

Abstract
Objective
Describe the clinical features of a Brazilian C9orf72 frontotemporal dementia – amyotrophic lateral sclerosis (FTD-ALS) kindred, and compare them to other reported C9orf72 families and FTD-ALS causing mutations.
Design
Report of a kindred.
Setting
Dementia center at an University hospital.
Patients
One kindred encompassing 3 generations.
Results
The presence of a hexanucleotide (GGGGCC) expansion in C9orf72 was confirmed by repeat-primed PCR and Southern blot. The observed phenotypes were behavioral variant FTD and ALS with dementia, with significant variability in age of onset and duration of disease. Parkinsonian features with focal dystonia, visual hallucinations and more posterior atrophy on neuroimaging than is typical for FTD were seen.
Conclusions
bvFTD due to C9orf72 expansions displays some phenotypic heterogeneity, and may be associated with hallucinations, parkinsonism, focal dystonia, and posterior brain atrophy. Personality changes may precede by many years the diagnosis of dementia and may be a distinguishing feature of this mutation.
Introduction
The familial co-occurrence of behavioral variant of frontotemporal dementia (bvFTD) and amyotrophic lateral sclerosis (ALS) has been acknowledged for decades.1 Since 2006, 14 autosomal dominant FTD-ALS families with linkage to chromosome 9p have been described.2 Those families had ALS and bvFTD as predominant phenotypes with some within- and inter-kindred phenotypic variation. Recently, a non-coding hexanucleotide repeat expansion was found in the C9orf72 (chromosome 9 open reading frame 72) gene implicated in those families.3,4 Other genes have been implicated in rare FTD-ALS families, such as TARDBP, FUS, CHMP2B, VCP, DCTN1, UBQLN2, MAPT and GRN.5 This report aims to describe a Brazilian C9orf72 kindred, and based on previous reports further characterize its distinguishing features.
Family characterization
We describe a Brazilian family of Italian and Portuguese origins (UCSFBR1, Table 1). Two patients were seen at the University of California, San Francisco, and other affected members’ data is summarized based on the information provided by the family and their neurologist (MLVP). The pedigree (Figure 1) was simplified and anonymized for confidentiality (at-risk individuals were omitted). This report is based on data collected in an IRB-approved project and family members (or their surrogates) signed informed consents. The diagnoses of bvFTD and ALS were made based on current diagnostic criteria.
Table 1.
Clinical features of the UCSFBR1 kindred
| Patient | Sex | Age of onset (years) |
Duratio n (years) |
bvFTD | ALS | Parkin sonism |
Dyston ia |
Halluci nations |
|---|---|---|---|---|---|---|---|---|
| I-2 | F | 40’s | >35 | dementia | − | N/A | N/A | N/A |
| II-1 | M | 30’s* | >40 | + | − | + | − | + |
| II-2 | F | 57 | Alive (>12) | + | − | + | − | − |
| II-3 | F | 55 | 9 | dementia | + | − | − | N/A |
| II-4 | F | 59 | Alive (>4) | + | − § | + | + | − |
| III-1 | M | 38 | Alive (>6) | + | − | + | + | − |
Diagnosis of bvFTD was made in his late sixties.
= Concerning signs for motor neuron disease.
N/A: Data not available.
Figure 1. UCSFBR1 kindred pedigree.
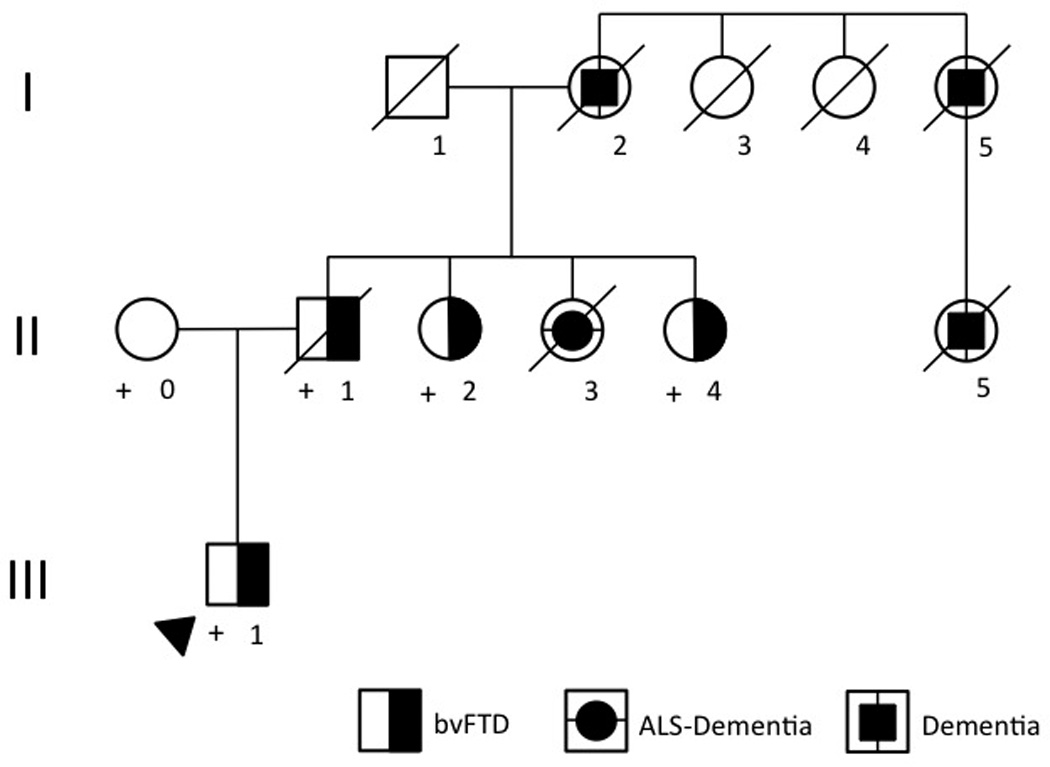
Legend: Open symbols represent unaffected individuals. Arrowhead denotes the proband. The symbol “+” indicates individuals who were tested for the mutation. Roman numerals denote the generation; Arabic numbers identify each individual in his/her generation. bvFTD= behavioral variant of frontotemporal dementia. ALS= amyotrophic lateral sclerosis.
Patient III-1
A right-handed man’s history of behavioral changes began at the age of 38, when he became obsessed with preparing for a course he planned to take the following year. Subsequently, his family noticed increased aggression, disinhibition and mood swings. Within two years he was laid off work due to poor judgment and inconsistent work performance. Four years after onset he developed a left hand resting tremor. Past medical history was relevant for chronic diarrhea (diagnosis unclear, possibly Crohn’s disease), diagnosed 2 years after onset of symptoms. By the time of his evaluation, he was 44-years-old and his family endorsed apathy and mental rigidity; psychotic symptoms were denied. Neuropsychological evaluation 6 revealed prominent executive function deficits and lesser, but significant, impairment on verbal and visual memory (eTable 1). His Clinical Dementia Rating (CDR) score was 1.7 On neurological examination, he had mild axial and limb parkinsonism (worse on the left) and dystonic posturing of his left hand during gait. Gait was also slow, with mild bradykinesia. Electromyography (EMG) did not show signs of MND. MRI showed mild atrophy in the dorsal and posterior aspects of the brain (Figure 2A). He was diagnosed with bvFTD.
Figure 2. Brain MRI – Patients III-1 and II-4.

Legend: Parasagittal and axial T1-weighted 3T brain MRI. 2-A: Patient III-1 - mild atrophy in the dorsal and posterior aspects of the brain; 2-B: Patient II-4 - mild atrophy of the anterior temporal lobes, insula and orbitofrontal region, as well as dorsal atrophy.
Patient II-4
A right-handed woman, whose history of behavioral changes began at the age of 59. She became obsessed with vitamins and religion, and her family noticed she was more short-tempered and aggressive. Food preference changes, loss of table manners and difficulty recognizing faces and objects were other symptoms endorsed by the family, although the patient had no insight into these changes. Her past medical history was relevant for a depressive episode at age 55, for which she had refused medical treatment, and long-standing anxiety. She had a hysterectomy at age 45, but her family later found out that she had faked symptoms of myoma so that she could rid herself of menstrual cycles.
She was examined at age 62 and her neuropsychological evaluation showed deficits on executive functions and naming (eTable 1). Her CDR score was 1. On neurological examination, a mildly increased tone with activation in her right arm and dystonic posturing of her left hand during gait were the only abnormalities observed. MRI of the brain revealed mild atrophy of the anterior temporal lobes, insula and orbitofrontal region, as well as dorsal atrophy (Figure 2B). EMG was normal (units were at the upper limit of normal in tongue EMG), but intermittent tongue fasciculations were noted. Her diagnosis was bvFTD, with concerning signs for concomitant MND.
Other patients
Patients I-1, I-3 and I-4: No history of cognitive or behavioral problems. Patient I-3 had Crohn’s disease.
Patient I-2: Was described as having memory problems and personality changes starting in her forties. She later developed movement problems and further cognitive decline, consistent with dementia. She died at the age of 76.
Patient I-5: History of unspecified late onset dementia (as did one of her daughters).
Patient II-1: Described as having “odd” behaviors beginning in his thirties, and being a cruel parent. He was diagnosed with bvFTD in his late sixties, with visual hallucinations and disinhibition as early manifestations, followed by memory, language and further behavioral decline. Later, he developed parkinsonian signs. He died at the age of 72.
Patient II-2: At the age of 57 she started having difficulty recognizing people and trouble with navigation, as well as being less interested in social interactions. Changes in empathy and eating habits three years later led to a diagnosis of bvFTD. Twelve years after the onset of symptoms she was bedridden and almost mute.
Patient II-3: Began with cognitive and behavioral problems at the age of 55 (although her family reported some degree of “social compromise” in her 40s) and was diagnosed as having late-onset schizophrenia. She died at the age of 64, five months after being diagnosed with ALS.
Genetic analyses
DNA was obtained from peripheral blood and expanded GGGGCC hexanucleotide repeats in the gene C9orf72 were detected in a stepwise fashion according to previously described methods.4 Samples from individuals II-0, II-1, III-1, II-2 and II-4 were first screened by fluorescent fragment-length PCR analysis to identify a potentially unamplifiable repeat expansion (Figure 3A). The fact that all patients showed a “homozygous” pattern (single peak), combined with the fact that the affected child (III-1) did not seem to inherit an allele from its affected parent (II-1), was considered suggestive evidence of a repeat expansion. Repeat-primed PCR analysis was then used to verify the presence of an expansion in all patients, confirmed by the observation of a stutter amplification pattern on electropherogram (Figure 3B). The presence of expanded alleles was further substantiated by Southern blot (Figure 3C), which showed additional expanded alleles at 5–23kb in affected individuals.
Figure 3. Molecular genetic analyses of C9orf72 repeat expansions in UCSFBR1 family.

Legend: (A) Fluorescent fragment length analysis of a PCR fragment containing the GGGGCC repeat in C9orf72 in 4 patients (II-1, III-1, II-2 and II-4) and one unaffected spouse (II-0). A lack of transmission from the affected parent (II-1) to the affected offspring (III-1) is seen. Numbers under the peaks indicate number of GGGGCC hexanucleotide repeats. (B) PCR products of repeat-primed PCR reactions separated on an ABI3730 DNA Analyzer and visualized by GENEMAPPER software. Electropherograms are zoomed to 2,000 relative fluorescence units to show stutter amplification. One expanded repeat carrier (II-1) and one healthy control are shown. (C) Southern blotting of 3 expanded repeat carriers and one unaffected spouse using genomic DNA extracted from blood. Patients with expanded repeats (II-1, III-1 and II-2) show additional alleles ranging from 5–23 kb, while the normal spouse (II-0) only shows the expected ~2.3 kb wild-type allele.
Discussion
Similar to previous reports, the main clinical syndromes seen in this C9orf72 family were bvFTD and ALS. The phenotypic heterogeneity observed in previously reported families thus far2,5 was also apparent in this kindred.
Age of onset with the C9orf72 mutation was difficult to precisely ascertain. In many members of the family, subtle personality changes started decades before dementia was diagnosed. In patient II-4, the history behind her hysterectomy - 14 years before the onset of symptoms - raises suspicion as to whether or not she was truly asymptomatic then. Cognitive impairments in presymptomatic mutation carriers have been described in MAPT mutations,8 but personality changes predating the diagnosis of dementia have not been reported and may be a distinguishing feature of C9orf72 expansions. Also, in generation III, there are at least three individuals carrying a diagnosis of depression and/or anxiety (not depicted in pedigree). Those conditions are prevalent in the general population, and also have familial aggregation,9 so it is currently unclear whether they represent a first symptom in the neurodegenerative process or if other confounding genetic factors are contributing to a higher frequency of psychiatric conditions in this family.
Anticipation is a characteristic of many repeat expansion disorders, although with the massive numbers of repeats present in all patients tested to date it is possible that anticipation will not be a strong feature of C9orf72. Patient III-1 was diagnosed with bvFTD at an earlier age than the previous generation; however, if the onset is considered as the earlier personality changes, his symptoms began at around the same age as his father’s. Further research is necessary to determine whether C9orf72 expansions are associated with anticipation, and developing ways to accurately measure the exact number of repeats may help with this understanding. Although in our study the presence of repeat expansions was confirmed by Southern blot, significant repeat size heterogeneity resulting in a smear on the Southern blot complicated accurate sizing of the repeat length.
Parkinsonian features, often a finding associated with C9orf72 mutations,2,10,11 were also observed in this family. In addition to parkinsonism appearing in clinical conjunction with either MND or FTD,1,12 nigral degeneration is a frequent finding in ALS with dementia,13 so this association is not surprising.
Dystonia has rarely been described as an additional sign, and only briefly in a previous chromosome 9p family.11 Parkinsonism with focal dystonia may be seen in atypical parkinsonism,14 but is only rarely observed within the bvFTD phenotype. Also, a previous report mentioned parkinsonism with dystonia in a MAPT FTD-ALS family.15 Further observations are needed to ascertain the significance of focal dystonia in C9orf72 mutations.
TARDBP mutations have also been associated with parkinsonian or dystonic features in ALS families.16 Parkinsonism was reported with dementia and “upper limb muscle weakness” in one individual with a VCP mutation, though it was not described at which point during the course of disease it appeared.17 DCTN1 has been associated with ALS, FTD and Perry syndrome, but its pathogenicity in FTD-ALS is still not entirely clear.18
Psychosis has been reported in four chromosome 9p families 2,10,19,20; hallucinations may be distinguishing symptoms,2,10 as they are considered rare in sporadic bvFTD.12 Visual hallucinations are the most commonly reported type (as in patient II-1), but the description of auditory hallucinations in three previous reports 2,19,20 suggests they could also be a marker for this mutation. Psychosis was reported in one patient with a VCP mutation, though it was not described in detail.17
In this report, a somewhat more posterior pattern of atrophy on neuroimaging was observed. This is similar to a previous report in which C9orf72 bvFTD demonstrated more parietal and occipital and less temporal atrophy as compared to sporadic bvFTD.10 This pattern is in line with the finding of more significant visuospatial dysfunction in a subset of C9orf72 patients (such as patient II-3),2 which is unusual for bvFTD.12
Finally, the occurrence of inflammatory bowel disease in two members of this family is intriguing. Even though no direct associations can be made based on the limited knowledge gathered so far on the C9orf72 gene, there is increasing evidence linking neurodegenerative processes and inflammation.21 Even though no other previous reports have mentioned inflammatory diseases occurring in C9orf72 families, it is possible they have been overlooked.
Supplementary Material
Acknowledgements
We would like to thank this family for participating in research. BLM is funded by P50AG023501, P01AG019724, the Larry Hillblom Foundation and the State of CA and P50 AG1657303. This research was funded in part by NIH grants P50 AG016574, R01 NS065782 and R01 AG026251 and the ALS Association (to RR).
Contributor Information
Leonel T. Takada, Email: ltakada@memory.ucsf.edu, Memory and Aging Center - University of California, San Francisco.
Maria Lucia V. Pimentel, Email: mlvpimentel@gmail.com, Santa Casa da Misericordia, Pontifical Catholic University and Gama Filho University, Rio de Janeiro, Brazil.
Mariely DeJesus-Hernandez, Email: deJesusHernandez.Mariely@mayo.edu, Department of Neuroscience, Mayo Clinic, Jacksonville, Florida.
Jamie C. Fong, Email: jfong@memory.ucsf.edu, Memory and Aging Center - University of California, San Francisco.
Jennifer S. Yokoyama, Email: jyokoyama@memory.ucsf.edu, Memory and Aging Center - University of California, San Francisco.
Anna Karydas, Email: akarydas@memory.ucsf.edu, Memory and Aging Center - University of California, San Francisco.
Marie-Pierre Thibodeau, Email: mp_thibodeau@hotmail.com, Centre Hospitalier de l'Université de Montréal-Hopital Notre-Dame, University of Montreal, Montreal, Quebec, Canada.
Nicola J. Rutherford, Email: Rutherford.Nicola@mayo.edu, Department of Neuroscience, Mayo Clinic, Jacksonville, Florida.
Matthew C. Baker, Email: Baker.Matt@mayo.edu, Department of Neuroscience, Mayo Clinic, Jacksonville, Florida.
Catherine Lomen-Hoerth, Email: catherine.lomen-hoerth@ucsf.edu, ALS Center – University of California, San Francisco.
Rosa Rademakers, Email: Rademakers.Rosa@mayo.edu, Department of Neuroscience, Mayo Clinic, Jacksonville, Florida.
Bruce L. Miller, Email: bmiller@memory.ucsf.edu, Memory and Aging Center - University of California, San Francisco.
References
- 1.Hudson AJ. Amyotrophic lateral sclerosis and its association with dementia, parkinsonism and other neurological disorders: a review. Brain. 1981;104(2):217–247. doi: 10.1093/brain/104.2.217. [DOI] [PubMed] [Google Scholar]
- 2.Pearson JP, Williams NM, Majounie E, et al. Familial frontotemporal dementia with amyotrophic lateral sclerosis and a shared haplotype on chromosome 9p. J Neurol. 2011;258(4):647–655. doi: 10.1007/s00415-010-5815-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Renton AE, Majounie E, Waite A, et al. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron. 2011;72(2):257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron. 2011;72(2):245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fecto F, Siddique T. Making Connections: Pathology and Genetics Link Amyotrophic Lateral Sclerosis with Frontotemporal Lobe Dementia. J Mol Neurosci. 2011;45(3):663–675. doi: 10.1007/s12031-011-9637-9. [DOI] [PubMed] [Google Scholar]
- 6.Kramer JH, Jurik J, Sha SJ, et al. Distinctive neuropsychological patterns in frontotemporal dementia, semantic dementia, and Alzheimer disease. Cogn Behav Neurol. 2003;16(4):211–218. doi: 10.1097/00146965-200312000-00002. [DOI] [PubMed] [Google Scholar]
- 7.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43(11):2412–2414. doi: 10.1212/wnl.43.11.2412-a. [DOI] [PubMed] [Google Scholar]
- 8.Geschwind DH, Robidoux J, Alarcón M, et al. Dementia and neurodevelopmental predisposition: cognitive dysfunction in presymptomatic subjects precedes dementia by decades in frontotemporal dementia. Ann Neurol. 2001;50(6):741–746. doi: 10.1002/ana.10024. [DOI] [PubMed] [Google Scholar]
- 9.Lohoff FW. Overview of the Genetics of Major Depressive Disorder. Curr Psychiatry Rep. 2010;12(6):539–546. doi: 10.1007/s11920-010-0150-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boxer AL, Mackenzie IR, Boeve BF, et al. Clinical, neuroimaging and neuropathological features of a new chromosome 9p-linked FTD-ALS family. J Neurol Neurosurg Psychiatry. 2011;82(2):196–203. doi: 10.1136/jnnp.2009.204081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Le Ber I, Camuzat A, Berger E, et al. Chromosome 9p-linked families with frontotemporal dementia associated with motor neuron disease. Neurology. 2009;72(19):1669–1676. doi: 10.1212/WNL.0b013e3181a55f1c. [DOI] [PubMed] [Google Scholar]
- 12.Piguet O, Hornberger M, Mioshi E, Hodges JR. Behavioural-variant frontotemporal dementia: diagnosis, clinical staging, and management. Lancet Neurol. 2011;10(2):162–172. doi: 10.1016/S1474-4422(10)70299-4. [DOI] [PubMed] [Google Scholar]
- 13.Al-Sarraj S, Maekawa S, Kibble M, Everall I, Leigh N. Ubiquitin-only intraneuronal inclusion in the substantia nigra is a characteristic feature of motor neurone disease with dementia. Neuropathol Appl Neurobiol. 2002;28(2):120–128. doi: 10.1046/j.1365-2990.2002.00384.x. [DOI] [PubMed] [Google Scholar]
- 14.Pont-Sunyer C, Martí MJ, Tolosa E. Focal limb dystonia. Eur J Neurol. 2010;17:22–27. doi: 10.1111/j.1468-1331.2010.03046.x. [DOI] [PubMed] [Google Scholar]
- 15.Zarranz JJ, FERRER I, Lezcano E, et al. A novel mutation (K317M) in the MAPT gene causes FTDP and motor neuron disease. Neurology. 2005;64(9):1578–1585. doi: 10.1212/01.WNL.0000160116.65034.12. [DOI] [PubMed] [Google Scholar]
- 16.Borghero G, Floris G, Cannas A, et al. A patient carrying a homozygous p.A382T TARDBP missense mutation shows a syndrome including ALS, extrapyramidal symptoms, and FTD. Neurobiol Aging. 2011;32(12):2327.e1–2327.e5. doi: 10.1016/j.neurobiolaging.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson JO, Mandrioli J, Benatar M, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010;68(5):857–864. doi: 10.1016/j.neuron.2010.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vilariño-Güell C, Wider C, Soto-Ortolaza AI, et al. Characterization of DCTN1 genetic variability in neurodegeneration. Neurology. 2009;72(23):2024–2028. doi: 10.1212/WNL.0b013e3181a92c4c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vance C, Al-Chalabi A, Ruddy D, et al. Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2-21.3. Brain. 2006;129(Pt 4):868–876. doi: 10.1093/brain/awl030. [DOI] [PubMed] [Google Scholar]
- 20.Luty AA, Kwok JB, Thompson EM, et al. Pedigree with frontotemporal lobar degeneration – motor neuron disease and Tar DNA binding protein-43 positive neuropathology: genetic linkage to chromosome 9. BMC Neurol. 2008;8(1):32. doi: 10.1186/1471-2377-8-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lucin KM, Wyss-Coray T. Immune Activation in Brain Aging and Neurodegeneration: Too Much or Too Little? Neuron. 2009;64(1):110–122. doi: 10.1016/j.neuron.2009.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.