

Abstract
Therapeutic vaccines for the treatment of cancer are an attractive alternative to some of the conventional therapies that are currently used. More importantly, vaccines could be very useful to prevent recurrences when applied after primary therapy. Unfortunately, most therapeutic vaccines for cancer have performed poorly due to the low level of immune responses that they induce. Previous work done in our laboratory in cancer mouse models demonstrated that vaccines consisting of synthetic peptides representing minimal CD8 T-cell epitopes administered i.v. mixed with poly-IC and anti-CD40 antibodies (TriVax) were capable of inducing massive T cell responses similar to those found during acute infections. We now report that some peptides are capable of inducing similarly large T cell responses after vaccination with poly-IC alone (BiVax). The results show that amphiphilic peptides are more likely to function as strong immunogens in BiVax and that systemic immunizations (i.v. or i.m.) were more effective than local (s.c.) vaccine administration. The immune responses induced by BiVax were found to be effective against established tumors in two mouse cancer models. The roles of various immune-related pathways such as type-I IFN, CD40 costimulation, CD4 T cells, TLRs and the MDA5 RNA helicase were examined. The present findings could facilitate the development of simple and effective subunit vaccines for diseases where CD8 T cells provide a therapeutic benefit.
Electronic supplementary material
The online version of this article (doi:10.1007/s00262-012-1382-6) contains supplementary material, which is available to authorized users.
Keywords: Peptide vaccines, Immune adjuvants, Immunotherapy, CD8 T cells, Type-I interferon
Précis
A novel vaccine consisting of amphiphilic peptides mixed with poly-IC when given i.v. activates high numbers of antigen-specific, tumor-reactive CD8 T cells and provides significant anti-tumor effects.
Introduction
CD8 T cells play an important role in the control of intracellular infectious agents and have the potential to mitigate malignant diseases. The CD8 TCR recognizes small peptides bound to MHC class I (MHC-I) products on APCs. These peptides known as CD8 T-cell epitopes are usually derived from processed proteins corresponding to microbial components or tumor-associated antigens (TAAs). The identification of these peptides has lead to developing epitope-based vaccines to induce antigen-specific CD8 T-cell responses for the prevention or treatment of various infections and malignancies [1]. Synthetic peptides containing defined CD8 T-cell epitopes constitute an attractive approach for vaccine development due to their ease of manufacturing and safety as compared to other vaccine types such as recombinant DNA-derived proteins, plasmids, viruses, or genetically engineered cells [2]. However, most peptide vaccines generate minute CD8 T-cell responses as compared to the T cell levels observed during acute infections. Without a doubt, the vaccine’s poor immunogenicity results in suboptimal clinical benefit against an established/advanced disease and has enticed many clinical researchers to seek other immunotherapy alternatives [3]. These disappointing results could be explained in part by the use of weak immunological adjuvants (e.g., incomplete Freund’s adjuvant), suboptimal peptide formulations, and inappropriate routes of vaccine administration.
For some time, our laboratory has been involved in the optimization of peptide vaccines for the induction of anti-tumor CD8 T-cell responses [4, 5]. We have recently proposed that in order to have an impact against established tumors, the vaccines must elicit a CD8 T-cell response resembling the magnitude and duration of the responses observed during acute viral infections, where more than one-third of the circulating CD8 T cells show specificity for the offending microorganism [6]. We have reported that synthetic peptides corresponding to the minimal CD8 T-cell epitope administered intravenously mixed with poly-IC and costimulatory anti-CD40 antibodies resulted in the induction of vast numbers of antigen-specific CD8 T cells in mice, resembling the levels observed during acute infections [7]. Furthermore, experiments performed in several mouse cancer models demonstrated that this vaccination strategy (TriVax) was highly effective against established tumors resulting in many instances in complete disease eradication [8, 9]. Although these results were highly encouraging for developing therapeutic peptide vaccines for humans, there are serious concerns regarding the systemic use of agonistic anti-CD40 antibodies due to potential deleterious effects such as cytokine storm and or liver toxicity [10, 11].
We report here a novel vaccination strategy (BiVax) that allows synthetic peptides to induce high levels of antigen-specific CD8 T cells, when administered systemically (i.v.) in combination with poly-IC without the use of costimulatory anti-CD40 antibodies. Immune responses produced by BiVax were highly dependent on the simultaneous administration of peptide and poly-IC, on the peptide composition, vaccine formulation, and route of administration. As expected, the magnitude of the response was dependent on the expression of the poly-IC receptors TLR3 and MDA5. Peptide combinations with supposedly potent agonists to other TLRs (CpG, Pam3CSK4) were not able to generate the strong CD8 T-cell responses. Interestingly, the magnitude and duration of the CD8 T-cell responses generated by peptide and poly-IC mixtures did not rely on the presence of CD4 T cells, scavenger receptor-A (SR-A), or type-I IFN signals and were minimally affected by the absence of CD40 signaling. The present findings may help to clarify some of the mechanisms involved in the generation of massive and lasting CD8 T-cell responses by peptide epitope vaccines and could facilitate the development of more effective immunotherapies for cancer.
Materials and methods
Mice and cell lines
Six- to eight-week-old female C57BL/6 (B6) mice were obtained from the National Cancer Institute/Charles River Program (Wilmington, MA). CD40-deficient (B6.129P2-Tnfrsf5 tm1Kik/J) mice and TLR3-deficient (B6;129S1-Tlr3 tm1Flv/J) mice in a B6 genetic background were purchased from The Jackson Laboratory (Bar Harbor, ME). Breeder mice deficient on MDA5 (Ifih1) and interferon alpha/beta receptor (IFNαβR,) both on a B6 background were kindly provided by Dr. Marco Colonna (Washington University School, of Medicine, St. Louis MO) and Philippa Marrack (National Jewish Medical and Research Center, Denver CO), respectively. Mice deficient for the expression of both scavenger receptors-A (SR-A) and MARCO (double knockouts on a B6 background) were a gift from Drs. Jim Mulé and Shari Pilon-Thomas (Moffitt Cancer Center). EL4 thymoma cells were purchased from the American Type Culture Collection (Manassas, VA). The TC-1 tumor cell line, expressing HPV16-E7 antigen was obtained from Dr. T-C Wu (Johns Hopkins University, Baltimore, MD). The B16-F10 mouse melanoma cell line was provided by Dr. Alan Houghton (Memorial Sloan Kettering Cancer Center, New York, NY).
Peptides, antibodies, and reagents
All synthetic peptides were purchased from A&A Labs (San Diego, CA). Lipopeptides were synthesized by attachment of 2 palmitic acid (Pam) chains via the 2 amino groups of an N-terminal lysine. The purity and identity of peptides were determined by high-performance liquid chromatography and mass spectrometry analysis by the vendor. Amino acid sequences of the peptides used in this study are shown throughout the text and in the corresponding figures. All peptides were solubilized at 20 mg/ml in DMSO-TFA (99.9 %/0.1 %) and stored in aliquots at −80 °C. Polyinosinic:polycytidylic acid (poly-IC) stabilized with poly-lysine and carboxy-methylcellulose (Poly-ICLC/Hiltonol) was kindly provided by Dr. Andres Salazar (Oncovir, Inc., Washington, DC). Synthetic oligodeoxynucleotides containing CpG motifs, CpG-1826 (Class-B; 5′-tccatgacgttcctgacgtt-3′) and CpG-1585 (Class-A; 5′-ggGGTCAACGTTGAgggggg-3′), were prepared at the Mayo Clinic Molecular Biology Core and were kindly provided by Dr. R. Vile (Mayo Clinic, Rochester, MN). Poly-AU and Pam3CSK4 were purchased from InvivoGen (San Diego, CA). Antibodies for in vivo use in mice, anti-PD-L1 (clone 10F.9G2), anti-CD4 (clone GK1.5), and anti-CD8 (clone 2.43), were purchased from BioXCell (West Lebanon, NH). Fluorochrome-labeled antibodies were obtained from eBioscience, Inc (San Diego, CA). Fluorescence-labeled MHC-I/peptide tetramers were kindly provided by the National Institute of Allergy and Infectious Disease Tetramer Facility at the Emory University (Atlanta, GA from NIH).
Immunizations and assessment of immune responses
Vaccines were freshly prepared by diluting and mixing the peptides and TLR agonists in PBS to the appropriate concentration in order to inject 30–200 μg peptide (depending on the peptide) and 50–100 μg TLR agonist in 50–200 μl per mouse (depending in the route of administration). In most instances, vaccines were administered intravenously (200 μl/injection in the tail vein). In some experiments, mice were vaccinated subcutaneously or intramuscularly (50 μl/injection in 2 sites per mouse). Mice received 1–3 booster immunizations every 5–7 days. In some experiments, mice were depleted of CD4 T cells using anti-CD4 monoclonal antibodies administered i.p. 300 μg twice, on days −2 and 0 before each immunization. Immune responses were mostly assessed by tetramer staining using peripheral blood samples obtained at various time points or in spleen cells at the termination of the experiments. Fluorescence was measured using either a FACSCalibur or an LSRII flow cytometer (BD Biosciences, San Jose, CA) and analyzed using FlowJo software (Ashland, OR). Immune responses were also determined in spleens with EliSpot assays as described [8].
Evaluation of vaccine therapeutic anti-tumor effect
Mice received 3 × 105/mouse tumor cells (TC-1 or B16-F10) s.c. in a shaved rear flank 5–6 days (as noted) before their primary immunization. In some instances, mice received 200 μg anti-PD-L1 antibodies i.p. on days 1 and 3 post-immunization. Tumor growth was monitored every 2–4 days in individual tagged mice by measuring 2 opposing diameters with a set of calipers. Mice were euthanized when the tumors area reached 400 mm2. Results are presented as the mean tumor size (area in mm2) ± SD for every treatment group at various time points until the termination of the experiment.
Statistical analyses
Unpaired Student’s t-test was used to determine statistical significance of differences in numbers of antigen-specific CD8 T cells. Tumor sizes between 2 populations throughout time were analyzed for significance using 2-way ANOVA tests. All analysis and graphics were done using GraphPad Prism 5.01 (GraphPad Software, San Diego, CA).
Results
Immunogenicity and anti-tumor effects of HPV-BiVax
We recently reported that two sequential peptide immunizations (prime and boost, 2-weeks apart) using the minimal CD8 T-cell epitope HPV16-E749–57 (RAHYNIVTF) combined with poly-IC and anti-CD40 monoclonal antibodies (TriVax) resulted in huge CD8 T-cell responses capable of eradicating 100 % established TC1 tumors in mice [9]. Interestingly, vaccination with a mixture of HPV16-E749–57 and poly-IC, without the αCD40 mAb (BiVax), was able to induce a CD8 T-cell response, but the magnitude was lower (~50 % compared to TriVax), and only 30–50 % of the vaccinated mice were able to completely reject their tumors. To further optimize BiVax in this tumor model, we first assessed the possibility of reducing the time interval between prime and booster immunizations and adding another booster to increase the speed and intensity of immune response and enhance the anti-tumor effect. As shown in Fig. 1a, a time interval between prime and boost of 7 or 5 days resulted in ~40 % antigen-specific CD8 T cells, which is identical to the levels observed when prime and boost were administered 13–21 days apart (data not shown and [9]). On the other hand, a substantially lower response was observed when the booster immunization was given 3 days after the prime. Next, we assessed whether an additional booster immunization (prime and 2 boosts, 5 days apart) would further augment the numbers of antigen-specific CD8 T cells and would improve the outcome in TC-1 tumor-bearing mice. While an additional BiVax booster immunization appeared to increase the levels of antigen-specific CD8 T cells in tumor-free mice, this difference was not significant (Fig. 1b). On the other hand, an additional booster in tumor-bearing mice increased significantly to ~60 % the levels of antigen-specific CD8 T cells. The tumor-bearing mice that were not vaccinated did not exhibit antigen-specific CD8 T cells in their blood at the same time points. Most importantly, this experiment showed that all the mice (6/6) that received the prime +2 booster BiVax therapy rejected their tumors (Fig. 1c). The tumor-cured mice were depleted of CD8 T cells using antibodies (as described in the legend of Fig. 1c) from day 43 to 65 after the vaccine prime and no tumor recurrences were observed, indicating that most, if not all the tumor cells had been eliminated. These results indicate that an additional booster immunization helps maintain high levels of antigen-specific CD8 T cells in tumor-bearing mice, which may be critical for completely eradicating disease and prolonging overall survival.
Fig. 1.

Additional BiVax boosters improve vaccine immunogenicity and correlate with enhanced anti-tumor effect. a B6 mice were given a boost 3, 5, and 7 days after prime; 5 days later, the percentage of tetramer positive CD8 T cells was determined in blood (n = 3 mice per group). b Mice were inoculated with 3 × 105 TC-1 cells s.c.; 6 days later, they were primed with BiVax E749–57 and boosted 5 and 10 days later. The percentage of tetramer positive CD8 T cells was determined in blood 7 or 8 days after boost. A tumor-bearing non-vaccinated group and a tumor-free vaccinated group were included as controls. ns = no statistical significant differences between 1 and 2 boosts in tumor-free mice were observed. **Statistical significance between 1 and 2 boosts (P < .001) in tumor-bearing mice was observed. c Tumor sizes were assessed 2–3 times a week using automatic calipers. Mice were euthanized when tumors reached 2 cm in diameter on either side. After tumor rejection, mice were CD8-T-cell-depleted by i.p. inoculation with 500 μg of αCD8 antibody (days 43, 46, 49, 62, and 65 after prime), which was followed until day 80 and no tumors appeared. CD8 T-cell depletion was monitored by flow cytometry (n = 6 mice treatment group, 3 mice in the control groups)
Effect of peptide size in the immunogenicity of BiVax-HPV
It has been shown that vaccines using of long synthetic peptides containing within their sequence a defined CD8 T-cell epitope will result in superior immune responses as compared to the use of peptides corresponding to the minimal epitope when administered s.c. either emulsified in IFA or in combination with anti-CD40 antibodies or CpG adjuvant in PBS [12–14]. The rationale for the use of long peptides versus minimal peptides is that the former need to be presented to the CD8 T cells by professional APCs such as dendritic cells (DCs) that are capable of internalizing and processing the long peptides, while the short minimal peptides could be presented by any MHC-I expressing cell, potentially resulting in immune tolerance. In addition, some long peptides may contain MHC class II binding sequences, which would stimulate CD4 helper T cells, further enhancing CD8 T-cell activation end expansion. Thus, we assessed the immunogenicity of BiVax with two long peptides of 13 and 35 residues, HPV16-E745–57 (AEPDRAHYNIVTF) and HPV16-E743–77 (GQAEPDRAHYNIVTFCCKCDSTLRLCVQSTHVDIR), containing within their sequences the minimal 9-residue HPV16-E749–57 CD8 T-cell epitope. Both long peptides contain a CD4 T cell epitope, HPV16-E748–57 (DRAHYNIVTF) [15] that could potentially enhance the CD8 T-cell responses. Contrary to what was predicted, the magnitude of the CD8 T-cell responses observed in blood generated by the long peptides was considerably lower as compared to the minimal CD8 T-cell epitope (Fig. 2a). The differences in immunogenicity between short and long peptides were even more evident when quantifying of the total numbers of antigen-specific CD8 T cells in spleens, where the minimal epitope HPV16-E749–57 was found to generate vastly more antigen-specific CD8 T cells as compared to the long peptides (Fig. 2b). The lower performance of the long peptides compared to the minimal epitope could be attributed in part to their suboptimal boosting capacity, since mice that were primed with the long HPV16-E743–77 peptide and were later boosted with the minimal HPV16-E749–57 peptide generated high levels of antigen-specific CD8 T cells, similar to those observed in mice that were primed and boosted with the minimal peptide epitope (Fig. 2c). The levels of antigen-specific CD8 T cells induced by all three peptides HPV16-E749–57, HPV16-E745–57, and HPV16-E743–77 after the BiVax prime were quite low (<1 %, data not shown), which also suggests that in this vaccine model, the vast T cell expansion takes place after the secondary immunization and requires the minimal peptide epitope.
Fig. 2.

Effect of peptide size on BiVax HPV16-E749–57 immunogenicity. Mice were immunized with equimolar amounts of the E749–57, E745–57, and E743–77 peptides, a booster shot was given 7 days later, and the percentage of tetramer positive CD8 T cells was determined in blood (a) and spleens (b) 14 and 22 days after prime, respectively. c Mice were primed (p) with either BiVax E749–57 or BiVax E743–77 (as indicated), 7 days later received a boost (b) with BiVax E749–57 and 7 days after the boost the % tetramer positive cells were measured in blood. ns = no statistical significant difference between the two groups was observed (n = 3 mice per group for all experiments)
Applicability of BiVax to other CD8 T-cell epitopes
Next, we wished to extend the observations of the BiVax immunization strategy to other MHC-I binding peptides known to function as strong CD8 T-cell epitopes. For these experiments, we selected the well-known H-2 Kb-restricted epitope Ova257–264 (SIINFEKL) and 2 epitopes derived from the melanosomal antigens tyrosine-related proteins 1 and 2 (Trp1 and Trp2), which function as tumor-associated antigens for the mouse B16 melanoma. While Trp2180–188 (SVYDFFVWL) is restricted by H-2 Kb [16], the heteroclitic epitope Trp1455–463/9M (TAPDNLGYM) is restricted by H-2Db and has a substitution at position 9 (M for A) to improve MHC-I binding and immunogenicity [17, 18]. After a prime-boost BiVax with Trp2180–188, mice generated approximately 25 % tetramer positive CD8 T cells in blood (Supplemental Fig. 1). In contrast, mice that received BiVax using Ova257–264 or Trp1455–463/9M produced a much lower T cell response (<5 %; Supplemental Fig. 1). Comparing the sequences of the 2 epitopes that performed relatively well in BiVax (HPV16-E749–57 and Trp2180–188) with the 2 peptides that did not functioned well (Ova257–264 and Trp1455–463/9M), it became apparent that immunogenicity could not be clearly attributed to the overall hydrophobicity/hydrophilicity nature of the peptides, which could affect pharmacokinetics and antigen persistence after immunization. Ranking the peptides by the Kyte-Doolittle hydrophobicity scale [19] showed that the two most immunogenic peptides ranked 1st (Trp2180–188) and 3rd (HPV16-E749–57). However, when examining the hydrophobic and hydrophilic regions within each peptide it became evident that the 2 immunogenic peptides had an amphiphilic feature, where approximately one half of the molecule was highly hydrophobic while the other half was relatively hydrophilic (Supplemental Fig. 2).
Enhancing immunogenicity of Trp1-BiVax by increasing peptide amphiphilicity
In view of the above, we examined whether the immunogenicity of Trp1455–463/9M using BiVax could be improved by increasing the peptide’s amphiphilic nature, which was achieved by the addition of 2 palmitic acid chains (Pam2) to the amino terminus end using a KMFV linker (the positive charges of K are eliminated with Pam conjugation). It should be noted that lipopeptides prepared by attaching Pam2 to a N-terminal lysine have not been reported to function as TLR agonists as opposed to other lipopeptides where Pam2 are attached via an N-terminal cysteine and a SKKKK linker (Pam2CSKKKK-), which have been shown to stimulate TLR2/6 [20, 21]. The results in Fig. 3a demonstrate that peptide Pam2KMFVTAPDNLGYM was substantially more immunogenic than the minimal epitope TAPDNLGYM after a BiVax prime-boost protocol. Interestingly, the minimal epitope was highly effective in boosting responses in mice that were generated by priming with Pam2KMFVTAPDNLGYM (Fig. 3b). The enhancement of immunogenicity was also achieved with the addition of 6–7 hydrophobic residues to the amino terminus end of the peptide (Fig. 3c). On the other hand, adding only 3 hydrophobic residues did not affect the immunogenicity of the peptide. Extending the natural sequence of the epitope at the amino end by 7 residues (TNTEMFV) or adding the palmitic acid chains directly to the minimal T cell epitope (without the hydrophobic MFV linker) somewhat increased the immunogenicity as compared to the minimal epitope but to a much lower extent as compared to the peptide constructs that contained 6–7 hydrophobic residues or the Pam2KMFV extension. Lastly, extending the Trp1 peptide using charged or hydrophilic residues did not substantially improve the immunogenicity of the peptides using BiVax (Supplemental Fig. 3). The strategy to enhance the peptide immunogenicity by generating an amphiphilic construct also worked well with Ova257–264 (Supplemental Fig. 4).
Fig. 3.
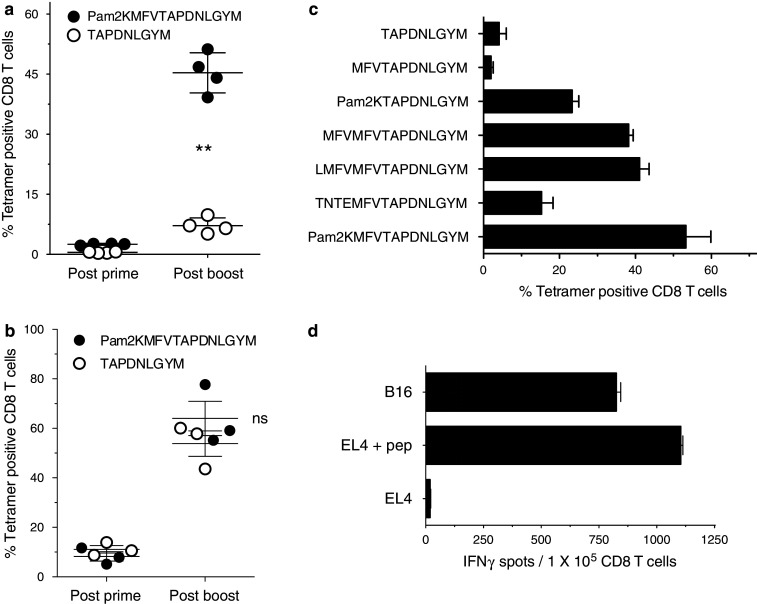
Increasing peptide amphiphilicity enhances immunogenicity of BiVax. B6 mice were vaccinated i.v. on days 0 and 12 with BiVax composed of 150 μg of peptide and 50 μg of poly-IC. a Mice (4 per group) received identical BiVax immunizations (prime/boost) with either minimal peptide (TAPDNLGYM) or palmitilated peptide (Pam2KMFVTAPDNLGYM). Immune responses were measured in blood 7 days after the boost. **Statistical significance (P < .001) between the 2 peptides was observed after the boost. b Mice (3 per group) were primed with palmitilated peptide and boosted with either the palmitilated peptide or the minimal peptide. On day 7 and 19, antigen-specific CD8 T cells were evaluated in blood by tetramer analysis. ns = no statistical significant difference was observed between the 2 peptides after the boost. c Mice (3 per group) received two identical BiVax immunizations (prime-boost) with indicated peptides as described above. 7 days after the boost the antigen-specific CD8 T cells in blood were evaluated by tetramer analysis. Results represent the average percentage of tetramer positive CD8 T cells from 3 mice per group with SD (bars) of the means. d Purified spleen CD8 T cells from the experiment presented in panel (a) from the mice vaccinated with palmitilated peptide were evaluated for their ability to recognize tumor cells using an IFN-γ secretion EliSpot assay. APCs used: Trp1455–463-pulsed EL4 (EL4 + pep), B16F10 melanoma, and un-pulsed EL4 cells (negative control). Results represent the average number of spots from triplicate wells with SD (bars) of the means
Therapeutic effects of BiVax-Trp1 against established melanomas
A significant concern raised with the use of synthetic peptides as immunogens is that these vaccines will mainly generate low avidity T cells incapable of recognizing tumor cells that may express low density of peptide/MHC complexes on their surface. Nevertheless, as shown in Fig. 3d, the CD8 T cells induced by BiVax with Pam2KMFVTAPDNLGYM (from hereafter simply referred to as “Pam2-Trp1”) were highly efficient in vitro in recognizing B16 melanoma cells. The therapeutic effect of BiVax immunization was evaluated against 5-day established subcutaneous B16-F10 melanoma tumors using Pam2-Trp1. Tumors grew at a significantly lower rate in mice that received 2 sequential BiVax immunizations as compared to the untreated group (Fig. 4a). Moreover, the inclusion of 2 additional booster immunizations further increased the therapeutic effect of BiVax with Pam2-Trp1 (Fig. 4b) and the addition of PD1 blockade (with anti-PD-L1 antibodies) resulted in a remarkable therapeutic effect, where the majority of the mice (4/5) were able to completely reject their tumors (Fig. 4c). Interestingly, the control groups that received an irrelevant peptide (Pam2-Ctrl) and poly-IC also showed a decrease in the rate of tumor growth but the therapeutic benefit was not as effective as compared to the use of the Pam2-Trp1 peptide. The therapeutic effects of Pam2-Trp1 BiVax and the control Pam-Ctrl BiVax disappeared when mice were depleted of CD8 T cells (data not presented).
Fig. 4.

Therapeutic effects of BiVax against established B16 melanoma. B6 mice (4 per group) were inoculated s.c. on day 0 with 3 × 105 live B16-F10 cells and vaccinated i.v. with Pam-Trp1 or control Pam2-Ctrl BiVax. Tumor-bearing mice received 2 (a) or 4 (b) immunizations (arrows). (c) PD1 blockade was included in an experiment using 4 BiVax immunizations. Anti-PD-L1 mAb (200 μg/dose) was administered i.p. on days 1 and 3 post-immunization, and two more antibody injections (on days 5 and 7) were added after the 4th BiVax immunization. Non-vaccinated mice (No Vax) and palmitilated irrelevant peptide BiVax (Pam2-Ctrl) were included as controls. Tumor sizes were determined in individual mice by measuring 2 opposing diameters and are presented as tumor areas in square millimeters. Points, mean for each group of mice; gray bars, period of anti-PD-L1 mAb treatment; bars, SD. P values were calculated using 2-way ANOVA tests. *, 4 of 5 mice in this group completely rejected their tumors and survived until day 60 (not shown)
Mechanisms involved in the immunogenicity of BiVax
Next, we examined some of the mechanisms that could play a role in the generation of the strong CD8 T-cell responses observed with BiVax. First, we assessed whether other TLR agonists also being considered as immune adjuvants performed with Pam2-Trp1. The results in Fig. 5a demonstrate that only poly-IC was capable of generating strong CD8 T-cell responses with Pam2-Trp1. The use of poly-AU (another TLR3 agonist) did not lead to the generation of the large CD8 T-cell response observed with poly-IC. Furthermore, CpG containing oligodeoxynucleotides of either type-A (CpG-1585) or type-B (CpG-1826), which function as TLR9 agonists or the TLR2/TLR1 agonist Pam3CSK4, failed to generate substantial T cell responses when administered mixed with Pam2-Trp1. Interestingly, combining CpG-1826 with poly-IC reduced the magnitude of the CD8 T-cell response as compared to the use of poly-IC alone. On the other hand, the low T cell response generated by the administration of Pam2-Trp1 peptide with CpG-1826 could be dramatically increased with a BiVax booster containing poly-IC (Fig. 5b). The results so far presented were obtained with poly-IC formulation, known as poly-ICLC containing poly-lysine and carboxy-methyl cellulose to stabilize the compound and protect it from RNAse degradation, which occurs mostly in primates [22]. Nevertheless, identical results were obtained using several commercially available non-stabilized poly-IC formulations (data not presented).
Fig. 5.

Effect of various TLR agonists on BiVax immunization. a Mice (3 per group) were immunized i.v. on days 0 and 12 with Pam2-Trp1 peptide and one of the following TLR agonists: 50 μg poly-IC, 100 μg CpG-1826, 100 μg CpG-1585, 50 μg poly-AU, 100 μg Pam3CSK4, and a mixture of poly-IC + CpG-1826. On day 7 (post-prime) and 19 (post-boost), blood samples were evaluated by tetramer analysis. b Mice (3 per group) were immunized i.v. on days 0 and 12 with BiVax/CpG-1826, and boosted one more time with BiVax/poly-IC on day 24. The presence of antigen-specific CD8 T cells in blood was evaluated by tetramer analysis on days 7 (post-prime), 19 (post-boost), and 31 (post-2nd boost). Points, the value for each individual mouse; horizontal line, the average value of the group; bars, SD
CD4 T cells may be important in the generation and long-term maintenance of CD8 T-cell responses by functioning as helper T cells. On the other hand, another CD4 T cell subset known as T regulatory cells are known to inhibit CD8 T-cell responses [23, 24], in particular those directed against self-antigens such as Trp1. Thus, we assessed whether depletion of CD4 T cells prior to each immunization would impact the magnitude and duration of the CD8 T-cell response generated by BiVax with the Pam2-Trp1 peptide. As shown in Fig. 6a, the level of the CD8 T-cell response after the BiVax prime (day 6 post-vaccine priming) was almost twofold higher when CD4 T cells were depleted as compared to the untreated mice. However, the magnitude of the immune response increased after the booster immunization regardless of whether CD4 T cells had been depleted or not. Also, the duration of the Trp1 CD8 T-cell response to BiVax was not affected by the absence of CD4 T cells during the immunizations.
Fig. 6.
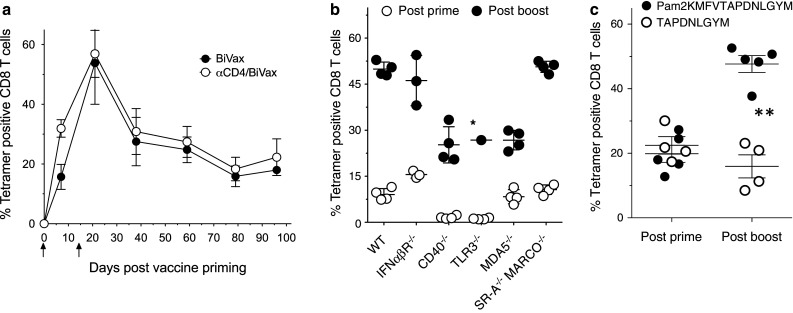
Immunological mechanisms involved in BiVax CD8 T-cell responses. a Effect of CD4 T cells in immune responses to BiVax. Untreated or CD4 T cell-depleted mice (3 per group) were immunized i.v. on days 0 and 14 with Pam2-Trp1-Bivax (arrows), and the presence of antigen-specific CD8 T cells in blood was measured at various time points using tetramer analysis. CD4 depletion was initiated 3 and 1 day before the BiVax prime and repeated in a similar way prior to the boost. CD4 T cell depletion was confirmed by flowcytometry. b Role of immune receptors in the immunogenicity of BiVax. WT B6 mice (WT), IFNαβR−/−, CD40−/−, TLR3−/−, MDA5−/−, and scavenger receptor SR-A and MARCO double knockout (SR-A−/−MARCO−/−) mice were evaluated for their immune responses to Pam2-Trp1 7 days after prime and boost. *, 3/4 TLR3−/− mice died after the boost. c IFNαβR−/− mice were primed with Pam2-Trp1 and boosted with either the Pam2-Trp1 or the minimal peptide. On day 7 (post-prime) and 19 (post-boost), antigen-specific CD8 T cells were evaluated in blood by tetramer analysis. **Statistical significance (P < .01) between the 2 peptide responses was observed after the boost
Next, using several genetically deficient mouse strains, we evaluated the role that various immune-related receptors could play in the generation of CD8 T-cell responses by BiVax. The absence of CD40 and TLR3 resulted in a marked reduction in the levels of antigen-specific CD8 T cells observed after the vaccine prime (Fig. 6b). On the other hand, in the absence of IFNαβR, MDA5, and scavenger receptors SR-A and MARCO, the primary immune responses to BiVax were comparable as those observed in wild-type (WT) mice. Strong secondary immune responses, that more than doubled the levels of antigen-specific CD8 T cells as compared to the prime, were observed in all instances. However, the absence of TLR3 resulted in severe toxicity where 3/4 vaccinated mice died by what appeared to be cytokine storm syndrome. In addition, the fold-increase of the secondary response was lower in mice deficient of the MDA5 RNA helicase, as compared to the WT and other genetically deficient mice. Although the CD40- and the TLR3-deficient mice exhibited lower primary CD8 T-cell responses, the booster immunization generated strong secondary responses when considered as the fold-increase, compared to the primary response for each mouse strain. Since the adjuvant effect of poly-IC is considered to depend in great part by its ability to induce high levels of type-I IFN, it was somewhat puzzling that the CD8 T-cell responses observed in IFNαβR-deficient mice were similar, if not identical to those observed in WT mice (Fig. 6b). However, very different results were observed when the minimal Trp1 peptide was used to boost the T cell response induced by priming with Pam2-Trp1. While as previously noted, the minimal Trp1 peptide was efficient in boosting the responses in WT mice (Fig. 3b), this was not the case with the IFNαβR-deficient mice where only the Pam2-Trp1 was capable of boosting the responses (Fig. 6c). These results indicate that type-I IFN may be critical only when antigen is presented by non-professional APCs, which occurs when the minimal peptide is used to expand the primed CD8 T cells.
Effects of the mode of administration in the immunogenicity of BiVax
So far, the results presented above were obtained using i.v. immunizations, which could be considered somewhat unconventional. Thus, we compared BiVax immunogenicity against Pam2-Trp1 using three different vaccination routes. While both the i.v. and i.m. routes were highly effective for the generation of strong responses, administration of BiVax via the s.c. route was clearly less effective (Fig. 7a). Lastly, we assessed whether the 2 components of BiVax, peptide and poly-IC needed to be administered simultaneously, or whether separate injections given at different times would provide similar results. Injecting Pam2-Trp1 5 h before poly-IC had no deleterious effect as compared to the simultaneous administration of both BiVax components (Fig. 7b). On the other hand, the level of the immune response was dramatically reduced when poly-IC was injected 5 h prior to the administration of the Pam2-Trp1 antigen. A somewhat different pattern of responses was observed with BiVax using the HPV16-E749–57 peptide. As with the previous result, the simultaneous administration of peptide and poly-IC resulted in generating the strongest immune response and administration of the poly-IC before peptide reduced to a great extent this response (Fig. 7c). However, in this case, HPV16-E749–57 injection before poly-IC reduced the response by ~50 %.
Fig. 7.
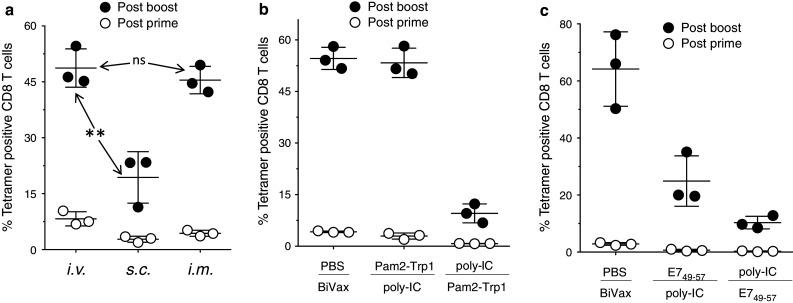
Effects of the mode of administration in BiVax vaccines. a Mice (3 per group) were immunized though different routes and the presence of antigen-specific CD8 T cells in blood was analyzed by tetramer analyses 7 days after the prime and the boost. **Statistical significance (P < .01) between i.v. and s.c. induced responses was observed after the boost. ns = no statistical significant difference was observed between i.v. and i.m. after the boost. b Mice (3 per group) were pretreated i.v. with either PBS (control), Pam2-Trp1 or poly-IC, and 5 h later received i.v. BiVax (poly-IC and Pam2-Trp1) poly-IC or Pam2-Trp1 (as indicated for each group). CD8 T-cell responses were measured by tetramer analysis on days 7 (post-prime) and 19 (post-boost). c Mice were injected i.v. with PBS, E749–57, or poly-IC and 5 h later received a second i.v. injection with BiVax (E749–57 + poly-IC), poly-IC or the E749–57 (as indicated for each group) and were boosted the same way 16 days later. The percentage of tetramer positive CD8 T cells was determined in blood 6 days after prime and boost (n = 3 mice per group)
Discussion
There is little doubt that the effectiveness of a vaccine will depend in great part on its ability to elicit high levels of long-lasting antigen-specific, pathogen-reactive CD8 T cells. Our group has proposed that these levels and the duration of the response should resemble those observed during acute infections [6]. The failure of many vaccination strategies can be attributed in many cases to the inability of the vaccines to generate potent and persistent immune responses. Other instances of vaccine failures, when strong immune responses have been observed, could be related to the inability of the T cells to recognize the pathogen-infected or tumor cells due to low avidity of the T cells for their antigen. Particularly, in the case of TAAs that are also expressed by normal cells such as Trp1, it is likely that the highest avidity T cells are eliminated via immunological tolerance and that vaccines would recruit T cells of insufficient avidity to recognize tumor cells. Additional concerns have been brought up on the use of vaccines that utilize synthetic peptides corresponding to the minimal CD8 T-cell epitopes, including the induction of low avidity T cells due to the generation of supraoptimal levels of peptide/MHC-I complexes on APCs. In addition, some vaccines prepared with short peptide epitopes have been reported to induce T cell deletion [25], presumably because these peptides can be presented by non-professional APCs. In view of this, it has been advocated that peptide vaccines should be prepared using long peptides that would require capture and antigen processing by professional APCs [14]. Indeed, when long peptides were administered s.c. using suboptimal adjuvants such as IFA or CpG, they were found to be more immunogenic than their short peptide counterparts [12, 13].
We present here an improved vaccination strategy called BiVax that was designed to mimic a viral infection, inducing high levels of CD8 T cells with sufficient avidity to recognize foreign and self-TAAs and capable of persisting for a long time period. Our results show that a synthetic peptide corresponding to a minimal CD8 T-cell epitope, HPV16-E749–57, mixed with poly-IC and administered systemically (i.v.) in an aqueous formulation generated after a short prime-boost protocol (5–7 days apart), huge numbers of antigen-specific, tumor-reactive T cells capable of rejecting established tumors (Fig. 1). BiVax immunization using Pam2-Trp1 construct was also able to generate very large numbers of antigen-specific CD8 T cells capable of recognizing B16 tumor cells (Fig. 3) but the anti-tumor effects were less impressive (Fig. 4a) as compared to the results in the HPV tumor model. However, administration of two additional booster immunizations and anti-PDL-L1 antibodies resulted in a remarkable anti-tumor effect where the majority of the mice rejected their tumors (Fig. 4c). These results indicate that for some tumors such as B16 melanoma where PD-L1 exhibits a strong inhibitory function, large numbers of antigen-specific CD8 T cells by themselves are not sufficient to eliminate the disease and that additional manipulations such as PD1 blockade will be necessary.
In contrast to results from others obtained with vaccines administered s.c. in oil:water emulsions, the long peptides bearing the HPV16-E749–57 epitope were markedly less immunogenic than the minimal epitope, when administered using the BiVax strategy (Fig. 2). In a report by Wick et al., a long HPV peptide (19mer) administered s.c. in PBS mixed with poly-IC in 4 sequential doses was shown to be more effective in eliciting CD8 T-cell responses than the minimal (9mer) epitope [26]. However, the magnitude of the CD8 T-cell responses achieved in these studies by this vaccination mode was several orders of magnitude lower as to what BiVax achieved in the present studies after 2 sequential immunizations using the minimal 9mer HPV epitope. The disparities in results between both studies could be due to differences in peptide and poly-IC doses and the route of vaccine administration. It remains unknown the degree of participation of professional APCs in the response to BiVax with the minimal HPV16-E749–57 epitope and whether non-professional APCs could be presenting antigen to the CD8 T cells to facilitate clonal expansion. Also, we do not know the reason(s) why the elongated HPV peptides were not as effective as the short peptide in generating the vast CD8 T-cell responses observed with BiVax. We propose that to be highly immunogenic, a peptide must be able to both prime and boost effectively and so far only the amphiphilic peptides, no matter of what size can accomplish both tasks. In some instances, peptides such as the long non-amphiphilic HPV constructs could prime but were unable to boost (Fig. 2). In other instances, the small non-amphiphilic peptides such as Trp1455–463/9M and Ova257–264 could not prime but were able to boost (Fig. 3b, Supplemental Fig. 4 and data not shown). More extensive studies using additional CD8 epitopes and corresponding peptide constructs will be needed in order to establish whether these observations are broadly applicable.
One could assume that the extensive CD8 T-cell responses observed in BiVax with HPV16-E749–57 (and amphiphilic Trp1 peptides) require the participation of both professional APCs such as DCs (for the initial priming) and non-professional APCs (for facilitating clonal expansion of the activated T cells). Thus, the long HPV peptides would be inefficient because they rely solely in professional APCs, which exist at much lower frequencies than non-professional APCs. Our results agree with the above-stated assumption since a long HPV16-E7 peptide was found to be effective in priming but not boosting the CD8 T-cell response, while the short HPV16-E7 peptide was effective in boosting responses induced by priming with either the short or the long peptide (Fig. 2). Also, the minimal Trp1 peptide was found to be inefficient in priming CD8 T cells (Fig. 3a) but was highly effective in expanding the T cells generated by the Pam2-Trp1 peptide (Fig. 3b). Nevertheless, the ability of the Pam2-Trp1 and elongated hydrophobic Trp1 peptide constructs not only to effectively prime the CD8 T cells, but also to successfully expand these responses needs to be addressed since these peptides presumably would require to be presented by non-professional APCs. The possibility exists that amphiphilic, lipidated, and hydrophobic elongated peptides may be able to be captured and undergo processing by both professional and non-professional APCs, while other long, not-so-amphiphilic peptides such as HPV16-E745–57 and HPV16-E743–77 may not be so effective in being captured by non-professional APCs. In support of this possibility is the observation that amphiphilic peptides can self-assemble into well-defined nanostructures [27], which presumably could be taken up quite effectively by APCs. The possibility also exists that short peptides such as HPV16-E749–57 and elongated amphiphilic Trp1 peptides may form complexes with poly-IC, which are more effectively internalized by APCs via some type of nucleic acid-binding scavenger receptor (SR). Once inside of the cell, the peptide/poly-IC complexes within endosomal compartments will stimulate TLR3 and may spill the contents to the cytoplasm due to the proton sponge effect generated by the endosomal nucleic acid content. Although all these possibilities remain to be studied, the necessity for the formation of peptide/poly-IC complexes is somewhat supported by the observations that administration of poly-IC prior to the peptide did not lead to the generation of the strong immune responses (Fig. 7b, c). One would expect that poly-IC would quickly be cleared from circulation by binding to the numerous SRs whose function is to rapidly eliminate foreign and self-free nucleic acids. The injection of the peptide before poly-IC could work if the persistence of the peptide in circulation allows the in vivo formation of complexes with poly-IC. The formation of peptide/poly-IC complexes has been suggested due to the formation of co-precipitates when HPV16-E749–57 was mixed with poly-IC [28]. Members of the SR class-A (SR-A) family are known to bind nucleic acids. Our results indicate that BiVax generated levels of CD8 T cells in mice deficient in SR-AI/II/III and MARCO that were equivalent to those observed in WT mice. Thus, it is likely that other class of SRs may be capable of interacting with poly-IC. The redundancy in the specificity of SRs makes it difficult to pinpoint exactly those receptors in professional and non-professional APCs that would be involved in the capture of peptide/poly-IC complexes.
With respect to the differences observed with the various routes of administration, we initially reasoned that a systemic administration (i.v.) of the vaccine would be able to deliver antigen throughout most lymphoid organs recruiting larger numbers of antigen-specific T cells as compared to a local (s.c.) immunization, where only draining lymph nodes would be involved. Indeed, with the Pam-Trp1 peptide, BiVax given i.v. was far superior as compared to BiVax s.c. (Fig. 7a). Interestingly, with Pam-Trp1, BiVax given i.m. was as effective as BiVax i.v. Thus, it is possible that the peptide, poly-IC mixture (or complexes) can effectively diffuse from the muscle into the general circulation to reach proximal and distal lymphoid organs. However, BiVax i.m. with HPV16-E749–57 was less effective (~50 %) as compared to BiVax i.v., but the levels of antigen-specific CD8 T cells could be further increased to similar levels as those observed with BiVax i.v. with an additional i.m. booster immunization (data not shown). It has been advocated that most vaccines should be administered i.m. and not s.c. because of higher vascularity of the muscle as compared to the subcutaneous fat layers, which will influence antigen diffusion into the bloodstream [29].
Poly-IC has been considered to function as a potent immune adjuvant due to its ability to stimulate endosomal TLR3 and cytoplasmic retinoic acid-inducible gene (RIG)-1-like receptor (RLR) family member, MDA5. As a result of TLR3 and MDA5 activation, cells produce high amounts of type-I IFN and other proinflammatory cytokines such as IL-1, TNFα, IL-6, and IL-12. Our results showed that the levels of immune responses induced by BiVax were lower in mice deficient for either TLR3 or MDA5 as compared to WT mice (Fig. 6b). The levels of CD8 T cells induced after priming were substantially lower in TLR3-KO but not in MDA5-KO as compared to the WT mice. On the other hand, the fold-increase in the level of the immune response observed in MDA5-KO was decreased as compared to what was observed in WT mice. There was some indication that the secondary response induced by booster immunization in TLR3-KO mice was effective, as evident by the large difference between primary and secondary responses, but this could only be verified in 1/4 mice because of severe toxicity of the vaccine in these mice. Mice deficient in CD40 responded poorly to the primary immunization but exhibited a large secondary response after the booster. Putting together all of these results, one could speculate that during the primary immune response in which professional APCs may play a more critical role, that TLR3 activation and CD40 costimulation play an important role in the activation of the naïve T cells. On the other hand, during the secondary response, the stimulation of the previously activated T cells can be effectively carried out by non-professional APCs that are activated by poly-IC mostly through MDA5 and that do not require the participation of TLR3 and CD40.
The elimination of CD4 T cells resulted in an increased primary CD8 T-cell response as compared to the normal mice controls. It is possible that eliminating CD4+/CD25+/Foxp3+ T regulatory cells with anti-CD4 antibody treatment could increase the effectiveness of BiVax priming. However, the use of anti-CD25 antibodies to reduce CD4 T regulatory cell function did not enhance the potency of BiVax using various peptide epitopes (data not shown). More importantly, the results demonstrating that CD40 was required for effective priming, suggest that other cells besides CD4 T cells that express CD40L (CD154) such as platelets, granulocytes, subsets of CD8 or gamma-delta T cells may function as helpers during the BiVax primary response. As mentioned above, poly-IC is known to induce the production of high levels of type-I IFN, which has been demonstrated to function as “signal 3” for the activation and expansion of CD8 T cells [30]. In view of this, we assumed that the adjuvant effect of poly-IC in BiVax could be due to the signal 3 effects of type-I IFN on the peptide-stimulated CD8 T cells. Thus, we were surprised that BiVax using Pam2-Trp1 functioned well in IFNαβR-deficient mice (Fig. 6b), indicating that type-I IFN signals are not critical for the activation and expansion of CD8 T cells in this vaccination strategy. Interestingly, in contrast to what was observed in WT mice, that the minimal Trp1 peptide was effective in boosting the CD8 T-cell responses of Pam2-Trp1 primed mice (Fig. 3b), in IFNαβR-deficient mice, only the Pam2-Trp1 was able to boost while the minimal Trp1 peptide did not (Fig. 6c). These findings suggest that type-I IFN may play a critical role when non-professional APCs present antigen during the expansion phase to the previously activated CD8 T cells. Thus, type-I IFN may serve as an endogenous danger signal that would allow proliferation of T cells to antigens presented by infected cells that are not professional APCs. On the other hand, T cells activated by professional APCs reactive to self-antigens, when presented by antigen by non-professional APCs in the absence of type-I IFN would not continue to proliferate, limiting an autoimmune response. These results indicate that poly-IC stimulation of professional APCs results in a TLR3/CD40-dependent and type-I IFN/MDA5-independent primary activation of CD8 T cells where alternative cytokines such as IL2, IL12, and IL15 may serve as signal 3 leading to activation and an initial somewhat restrained clonal T cell expansion. However, soon after (5–7 days) the initial activation of the CD8 T cells by professional APCs when non-professional APCs present antigen to the T cells in circumstances where type-I IFN is being produced via MDA5 stimulation by poly-IC (or viral dsRNA), a massive T cell expansion will be generated. We believe that many of vaccines designed to elicit CD8 T-cell responses such as peptide (long or short) vaccines formulated in IFA, peptide-pulsed DCs, and plasmid DNA immunization are probably effective in accomplishing the primary T cell activation and initial limited T cell expansion but fail to induce the secondary massive T cell expansion, observed in natural immune responses during acute infections, which may be necessary to obtain effective anti-tumor effects. In contrast, BiVax using amphiphilic peptides and poly-IC is highly effective in both the priming (activation/initial expansion) and massive expansion after the secondary immunization.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
This work was supported by NIH grants R01CA136828 and R01CA157303. We thank Andres Salazar (Oncovir, Inc.) for kindly providing Poly-ICLC (Hiltonol™) and the NIH Tetramer Core Facility for providing several peptide/MHC tetramers.
Conflict of interest
Esteban Celis has filed a patent application based on the use of synthetic peptides and poly-IC combinatorial vaccines. The rights of the patent application have been transferred to the Moffitt Cancer Center. The remaining authors (HC, KB, YL and AL) declare no conflict of interest.
Footnotes
Hyun-Il Cho and Kelly Barrios contributed equally to the work.
References
- 1.Van Der Bruggen P, Zhang Y, Chaux P, Stroobant V, Panichelli C, Schultz ES, Chapiro J, Van Den Eynde BJ, Brasseur F, Boon T. Tumor-specific shared antigenic peptides recognized by human T cells. Immunol Rev. 2002;188:51–64. doi: 10.1034/j.1600-065X.2002.18806.x. [DOI] [PubMed] [Google Scholar]
- 2.Melief CJ, Offringa R, Toes RE, Kast WM. Peptide-based cancer vaccines. Curr Opin Immunol. 1996;8:651–657. doi: 10.1016/S0952-7915(96)80081-1. [DOI] [PubMed] [Google Scholar]
- 3.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Appella E, Loftus DJ, Sakaguchi K, Sette A, Celis E. Synthetic antigenic peptides as a new strategy for immunotherapy of cancer. Biomed Pept Proteins Nucleic Acids. 1995;1:177–184. [PubMed] [Google Scholar]
- 5.Buteau C, Markovic SN, Celis E. Challenges in the development of effective peptide vaccines for cancer. Mayo Clinic Proceed. 2002;77:339–349. doi: 10.4065/77.4.339. [DOI] [PubMed] [Google Scholar]
- 6.Cho HI, Celis E. Overcoming doubts and other obstacles in the development of effective peptide-based therapeutic vaccines against cancer. Expert Rev Vaccines. 2010;9:343–345. doi: 10.1586/erv.10.13. [DOI] [PubMed] [Google Scholar]
- 7.Assudani D, Cho HI, DeVito N, Bradley N, Celis E. In vivo expansion, persistence, and function of peptide vaccine-induced CD8 T cells occur independently of CD4 T cells. Cancer Res. 2008;68:9892–9899. doi: 10.1158/0008-5472.CAN-08-3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cho HI, Celis E. Optimized peptide vaccines eliciting extensive CD8 T-cell responses with therapeutic antitumor effects. Cancer Res. 2009;69:9012–9019. doi: 10.1158/0008-5472.CAN-09-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barrios K, Celis E. TriVax-HPV: an improved peptide-based therapeutic vaccination strategy against human papillomavirus-induced cancers. Cancer Immunol Immunother. 2012;61:1307–1317. doi: 10.1007/s00262-012-1259-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hixon JA, Blazar BR, Anver MR, Wiltrout RH, Murphy WJ. Antibodies to CD40 induce a lethal cytokine cascade after syngeneic bone marrow transplantation. Biol Blood Marrow Transplant. 2001;7:136–143. doi: 10.1053/bbmt.2001.v7.pm11302547. [DOI] [PubMed] [Google Scholar]
- 11.Kimura K, Moriwaki H, Nagaki M, Saio M, Nakamoto Y, Naito M, Kuwata K, Chisari FV. Pathogenic role of B cells in anti-CD40-induced necroinflammatory liver disease. Am J Pathol. 2006;168:786–795. doi: 10.2353/ajpath.2006.050314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zwaveling S, Ferreira Mota SC, Nouta J, Johnson M, Lipford GB, Offringa R, van der Burg SH, Melief CJ. Established human papillomavirus type 16-expressing tumors are effectively eradicated following vaccination with long peptides. J Immunol. 2002;169:350–358. doi: 10.4049/jimmunol.169.1.350. [DOI] [PubMed] [Google Scholar]
- 13.Bijker MS, van den Eeden SJ, Franken KL, Melief CJ, Offringa R, van der Burg SH. CD8+ CTL priming by exact peptide epitopes in incomplete Freund’s adjuvant induces a vanishing CTL response, whereas long peptides induce sustained CTL reactivity. J Immunol. 2007;179:5033–5040. doi: 10.4049/jimmunol.179.8.5033. [DOI] [PubMed] [Google Scholar]
- 14.Melief CJ, van der Burg SH. Immunotherapy of established (pre)malignant disease by synthetic long peptide vaccines. Nat Rev Cancer. 2008;8:351–360. doi: 10.1038/nrc2373. [DOI] [PubMed] [Google Scholar]
- 15.Tindle RW, Fernando GJ, Sterling JC, Frazer IH. A “public” T-helper epitope of the E7 transforming protein of human papillomavirus 16 provides cognate help for several E7 B-cell epitopes from cervical cancer-associated human papillomavirus genotypes. Proc Natl Acad Sci USA. 1991;88:5887–5891. doi: 10.1073/pnas.88.13.5887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bloom MB, Perry-Lalley D, Robbins PF, Li Y, el-Gamil M, Rosenberg SA, Yang JC. Identification of tyrosinase-related protein 2 as a tumor rejection antigen for the B16 melanoma. J Exp Med. 1997;185:453–459. doi: 10.1084/jem.185.3.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guevara-Patino JA, Engelhorn ME, Turk MJ, Liu C, Duan F, Rizzuto G, Cohen AD, Merghoub T, Wolchok JD, Houghton AN. Optimization of a self antigen for presentation of multiple epitopes in cancer immunity. J Clin Invest. 2006;116:1382–1390. doi: 10.1172/JCI25591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cho HI, Lee YR, Celis E. Interferon gamma limits the effectiveness of melanoma peptide vaccines. Blood. 2011;117:135–144. doi: 10.1182/blood-2010-08-298117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 20.Braun V. Covalent lipoprotein from the outer membrane of Escherichia coli . Biochim Biophys Acta. 1975;415:335–377. doi: 10.1016/0304-4157(75)90013-1. [DOI] [PubMed] [Google Scholar]
- 21.Takeda K, Takeuchi O, Akira S. Recognition of lipopeptides by Toll-like receptors. J Endotoxin Res. 2002;8:459–463. doi: 10.1179/096805102125001073. [DOI] [PubMed] [Google Scholar]
- 22.Levine AS, Levy HB. Phase I-II trials of poly IC stabilized with poly-l-lysine. Cancer Treat Rep. 1978;62:1907–1912. [PubMed] [Google Scholar]
- 23.Shevach EM, McHugh RS, Piccirillo CA, Thornton AM. Control of T-cell activation by CD4+ CD25+ suppressor T cells. Immunol Rev. 2001;182:58–67. doi: 10.1034/j.1600-065X.2001.1820104.x. [DOI] [PubMed] [Google Scholar]
- 24.Yamaguchi T, Sakaguchi S. Regulatory T cells in immune surveillance and treatment of cancer. Semin Cancer Biol. 2006;16:115–123. doi: 10.1016/j.semcancer.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 25.Toes RE, van der Voort EI, Schoenberger SP, Drijfhout JW, van Bloois L, Storm G, Kast WM, Offringa R, Melief CJ. Enhancement of tumor outgrowth through CTL tolerization after peptide vaccination is avoided by peptide presentation on dendritic cells. J Immunol. 1998;160:4449–4456. [PubMed] [Google Scholar]
- 26.Wick DA, Martin SD, Nelson BH, Webb JR. Profound CD8+ T cell immunity elicited by sequential daily immunization with exogenous antigen plus the TLR3 agonist poly(I:C) Vaccine. 2011;29:984–993. doi: 10.1016/j.vaccine.2010.11.036. [DOI] [PubMed] [Google Scholar]
- 27.Cavalli S, Albericio F, Kros A. Amphiphilic peptides and their cross-disciplinary role as building blocks for nanoscience. Chem Soc Rev. 2010;39:241–263. doi: 10.1039/b906701a. [DOI] [PubMed] [Google Scholar]
- 28.Cui Z, Qiu F. Synthetic double-stranded RNA poly(I:C) as a potent peptide vaccine adjuvant: therapeutic activity against human cervical cancer in a rodent model. Cancer Immunol Immunother. 2006;55:1267–1279. doi: 10.1007/s00262-005-0114-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zuckerman JN. The importance of injecting vaccines into muscle. Different patients need different needle sizes. BMJ. 2000;321:1237–1238. doi: 10.1136/bmj.321.7271.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Curtsinger JM, Valenzuela JO, Agarwal P, Lins D, Mescher MF. Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J Immunol. 2005;174:4465–4469. doi: 10.4049/jimmunol.174.8.4465. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.