

Abstract
Congenital disorders of glycosylation comprise most of the nearly 70 genetic disorders known to be caused by impaired synthesis of glycoconjugates. The effects are expressed in most organ systems, and most involve the nervous system. Typical manifestations include structural abnormalities, (eg, rapidly progressive cerebellar atrophy), myopathies (including congenital muscular dystrophies and limb-girdle dystrophies), strokes and stroke-like episodes, epileptic seizures, developmental delay, and demyelinating neuropathy. Patients can have neurological symptoms associated with coagulopathies, immune dysfunction with or without infections, and cardiac, renal, or hepatic failure, which are common features of glycosylation disorders. The diagnosis of congenital disorders of glycosylation should be considered for any patient with multisystem disease and in those with more specific phenotypic features. Measurement of concentrations of selected glycoconjugates can be used to screen for many of these disorders, and molecular diagnosis is becoming more widely available in clinical practice. Disease-modifying treatments are available for only a few disorders, but all affected individuals benefit from early diagnosis and aggressive management.
Introduction
A hypotonic child presents with seizures, hypoglycaemia, mild liver fibrosis and high transaminase concentrations. Another child with intrauterine growth retardation and dysmorphic features, including a long, thin face with large protruding ears and a micropenis, presents with delayed speech and motor development. A third child is referred with retinitis pigmentosa and stroke-like episodes in the context of heart failure secondary to cardiomyopathy. An active athletic adult aged 25 years has recently developed peripheral neuropathy and progressive foot drop. These patients have very different phenotypes, but they all have inherited defects in glycosylation—the process of adding complex sugar chains to proteins and lipids. Nearly 70 genetic disorders of glycosylation have been discovered, mostly within the past 15 years, and the catalogue continues to grow. A simple biochemical test can confirm the general diagnosis in most cases, although a few disorders require more invasive procedures, and all require definitive genetic confirmation. We present an overview of these diseases, with an emphasis on phenotypes, diagnostic approaches, and treatment.
Common features of glycosylation pathways
The glycome—all the sugar chain structures in a cell or organism—is orders of magnitude larger than the expressed genome. Its daunting complexity claims 1–2% of the genome to encode the known glycosylation machinery.1 Sugar chains (glycans) are added to mammalian proteins and lipids through eight distinct pathways (table 1, figure 1). Each pathway requires a different enzyme or transferase complex to initiate glycosylation. The first sugar unit (monosaccharide) linked to the protein or lipid defines the pathway, to which a single sugar or a preformed sugar chain might be added (figure 1). All pathways require activated monosaccharides in the form of nucleotide sugars, which are delivered to correct locations in the endoplasmic reticulum (ER) or Golgi apparatus to enable glycan biosynthesis. Because pathway precursors are shared, low concentrations or inefficient delivery could affect several pathways, although such effects have generally been studied in only one pathway at a time. Effects on multiple pathways have been reported in some instances and might be more common than is appreciated at present. Most of the effects of defects in the early steps of glycosylation are highly pathway specific, whereas those later in the process can affect multiple pathways. One protein can carry multiple glycans from different pathways. The outcome is not pathway driven, and many factors determine the spectrum of glycan structures. Examples include protein structure, availability of donor substrates, and the amounts of different sugar transferases and their kinetic constants. The effects of these factors on a particular glycan can exclude or enhance subsequent extension, or can place proteins in an environment where other transferases compete for a single glycan. Transferases are transcriptionally regulated, but their localisation and efficiency of recycling through the dynamic ER-Golgi network is crucial.2 The effect of faulty glycosylation on the function of any individual protein is unpredictable and ranges from trivial to essential. Effects must, therefore, be determined for each protein and for each function.3
Table 1.
Overview of glycan types by pathway
| Number of disorders | Typical clients | Functions | |
|---|---|---|---|
| N-linked | 38 | Nearly all cell-surface receptors, ECM, and secreted proteins | Assistance in folding, stabilisation of target proteins, signalling |
|
| |||
| O-linked | |||
| O-xylose | 10 | ECM proteins, heparan and chondroitin sulphate | Growth-factor binding, structure of ECM |
| O-mannose | 6 | α-Dystroglycan | Bridging neuromuscular 1α-receptor junction |
| O-fucose | 2 | Notch and notch ligands | Notch signalling, developmental patterning |
| O-GalNAc | 2 | Mucins, leucocyte receptors | Pathogen decoys, lubrication, protection of cell surface |
| O-glucose | 0 | Notch and Notch ligands | Notch signalling, developmental patterning |
|
| |||
| GPI-anchor glycans | 5 | Proteins in lipid rafts, signalling molecules | Organisation of plasma-membrane domains |
|
| |||
| Glycosphingolipid | 1 | Brain (highest concentration), synapses | Membrane organisation, signalling |
|
| |||
| Multiple | 13 | Soluble and membrane-bound molecules to provide substrates or Golgi-ER homoeostasis | Trafficking of Golgi resident proteins, synthesis of precursors |
ECM=extracellular matrix. GalNAc=N-acetylgalactosamine. GPI=glycophosphatidylinositol. ER=endoplasmic reticulum.
Figure 1. Pathways of glycosylation in the endoplasmic reticulum-Golgi network of mammalian cells.

The main types of glycosylation are shown. Various representative sugar-chain structures are given as examples. The grey shaded areas denote common core regions. Most of the glycosylation disorders that affect the nervous system involve alterations in N-linked and O-mannosylated glycoproteins. Some glycophosphatidylinositol-anchor and glycosphingolipid disorders also involve these alterations, but most proteoglycan and O-GalNAc defects do not. Other types of glycosylation exist, such as cytoplasmic O-GlcNAc and C-mannosylation, but are not shown. Glc=glucose. Gal=galactose. Man=mannose. GlcNAc=N-acetylglucosamine. GalNAc=N-acetylgalactosamine. GlcA=glucuronic acid. IdoA=Iduronic acid. Fuc=fucose. Xyl=xylose. Sia=sialic acid. S=sulphation. P=phosphorylation. Ac=acetylation. S/T=serine or threonine. Adapted from Stanley and colleagues,5 by permission of the Consortium of Glycobiology Editors, La Jolla, CA, USA.
The most recent nomenclature for glycosylation disorders proposes using the gene name followed by CDG to denote a congenital disorder of glycosylation.4 While this system is not the only one in use, we find it useful and convenient, and we use it in this Review. Where relevant we also provide other common or traditional names, such as CDG-I or CDG-II.
Specific glycosylation pathways
Genetic defects are known to occur in seven of the eight major ER-Golgi network glycan-generating pathways (table 1). The best known, and by far the most studied, is the N-linked glycosylation pathway, especially in terms of defects located in the ER (figure 1). Protein O-linked glycosylation is more diverse than N-linked glycosylation. Serine or threonine residues are linked to glycans through N-acetylgalactosamine, xylose, mannose or fucose (figure 1). Chains are extended by specific glycosyltransferases, whereas terminal sugars are in many cases added by non-specific transferases that service multiple pathways.
N-linked glycosylation
N-acetylglucosamine is bound to asparagine on nascent proteins in the ER lumen, but it is not added as a single sugar. Rather, a universal 14-sugar precursor containing two N-acetylglucosamine, nine mannose, and three glucose units is assembled on a lipid carrier (dolichol) to form a lipid-linked oligosaccharide.5 The entire glycan is transferred to asparagine by the multisubunit oligosaccharyl transferase complex.6,7 The glucose units and up to six of the mannose units are often removed after transfer, and N-acetylglucosamine, galactose, sialic acid, and fucose are added on to multiple branches. The order of remodelling of the chains is prescribed, but, as with all glycosylation, it is not template driven.
Essentially, all proteins (except albumin) that travel through the ER-Golgi network undergo N-linked glycosylation. Glycans promote protein folding, stability, trafficking, localisation, and oligomerisation.5 They play vital parts in cell–cell interactions and intracellular signalling.8
The dolichol carrier lipid also carries mannose, which serves three other pathways: glycophosphatidylinositol anchors, O-mannosylation, and C-mannosylation. The first two are described below. No C-mannosylation disorders have been reported.9,10
O-linked glycosylation
O-linked protein glycosylation involves initial linkage between serine or threonine residues and mannose, xylose, N-acetylgalactosamine, fucose, or glucose (figure 1). The O-linked α-mannose glycans contain N-acetylglucosamine, galactose, N-acetylgalactosamine, and sialic acid.11,12 α-Dystroglycan, which has a crucial role as a link between the extracellular matrix and the cytoskeleton in skeletal muscle cells, is the major identified carrier for O-linked α-mannose glycans. Other proteins certainly carry them, but are yet to be identified. Defects in this pathway often cause neurological deficits.13
O-linked α-N-acetylgalactosamine-based glycans link to serine or threonine, after which one of four sugars is added to form a disaccharide. Sequential addition of galactose, N-acetylglucosamine, fucose, and sialic acid generates linear or multibranched chains14 that are found on secreted or cell-surface mucins of epithelial cells. These chains can lubricate and are effective pathogen decoys. None causes neurological pathology.
O-linked β-xylose glycans on serine generate glycosaminoglycans, such as heparan sulphate, heparin, chondroitin, and dermatan sulphates.15 Long repeating disaccharides of glucuronic acid-N-acetylgalactosamine (chondroitin and dermatan sulphates) or glucuronic acid-N-acetylglucosamine (heparin and heparan sulphate) are extended from a small core glycan.13 Some glucuronic acid is epimerised to iduronic acid. Sulphation occurs on de-N-acetylated N-acetylglucosamine NH2 groups or OH groups. Proteins carrying chondroitin are used for physical integrity and cushioning. Cell-surface heparin-sulphate chains bind growth factors (eg, fibroblast growth factors), cytokines, and morphogens during development to establish gradients of these molecules.16,17
O-linked α-fucose-based glycans occur in selected epidermal growth factor modules in Notch and Notch ligands, and are extended by Fringe family N-acetylglucosamine transferases and other glycosyl-transferases.18,19 The presence of these glycans has a strong effect on Notch signalling. Thrombospondin type I repeats can be O-fucosylated by a different transferase, and fucose is extended with one or two glucose units.20
Lipid-bearing glycans
Glycosphingolipids link glucose to ceramide. If galactose is also added, lactosylceramide is formed. This core can be variably extended to more complex glycosphingolipids, including the sialylated gangliosides.21 The greatest diversities in types and concentrations of glycosphingolipids occur in the brain and peripheral nervous system. Glycosphingolipids bind to each other or to proteins, such as integrins, which enables them to affect signalling.22 Glycophosphatidylinositol-anchor glycans substitute for transmembrane regions of many proteins. They contain mannose and glucosamine, and are assembled in the ER on a phosphatidylinositol backbone. The entire glycolipid is transferred to C-terminal regions of proteins.23 Glycophosphatidylinositol anchors also have roles in membrane diffusion, intracellular protein sorting, and signalling.24,25 Defects in glycosphingolipid and glycophosphatidylinositol pathways can cause neurological complications.26–29
Trafficking and homoeostasis
Client proteins travel through the dynamic ER-Golgi network, where glycosylation occurs. Genetic defects in proteins needed to recycle the glycosylation machinery between the ER and the Golgi apparatus can affect the process.2 Most of the known genes encode soluble cytoplasmic proteins that transiently associate with the Golgi apparatus and help to guide vesicles containing the glycosylation machinery to their location. Most patients with these defects have neurological deficits that, along with skeletal abnormalities and dysmorphic features, are probably due to their effects on multiple glycosylation pathways.30
Selected specific defects
Space limitations prevent description of all 66 glycosylation disorders in this Review. In tables 2–4 we list the glycosylation disorders with neurological manifestations and their most common symptoms. A list of disorders without or with minor neurological complications is available online (appendix). Many of the listed congenital disorders of glycosylation, such as deficiencies in sulphation, primarily affect glycosaminoglycan structure, and their classification as glycosylation disorders is the subject of debate. In this section we highlight disorders that have been documented in at least ten patients.
Table 2.
Neurological features of glycosylation disorders detectable by measurement of transferrin glycosylation
| Enzymatic defect | Gene | OMIM code | Number of known patients | Neurological abnormalities | ||||
|---|---|---|---|---|---|---|---|---|
| ID | Brain | Seizures | Ocular | |||||
| PMM2-CDG (CDG-Ia)* | Phosphomannomutase conversion of Man-6-phosphate to Man-1-phosphate | PMM3 | 212065 | >600 | Yes | Yes | Yes | Yes |
| ALG6-CDG (CDG-Ic)* | α-1,3-glucosyltransferase | ALG3 | 603147 | 57 | Yes | Yes | Yes | Yes |
| ALG3-CDG (CDG-Id)* | α-1,3-mannosyltransferase | ALG3 | 601110 | ~10 | Yes | Yes | Yes | Yes |
| DPM1-CDG (CDG-Ie)* | Dolichol-phosphate-Man synthase | DPM3 | 603503 | 14 | Yes | Yes | Yes | Yes |
| MPDU1-CDG (CDG-If)* | Dolichol-phosphate sugar utilisation protein | MPDU3 | 609180 | 7 | Yes | Yes | Yes | Yes |
| ALG12-CDG (CDG-Ig)* | α-1,6-mannosyltransferase | ALG33 | 607143 | 11 | Yes | Yes | Yes | Yes |
| ALG8-CDG (CDG-Ih)* | α-1,3-glucosyltransferase | ALG3 | 608104 | 8 | Yes | Yes | Yes | Yes |
| ALG2-CDG (CDG-Ii)* | α-1,3-mannosyltransferase | ALG3 | 607906 | 1 | Yes | Yes | Yes | Yes |
| DPAGT1-CDG (CDG-Ij)* | GlcNAc-1-P transferase | DPAGT3 | 608093 | 11 | Yes | Yes | Yes | Yes |
| ALG1-CDG (CDG-Ik)* | β-1,4-mannosyltransferase | ALG3 | 608540 | 14 | Yes | Yes | Yes | Yes |
| ALG9-CDG (CDG-IL)* | α-1,2-mannosyltransferase | ALG3 | 608776 | 3 | Yes | Yes | Yes | Yes |
| DOLK-CDG (CDG-Im)* | Dolichol kinase | DOLK | 610768 | 16 | Yes | Yes | Yes | Yes |
| RFT1-CDG (CDG-In)* | Man5GlcNAc2 flippase | RFT3 | 612015 | 6 | Yes | Yes | Yes | Yes |
| TUSC3-CDG (MRT7)* | Subunit of the OST complex | TUSC3 | 611093 | 4 | Yes | NR | NR | NR |
| MAGT1-CDG (MRX95)* | Subunit of the OST complex | MAGT3 | 300716 | 14 | Yes | NR | NR | NR |
| ALG11-CDG (CDG-Ip)* | α-1,2-mannosyltransferase | ALG33 | 613661 | 1 | Yes | Yes | Yes | NR |
| SRD5A3-CDG (CDG-Iq)* | 5-α steroid reductase (polyprenol reductase) | SRD3A3 | 612379 | 14 | Yes | Yes | Yes | Yes |
| DDOST-CDG (CDG-Ir)* | Subunit of the OST complex | DDOST | 614507 | 1 | Yes | NR | Yes | NR |
| MGAT2-CDG (CDG-IIa)* | N-acetylglucosaminyltransferase II | MGAT3 | 212066 | 3 | Yes | NR | NR | NR |
| GCS1-CDG (CDG-IIb)* | α-1,2-glucosidase | GCS3 | 606056 | 1 | NR | NR | Yes | NR |
| SLC35C1-CDG (CDG-IIc)* | GDP-fucose transporter | FUCTI | 266265 | 7 | Yes | Yes | Yes | NR |
| B4GALT1-CDG (CDG-IId)* | β-1,4-galactosyltransferase | B3GALTI | 607091 | 2 | Yes | Yes | Yes | NR |
| COG1-CDG (CDG-IIg)* | Conserved oligomeric Golgi subunit 1 | COG3 | 611209 | 3 | Yes | Yes | Yes | NR |
| COG4-CDG (CDG-IIj)* | Conserved oligomeric Golgi subunit 4 | COG3 | 613489 | 2 | Yes | Yes | Yes | NR |
| COG5-CDG (CDG-Iii)* | Conserved oligomeric Golgi subunit 5 | COG3 | 613612 | 4 | Yes | Yes | Yes | NR |
| COG6-CDG (CDG-IIL)* | Conserved oligomeric Golgi subunit 6 | COG3 | ·· | 1 | Yes | Yes | Yes | NR |
| COG7-CDG (CDG-IIe)* | Conserved oligomeric Golgi subunit 7 | COG3 | 608779 | 9 | Yes | Yes | Yes | NR |
| COG8-CDG (CDG-IIh)* | Conserved oligomeric Golgi subunit 8 | COG3 | 611182 | 2 | Yes | Yes | Yes | NR |
| ATP6V0A2-CDG | Golgi pH regulator | ATP3V3A3 | 219200 | >20 | Yes | Yes | Yes | NR |
OMIM=Online Mendelian Inheritance in Man database. ID=intellectual development is impaired. Brain=brain malformations, such as cerebellar atrophy or hypoplasia, or thinning of the corpus callosum. Ocular=ocular abnormalities, such as cataracts, optic-nerve atrophy, or coloboma. Man=mannose. GlcNAc=N-acetylglucosamine. OST=oligosaccharyltransferase. NR=not reported.
Previous classification provided in brackets.
Table 4.
Neurological features of glycosylation disorders detectable by methods other than measurement of transferrin glycosylation
| Enzymatic defect | Gene | OMIM code | Neurological abnormalities
|
||||
|---|---|---|---|---|---|---|---|
| ID | Brain | Seizures | Ocular | ||||
|
GPI-anchor disorders*
| |||||||
| Autosomal recessive GPI-anchor deficiency | Synthesis of N-acetylglucosaminyl phosphatidylinositol | PIGA | 300868 | NR | Yes | Yes | NR |
| CHIME syndrome | De-N-acetylation of N-acetylglucosaminyl phosphatidylinositol | PIGL | 280000 | Yes | Yes | Yes | Yes |
| Autosomal recessive GPI-anchor deficiency | First α-mannosyltransferase in GPI biosynthesis | PIGM | 610293 | Yes | NR | Yes | NR |
| Autosomal recessive GPI-anchor deficiency | Transfers phosphoethanolamine to first mannose of GPI anchor | PIGN | 614080 | Yes | NR | Yes | NR |
| Hyperphosphatasia mental retardation syndrome | Second α-mannosyltransferase in GPI biosynthesis | PIGV | 239300 | Yes | NR | Yes | NR |
|
| |||||||
|
α-Dystroglycanopathies†
| |||||||
| Walker-Warburg syndrome (MDDGA1 and MDDGA2)‡ | O-mannosyltransferase | POMT3-POMT3 | 236670 | Yes | Yes | Yes | Yes |
| Muscle-eye-brain disease (MDDGA3)‡ | O-mannosyl glycan GlcNAc transferase | POMGNT3 | 253280 | Yes | Yes | Yes | Yes |
| Fukuyama-type congenital muscular dystrophy (MDDGA4, some MDDGB4)‡ | Putative glycosyltransferase | FKTN | 253800 | Yes | Yes | Yes | Yes |
| Congenital muscular dystrophy type 1C (MDDGA5)‡ | Fukutin-related protein, putative glycosyltransferase | FKRP | 606612 | Yes | Yes | NR | Yes |
| Congenital muscular dystrophy type 1D (MDDGA6)‡ | Glycosyltransferase | LARGE | 608840 | Yes | Yes | NR | NR |
|
| |||||||
|
Disorders without biochemical markers
| |||||||
| Amish infantile epilepsy | Sia2,3Galβ1,4Glc-Cer synthase (GM3) | SIAT3 | 609056 | Yes | NR | Yes | NR |
| Non-syndromic intellectual disability | α-1,2-mannosidase | MAN3B3 | 614202 | Yes | NR | NR | NR |
| Peters plus syndrome | β-1,3-glucosyltransferase specific for O-linked fucose on thrombospondin type 1 repeats | B3GALTL | 261540 | Yes | NR | NR | NR |
| I-cell disease | GlcNAc-1-P transferase | GNPTA | 252500 | Yes | Yes | NR | Yes§ |
OMIM=Online Mendelian Inheritance in Man database. ID=intellectual development is impaired. Brain=brain malformations, such as cerebellar atrophy or hypoplasia, or thinning of the corpus callosum. Ocular=ocular abnormalities, such as cataracts, optic-nerve atrophy, or coloboma. GPI=glycophosphatidylinositol. NR=not reported. CHIME=coloboma, heart defects, ichthyosis, mental insufficiency and ear defects. MDDG=muscular dystrophy-dystroglycanopathy. GlcNAc=N-acetylglucosamine.
Detectable by measurement of CD59 and fluorescently labelled aerolysin.
Detectable by measurement of antibodies to glycosylated α-dsytroglycans.
In the MDDG nomenclature system, the letter relates to severity (A, severe; B, intermediate; C, mild) and the number to the gene involved.
Cataracts.
PMM2-CDG (CDG-Ia)
PMM2-CDG is the most frequent N-linked congenital disorder of glycosylation and accounts for around 80% of all diagnosed cases.31 Mutations in PMM2, which encodes the phosphomannomutase 2 enzyme that catalyses conversion of mannose-6-phosphate to mannose-1-phosphate, cause the disorder. The CNS and peripheral nervous system are prime targets, but multisystem abnormalities cause substantial residual neurological deficits and 20% of mortality in infancy. Milder phenotypes have been recognised.
Clinical presentations
The classic presentation in infancy includes failure to thrive, inverted nipples, subcutaneous fat pads and other forms of lipodystrophy (figure 2). Affected children are hypotonic, developmentally delayed, and show evolving facial hypotonia and esotropia, which might progress to complete sixth-nerve paralysis.32–34 Roving eye movements are typical, and delayed or absent fixation might be present. Overt pigmentary retinopathy might be seen in adolescent and adult patients, but is generally not apparent in children. The subsequent evolution of this disease, which was initially described by Hagberg and colleagues,35 has four stages.
Figure 2. Clinical features of PMM2-CDG (CDG-1a).
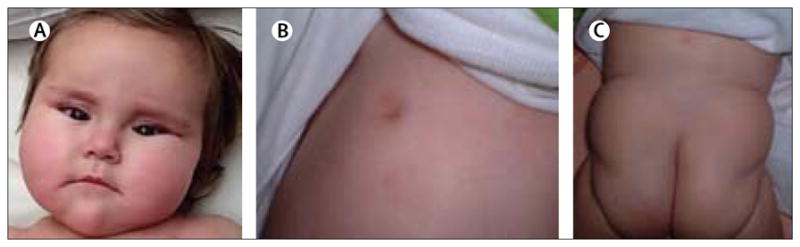
Children frequently present with (A) facies and esotropia, (B) nipple inversion, and (C) supragluteal fat pads.
In the first stage, during infancy, systemic symptoms dominate. These include susceptibility to infection, episodes of hepatic impairment or overt failure, episodes of bleeding or thrombosis, and hypertrophic cardiomyopathy, with or without pericardial effusion.36
The second phase occurs in early childhood and is dominated by seizures and stroke-like episodes. These symptoms might be seen in the context of intercurrent illness and are sometimes misdiagnosed as febrile seizures, with or without Todd’s paralysis.34 Studies suggest that the stroke-like episodes represent ischaemic stroke, but can also be mimicked by periods of otherwise subclinical focal status epilepticus or localised oedema after a seizure.37 The distinction cannot readily be made clinically, and MRI is required. Coagulation studies and MR angiography might also be useful. Seizures can be partial or generalised and persist beyond the stroke-like episode. They can be readily controlled in most cases.
The third stage is characterised by slowly progressive limb atrophy (reflecting demyelinating peripheral neuropathy) combined with severe ataxia and intellectual deficiency. Esotropia evolves into bilateral sixth-nerve palsies and visual loss associated with progressive pigmentary degeneration of the retina. In a series of 23 patients aged 10 months to 20 years, visual maturation was delayed in most, and at the last follow-up visit 16 had low vision and five were legally blind and showed pallor of the optic discs; 18 had myopia.38
The final stage occurs in adolescence and adulthood, when patients have fixed neurological deficits, including intellectual disability, severe cerebellar ataxia, peripheral neuropathy with distal wasting, and areflexia. Patients who survive to adulthood commonly have pigmentary retinopathy, kyphoscoliosis, and endocrinopathies.39 Some patients have hypergonadotropic hypogonadism, and skeletal deformities have also been described.
Screening has been used increasingly and has identified milder phenotypes. Two infants with absent or mild lipodystrophy were reported to have only subtle biochemical alterations and one of these children showed loss of purposeful hand movements and stereotypes similar to those reported in Rett’s disorder, plus intention tremor.40 Patients with mild phenotypes who survive to adulthood might have borderline cognitive impairment, with or without strabismus. In one report, all three affected adults were in full-time employment.41 One individual has been described who had only gastrointestinal dysfunction in childhood and normal neurodevelopment.42
Pathology
Several reports have described the neuropathology of PMM2-CDG. Antoun and colleagues43 described a patient who presented in early infancy with dysmorphism (high nasal bridge and large jaw and ears), failure to thrive, hypotonia, lipodystrophy, acquired microcephaly, and abnormal (roving) eye movements. The child died at age 7 months from haematemesis complicating hepatic failure. Total brain weight was normal, but the cerebellum was very small, particularly the anterior vermis and adjacent hemispheres. Microscopic examination showed substantial neuronal loss in the olives and internal granular and Purkinje-cell layers, accompanied by reactive gliosis. Slight neuronal loss was seen in the pontine nuclei, but the dentate nuclei were preserved. The cerebrum was spared. An earlier report had described two siblings who had neonatal olivopontocerebellar atrophy with similar clinical course and autopsy findings, although those patients exhibited macrocephaly.44
Sural-nerve pathology has been described in nine patients aged 6 months to 15 years.45 The chief findings included thin myelin sheaths, distorted periaxonal Schmidt-Lanterman incisures, and periaxonal dense bodies. Schwann cells contained multivacuolar bodies, and in some cases foamy inclusions. Fibrillary inclusions were noted in fibroblasts. These effects were seen at presentation in all patients except the youngest, in whom they appeared later and became more prominent with increasing age. Onion bulb formation and axonal regeneration was also noted in most patients. Nordborg and colleagues45 speculated that myelin-associated glycoprotein dysfunction had a crucial role because its distribution coincided with abnormalities seen in the region of the Schmidt-Lanterman incisures.
Neurophysiology
Detailed neurophysiology was reported in a boy aged 8 years with PMM2-CDG.46 Motor-nerve conduction gradually slowed over time, with a concomitant increase in latency, a decrease in amplitude of somatosensory evoked potentials, and progressive impairment of response on electroretinography and of visual evoked potentials that were consistent with pigmentary retinopathy. Selective rod involvement was noted on electroretinography.38 Auditory brainstem responses showed late changes suggestive of sensorineural hearing loss, and electroencephalography showed progressive slowing of background activity and the presence of sharp waves. A study of seven children with PMM2-CDG confirmed the slowing of motor-nerve conduction with sparing of sensory-nerve conduction.47
Neuroimaging
Most children with PMM2-CDG have notable atrophy of the superior vermis and cerebellar hemispheres at birth, but in mild cases initial MRI findings might be normal, with subsequent progressive atrophy (figure 3).48 Pathological findings suggest a very-early-onset atrophic process, rather than primary hypoplasia.43,44 PMM2-CDG is an important cause of cerebellar ataxia, as was shown in a study of posterior fossa imaging in 158 children with ataxia.49 Of 84 children with global cerebellar atrophy, 21 had PMM2-CDG, compared with 18 who had respiratory-chain defects. In general, the cerebral hemispheres are spared in PMM2-CDG.
Figure 3. Cranial MRI in a patient with PMM2-CDG (CDG-1a).
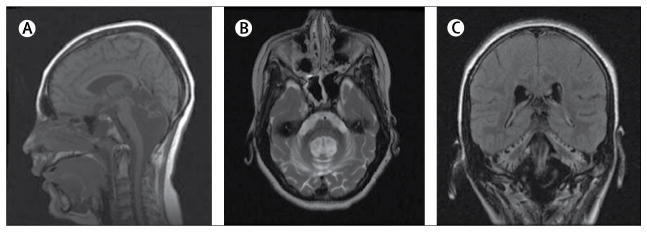
Images were obtained from a girl aged 17 years. Notable atrophy of the cerebellar vermis and hemispheres can be seen in (A) sagittal T1, (B) axial T2, and (C) coronal fluid-attenuated inversion recovery images.
Management
No definitive therapy is yet available for PMM2-CDG, but symptomatic management of seizures, stroke-like episodes, and systemic manifestations improves quality of life and can be life-saving.
ALG6-CDG (CDG-Ic)
ALG6-CDG, is the second most common N-linked congenital disorder of glycosylation. More than 50 patients have been identified.31 An initial report of eight patients showed that the phenotype of ALG6-CDG is milder than that of PMM2-CDG.50 Like PMM2-CDG, clinical features include developmental delay, axial hypotonia, strabismus, and seizures. Ataxia is less common in ALG6-CDG than in PMM2-CDG, and usually the cerebellum is spared. Dysmorphic features are seldom seen in patients with ALG6-CDG, episodes are typically not life-threatening, and hepatic and renal involvement is rare. Patients with ALG6-CDG might, however, have severe coagulopathy, which is thought to have contributed to or caused a grade III intraventricular haemorrhage in one neonate.51 The overall neurological outcome in ALG6-CDG is generally better than in typical PMM2-CDG, although at least five children have died from complications.31
Additional features have included severe protein-losing enteropathy during rotavirus infections and in the context of gastroenteritis and skeletal dysplasia.52,53 One woman with confirmed ALG6 deficiency has skeletal anomalies, virilisation, and intellectual disability, and has experienced deep-vein thrombosis and benign intracranial hypertension.54 By contrast, normal puberty was reported in another woman with ALG6-CDG.55 Benign intracranial hypertension has been reported in another case, in combination with optic atrophy and pigmentary retinopathy.56 Furthermore, one patient with ALG6-CDG presented with dilated cardiomyopathy.57
Most patients are compound heterozygotes for point mutations, but a Japanese child with sagittal craniosynostosis, a hypoplastic corpus callosum, absent septum pellucidum, and grade III intraventricular haemorrhage had a novel point mutation in the maternal allele, and a deletion that included the ALG6 locus in the paternal allele.51
SRD5A3-CDG (CDG-Iq)
In 2010, Cantagrel and colleagues58 used homozygosity mapping of a consanguineous family and identified a candidate reductase suspected of being involved in testosterone metabolism. The “long-sought polyprenol reductase”, SRD5A3, was shown to be encoded by SRD5A3 and to be crucial to proper glycosylation. Interestingly, most of the mutations identified in SRD5A3 led to protein truncations and probably to complete loss of function. Additional proteins might be involved in the reduction of polyprenol, as Srd5a3 null mice can synthesise small amounts of dolichol.58
Patients with SRD5A3-CDG reported so far have had similar clinical presentations. All patients have shown intellectual disability, neurological manifestations (brain malformations or structural defects), including cerebellar vermis atrophy or hypoplasia and cerebellar ataxia. Ophthalmological defects have included forms of coloboma, nystagmus, optic nerve atrophy or hypoplasia, cataracts, and glaucoma.59
ALG1-CDG (CDG-lk)
ALG1 encodes an enzyme that acts early in the lipid-linked oligosaccharide biosynthesis pathway. Seven cases of ALG1-CDG had been described by 2009,29 Dupré and colleagues60 later identified an additional five cases, and a further ten cases were indentified in eight families by a targeted analysis of unsolved cases in the USA (Freeze HH, unpublished). The latter finding emphasises the usefulness of a targeted approach.61
The patients described so far have all had severe neurological phenotypes. The children presented with muscular hypotonia, usually accompanied by facial dysmorphisms. Half of the patients had microcephaly and severe intellectual impairment, and all had epileptic seizures that were frequently intractable. MRI revealed cerebellar hypoplasia and cortical and subcortical atrophy in some cases, although results were normal in a few patients. Several patients were blind at presentation. In the initial reports, four of seven patients died in early childhood. Of the 15 identified since 2009, however, 13 survived beyond the first year of life. Thus, ALG1-CDG is more common and has a much broader clinical spectrum than was initially suspected.
Defects of the conserved oligomeric Golgi complex
Proper glycosylation depends on a bi-directional endosomal transportation system (secretory pathway) between the ER and the Golgi apparatus. Several protein complexes regulate transportation within and between these organelles, such as the conserved oligomeric Golgi (COG) complex, which is an eight-subunit complex believed to regulate late and early endosomal retrograde transport to the trans-Golgi network.62,63 In 2004, mutations detected in COG complex subunit 7 led to the discovery of a new subgroup of congenital disorders of glycosylation, termed the COG-CDGs.64 Two siblings presented with multiple facial dysmorphias, generalised hypotonia, hepatomegaly, seizures, and cardiac insufficiency. Biochemical analysis showed combined N-linked and O-linked deficiencies, with hypoglycosylation of transferrin and of apolipoprotein C-III, which is one of the few serum proteins with exclusively O-linked glycosylation. Deficiencies in COGs 1, 4, 5, 6, and 8 have since been described.65–70
The severity of disease and degree of organ involvement differ between patients, but psychomotor developmental delay (mild to severe) has been seen in all, along with microcephaly and hypotonia, and often with epilepsy. Facial dysmorphias and failure to thrive are common, as are hepatomegaly and cardiac involvement (heart failure, hypertrophy, or congenital heart defects). Brain imaging can be normal, but some patients have cerebral and cerebellar atrophy. The diagnostic procedure for COG-CDGs involves analysis of transferrin and apolipoprotein C-III western blot analysis of the subunits and sequencing.
Cutis laxa syndromes
Cutis laxa is a dermatological disorder that can be localised or generalised, and is characterised by loose, sagging, inelastic skin. This disorder was seen in the first COG7-CDG patient and was accompanied by severe neurological deficits.64 Kornak and colleagues71 found that some patients with autosomal recessive cutis laxa II, or wrinkled skin syndrome, had mutations in ATP6V0A2, which encodes the α2 subunit of vacuolar-type proton-ATPase. Patients showed combined N-linked and O-linked glycosylation deficiencies, which led to the creation of the classification ATP6V0A2-CDG.71
20 patients with ATP6V0A2-CDG have been described. All have wrinkly skin and, typically, a large fontanelle with delayed closure, down-slanting palpebral fissures, joint laxity, muscular hypotonia, and eye anomalies (strabismus and myopia). Many patients have microcephaly. Intellectual disability and developmental delay are variable. Half of the patients have brain abnormalities, such as partial pachygyria or cobblestone-like lissencephaly, but are less severe than in the α-dystroglycanopathies, which are discussed below. Seizures are uncommon, but have been reported in older children. The early feeding difficulties, growth retardation and skin condition resolves with age in most patients.72,73
Although most patients have abnormal transferrin glycosylation, this feature was normal in some patients who presented at young ages, whereas O-linked glycosylation was abnormal.74 Nevertheless, some patients were only positive for deficient N-linked glycosylation.72 Testing for both N-linked and O-linked hypoglycosylation is recommended if ATP6V0A2-CDG is suspected.
α-Dystroglycanopathies
O-mannosylation has been proven to occur on α-dystroglycan, but other O-mannosylated proteins probably exist in the brain and in skeletal muscle. Defective O-mannosylation of α-dystroglycan causes a set of heterogeneous disorders called α-dystroglycanopathies. Manifestations range from early-onset muscular dystrophy, to severe brain and eye malformations, to late-onset muscle weakness with normal intelligence. The most severe disorders include muscle-eye-brain disease, Walker-Warburg syndrome, and Fukuyama congenital muscular dystrophy. Milder forms include limb-girdle muscular dystrophies. Intermediate presentations might present with or without intellectual disability (eg, congenital muscular dystrophy type 1D).13 Mutations in several genes have been associated with the traditional clinical syndromes, termed muscular dystrophy-dystroglycanopathies(MDDGs; table 4), which has led to anomenclature based on clinical severity and genetic cause. The severity classifications are A (severe), B (intermediate), and C (mild). The subtypes are numbered one to six according to the genetic cause, in the following order: POMT1, POMT2, POMGNT1, FKTN, FKRP, and LARGE. These genes encode known (POMT1, POMT2, POMGNT1, LARGE) or putative (FKTN and FKRP) glycosyltransferases.75 Thus, for example, Walker-Warburg syndrome due to POMT1 mutations is referred to as MDDGA1.76 More genes are likely to be discovered in this pathway.12
Patients with muscle-eye-brain disease (mainly MDDGA3) have generalised muscle weakness from birth, intellectual disability, and eye disorders, such as congenital myopia, pallor of the optic discs, and glaucoma. CNS malformations include frontoparietal pachygyria, polymicrogyria, cerebellar hypoplasia, and a flattened pons and brain stem. The disease progresses slowly and most patients survive into adulthood. Most of the patients reported so far have been Finnish.77
Walker-Warburg syndrome (mainly MDDGA1 and MDDGA2) progresses rapidly and patients generally succumb by age 3 years.78 Muscular dystrophy is severe, with no motor development, as are ophthalmopathies (congenital cataracts, microphthalmia, and buphthalmos). Imaging often reveals severe hydrocephalus, agenesis of the corpus callosum, agyria or cobblestone lissencephaly, and cerebellar hypoplasia.77
Fukuyama congenital muscular dystrophy (mainly MDDGA4) overlaps in phenotype with muscle-eye-brain disease. Patients present with generalised muscle weakness and hypotonia from early infancy. They have intellectual deficits and some have seizures. The brain malformations include cerebral and cerebellar micropolygyria, hydrocephalus, and hypoplasia of the corticospinal tracts. Milder forms (MDDGB4) are well known. Most patients with Fukuyama congenital muscular dystrophy are of Japanese ancestry.77
The N-linked glycan and O-linked mannose pathways require dolichol phosphate-mannose, and deficiency of this carrier might affect both pathways. Discovery of a patient with DPM3-CDG, who had exclusively late-onset muscular dystrophy and cardiomyopathy underscores this point.79 The DPM3 mutation reduces concentrations of functional dolichol phosphate-mannose, which leads to hypoglycosylation of α-dystroglycan and muscle symptoms. Furthermore, 11 patients with dilated cardiomyopathy as their major symptom had DOLK-CDG.80 This congenital disorder of glycosylation is normally a severe multisystem disorder,81 but in this subgroup, heart biopsies revealed deficient α-dystroglycan O-mannosylation. Thus, a transferrin test should be done in dystroglycanopathies where mutations cannot be found in the six known MDDG genes.
Glycosylphosphatidylinositol-anchor defects
Typical of glycosylation pathway disorders, glycosylphosphatidylinositol-anchor defects display a broad clinical spectrum. The first defect to be identified causes paroxysmal nocturnal haemoglobinuria owing to somatic mutations of the PIGA gene, exclusively in haemopoietic precursors.82 Inherited mutations in PIGA, PIGM, PIGN, PIGV, and PIGL systemically disrupt this pathway. Mutations in PIGM cause venous thrombosis and seizures; however, in one patient targeted butyrate treatment reversed intractable seizures because the mutations occur in an Sp1-promoter binding site.27,83
Mutations in PIGN led to severe neurological impairment, dysmorphia, chorea, seizures, and early death in one consanguineous family with seven affected children.28 Whole-exome sequencing showed that mutations in PIGV caused hyperphosphatasia mental retardation syndrome, with seizures of variable severity and muscular involvement.29 These patients have increased serum alkaline phosphatase concentrations with normal calcium and phosphate levels, brachytelephalangy, and facial dysmorphism. A germ-line mutation in PIGA resulted in a lethal disorder in three infants in one family. The children had neonatal seizures, thin corpus callosum, white-matter immaturity, no septum pellucidum, and a small cerebellum.84 Defects in PIGL have recently been shown to cause CHIME (coloboma, heart defects, ichthyosis, mental insufficiency and ear defects) syndrome (also called Zunich-Kaye syndrome).85
Diagnosis is challenging owing to the variable clinical presentations. All patients exhibit deficiencies of glycophosphatidylinositol-anchored proteins on the cell surface. CD59 and fluorescently labelled aerolysin are reliable surface markers on leucocytes that can quickly and easily identify glycophosphatidylinositol deficiencies (table 4). At least 11 separate steps and more than 20 known proteins are involved in the formation of a functional glycophosphatidylinositol anchor on proteins and, therefore, other inherited disorders that involve glycophosphatidylinositol biosynthesis will doubtless be found.23,86
Disorders in general substrates
Mutations in genes encoding proteins at the neuromuscular junction cause congenital myasthenic syndromes by impairing the safety margin for neuro-muscular transmission.87 In a study that exploited genetic linkage, Senderek and colleagues88 found 18 different biallelic mutations in GFPT1 in 13 unrelated Pakistani families with myasthenic syndromes. The gene encodes glutamine-fructose-6-phosphate transaminase 1.88 This enzyme catalyses the first step of hexosamine biosynthesis and has a rate-limiting effect that controls the flux of glucose into the pathway, which ultimately produces uridine diphosphate N-acetylglucosamine—a donor in nearly all glycosylation pathways. Atlhough no specific glycosylation pathways have been shown to cause the pathology, knock down of the GFPT1 orthologue in zebrafish embryos altered muscle fibre morphology and impaired neuromuscular-junction development. Some mutations reduced enzymatic activity, but many did not, which suggests that the mutations could alter binding of the enzyme to other proteins to efficiently direct substrate use.
Diagnosis
Current status
Biochemical and genetic approaches are both required to identify a defective gene and to show that the mutations impair function. Biochemical analysis initially guided gene identification, but high-capacity, low-cost DNA sequencing and powerful informatics computer programs can now identify possible candidate genes. Until all known and likely candidates have been queried, however, each candidate must be proven to cause a biochemical or biological abnormality.89,90
The discovery of many glycosylation disorders in the past 15 years is largely due to the ease of measurement of glycosylated transferrin concentrations in serum as a marker of N-linked glycosylation fidelity (table 2). Mass spectrometry is the preferred method and can indicate the absence of entire N-linked glycan chains, incomplete sculpting of those chains, or both. Isoelectric focusing analysis of transferrin measures loss of sialic acids and can, therefore, serve as a less expensive method for assessment of N-linked glycosylation disorders. Other diagnostic methods that measure glycosylated transferrin concentrations include capillary electrophoresis, high-performance liquid chromatography, and ion-exchange chromatography. Repeated analyses, sialidase digestion, or analysis of other serum glycoproteins might be required to confirm results. In the traditional nomenclature for congenital disorders of glycosylation, absence of entire glycans was designated type I, and loss of one or more monosaccharides as type II. These patterns are not gene specific, but they help to identify the defective gene by localisation in the relevant pathway.
False-positive results occur in the context of uncontrolled hereditary fructose intolerance, galactosaemia, heavy alcohol consumption, certain hepatic pathologies, and rare mutations at the transferrin glycosylation site.91 Conversely, preterm infants might have normal patterns of transferrin glycosylation at birth that later become abnormal. In patients who present with N-linked glycosylation deficiencies, initially abnormal transferrin glycosylation sometimes normalises without improvements in symptoms, and initially normal transferrin glycosylation has been reported in patients who were later proved to have glycosylation disorders.39,91–93 These findings suggest that a window of opportunity exists for diagnosis made on the basis of transferrin measurement. Data from thousands of transferrin tests done at the Mayo Clinic, Rochester, MN, USA, clearly show that the period between the ages of 6 months and 18 months is optimum for diagnosis (unpublished). Moreover, abnormal transferrin glycosylation is detected three to four times as often as are aminoacid, organic acid, fatty acid, mitochondrial and peroxisomal disorders (Raymond K, Mayo Clinic, Rochester, MN, USA, unpublished), which suggests that the frequency of glycosylation disorders is notably higher than is currently appreciated. Genetic confirmation of diagnoses was not available and, therefore, we cannot distinguish prevalence from a phenotypic clinical bias. A congenital disorder of glycosylation should be suspected in any patient with developmental delay of undetermined cause, particularly when associated with symptoms and signs presented in table 2 and table 3. Measurement of additional markers, such as transaminases, creatine kinase, thyroxine-binding globulin, coagulation factors, and lysosomal enzymes can be useful, along with liver, muscle, and kidney function tests. These tests are frequently clinically indicated for other reasons.
Table 3.
Non-neurological and muscular features of glycosylation disorders with neurological involvement detectable by measurement of transferrin glycosylation
| Facial dys- morphisms |
Inverted nipples or lipodys- trophy |
Skeletal defects |
Failure to thrive |
Liver dysfunction/ raised ALT |
Renal pathology |
Ichthyosis or other skin disorder |
Coagu- lopathy |
Recurrent infection or low immuno- globulin |
Cardio- myopathy |
Deafness | Endocrin- opathy or micropenis |
Hypogly- caemia |
PLE or intestinal dysfunction |
|
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PMM2-CDG (CDG-Ia)* | ++ | ++ | ++ | ++ | ++ | ++ | NR | ++ | (+) | (+) | (+) | ++ | (+) | (+) |
| ALG6-CDG (CDG-Ic)* | ++ | ++ | + | + | ++ | NR | NR | ++ | NR | (+) | NR | + | NR | (+) |
| ALG3-CDG (CDG-Id)* | ++ | (+) | (+) | ++ | + | NR | NR | ++ | (+) | NR | NR | (+) | (+) | + |
| DPM1-CDG (CDG-Ie)* | ++ | (+) | + | ++ | ++ | NR | NR | ++ | (+) | NR | NR | NR | NR | NR |
| MPDU1-CDG (CDG-If)* | ++ | NR | NR | (+) | NR | (+) | ++ | + | NR | NR | NR | NR | NR | (+) |
| ALG12-CDG (CDG-Ig)* | ++ | ++ | + | ++ | ++ | NR | NR | ++ | ++ | (+) | (+) | ++ | ++ | NR |
| ALG8-CDG (CDG-Ih)* | ++ | NR | NR | (+) | ++ | + | NR | ++ | NR | NR | NR | + | NR | ++ |
| ALG2-CDG (CDG-Ii)* | (+) | NR | NR | NR | (+) | NR | NR | (+) | NR | NR | NR | NR | NR | NR |
| DPAGT1-CDG (CDG-Ij)* | ++ | (+) | (+) | NR | (+) | NR | NR | NR | NR | NR | NR | NR | NR | NR |
| ALG1-CDG (CDG-Ik)* | ++ | NR | NR | + | NR | NR | NR | (+) | (+) | (+) | NR | (+) | NR | NR |
| ALG9-CDG (CDG-IL)* | ++ | ++ | NR | ++ | ++ | (+) | NR | (+) | NR | NR | NR | NR | NR | (+) |
| DOLK-CDG (CDG-Im)* | (+) | NR | NR | + | (+) | NR | ++ | NR | NR | ++ | NR | NR | (+) | NR |
| RFT1-CDG (CDG-In)* | ++ | ++ | NR | ++ | (+) | NR | NR | ++ | NR | NR | ++ | NR | NR | (+) |
| ALG11-CDG (CDG-Ip)* | (+) | ++ | NR | (+) | NR | NR | NR | (+) | NR | NR | (+) | (+) | NR | NR |
| SRD5A3-CDG (CDG-Iq)* | ++ | (+) | NR | + | ++ | NR | ++ | ++ | NR | NR | NR | (+) | NR | NR |
| DDOST-CDG (CDG-Ir)* | (+) | NR | (+) | (+) | (+) | NR | NR | (+) | NR | NR | NR | NR | NR | NR |
| MGAT2-CDG (CDG-IIa)* | ++ | NR | ++ | (+) | NR | NR | NR | ++ | (+) | NR | NR | ++ | NR | ++ |
| GCS1-CDG | (+) | NR | (+) | (+) | (+) | NR | NR | (+) | (+) | NR | NR | NR | NR | NR |
| SLC35C1-CDG | ++ | NR | ++ | NR | NR | NR | NR | NR | ++ | NR | NR | NR | NR | NR |
| B4GALT1-CDG (CDG-IId)* | NR | NR | NR | NR | (+) | NR | NR | (+) | NR | NR | NR | (+) | NR | NR |
| COG1-CDG (CDG-IIg)* | ++ | NR | ++ | (+) | (+) | (+) | NR | NR | (+) | NR | NR | NR | NR | NR |
| COG4-CDG (CDG-IIj)* | (+) | NR | NR | (+) | (+) | NR | NR | (+) | (+) | NR | NR | NR | NR | NR |
| COG5-CDG (CDG-Iii)* | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR |
| COG6-CDG (CDG-IIL)* | NR | NR | NR | NR | (+) | NR | NR | (+) | NR | NR | NR | NR | NR | NR |
| COG7-CDG (CDG-IIe)* | ++ | (+) | ++ | ++ | ++ | (+) | ++ | (+) | + | NR | NR | NR | NR | + |
| COG8-CDG (CDG-IIh)* | (+) | NR | (+) | (+) | (+) | NR | NR | NR | NR | NR | NR | NR | NR | (+) |
| ATP6V0A2-CDG | ++ | NR | NR | NR | NR | NR | ++ | NR | NR | NR | NR | NR | NR | NR |
Non-neurological features are presented as a diagnostic aid. ++=very common finding (≥50 % of patients). +=common finding (<50% of patients). (+)=described but uncommon finding (<3 known patients).
ALT=alanine aminotransferase. PLE=protein-losing enteropathy. NR=not reported.
Previous classifications provided in brackets.
As mentioned above, a normal result in transferrin glycosylation testing does not exclude a glycosylation disorder. For instance, normal results have been reported in patients with genetic disorders involving the oligosaccharyltransferase complex subunits encoded by TUSC3 and MAGT1.94,95 By contrast, mutations in another subunit, encoded by DDOST, have led to abnormal transferrin glycosylation.85,96 The reason for this difference is unclear, but additional disorders involving the other subunits of the complex are likely to be identified.
Not all suspected congenital disorders of glycosylation have convenient markers, although some tests can indicate pathways, albeit without identification of specific genes: the α-dystroglycanopathies can be investigated by measurement of monoclonal antibodies to the O-mannosylated glycan in muscle biopsy samples,97 and measurement of CD59 concentrations can be useful to detect glycophosphatidylinositol-anchor deficiencies (table 4).27,29 Testing of targeted genes that is approved through Clinical Laboratory Improvement Amendments is available.61 An analysis of about 35 patients with abnormal transferrin glycosylation showed damaging mutations in only seven known genes (Freeze H, Ng B, unpublished). Barring the unlikely event that these patients have mutations in the promoter, deep intronic regions, or exons that were not amplified, the results suggest that many glycosylation disorders result from mutations in as yet unidentified genes.
Next-generation sequencing
Discovery of most congenital disorders of glycosylation has relied on initial biochemical analysis of fibroblasts and serum glycans to identify mutated genes. Many of the expected candidate genes were identified, but unexpected genes were found, as some of the examples above show, and more will certainly be found in the future. Genetic mapping techniques have been very useful, especially in consanguineous families, but the availability of whole-exome (or entire-genome) sequencing is the future for diagnosis of glycosylation and other rare disorders. Falling costs, improvements in informatics analysis, and increased availability of technology puts such approaches within reach. However, findings must be considered in conjunction with initial biochemical analysis and evidence of the glycosylation-related pathology for the optimum paradigm. In the past 3 years, five defects were solved with traditional biochemical analysis and ten by homozygosity mapping. Within the past year, six defects have been assessed with state-of-the-art whole-exome analysis.
The coming years will require the dedicated collaboration of physicians and scientists to confirm the putative candidate genes in patients with glycosylation disorders. This approach will be extremely important to enable translation of the results into useful information for families, who are the biggest stakeholders in the field of rare diseases.
Prospects for therapy
Although early in-vitro studies showed that PMM2 deficiency could be corrected in patient donor cells by the addition of mannose to the culture medium, this treatment was not successful in vivo for PMM2-CDG patients.98,99 In-vitro studies aimed at redirecting exogenous mannose from catabolism towards glycosylation have shown positive results in some patient-derived cells, but not in others.100,101 A recent study in Pmm2 hypomorphic mice is noteworthy.102 Compound heterozygous mice carrying the two most common human mutations in Pmm2 all die in utero at about 10·5 days’ gestation. Provision to pregnant dams of small amounts of mannose in their drinking water during gestation prevented embryonic death. Pups did not require mannose after weaning and seemed to be healthy and have a normal lifespan. This remarkable finding suggests that only slight increases in mannose availability and use can have substantial benefits for this specific genotype. Whether this result will be applicable to PMM2-CDG patients remains to be seen and we believe it would be premature to view mannose intake by at-risk pregnant mothers as a potential benefit for a PMM2-CDG fetus.
The antidiabetic drug, metformin, has been proposed as a glycosylation enhancer.103 MPI-CDG patients who have a non-neurological phenotype (appendix) have shown almost complete remission with dietary mannose supplementation.104,105 Fucose therapy is effective for a few patients with CDG-IIc, which is a deficiency of Golgi GDP-fucose transporter, and butyrate is effective in patients with PIGM mutations.83,106 Whether this approach can be extended to other disorders is unknown, but provision of N-acetylglucosamine (although not glucosamine) has shown some efficacy in a mouse model of multiple sclerosis.107,108 Sialic acid and N-acetylmannosamine supplements might be useful in the treatment of hereditary inclusion-body myopathy, which has adult onset.109,110 These examples show that simple monosaccharide therapy can be useful for treating selected disorders.
The repurposing of approved drugs has not been tried to any great extent. This approach has obvious advantages, but there is currently insufficient information to aid selection of candidate drugs. Another approach is to identify small molecules that enhance substrate flux into depleted pathways or stabilise mutated proteins.111,112 The former approach has been used to inhibit the activity of mannose-6-phosphate isomerase and direct the flux of mannose through PMM2 to increase glycosylation. The latter approach, in the form of enzyme enhancement or chaperone therapy, should be applicable to most disorders that involve hypomorphic mutations, and has received substantial attention,113,114 although it has not been applied to glycosylation disorders. Stem-cell therapy might ultimately provide a therapeutic solution, but is not yet a viable option. Gene therapy was used in a case of adult-onset inclusion-body myopathy with promising results.115
Even in the absence of definitive disease-modifying therapies for congenital disorders of glycosylation, substantial benefits can be expected from early recognition and management. Early diagnosis saves patients and their families from a diagnostic odyssey, enables accurate genetic counselling for members of the extended family, and patients can receive appropriate treatment for symptoms and proactive management in view of the types of complications that are likely to occur.
Aggressive symptomatic therapy can yield impressive benefits in disorders for which there are no definitive treatments. The best example is cystic fibrosis. In the past 50 years, the lifespan of patients with cystic fibrosis has increased substantially, despite the absence of disease-modifying therapy.116 The institution of treatment guidelines based on evidence and experience, and the establishment of specialist clinical centres, combined with aggressive symptomatic therapy explain this impressive result. There is no reason why this approach should not be employed with equal efficacy in other rare diseases, including congenital disorders of glycosylation.
Conclusions and future perspectives
In the past decade the number of genes identified as having causal roles in congenital disorders of glycosylation has far surpassed that for any other subgroup of congenital errors of metabolism. The classic PMM2-CDG phenotype—the first to be recognised—describes only a portion of the patients, and defective glycosylation should be suspected in any patient with symptoms that involve many organs. Because many of these disorders can be detected with a simple and inexpensive blood test, there is every incentive to exclude or confirm the diagnosis. Despite vast growth of diagnostic knowledge for congenital disorders of glycosylation, treatment options are non-existent in most cases, and substantially more research needs to be done.
In the next few years, inexpensive whole-exome and whole-genome sequencing are likely to reveal candidate genes for scores, if not hundreds, of rare disorders with neurological consequences. Confirmation of the pathogenicity of candidate genes might be straightforward in cases where no gene product is made, but is not typically the case. More often, to determine the pathogenicity of mutations, cooperation will be required between clinicians and basic scientists. For glycosylation disorders, participation of a consultant glycobiologist will also be important.
Search strategy and selection criteria.
We searched PubMed for papers published from 1978 to Feb 10, 2012, with the terms “glycosylation”, “carbohydrate deficient glycoprotein syndrome”, “congenital disorder(s) of glycosylation”, “glycosaminoglycan”, “glycosylphosphatidylinositol anchors”, “glycolipid”, “glycosphingolipid”, and “transferrin”. Articles were also identified by consultation with other experts and by manual searches of the reference lists of retrieved articles. We only reviewed papers published in English. The final reference list was generated on the basis of relevance to the topics covered in this Review. In addition, we reviewed Online Mendelian Inheritance in Man (OMIM) for each of the disorders mentioned. We placed emphasis on congenital disorders of glycosylation discovered in the past 2–3 years, those for which ten or more patients have been described, and those being seen with increasing frequency in the clinic.
Acknowledgments
HHF is supported by The Rocket Fund, National Institutes of Health (R01DK55615), and a Sanford Professorship; EAE is supported by Avtal om läkarutbildning och forskning and Crafoordska stiftelsen; and MCP is supported by NINDS (U54 NS065768), National MS Society, Actelion Pharmaceuticals, Merck-Serono. We thank Kimiyo Raymond, for allowing us to cite her unpublished work, Ping He, Mariam Rodriguez Lee, Marie-Estelle Losfeld, and Vandana Sharma for critical review of the paper, and Amy Zimmon for her expert assistance in preparation of the Review. We also thank Gert Matthijs, Rafael Artuch Iriberri, Donna Krasnewich, Flemming Skovby, and Nathalie Seta providing information on the numbers of patients with specific congenital disorders of glycosylation.
Footnotes
Contributors
HHF originated and organised the paper, EAE and BGN prepared tables, and MCP planned the Review. All authors wrote portions of the article and reviewed drafts of the paper.
Conflicts of interest
MCP acts as a consultant for Shire HGT and is Chair of the data monitoring committee for Stem Cells Inc. He has received travel expenses from the National Niemann-Pick Disease Foundation and the US Institute of Medicine (as a member of the Committee on Adverse Effects of Vaccines), and is a member of the WHO Topic Advisory Group (Neurology) – Revision of ICD-10, for which he receives no compensation or expenses. The other authors declare that they have no conflicts of interest.
Contributor Information
Prof HH Freeze, Sanford-Burnham Medical Research Institute, La Jolla, CA, USA.
E A Eklund, Section of Experimental Paediatrics, Department of Clinical Sciences, Lund University, Lund, Sweden.
BG Ng, Sanford-Burnham Medical Research Institute, La Jolla, CA, USA.
Prof M C Patterson, Division of Child and Adolescent Neurology, Mayo Clinic, Rochester, MN, USA.
References
- 1.Freeze HH. Genetic defects in the human glycome. Nat Rev Genet. 2006;7:537–51. doi: 10.1038/nrg1894. [DOI] [PubMed] [Google Scholar]
- 2.Ungar D. Golgi linked protein glycosylation and associated diseases. Semin Cell Dev Biol. 2009;20:762–69. doi: 10.1016/j.semcdb.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 3.Varki A. Biological roles of oligosaccharides: all of the theories are correct. Glycobiology. 1993;3:97–130. doi: 10.1093/glycob/3.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jaeken J, Hennet T, Freeze HH, Matthijs G. On the nomenclature of congenital disorders of glycosylation (CDG) J Inherit Metab Dis. 2008;31:669–72. doi: 10.1007/s10545-008-0983-x. [DOI] [PubMed] [Google Scholar]
- 5.Stanley P, Schachter H, Taniguchi N. N-Glycans. In: Varki A, Cummings RD, Esko JD, et al., editors. Essentials of glycobiology. 2. New York, NY: Cold Spring Harbor Laboratory Press; 2009. [Google Scholar]
- 6.Zielinska DF, Gnad F, Wisniewski JR, Mann M. Precision mapping of an in vivo N-glycoproteome reveals rigid topological and sequence constraints. Cell. 2010;141:897–907. doi: 10.1016/j.cell.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 7.Li H, Chavan M, Schindelin H, Lennarz WJ. Structure of the oligosaccharyl transferase complex at 12 A resolution. Structure. 2008;16:432–40. doi: 10.1016/j.str.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 8.Dennis JW, Nabi IR, Demetriou M. Metabolism, cell surface organization, and disease. Cell. 2009;139:1229–41. doi: 10.1016/j.cell.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Furmanek A, Hofsteenge J. Protein C-mannosylation: facts and questions. Acta Biochim Pol. 2000;47:781–89. [PubMed] [Google Scholar]
- 10.Ihara Y, Manabe S, Ikezaki M, et al. C-Mannosylated peptides derived from the thrombospondin type 1 repeat interact with Hsc70 to modulate its signaling in RAW264. 7 cells. Glycobiology. 2010;20:1298–310. doi: 10.1093/glycob/cwq096. [DOI] [PubMed] [Google Scholar]
- 11.Yoshida-Moriguchi T, Yu L, Stalnaker SH, et al. O-mannosyl phosphorylation of alpha-dystroglycan is required for laminin binding. Science. 2010;327:88–92. doi: 10.1126/science.1180512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stalnaker SH, Aoki K, Lim JM, et al. Glycomic analyses of mouse models of congenital muscular dystrophy. J Biol Chem. 2011;286:21180–90. doi: 10.1074/jbc.M110.203281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang CH, Bonnemann CG, Rutkowski A, et al. Consensus statement on standard of care for congenital muscular dystrophies. J Child Neurol. 2010;25:1559–81. doi: 10.1177/0883073810381924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brockhausen I, Schachter H, Stanley P. O-GalNAc glycans. In: Varki A, Cummings RD, Esko JD, et al., editors. Essentials of glycobiology. 2. New York, NY: Cold Spring Harbor Laboratory Press; 2009. [PubMed] [Google Scholar]
- 15.Esko JD, Kimata K, Lindahl U. Proteoglycans and sulfated glycosaminoglycans. In: Varki A, Cummings RD, Esko JD, et al., editors. Essentials of glycobiology. 2. New York, NY: Cold Spring Harbor Laboratory Press; 2009. [PubMed] [Google Scholar]
- 16.Lander AD. Morpheus unbound: reimagining the morphogen gradient. Cell. 2007;128:245–56. doi: 10.1016/j.cell.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 17.Zhang YT, Lander AD, Nie Q. Computational analysis of BMP gradients in dorsal-ventral patterning of the zebrafish embryo. J Theor Biol. 2007;248:579–89. doi: 10.1016/j.jtbi.2007.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stanley P, Okajima T. Roles of glycosylation in Notch signaling. Curr Top Dev Biol. 2010;92:131–64. doi: 10.1016/S0070-2153(10)92004-8. [DOI] [PubMed] [Google Scholar]
- 19.Takeuchi H, Haltiwanger RS. Role of glycosylation of Notch in development. Semin Cell Dev Biol. 2010;21:638–45. doi: 10.1016/j.semcdb.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Freeze HH, Haltiwanger RS. Other classes of ER/Golgi-derived glycans. In: Varki A, Cummings RD, Esko JD, et al., editors. Essentials of glycobiology. 2. New York, NY: Cold Spring Harbor Laboratory Press; 2009. [PubMed] [Google Scholar]
- 21.Schnaar RL, Suzuki A, Stanley P. Glycosphingolipids. In: Varki A, Cummings RD, Esko JD, et al., editors. Essentials of glycobiology. 2. New York, NY: Cold Spring Harbor Laboratory Press; 2009. [PubMed] [Google Scholar]
- 22.Hakomori S. Carbohydrate-to-carbohydrate interaction, through glycosynapse, as a basis of cell recognition and membrane organization. Glycoconj J. 2004;21:125–37. doi: 10.1023/B:GLYC.0000044844.95878.cf. [DOI] [PubMed] [Google Scholar]
- 23.Ferguson MAJ, Kinoshita T, Hart GW. Glycosylphosphatidylinositol anchors. In: Varki A, Cummings RD, Esko JD, et al., editors. Essentials of glycobiology. New York, NY: Cold Spring Harbor Laboratory Press; 2009. [PubMed] [Google Scholar]
- 24.Hwa KY. Glycosyl phosphatidylinositol-linked glycoconjugates: structure, biosynthesis and function. Adv Exp Med Biol. 2001;491:207–14. doi: 10.1007/978-1-4615-1267-7_15. [DOI] [PubMed] [Google Scholar]
- 25.Hancock JF. GPI-anchor synthesis: Ras takes charge. Dev Cell. 2004;6:743–45. doi: 10.1016/j.devcel.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 26.Simpson MA, Cross H, Proukakis C, et al. Infantile-onset symptomatic epilepsy syndrome caused by a homozygous loss-of-function mutation of GM3 synthase. Nat Genet. 2004;36:1225–29. doi: 10.1038/ng1460. [DOI] [PubMed] [Google Scholar]
- 27.Almeida AM, Murakami Y, Layton DM, et al. Hypomorphic promoter mutation in PIGM causes inherited glycosylphosphatidylinositol deficiency. Nat Med. 2006;12:846–51. doi: 10.1038/nm1410. [DOI] [PubMed] [Google Scholar]
- 28.Maydan G, Noyman I, Har-Zahav A, et al. Multiple congenital anomalies-hypotonia-seizures syndrome is caused by a mutation in PIGN. J Med Genet. 2011;48:383–89. doi: 10.1136/jmg.2010.087114. [DOI] [PubMed] [Google Scholar]
- 29.Krawitz PM, Schweiger MR, Rodelsperger C, et al. Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nat Genet. 2010;42:827–29. doi: 10.1038/ng.653. [DOI] [PubMed] [Google Scholar]
- 30.Freeze HH, Ng BG. Golgi glycosylation and human inherited diseases. Cold Spring Harb Perspect Biol. 2011;3:a005371. doi: 10.1101/cshperspect.a005371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haeuptle MA, Hennet T. Congenital disorders of glycosylation: an update on defects affecting the biosynthesis of dolichol-linked oligosaccharides. Hum Mutat. 2009;30:1628–41. doi: 10.1002/humu.21126. [DOI] [PubMed] [Google Scholar]
- 32.Pearl PL, Krasnewich D. Neurologic course of congenital disorders of glycosylation. J Child Neurol. 2001;16:409–13. doi: 10.1177/088307380101600604. [DOI] [PubMed] [Google Scholar]
- 33.Miossec-Chauvet E, Mikaeloff Y, Heron D, et al. Neurological presentation in pediatric patients with congenital disorders of glycosylation type Ia. Neuropediatrics. 2003;34:1–6. doi: 10.1055/s-2003-38614. [DOI] [PubMed] [Google Scholar]
- 34.Grunewald S. The clinical spectrum of phosphomannomutase 2 deficiency (CDG-Ia) Biochim Biophys Acta. 2009;1792:827–34. doi: 10.1016/j.bbadis.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 35.Hagberg BA, Blennow G, Kristiansson B, Stibler H. Carbohydrate-deficient glycoprotein syndromes: peculiar group of new disorders. Pediatr Neurol. 1993;9:255–62. doi: 10.1016/0887-8994(93)90060-p. [DOI] [PubMed] [Google Scholar]
- 36.de Lonlay P, Seta N, Barrot S, et al. A broad spectrum of clinical presentations in congenital disorders of glycosylation I: a series of 26 cases. J Med Genet. 2001;38:14–19. doi: 10.1136/jmg.38.1.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ishikawa N, Tajima G, Ono H, Kobayashi M. Different neuroradiological findings during two stroke-like episodes in a patient with a congenital disorder of glycosylation type Ia. Brain Dev. 2009;31:240–43. doi: 10.1016/j.braindev.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 38.Jensen H, Kjaergaard S, Klie F, Moller HU. Ophthalmic manifestations of congenital disorder of glycosylation type 1a. Ophthalmic Genet. 2003;24:81–88. doi: 10.1076/opge.24.2.81.13994. [DOI] [PubMed] [Google Scholar]
- 39.Krasnewich D, O’Brien K, Sparks S. Clinical features in adults with congenital disorders of glycosylation type Ia (CDG-Ia) Am J Med Genet C Semin Med Genet. 2007;145C:302–06. doi: 10.1002/ajmg.c.30143. [DOI] [PubMed] [Google Scholar]
- 40.Casado M, O’Callaghan MM, Montero R, et al. Mild clinical and biochemical phenotype in two patients with PMM2-CDG (congenital disorder of glycosylation Ia) Cerebellum. 201 doi: 10.1007/s12311-011-0313-y. published online Oct 20. [DOI] [PubMed] [Google Scholar]
- 41.Coman D, McGill J, MacDonald R, et al. Congenital disorder of glycosylation type 1a: three siblings with a mild neurological phenotype. J Clin Neurosci. 2007;14:668–72. doi: 10.1016/j.jocn.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 42.Perez-Duenas B, Garcia-Cazorla A, Pineda M, et al. Long-term evolution of eight Spanish patients with CDG type Ia: typical and atypical manifestations. Eur J Paediatr Neurol. 2009;13:444–51. doi: 10.1016/j.ejpn.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 43.Antoun H, Villeneuve N, Gelot A, Panisset S, Adamsbaum C. Cerebellar atrophy: an important feature of carbohydrate deficient glycoprotein syndrome type 1. Pediatr Radiol. 1999;29:194–98. doi: 10.1007/s002470050571. [DOI] [PubMed] [Google Scholar]
- 44.Horslen SP, Clayton PT, Harding BN, Hall NA, Keir G, Winchester B. Olivopontocerebellar atrophy of neonatal onset and disialotransferrin developmental deficiency syndrome. Arch Dis Child. 1991;66:1027–032. doi: 10.1136/adc.66.9.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nordborg C, Hagberg BA, Kristiansson B. Sural nerve pathology in the carbohydrate-deficient glycoprotein syndrome. Acta Paediatr. 1991;80:39–49. [Google Scholar]
- 46.Veneselli E, Biancheri R, Di Rocco M, Tortorelli S. Neurophysiological findings in a case of carbohydrate-deficient glycoprotein (CDG) syndrome type I with phosphomannomutase deficiency. Eur J Paediatr Neurol. 1998;2:239–44. doi: 10.1016/s1090-3798(98)80037-8. [DOI] [PubMed] [Google Scholar]
- 47.Pascual-Castroviejo I, Pascual-Pascual SI, Quijano-Roy S, et al. Cerebellar ataxia of Norman-Jaeken. Presentation of seven Spanish patients. Rev Neurol. 2006;42:723–28. (in Spanish) [PubMed] [Google Scholar]
- 48.Mader I, Dobler-Neumann M, Kuker W, Stibler H, Krageloh-Mann I. Congenital disorder of glycosylation type Ia: benign clinical course in a new genetic variant. Childs Nerv Syst. 2002;18:77–80. doi: 10.1007/s003810100493. [DOI] [PubMed] [Google Scholar]
- 49.Boddaert N, Desguerre I, Bahi-Buisson N, et al. Posterior fossa imaging in 158 children with ataxia. J Neuroradiol. 2010;37:220–30. doi: 10.1016/j.neurad.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 50.Imbach T, Grunewald S, Schenk B, et al. Multi-allelic origin of congenital disorder of glycosylation (CDG)-Ic. Hum Genet. 2000;106:538–45. doi: 10.1007/s004390000293. [DOI] [PubMed] [Google Scholar]
- 51.Eklund EA, Sun L, Yang SP, Pasion RM, Thorland EC, Freeze HH. Congenital disorder of glycosylation Ic due to a de novo deletion and an hALG-6 mutation. Biochem Biophys Res Commun. 2006;339:755–60. doi: 10.1016/j.bbrc.2005.11.073. [DOI] [PubMed] [Google Scholar]
- 52.Westphal V, Schottstadt C, Marquardt T, Freeze HH. Analysis of multiple mutations in the hALG6 gene in a patient with congenital disorder of glycosylation Ic. Mol Genet Metab. 2000;70:219–23. doi: 10.1006/mgme.2000.3017. [DOI] [PubMed] [Google Scholar]
- 53.Drijvers JM, Lefeber DJ, de Munnik SA, et al. Skeletal dysplasia with brachytelephalangy in a patient with a congenital disorder of glycosylation due to ALG6 gene mutations. Clin Genet. 2010;77:507–09. doi: 10.1111/j.1399-0004.2009.01349.x. [DOI] [PubMed] [Google Scholar]
- 54.Sun L, Eklund EA, Van Hove JL, Freeze HH, Thomas JA. Clinical and molecular characterization of the first adult congenital disorder of glycosylation (CDG) type Ic patient. Am J Med Genet A. 2005;137:22–26. doi: 10.1002/ajmg.a.30831. [DOI] [PubMed] [Google Scholar]
- 55.Miller BS, Freeze HH, Hoffmann GF, Sarafoglou K. Pubertal development in ALG6 deficiency (congenital disorder of glycosylation type Ic) Mol Genet Metab. 2011;103:101–03. doi: 10.1016/j.ymgme.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kahook MY, Mandava N, Bateman JB, Thomas JA. Glycosylation type Ic disorder: idiopathic intracranial hypertension and retinal degeneration. Br J Ophthalmol. 2006;90:115–16. doi: 10.1136/bjo.2005.080648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Al-Owain M, Mohamed S, Kaya N, Zagal A, Matthijs G, Jaeken J. A novel mutation and first report of dilated cardiomyopathy in ALG6-CDG (CDG-Ic): a case report. Orphanet J Rare Dis. 2010;5:7. doi: 10.1186/1750-1172-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cantagrel V, Lefeber DJ, Ng BG, et al. SRD5A3 is required for converting polyprenol to dolichol and is mutated in a congenital glycosylation disorder. Cell. 2010;142:203–17. doi: 10.1016/j.cell.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Morava E, Wevers RA, Cantagrel V, et al. A novel cerebello-ocular syndrome with abnormal glycosylation due to abnormalities in dolichol metabolism. Brain. 2010;133:3210–20. doi: 10.1093/brain/awq261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dupré T, Vuillaumier-Barrot S, Chantret I, et al. Guanosine diphosphate-mannose:GlcNAc2-PP-dolichol mannosyltransferase deficiency (congenital disorders of glycosylation type Ik): five new patients and seven novel mutations. J Med Genet. 2010;47:729–35. doi: 10.1136/jmg.2009.072504. [DOI] [PubMed] [Google Scholar]
- 61.Jones MA, Bhide S, Chin E, et al. Targeted polymerase chain reaction-based enrichment and next generation sequencing for diagnostic testing of congenital disorders of glycosylation. Genet Med. 2011;13:921–32. doi: 10.1097/GIM.0b013e318226fbf2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Reynders E, Foulquier F, Annaert W, Matthijs G. How Golgi glycosylation meets and needs trafficking: the case of the COG complex. Glycobiology. 2011;21:853–63. doi: 10.1093/glycob/cwq179. [DOI] [PubMed] [Google Scholar]
- 63.Laufman O, Hong W, Lev S. The COG complex interacts directly with Syntaxin 6 and positively regulates endosome-to-TGN retrograde transport. J Cell Biol. 2011;194:459–72. doi: 10.1083/jcb.201102045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu X, Steet RA, Bohorov O, et al. Mutation of the COG complex subunit gene COG7 causes a lethal congenital disorder. Nat Med. 2004;10:518–23. doi: 10.1038/nm1041. [DOI] [PubMed] [Google Scholar]
- 65.Foulquier F, Vasile E, Schollen E, et al. Conserved oligomeric Golgi complex subunit 1 deficiency reveals a previously uncharacterized congenital disorder of glycosylation type II. Proc Natl Acad Sci USA. 2006;103:3764–69. doi: 10.1073/pnas.0507685103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Reynders E, Foulquier F, Leao Teles E, et al. Golgi function and dysfunction in the first COG4-deficient CDG type II patient. Hum Mol Genet. 2009;18:3244–56. doi: 10.1093/hmg/ddp262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Paesold-Burda P, Maag C, Troxler H, et al. Deficiency in COG5 causes a moderate form of congenital disorders of glycosylation. Hum Mol Genet. 2009;18:4350–56. doi: 10.1093/hmg/ddp389. [DOI] [PubMed] [Google Scholar]
- 68.Lubbehusen J, Thiel C, Rind N, et al. Fatal outcome due to deficiency of subunit 6 of the conserved oligomeric Golgi complex leading to a new type of congenital disorders of glycosylation. Hum Mol Genet. 2010;19:3623–33. doi: 10.1093/hmg/ddq278. [DOI] [PubMed] [Google Scholar]
- 69.Foulquier F, Ungar D, Reynders E, et al. A new inborn error of glycosylation due to a Cog8 deficiency reveals a critical role for the Cog1-Cog8 interaction in COG complex formation. Hum Mol Genet. 2007;16:717–30. doi: 10.1093/hmg/ddl476. [DOI] [PubMed] [Google Scholar]
- 70.Kranz C, Ng BG, Sun L, et al. COG8 deficiency causes new congenital disorder of glycosylation type IIh. Hum Mol Genet. 2007;16:731–41. doi: 10.1093/hmg/ddm028. [DOI] [PubMed] [Google Scholar]
- 71.Kornak U, Reynders E, Dimopoulou A, et al. Impaired glycosylation and cutis laxa caused by mutations in the vesicular H+-ATPase subunit ATP6V0A2. Nat Genet. 2008;40:32–34. doi: 10.1038/ng.2007.45. [DOI] [PubMed] [Google Scholar]
- 72.Hucthagowder V, Morava E, Kornak U, et al. Loss-of-function mutations in ATP6V0A2 impair vesicular trafficking, tropoelastin secretion and cell survival. Hum Mol Genet. 2009;18:2149–65. doi: 10.1093/hmg/ddp148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Morava E, Lefeber DJ, Urban Z, et al. Defining the phenotype in an autosomal recessive cutis laxa syndrome with a combined congenital defect of glycosylation. Eur J Hum Genet. 2008;16:28–35. doi: 10.1038/sj.ejhg.5201947. [DOI] [PubMed] [Google Scholar]
- 74.Morava E, Guillard M, Lefeber DJ, Wevers RA. Autosomal recessive cutis laxa syndrome revisited. Eur J Hum Genet. 2009;17:1099–110. doi: 10.1038/ejhg.2009.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hewitt JE. Abnormal glycosylation of dystroglycan in human genetic disease. Biochim Biophys Acta. 2009;1792:853–61. doi: 10.1016/j.bbadis.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 76.Amberger J, Bocchini C, Hamosh A. A new face and new challenges for Online Mendelian Inheritance in Man (OMIM®) Hum Mutat. 2011;32:564–67. doi: 10.1002/humu.21466. [DOI] [PubMed] [Google Scholar]
- 77.Clement E, Mercuri E, Godfrey C, et al. Brain involvement in muscular dystrophies with defective dystroglycan glycosylation. Ann Neurol. 2008;64:573–82. doi: 10.1002/ana.21482. [DOI] [PubMed] [Google Scholar]
- 78.Vajsar J, Schachter H, Orphanet J. Walker-Warburg syndrome. Rare Dis. 2006;1:29. doi: 10.1186/1750-1172-1-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lefeber DJ, Schonberger J, Morava E, et al. Deficiency of Dol-P-Man synthase subunit DPM3 bridges the congenital disorders of glycosylation with the dystroglycanopathies. Am J Hum Genet. 2009;85:76–86. doi: 10.1016/j.ajhg.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lefeber DJ, de Brouwer AP, Morava E, et al. Autosomal recessive dilated cardiomyopathy due to DOLK mutations results from abnormal dystroglycan O-mannosylation. PLoS Genet. 2011;7:e1002427. doi: 10.1371/journal.pgen.1002427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kranz C, Jungeblut C, Denecke J, et al. A defect in dolichol phosphate biosynthesis causes a new inherited disorder with death in early infancy. Am J Hum Genet. 2007;80:433–40. doi: 10.1086/512130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Miyata T, Yamada N, Iida Y, et al. Abnormalities of PIG-A transcripts in granulocytes from patients with paroxysmal nocturnal hemoglobinuria. N Engl J Med. 1994;330:249–55. doi: 10.1056/NEJM199401273300404. [DOI] [PubMed] [Google Scholar]
- 83.Almeida AM, Murakami Y, Baker A, et al. Targeted therapy for inherited GPI deficiency. N Engl J Med. 2007;356:1641–47. doi: 10.1056/NEJMoa063369. [DOI] [PubMed] [Google Scholar]
- 84.Johnston JJ, Gropman AL, Sapp JC, et al. The phenotype of a germline mutation in PIGA: the gene somatically mutated in paroxysmal nocturnal hemoglobinuria. Am J Hum Genet. 2012;90:295–300. doi: 10.1016/j.ajhg.2011.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ng BG, Hackmann K, Jones MA, et al. Mutations in the glycosylphosphatidylinositol gene PIGL cause CHIME syndrome. Am J Hum Genet. doi: 10.1016/j.ajhg.2012.02.010. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Maeda Y, Kinoshita T. Structural remodeling, trafficking and functions of glycosylphosphatidylinositol-anchored proteins. Prog Lipid Res. 2011;50:411–24. doi: 10.1016/j.plipres.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 87.Farrugia ME. Myasthenic syndromes. J R Coll Physicians Edinb. 2011;41:43–47. doi: 10.4997/JRCPE.2011.111. quiz 48. [DOI] [PubMed] [Google Scholar]
- 88.Senderek J, Muller JS, Dusl M, et al. Hexosamine biosynthetic pathway mutations cause neuromuscular transmission defect. Am J Hum Genet. 2011;88:162–72. doi: 10.1016/j.ajhg.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zelinger L, Banin E, Obolensky A, et al. A missense mutation in DHDDS, encoding dehydrodolichyl diphosphate synthase, is associated with autosomal-recessive retinitis pigmentosa in Ashkenazi Jews. Am J Hum Genet. 2011;88:207–15. doi: 10.1016/j.ajhg.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gahl WA, Markello TC, Toro C, et al. The National Institutes of Health Undiagnosed Diseases Program: insights into rare diseases. Genet Med. 2012;14:51–59. doi: 10.1038/gim.0b013e318232a005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Perez-Cerda C, Quelhas D, Vega AI, Ecay J, Vilarinho L, Ugarte M. Screening using serum percentage of carbohydrate-deficient transferrin for congenital disorders of glycosylation in children with suspected metabolic disease. Clin Chem. 2008;54:93–100. doi: 10.1373/clinchem.2007.093450. [DOI] [PubMed] [Google Scholar]
- 92.Vermeer S, Kremer HP, Leijten QH, et al. Cerebellar ataxia and congenital disorder of glycosylation Ia (CDG-Ia) with normal routine CDG screening. J Neurol. 2007;254:1356–58. doi: 10.1007/s00415-007-0546-3. [DOI] [PubMed] [Google Scholar]
- 93.Freeze HH. Congenital disorders of glycosylation: CDG-I, CDG-II, and beyond. Curr Mol Med. 2007;7:389–96. doi: 10.2174/156652407780831548. [DOI] [PubMed] [Google Scholar]
- 94.Garshasbi M, Hadavi V, Habibi H, et al. A defect in the TUSC3 gene is associated with autosomal recessive mental retardation. Am J Hum Genet. 2008;82:1158–64. doi: 10.1016/j.ajhg.2008.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Molinari F, Foulquier F, Tarpey PS, et al. Oligosaccharyl transferase-subunit mutations in nonsyndromic mental retardation. Am J Hum Genet. 2008;82:1150–57. doi: 10.1016/j.ajhg.2008.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jones MA, Ng BG, Bhide S, et al. DDOST mutations identified by whole-exome sequencing are implicated in congenital disorders of glycosylation. Am J Hum Genet. 2012;90:363–68. doi: 10.1016/j.ajhg.2011.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Muntoni F, Torelli S, Wells DJ, Brown SC. Muscular dystrophies due to glycosylation defects: diagnosis and therapeutic strategies. Curr Opin Neurol. 2011;24:437–42. doi: 10.1097/WCO.0b013e32834a95e3. [DOI] [PubMed] [Google Scholar]
- 98.Panneerselvam K, Freeze HH. Mannose corrects altered N-glycosylation in carbohydrate-deficient glycoprotein syndrome fibroblasts. J Clin Invest. 1996;97:1478–87. doi: 10.1172/JCI118570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mayatepek E, Schröder M, Kohlmüller D, Bieger WP, Nützenadel W. Continuous mannose infusion in carbohydrate-deficient glycoprotein syndrome type I. Acta Paediatr. 1997;86:1138–40. doi: 10.1111/j.1651-2227.1997.tb14825.x. [DOI] [PubMed] [Google Scholar]
- 100.Sharma V, Ichikawa M, He P, et al. Phosphomannose isomerase inhibitors improve N-glycosylation in selected phosphomannomutase deficient fibroblasts. J Biol Chem. 2011;286:39431–38. doi: 10.1074/jbc.M111.285502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dahl R, Bravo Y, Sharma V, et al. Potent, selective, and orally available benzoisothiazolone phosphomannose isomerase inhibitors as probes for congenital disorder of glycosylation Ia. J Med Chem. 2011;54:3661–68. doi: 10.1021/jm101401a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Schneider A, Thiel C, Rindermann J, et al. Successful prenatal mannose treatment for congenital disorder of glycosylation-Ia in mice. Nat Med. 2011;18:71–73. doi: 10.1038/nm.2548. [DOI] [PubMed] [Google Scholar]
- 103.Shang J, Lehrman MA. Metformin-stimulated mannose transport in dermal fibroblasts. J Biol Chem. 2004;279:9703–12. doi: 10.1074/jbc.M310837200. [DOI] [PubMed] [Google Scholar]
- 104.de Lonlay P, Seta N. The clinical spectrum of phosphomannose isomerase deficiency, with an evaluation of mannose treatment for CDG-Ib. Biochim Biophys Acta. 2009;1792:841–43. doi: 10.1016/j.bbadis.2008.11.012. [DOI] [PubMed] [Google Scholar]
- 105.Harms HK, Zimmer KP, Kurnik K, Bertele-Harms RM, Weidinger S, Reiter K. Oral mannose therapy persistently corrects the severe clinical symptoms and biochemical abnormalities of phosphomannose isomerase deficiency. Acta Paediatr. 2002;91:1065–72. doi: 10.1080/080352502760311566. [DOI] [PubMed] [Google Scholar]
- 106.Eklund EA, Freeze HH. The congenital disorders of glycosylation: a multifaceted group of syndromes. NeuroRx. 2006;3:254–63. doi: 10.1016/j.nurx.2006.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mkhikian H, Grigorian A, Li CF, et al. Genetics and the environment converge to dysregulate N-glycosylation in multiple sclerosis. Nat Commun. 2011;2:334. doi: 10.1038/ncomms1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Grigorian A, Araujo L, Naidu NN, Place D, Choudhury B, Demetriou M. N-acetylglucosamine inhibits T-helper 1 (Th1)/T-helper 17 (Th17) responses and treats experimental autoimmune encephalomyelitis. J Biol Chem. 2011;286:40133–41. doi: 10.1074/jbc.M111.277814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Malicdan MC, Noguchi S, Nishino I. A preclinical trial of sialic acid metabolites on distal myopathy with rimmed vacuoles/hereditary inclusion body myopathy, a sugar-deficient myopathy: a review. Ther Adv Neurol Disord. 2010;3:127–35. doi: 10.1177/1756285609359546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Galeano B, Klootwijk R, Manoli I, et al. Mutation in the key enzyme of sialic acid biosynthesis causes severe glomerular proteinuria and is rescued by N-acetylmannosamine. J Clin Invest. 2007;117:1585–94. doi: 10.1172/JCI30954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Freeze HH. Towards a therapy for phosphomannomutase 2 deficiency, the defect in CDG-Ia patients. Biochim Biophys Acta. 2009;1792:835–40. doi: 10.1016/j.bbadis.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hutt D, Balch WE. Cell biology. The proteome in balance. Science. 2010;329:766–67. doi: 10.1126/science.1194160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hutt DM, Herman D, Rodrigues AP, et al. Reduced histone deacetylase 7 activity restores function to misfolded CFTR in cystic fibrosis. Nat Chem Biol. 2010;6:25–33. doi: 10.1038/nchembio.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 2009;78:959–91. doi: 10.1146/annurev.biochem.052308.114844. [DOI] [PubMed] [Google Scholar]
- 115.Nemunaitis G, Jay CM, Maples PB, et al. Hereditary inclusion body myopathy: single patient response to intravenous dosing of GNE gene lipoplex. Hum Gene Ther. 2011;22:1331–41. doi: 10.1089/hum.2010.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cohen-Cymberknoh M, Shoseyov D, Kerem E. Managing cystic fibrosis: strategies that increase life expectancy and improve quality of life. Am J Respir Crit Care Med. 2011;183:1463–71. doi: 10.1164/rccm.201009-1478CI. [DOI] [PubMed] [Google Scholar]