemic lupus erythematosus and rheumatoid arthritis. Immunobiology. 2011;216:1010–1017. doi: 10.1016/j.imbio.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 58.D’Souza A, Kurien BT, Rodgers R, Shenoi J, Kurono S, Matsumoto H, Hensley K, Nath SK, Scofield RH. Detection of catalase as a major protein target of the lipid peroxidation product 4-HNE and the lack of its genetic association as a risk factor in SLE. BMC Med Genet. 2008;9:62. doi: 10.1186/1471-2350-9-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Franco R, Panayiotidis MI, Cidlowski JA. Glutathione depletion is necessary for apoptosis in lymphoid cells independent of reactive oxygen species formation. J Biol Chem. 2007;282:30452–30465. doi: 10.1074/jbc.M703091200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ballatori N, Krance SM, Notenboom S, Shi S, Tieu K, Hammond CL. Glutathione dysregulation and the etiology and progression of human diseases. Biol Chem. 2009;390:191–214. doi: 10.1515/BC.2009.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Perl A, Gergely P, Jr, Banki K. Mitochondrial dysfunction in T cells of patients with systemic lupus erythematosus. Int Rev Immunol. 2004;23:293–313. doi: 10.1080/08830180490452576. [DOI] [PubMed] [Google Scholar]
- 62.Circu ML, Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med. 2010;48:749–762. doi: 10.1016/j.freeradbiomed.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Circu ML, Aw TY. Glutathione and apoptosis. Free Radic Res. 2008;42:689–706. doi: 10.1080/10715760802317663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cordova EJ, Velazquez-Cruz R, Centeno F, Baca V, Orozco L. The NRF2 gene variant, -653G/A, is associated with nephritis in childhood-onset systemic lupus erythematosus. Lupus. 2010;19:1237–1242. doi: 10.1177/0961203310367917. [DOI] [PubMed] [Google Scholar]
- 65.Warchol T, Lianeri M, Wudarski M, Lacki JK, Jagodzinski PP. Catalase -262C>T polymorphism in systemic lupus erythematosus in Poland. Rheumatol Int. 2008;28:1035–1039. doi: 10.1007/s00296-008-0569-9. [DOI] [PubMed] [Google Scholar]
- 66.Kiyohara C, Washio M, Horiuchi T, Asami T, Ide S, Atsumi T, Kobashi G, Takahashi H, Tada Y. Risk modification by CYP1A1 and GSTM1 polymorphisms in the association of cigarette smoking and systemic lupus erythematosus in a Japanese population. Scand J Rheumatol. 2012;41:103–109. doi: 10.3109/03009742.2011.608194. [DOI] [PubMed] [Google Scholar]
- 67.Ollier W, Davies E, Snowden N, Alldersea J, Fryer A, Jones P, Strange R. Association of homozygosity for glutathione-S-transferase GSTM1 null alleles with the Ro+/La− autoantibody profile in patients with systemic lupus erythematosus. Arthritis Rheum. 1996;39:1763–1764. doi: 10.1002/art.1780391023. [DOI] [PubMed] [Google Scholar]
- 68.Tew MB, Ahn CW, Friedman AW, Reveille JD, Tan FK, Alarcon GS, Bastian HM, Fessler BJ, McGwin G, Jr, Lisse JR. Systemic lupus erythematosus in three ethnic groups. VIII. Lack of association of glutathione S-transferase null alleles with disease manifestations. Arthritis Rheum. 2001;44:981–983. doi: 10.1002/1529-0131(200104)44:4<981::AID-ANR158>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 69.Halliwelly B, Gutteridge JMC. Free Radical Biology and Medicine. 4. Oxford: Clarendon Press; 2006. [Google Scholar]
- 70.Sies H. Role of reactive oxygen species in biological processes. Klin Wochenschr. 1991;69:965–968. doi: 10.1007/BF01645140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Meister A. Glutathione metabolism and its selective modification. J Biol Chem. 1988;263:17205–17208. [PubMed] [Google Scholar]
- 72.Bremer HJ, Duran M, Kameling JP. Glutathione. In: Bremer HJ, Duran M, Kamerling JP, editors. Disturbances of amino acid metabolism: clinical chemistry and diagnosis. Urban & Schwarzenberg; Baltimore-Munich: 1981. pp. 80–82. [Google Scholar]
- 73.Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 74.Kemp M, Go YM, Jones DP. Nonequilibrium thermodynamics of thiol/disulfide redox systems: a perspective on redox systems biology. Free Radic Biol Med. 2008;44:921–937. doi: 10.1016/j.freeradbiomed.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Suthanthiran M, Anderson ME, Sharma VK, Meister A. Glutathione regulates activation-dependent DNA synthesis in highly purified normal human T lymphocytes stimulated via the CD2 and CD3 antigens. Proc Natl Acad Sci U S A. 1990;87:3343–3347. doi: 10.1073/pnas.87.9.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Huh YJ, Kim JM, Kim H, Song H, So H, Lee SY, Kwon SB, Kim HJ, Kim HH, Lee SH, Choi Y, Chung SC, Jeong DW, Min BM. Regulation of osteoclast differentiation by the redox-dependent modulation of nuclear import of transcription factors. Cell Death Differ. 2006;13:1138–1146. doi: 10.1038/sj.cdd.4401793. [DOI] [PubMed] [Google Scholar]
- 77.Sykes MC, Mowbray AL, Jo H. Reversible glutathiolation of caspase-3 by glutaredoxin as a novel redox signaling mechanism in tumor necrosis factor-alpha-induced cell death. Circ Res. 2007;100:152–154. doi: 10.1161/01.RES.0000258171.08020.72. [DOI] [PubMed] [Google Scholar]
- 78.Hassan MQ, Hadi RA, Al-Rawi ZS, Padron VA, Stohs SJ. The glutathione defense system in the pathogenesis of rheumatoid arthritis. J Appl Toxicol. 2001;21:69–73. doi: 10.1002/jat.736. [DOI] [PubMed] [Google Scholar]
- 79.Peterson JD, Herzenberg LA, Vasquez K, Waltenbaugh C. Glutathione levels in antigen-presenting cells modulate Th1 versus Th2 response patterns. Proc Natl Acad Sci U S A. 1998;95:3071–3076. doi: 10.1073/pnas.95.6.3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Messina JP, Lawrence DA. Cell cycle progression of glutathione-depleted human peripheral blood mononuclear cells is inhibited at S phase. J Immunol. 1989;143:1974–1981. [PubMed] [Google Scholar]
- 81.Murata Y, Shimamura T, Hamuro J. The polarization of T(h)1/T(h)2 balance is dependent on the intracellular thiol redox status of macrophages due to the distinctive cytokine production. Int Immunol. 2002;14:201–212. doi: 10.1093/intimm/14.2.201. [DOI] [PubMed] [Google Scholar]
- 82.McCord JM, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein) J Biol Chem. 1969;244:6049–6055. [PubMed] [Google Scholar]
- 83.Blum J, Fridovich I. Inactivation of glutathione peroxidase by superoxide radical. Arch Biochem Biophys. 1985;240:500–508. doi: 10.1016/0003-9861(85)90056-6. [DOI] [PubMed] [Google Scholar]
- 84.Johnson F, Giulivi C. Superoxide dismutases and their impact upon human health. Mol Aspects Med. 2005;26:340–352. doi: 10.1016/j.mam.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 85.McCord JM. Iron- and manganese-containing superoxide dismutases: structure, distribution, and evolutionary relationships. Adv Exp Med Biol. 1976;74:540–550. doi: 10.1007/978-1-4684-3270-1_45. [DOI] [PubMed] [Google Scholar]
- 86.Marklund SL. Properties of extracellular superoxide dismutase from human lung. Biochem J. 1984;220:269–272. doi: 10.1042/bj2200269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Forman HJ, Fisher AB. Antioxidant enzymes of rat granular pneumocytes. Constitutive levels and effect of hyperoxia. Lab Invest. 1981;45:1–6. [PubMed] [Google Scholar]
- 88.Jones DP, Eklow L, Thor H, Orrenius S. Metabolism of hydrogen peroxide in isolated hepatocytes: relative contributions of catalase and glutathione peroxidase in decomposition of endogenously generated H2O2. Arch Biochem Biophys. 1981;210:505–516. doi: 10.1016/0003-9861(81)90215-0. [DOI] [PubMed] [Google Scholar]
- 89.Aebi H. Catalase in vitro. Methods Enzymol. 1984;105:121–126. doi: 10.1016/s0076-6879(84)05016-3. [DOI] [PubMed] [Google Scholar]
- 90.Nilakantan V, Spear BT, Glauert HP. Liver-specific catalase expression in transgenic mice inhibits NF-kappaB activation and DNA synthesis induced by the peroxisome proliferator ciprofibrate. Carcinogenesis. 1998;19:631–637. doi: 10.1093/carcin/19.4.631. [DOI] [PubMed] [Google Scholar]
- 91.Gamble SC, Wiseman A, Goldfarb PS. Selenium-dependent glutathione peroxidase and other selenoproteins: their synthesis and biochemical roles. J Chem Technol Biotech. 1999;68:123–134. [Google Scholar]
- 92.Sen CK. Cellular thiols and redox-regulated signal transduction. Curr Top Cell Regul. 2000;36:1–30. doi: 10.1016/s0070-2137(01)80001-7. [DOI] [PubMed] [Google Scholar]
- 93.Lorenz HM, Herrmann M, Winkler T, Gaipl U, Kalden JR. Role of apoptosis in autoimmunity. Apoptosis. 2000;5:443–449. doi: 10.1023/a:1009692902805. [DOI] [PubMed] [Google Scholar]
- 94.Lorenz HM, Herrmann M, Kalden JR. The pathogenesis of autoimmune diseases. Scand J Clin Lab Invest Suppl. 2001;235:16–26. doi: 10.1080/003655101753352004. [DOI] [PubMed] [Google Scholar]
- 95.Dieker JW, van der Vlag J, Berden JH. Deranged removal of apoptotic cells: its role in the genesis of lupus. Nephrol Dial Transplant. 2004;19:282–285. doi: 10.1093/ndt/gfg485. [DOI] [PubMed] [Google Scholar]
- 96.Bijl M, Horst G, Limburg PC, Kallenberg CG. Fas expression on peripheral blood lymphocytes in systemic lupus erythematosus (SLE): relation to lymphocyte activation and disease activity. Lupus. 2001;10:866–872. doi: 10.1191/096120301701548517. [DOI] [PubMed] [Google Scholar]
- 97.Suzuki N, Ichino M, Mihara S, Kaneko S, Sakane T. Inhibition of Fas/Fas ligand-mediated apoptotic cell death of lymphocytes in vitro by circulating anti-Fas ligand autoantibodies in patients with systemic lupus erythematosus. Arthritis Rheum. 1998;41:344–353. doi: 10.1002/1529-0131(199802)41:2<344::AID-ART19>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 98.Theofilopoulos AN, Dixon FJ. Etiopathogenesis of murine SLE. Immunol Rev. 1981;55:179–216. doi: 10.1111/j.1600-065x.1981.tb00343.x. [DOI] [PubMed] [Google Scholar]
- 99.Takahashi T, Tanaka M, Brannan CI, Jenkins NA, Copeland NG, Suda T, Nagata S. Generalized lymphoproliferative disease in mice, caused by a point mutation in the Fas ligand. Cell. 1994;76:969–976. doi: 10.1016/0092-8674(94)90375-1. [DOI] [PubMed] [Google Scholar]
- 100.McNally J, Yoo DH, Drappa J, Chu JL, Yagita H, Friedman SM, Elkon KB. Fas ligand expression and function in systemic lupus erythematosus. J Immunol. 1997;159:4628–4636. [PubMed] [Google Scholar]
- 101.Gaipl US, Munoz LE, Grossmayer G, Lauber K, Franz S, Sarter K, Voll RE, Winkler T, Kuhn A, Kalden J, Kern P, Herrmann M. Clearance deficiency and systemic lupus erythematosus (SLE) J Autoimmun. 2007;28:114–121. doi: 10.1016/j.jaut.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 102.Fisher GH, Rosenberg FJ, Straus SE, Dale JK, Middleton LA, Lin AY, Strober W, Lenardo MJ, Puck JM. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell. 1995;81:935–946. doi: 10.1016/0092-8674(95)90013-6. [DOI] [PubMed] [Google Scholar]
- 103.Rieux-Laucat F, Le Deist F, Hivroz C, Roberts IA, Debatin KM, Fischer A, de Villartay JP. Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science. 1995;268:1347–1349. doi: 10.1126/science.7539157. [DOI] [PubMed] [Google Scholar]
- 104.Cheng J, Zhou T, Liu C, Shapiro JP, Brauer MJ, Kiefer MC, Barr PJ, Mountz JD. Protection from Fas-mediated apoptosis by a soluble form of the Fas molecule. Science. 1994;263:1759–1762. doi: 10.1126/science.7510905. [DOI] [PubMed] [Google Scholar]
- 105.Robak E, Sysa-Jedrzejowska A, Robak T, Smolewski P. Peripheral blood lymphocyte apoptosis and circulating dendritic cells in patients with systemic lupus erythematosus: correlation with immunological status and disease-related symptoms. Clin Rheumatol. 2006;25:225–233. doi: 10.1007/s10067-005-1163-0. [DOI] [PubMed] [Google Scholar]
- 106.Emlen W, Niebur J, Kadera R. Accelerated in vitro apoptosis of lymphocytes from patients with systemic lupus erythematosus. J Immunol. 1994;152:3685–3692. [PubMed] [Google Scholar]
- 107.Dhir V, Singh AP, Aggarwal A, Naik S, Misra R. Increased T-lymphocyte apoptosis in lupus correlates with disease activity and may be responsible for reduced T-cell frequency: a cross-sectional and longitudinal study. Lupus. 2009;18:785–791. doi: 10.1177/0961203309103152. [DOI] [PubMed] [Google Scholar]
- 108.Xue C, Lan-Lan W, Bei C, Jie C, Wei-Hua F. Abnormal Fas/FasL and caspase-3-mediated apoptotic signaling pathways of T lymphocyte subset in patients with systemic lupus erythematosus. Cell Immunol. 2006;239:121–128. doi: 10.1016/j.cellimm.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 109.Liphaus BL, Kiss MH, Carrasco S, Goldenstein-Schainberg C. Increased Fas and Bcl-2 expression on peripheral blood T and B lymphocytes from juvenile-onset systemic lupus erythematosus, but not from juvenile rheumatoid arthritis and juvenile dermatomyositis. Clin Dev Immunol. 2006;13:283–287. doi: 10.1080/17402520600877786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Di Stefano A, Frosali S, Leonini A, Ettorre A, Priora R, Di Simplicio FC, Di Simplicio P. GSH depletion, protein S-glutathionylation and mitochondrial transmembrane potential hyperpolarization are early events in initiation of cell death induced by a mixture of isothiazolinones in HL60 cells. Biochim Biophys Acta. 2006;1763:214–225. doi: 10.1016/j.bbamcr.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 111.Circu ML, Aw TY. Glutathione and modulation of cell apoptosis. Biochim Biophys Acta. 2012 doi: 10.1016/j.bbamcr.2012.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jones DP. Redefining oxidative stress. Antioxid Redox Signal. 2006;8:1865–1879. doi: 10.1089/ars.2006.8.1865. [DOI] [PubMed] [Google Scholar]
- 113.Anathy V, Roberson EC, Guala AS, Godburn KE, Budd RC, Janssen-Heininger YM. Redox-based regulation of apoptosis: S-glutathionylation as a regulatory mechanism to control cell death. Antioxid Redox Signal. 2012;16:496–505. doi: 10.1089/ars.2011.4281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pierce GB, Parchment RE, Lewellyn AL. Hydrogen peroxide as a mediator of programmed cell death in the blastocyst. Differentiation. 1991;46:181–186. doi: 10.1111/j.1432-0436.1991.tb00880.x. [DOI] [PubMed] [Google Scholar]
- 115.Devadas S, Hinshaw JA, Zaritskaya L, Williams MS. Fas-stimulated generation of reactive oxygen species or exogenous oxidative stress sensitize cells to Fas-mediated apoptosis. Free Radic Biol Med. 2003;35:648–661. doi: 10.1016/s0891-5849(03)00391-5. [DOI] [PubMed] [Google Scholar]
- 116.Perricone C, De Carolis C, Perricone R. Glutathione: a key player in autoimmunity. Autoimmun Rev. 2009;8:697–701. doi: 10.1016/j.autrev.2009.02.020. [DOI] [PubMed] [Google Scholar]
- 117.Dalton TP, Chen Y, Schneider SN, Nebert DW, Shertzer HG. Genetically altered mice to evaluate glutathione homeostasis in health and disease. Free Radic Biol Med. 2004;37:1511–1526. doi: 10.1016/j.freeradbiomed.2004.06.040. [DOI] [PubMed] [Google Scholar]
- 118.Dalton TP, Dieter MZ, Yang Y, Shertzer HG, Nebert DW. Knockout of the mouse glutamate cysteine ligase catalytic subunit (Gclc) gene: embryonic lethal when homozygous, and proposed model for moderate glutathione deficiency when heterozygous. Biochem Biophys Res Commun. 2000;279:324–329. doi: 10.1006/bbrc.2000.3930. [DOI] [PubMed] [Google Scholar]
- 119.Valverde M, Rojas E, Kala SV, Kala G, Lieberman MW. Survival and cell death in cells constitutively unable to synthesize glutathione. Mutat Res. 2006;594:172–180. doi: 10.1016/j.mrfmmm.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 120.Hammond CL, Lee TK, Ballatori N. Novel roles for glutathione in gene expression, cell death, and membrane transport of organic solutes. J Hepatol. 2001;34:946–954. doi: 10.1016/s0168-8278(01)00037-x. [DOI] [PubMed] [Google Scholar]
- 121.Ghibelli L, Fanelli C, Rotilio G, Lafavia E, Coppola S, Colussi C, Civitareale P, Ciriolo MR. Rescue of cells from apoptosis by inhibition of active GSH extrusion. FASEB J. 1998;12:479–486. doi: 10.1096/fasebj.12.6.479. [DOI] [PubMed] [Google Scholar]
- 122.Gouaze V, Andrieu-Abadie N, Cuvillier O, Malagarie-Cazenave S, Frisach MF, Mirault ME, Levade T. Glutathione peroxidase-1 protects from CD95-induced apoptosis. J Biol Chem. 2002;277:42867–42874. doi: 10.1074/jbc.M203067200. [DOI] [PubMed] [Google Scholar]
- 123.Liuzzi F, Fanelli C, Ciriolo MR, Cerella C, D’Alessio M, Denicola M, Magrini A, Bergamaschi A, Ghibelli L. Rescue of cells from apoptosis by antioxidants occurs downstream from GSH extrusion. Ann N Y Acad Sci. 2003;1010:441–445. doi: 10.1196/annals.1299.080. [DOI] [PubMed] [Google Scholar]
- 124.Sato T, Machida T, Takahashi S, Iyama S, Sato Y, Kuribayashi K, Takada K, Oku T, Kawano Y, Okamoto T, Takimoto R, Matsunaga T, Takayama T, Takahashi M, Kato J, Niitsu Y. Fas-mediated apoptosome formation is dependent on reactive oxygen species derived from mitochondrial permeability transition in Jurkat cells. J Immunol. 2004;173:285–296. doi: 10.4049/jimmunol.173.1.285. [DOI] [PubMed] [Google Scholar]
- 125.Armstrong JS, Jones DP. Glutathione depletion enforces the mitochondrial permeability transition and causes cell death in Bcl-2 overexpressing HL60 cells. FASEB J. 2002;16:1263–1265. doi: 10.1096/fj.02-0097fje. [DOI] [PubMed] [Google Scholar]
- 126.Varghese J, Khandre NS, Sarin A. Caspase-3 activation is an early event and initiates apoptotic damage in a human leukemia cell line. Apoptosis. 2003;8:363–370. doi: 10.1023/a:1024121017841. [DOI] [PubMed] [Google Scholar]
- 127.Guha P, Dey A, Sen R, Chatterjee M, Chattopadhyay S, Bandyopadhyay SK. Intracellular GSH depletion triggered mitochondrial Bax translocation to accomplish resveratrol-induced apoptosis in the U937 cell line. J Pharmacol Exp Ther. 2011;336:206–214. doi: 10.1124/jpet.110.171983. [DOI] [PubMed] [Google Scholar]
- 128.Pan Z, Voehringer DW, Meyn RE. Analysis of redox regulation of cytochrome c-induced apoptosis in a cell-free system. Cell Death Differ. 1999;6:683–688. doi: 10.1038/sj.cdd.4400544. [DOI] [PubMed] [Google Scholar]
- 129.Rudin CM, Yang Z, Schumaker LM, VanderWeele DJ, Newkirk K, Egorin MJ, Zuhowski EG, Cullen KJ. Inhibition of glutathione synthesis reverses Bcl-2-mediated cisplatin resistance. Cancer Res. 2003;63:312–318. [PubMed] [Google Scholar]
- 130.Kane DJ, Sarafian TA, Anton R, Hahn H, Gralla EB, Valentine JS, Ord T, Bredesen DE. Bcl-2 inhibition of neural death: decreased generation of reactive oxygen species. Science. 1993;262:1274–1277. doi: 10.1126/science.8235659. [DOI] [PubMed] [Google Scholar]
- 131.Voehringer DW, Meyn RE. Redox aspects of Bcl-2 function. Antioxid Redox Signal. 2000;2:537–550. doi: 10.1089/15230860050192314. [DOI] [PubMed] [Google Scholar]
- 132.Zimmermann AK, Loucks FA, Schroeder EK, Bouchard RJ, Tyler KL, Linseman DA. Glutathione binding to the Bcl-2 homology-3 domain groove: a molecular basis for Bcl-2 antioxidant function at mitochondria. J Biol Chem. 2007;282:29296–29304. doi: 10.1074/jbc.M702853200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bojes HK, Datta K, Xu J, Chin A, Simonian P, Nunez G, Kehrer JP. Bcl-xL overexpression attenuates glutathione depletion in FL5. 12 cells following interleukin-3 withdrawal. Biochem J. 1997;325 (Pt 2):315–319. doi: 10.1042/bj3250315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Suwannaroj S, Lagoo A, Keisler D, McMurray RW. Antioxidants suppress mortality in the female NZB x NZW F1 mouse model of systemic lupus erythematosus (SLE) Lupus. 2001;10:258–265. doi: 10.1191/096120301680416940. [DOI] [PubMed] [Google Scholar]
- 135.Perricone C, De Carolis C, Giacomelli R, Greco E, Cipriani P, Ballanti E, Novelli L, Perricone R. Inhibition of the complement system by glutathione: molecular mechanisms and potential therapeutic implications. Int J Immunopathol Pharmacol. 2011;24:63–68. doi: 10.1177/039463201102400108. [DOI] [PubMed] [Google Scholar]
- 136.Li KJ, Wu CH, Hsieh SC, Lu MC, Tsai CY, Yu CL. Deranged bioenergetics and defective redox capacity in T lymphocytes and neutrophils are related to cellular dysfunction and increased oxidative stress in patients with active systemic lupus erythematosus. Clin Dev Immunol. 2012;2012:548516. doi: 10.1155/2012/548516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lai ZW, Hanczko R, Bonilla E, Caza TN, Clair B, Bartos A, Miklossy G, Jimah J, Doherty E, Tily H, Francis L, Garcia R, Dawood M, Yu J, Ramos I, Coman I, Faraone SV, Phillips PE, Perl A. N-acetylcysteine reduces disease activity by blocking mammalian target of rapamycin in T cells from systemic lupus erythematosus patients: a randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2012;64:2937–2946. doi: 10.1002/art.34502. [DOI] [PMC free article] [PubMed] [Google Scholar]