

Abstract
Purpose
Secondary lymphedema is a frequent complication of breast cancer associated with surgery, chemotherapy, or radiation following breast cancer treatment. The potential contribution of genetic susceptibility to risk of developing secondary lymphedema following surgical trauma, radiation, and other tissue insults has not been studied.
Experimental Design
To determine if women with breast cancer and secondary lymphedema had mutations in candidate lymphedema genes, we undertook a case - control study of 188 women diagnosed with breast cancer recruited from the University of Pittsburgh Breast Cancer Program (http://www.upmccancercenter.com/breast/index.cfm) between 2000–2010.
Candidate lymphedema genes, GJC2 (encoding connexin 47 [Cx47]), FOXC2, HGF, MET, and FLT4 (encoding VEGFR3), were sequenced for mutation. Bioinformatics analysis and in vitro functional assays were used to confirm significance of novel mutations.
Results
Cx47 mutations were identified in individuals having secondary lymphedema following breast cancer treatment but not in breast cancer controls or normal women without breast cancer. These novel mutations are dysfunctional as assessed through in vitro assays and bioinformatics analysis, and provide evidence that altered gap junction function leads to lymphedema.
Conclusions
Our findings challenge the view that secondary lymphedema is solely due to mechanical trauma and support the hypothesis that genetic susceptibility is an important risk factor for secondary lymphedema. A priori recognition of genetic risk 1) raises the potential for early detection and intervention for a high risk group, and 2) allows the possibility of altering surgical approach and/or chemo- and radiation therapy, or direct medical treatment of secondary lymphedema with novel connexin modifying drugs.
Translational Relevance
Secondary lymphedema is a frequent and serious chronic complication of breast cancer treatment. Our finding of four independent mutations in Cx47, including one shared mutation previously reported in primary lymphedema, not only supports these mutations as a genetic risk to the development of secondary lymphedema but raises the likelihood that other genes may contribute to such a genetic risk to secondary lymphedema as well.
Introduction
Secondary lymphedema is frequent serious chronic complication of breast cancer treatment. The staging and treatment of other cancers involving removal and/or radiation of lymph nodes may also precede secondary lymphedema. Secondary lymphedema following breast cancer therapy occurs in approximately 30% of patients, but estimates range from 2% to 80%, depending on the timing and method of ascertainment of lymphedema (1). As many as 600,000 women may suffer from secondary lymphedema following breast cancer treatment (2). Risk factors for secondary lymphedema, although not found to be the same in all studies, include treatment related factors: extent of surgery, radiation and chemotherapy; disease related factors: stage at diagnosis, pathological nodal status and number of dissected lymph nodes; and patient related factors: age at diagnosis, body mass index and presence of a sedentary lifestyle (2, 3). As demonstrated by the nature of these risk factors, secondary lymphedema is viewed as the consequence of a traumatic event. This contrasts with familial or primary lymphedema which has a genetic etiology, and is viewed as a developmental abnormality often segregating within families and having mutations in one of several causal genes (4–7). The contribution of genetic susceptibility to the subsequent risk of developing secondary lymphedema following surgical trauma, radiation, and other tissue insults has not been evaluated.
Finegold et al.(6) reported a shared, rare mutation in the high affinity receptor for hepatocyte growth factor, MET, between a patient with primary lymphedema and an unrelated patient with breast cancer and secondary lymphedema. This observation supported our hypothesis that some cases of secondary lymphedema are conditioned by mutation in genes causing primary lymphedema influencing lymphatic development or function. This hypothesis is further supported by quantitative lymphoscintigraphy in women with breast cancer secondary lymphedema demonstrating abnormalities in the unaffected contra-lateral normal arm (8). The pre-symptomatic identification of individuals susceptible to secondary lymphedema would identify a subset of patients for preventive intervention or early therapy, with the potential of ameliorating the negative effects of secondary lymphedema (9). We studied women with breast cancer, post treatment, with and without secondary lymphedema to determine whether they carried mutations in known causal genes for primary lymphedema.
Materials and METHODS
Recruitment
This study was approved by the Institutional Review Board of the University of Pittsburgh and written informed consent was obtained from all participants. We studied 188 breast cancer patients recruited from the University of Pittsburgh Breast Cancer Program (http://www.upmccancercenter.com/breast/index.cfm) between 2000 and 2010; 80 cases with secondary lymphedema and 108 breast cancer controls. Subjects were recruited through an initial mailing as well as during prospective follow-up. Subjects were eligible if they had a defined diagnosis of breast cancer, were willing to participate, and were willing to provide a DNA sample. Population controls were derived from a western Pennsylvania sampling of healthy women.
Blood was obtained for DNA isolation and analysis. We classified participants as cases if diagnosed with secondary lymphedema by a physician, physical therapist, or had received therapeutic treatment for lymphedema. Those without lymphedema were treated as breast cancer controls. All participants returned a questionnaire to assess risk factors for secondary lymphedema (supplementary material). Comparisons of non-genetic risk factors between cases and controls were performed using a t-test for continuous measures and using Fisher's exact test for binary measures. Genotype frequency differences were also tested using Fisher's exact test. A p-value of less than 0.05 was considered statistically significant.
Sequencing
Each participant was sequenced for the candidate lymphedema genes FLT4 (encoding VEGFR3), FOXC2, HGF, MET, GJC2 (connexin 47 [Cx47]) as previously described (10). We previously reported numbering for amino acid sequence based on the first ATG start site for human GJC2 as originally published by Uhlenberg et al. (11). There is now sufficient evidence supporting the second ATG site for initiation of translation for human GJC2 (12–15) and we use this site for initiation of numbering the amino acid sequence. This results in the amino acid substitutions in this report being numbered 3 amino acids less than our original report of GJC2 mutation in primary lymphedema (16). Statistical comparisons of mutation frequencies in case and control groups were performed using Fisher’s exact test.
Functional Assays in HeLa Cells
The Cx47 mutations were transfected (transient and stable) into communication deficient HeLa cells (17) to determine functional changes in gap junction intercellular communication (GJIC) or connexin function. The four mutations were introduced by site directed mutagenesis into a vector containing wild type human Cx47 pIRES2-EGFP (WT-hCx47-EGFP), a gift from Dr. S. Scherer, and the fidelity of the wild type and all mutant constructs confirmed by bidirectional sequence analysis.
Immunofluorescence microscopy was used to determine the presence or absence of Cx47 gap junction plaques when the constructs are transiently expressed in HeLa cells. A human Cx47 antibody was obtained from the Scherer laboratory (18): we used human CNS tissue and positive oligodendrocyte staining as a positive control and primary antibody delete as a negative control (data not shown). Cultured HeLa cells were routinely fixed and stained with the primary antibodies against Cx47, along with a nuclear marker, and transfected cells were identified by their EGFP signal. Plaques were imaged using an Olympus Fluoview 1000 confocal microscope, 100× oil objective.
Electrophysiologic characteristics of GJIC were measured between HeLa cell pairs transfected with the mutant constructs (as indicated by EGFP expression) by dual whole cell patch clamp recording. Pipette series resistance averaged 7.2 + 0.5 MΩ (mean ± SEM, n=50). All experiments were carried out in a blinded manner. Coupling current was quantified by measuring peak current recorded in the pair when the neighboring cell received a 100 mV step membrane potential change (in both positive and negative directions). Step changes in membrane potential were delivered to each cell in the pair in sequence and the average current recorded in the neighboring cell, determined and divided by 100 to generate coupling conductance (nS). Untransfected HeLa cells and cells transfected with empty vector (i.e., no hCx47) were used as additional controls.
The wound assay measure of proliferation/migration used differential interference contrast time lapse imaging of over 24 hrs with confluent transfected HeLa cells. Analysis was done using TScratch (19), mean ± SEM of at least 10 positions along the wound; the scrape width was normalized to the first image for each position. In all cases, we tested for statistical significance with a two tailed Student’s t-test.
Functional assay in Human Lymphatic Endothelial Cells
Adult human dermal lymphatic microvascular endothelial cells (LECs) were cultured in EGM™ −2 MV (both from Lonza). Cells were electroporated with 2 µg cDNA of the EGFP tagged mutant constructs and then selected with G418 as described above. Cells were microinjected using a combination (1:4 ratio) of 70 kd Texas Red dextran (Invitrogen) to mark the injected cell for reference, and Lucifer yellow, a known gap junction permeable dye (m.w. 443, −2 charge), to assess change in extent of spread (20). All cells were injected using constant conditions and scored for dye spread in tiers from a reference cell, using constant exposure time and thresholding, and imaged using a Nikon TE2000 with a temperature controlled motorized stage and QImaging Retiga CCD camera. Images were obtained using differential interference contrast (DIC) and standard filters for EGFP (identify expression of mutation), DAPI (for Lucifer yellow) and dsRed (for dextran) preinjection, immediately post-injection and 2 min after injection. Results were calculated as mean ± SEM. Statistical significance was determined in comparison to WT-hCx47-EGFP expressing cells using a Mann-Whitney Test.
RESULTS
Patient characteristics and mutation analysis
The characteristics of the study subjects derived from the questionnaire responses are shown in Table 1. Few significant differences were seen in demographic, clinical, or treatment variables between women who developed secondary lymphedema and controls that did not. Women with secondary lymphedema were older, diagnosed at a slightly later age, and were more likely to have had radiation therapy as part of their treatment. No differences were seen between cases and controls in other variables associated with increased risk for secondary lymphedema. None of the cases or controls had amino acid substitutions in the lymphedema genes FLT4 (VEGFR3), FOXC2, or HGF. A single case had a mutation in MET previously reported (6).
Table 1.
Characteristics of Secondary Lymphedema Cases and Controls
| Cases | Controls | P | |
|---|---|---|---|
| N | 80 | 108 | |
| Age (years) Current | 60 (37–93) | 54 (22–78) | < 0.01 |
| Age at Diagnosis (BC) | 54 (30–77) | 51 (20–74) | 0.015 |
| Age at Diagnosis (LE) | 56 (37–82) | - | |
| Body Mass Index | 28·6 (19·6–48.4) | 27·5 (19·2–43·9) | 0.23 |
| Mastectomy | 33 (41%) | 51 (47%) | 0.46 |
| Radiation | 68 (85%) | 63 (58%) | <0.01 |
| Risk Factors | |||
| Blood draw | 7 (9%) | 21 (19%) | 0.06 |
| Blood pressure | 9 (11%) | 18 (17%) | 0.40 |
| Cat scratch | 7 (9%) | 20 (18%) | 0.09 |
| Cut | 24 (30%) | 47 (44%) | 0.07 |
| Insect bite | 22 (28%) | 24 (22%) | 0.49 |
| Manicure | 18 (22%) | 31 (29%) | 0.40 |
| Sun pain | 9 (11%) | 16 (15%) | 0.52 |
BC, breast cancer; LE, lymphedema. (mean,%, and range)
Among the 80 sequenced breast cancer patients with secondary lymphedema, we observed Cx47 mutations in four patients and no mutations among 108 sequenced breast cancer controls that did not develop secondary lymphedema (Table 2; p=0·03). Comparable Cx47 mutations were not seen among at least 298 population controls (596 alleles) (p=0·002). One secondary lymphedema patient had the same mutation (G146S) seen in a family with primary lymphedema recently reported by our group (16). The other three mutations (G183C, P381S and H409Y) are unique. The Cx47 mutations found in these secondary lymphedema patients all met the following criteria for relevance of mutation status (similar to the Cx47 mutations observed in our previously reported primary lymphedema patients). Each mutation causes a change in the amino acid sequence of Cx47, is not present in at least 298 sequenced, ethnically matched controls (0/596 alleles), and is well conserved in mammalian evolution. The second allele in each of these mutation patients was wild type.
Table 2.
Connexin 47 Variation Seen in Secondary Lymphedema
| cDNA | Δnt | AMINO ACID | PROTEIN LOCATION | COMMENT |
|---|---|---|---|---|
| bp 436 | G→A | G 146 S | Intracellular loop | identified in primary lymphedema also reported in PMLD |
| bp 547 | G→T | G 183 C | Intracellular loop | |
| bp 1141 | C→T | P 381 S | C terminal | |
| bp1225 | C→T | H 409 Y | C terminal | sister with secondary lymphedema following breast cancer |
| bp 585 | C→T | H195H | Intracellular loop | polymorphism |
None of the cases with these mutations reported a personal or family history of primary lymphedema. Two cases (P381S and H409Y) had sisters with breast cancer and one case (H409Y) reported her sister also having secondary lymphedema following breast cancer treatment. All four women with mutations were receiving therapy for the lymphedema including bandaging, compression garments, and in one case, exercise. None of them reported metastatic disease. Of note, all four women had prior surgeries including hysterectomy, cholecystectomy, knee surgery, and other procedures. They did not report lymphedema following these surgical procedures.
We identified a fifth synonymous Cx47 polymorphic mutation, H195H, occurring in secondary lymphedema patients, breast cancer controls, and Western Pennsylvania non-breast cancer population controls with essentially equal frequency (5/80 secondary lymphedema patients, 8/108 breast cancer controls, and 27/298 population controls).
The three mutations found in probands with breast cancer and secondary lymphedema, the shared G146S mutation (by probands with primary and secondary lymphedema), and our previously identified mutations in families with primary lymphedema are distributed throughout the Cx47 monomer (although no mutations have been found in the transmembrane domains) (Figure 1, Supplementary Figure 2). Mutations G146S and G183C are located within the intracellular loop domain while P381S and H409Y are located in the C-terminal domain.
Figure 1.
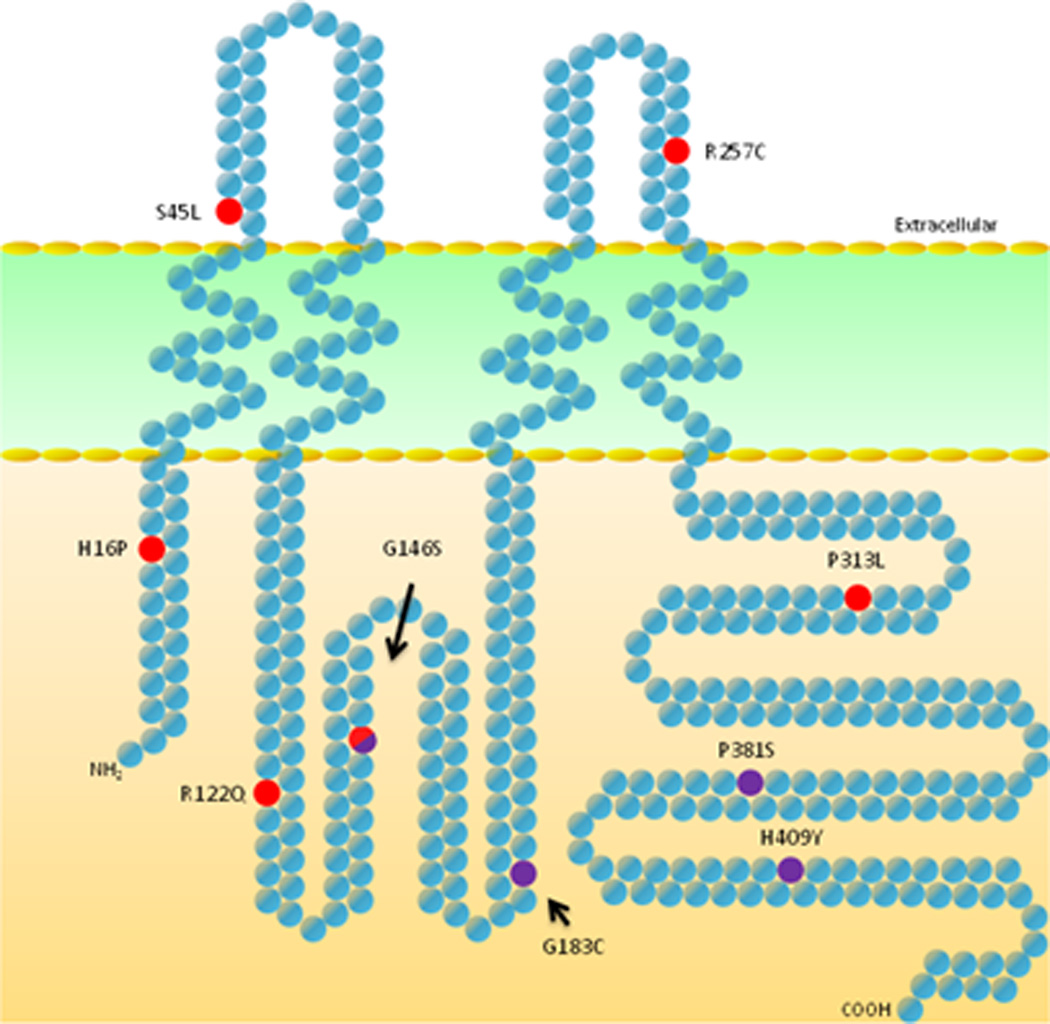
Schematic drawing of connexin 47 protein locating secondary lymphedema ( ), shared primary and secondary (
), shared primary and secondary ( ), and primary lymphedema- associated amino acid substitutions (
), and primary lymphedema- associated amino acid substitutions ( ). Domain break points and sequence are as described by Orthmann-Murphy et. al. (18).
). Domain break points and sequence are as described by Orthmann-Murphy et. al. (18).
No single functional assay is adequate to assess the complex spectrum of connexin physiology and the effect of connexin (Cx) mutations. We used a combination of frequently used assays in HeLa cells and another assay done in human dermal LECs, the cell type most likely to manifest the dysfunction causing clinical lymphedema. Each of the four mutations found in patients with secondary lymphedema demonstrate a phenotype different from that found in cells (HeLas and/or LECs) expressing WT- hCx47-EGFP.
Multiple assays for Cx function were performed in HeLa cells because they have little endogenous Cx expression, allowing the role of the specific Cx of interest to be isolated, and because of the ease of manipulation of these cells. The most common functional assays use immunofluorescent microscopy to demonstrate the presence or absence of Cx plaques along the cell membrane between adjoining cells, dye transfer studies to determine transport of gap junction permeable dyes between cells, and dual whole cell patch clamp recordings to measure electrical coupling between paired cells. Since there is increasing evidence that Cxs function independent of gap junctional communication (21–23), we also performed a wound healing assay to quantify the mutations’ effects on cell migration/proliferation.
Mutant transfected HeLa cells showed no significant change in plaque formation: by immunofluorescent confocal microscopy intercellular plaques were indistinguishable from those detected in WT-hCx7 transfected cells (Figure 2). However, both mutations in the intracellular loop domain showed significant functional differences as compared to WT- hCx47-EGFP transfected cells. G146S transfected cells showed faster wound closure in a conventional cell scratch assay than the WT- hCx47-EGFP transfected cells (Table 3). G183C transfected cells showed increased electrical coupling as compared to the WT- hCx47 transfected cells (This effect was statistically significant, but we recognize there are limitations in interpretation due to potential voltage errors in the whole cell patch clamp method when coupling currents are large, see Figure 2 and Table 3.). Hela cells transfected with the mutations located in the C-terminal domain (P381S and H409Y) were not functionally distinct from WT- hCx47-EGFP transfected HeLa cells.
Figure 2.
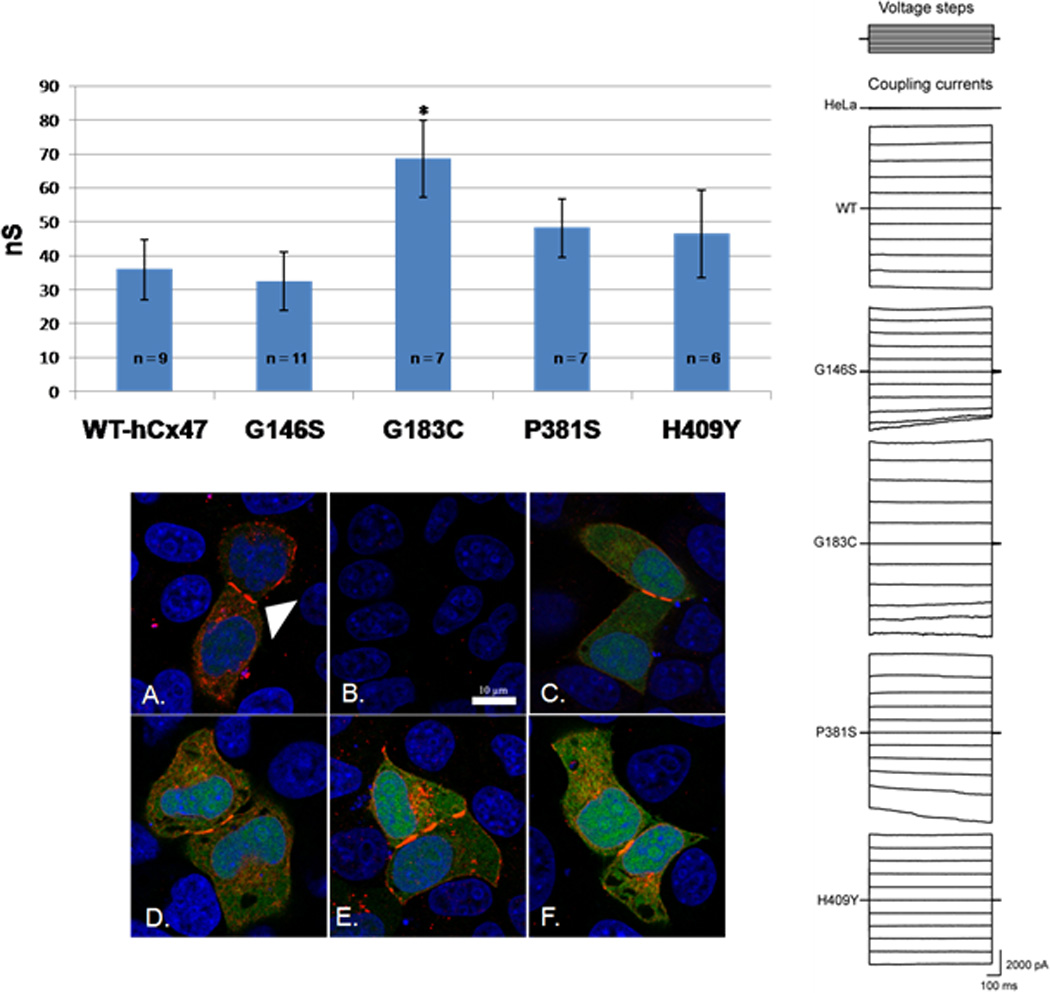
Top and right panel: representative transjunctional currents and average peak coupling current in HeLa cell pairs measured by dual whole cell patch clamping in response to a voltage step protocol, top right (−100 to +100 in 20 mV steps). p<0·05, two tailed Student’s t-test, *. Bottom panel: Immunofluorescent confocal microscopy of Cx47 intercellular plaques in near confluent HeLa cells transfected with WT-hCx47-EGFP (A), and mutants: G146S (C), G183C (D), P381S (E), and H409Y (F). Untransfected (B). White arrow head indicates sample plaques in WT-hCx47-EGFP expressing cells (A). Green indicative of EGFP and transfection, red Cx47, and blue nuclei. Scale bar, 10 µm
Table 3.
Summary of Functional Assessment of Mutations
| WT-Cx47 | G146S1 | G183C | P381S | H409Y | |
|---|---|---|---|---|---|
| LEC spread2,3 | 1·4±0·14 | 2·0±0·13 | 1·4±0·18 | 2·26±0·17# | 0·07 ±0·07# |
| electrical coupling2,4 | 35·9 ± 8·9 | 32·4 ± 8·6 | 68·6 ± 11·3* | 48·2 ± 8·6 | 46·4 ± 12·9 |
| wound assay2,5 | 0·72 ± 0·01 | 0·62 ± 0·03* | 0·68± 0·16 | 0·70±0·03 | 0·73± 0·02 |
-G146S mutation also found in patients with primary lymphedema (16).
- mean ± SEM.
-Dye spread to tiers of LECs after 2 min;
indicates significance determined by Mann Whitney test.
indicates significantly different than WT-hCx47 at p<0.05, two tailed Student’s t-test.
- in nS.
- expressed as fraction of original wound in HeLa cell monolayer after 24 hrs.
When dye spread was evaluated in LECs expressing the human mutations, C-terminal domain mutations were phenotypically distinguished from WT- hCx47-EGFP expressing LECs (Table 3, Supplementary Figure 1). The H409Y mutation showed dramatically impaired dye transfer of Lucifer yellow after microinjection. In contrast, the P381S mutation showed significantly enhanced dye transfer.
Discussion
Secondary lymphedema is a serious chronic complication of breast cancer treatment. Detection of increased risk of lymphedema is particularly important given the value of preoperative assessment and early postoperative intervention in reducing the impact of secondary lymphedema (9). Typically, studies of secondary lymphedema use patient specific information, like age and body mass index, in evaluating the risk of secondary lymphedema, but family history for lymphedema and genotype is not usually considered. A decision to intervene with treatment is usually based on the clinical burden of secondary lymphedema.
Recently Ferrell et al. (16) identified mutations in Cx47 encoded by GJC2 as a frequent cause of primary lymphedema. This finding was confirmed by Ostergaard et al. (24). Cxs are the major constituents of gap junctions which mediate intercellular communication. Increasingly there is evidence of Cxs’ role in a large signaling complex of associated proteins which serve to regulate coordination of conventional cell-cell communication in adhesion and motility, but also other basic cell processes including proliferation (22, 25). Gap junctions form as two apposing hexamers of Cx in adjoining cells. Gap junction communication mediates the propagation of spontaneous contractions in mesenteric lymphatics (26, 27).
Mutations leading to secondary lymphedema might be expected to result in fairly subtle dysfunction in vitro since, clinically, no lymphedema is observed until after some other significant insult in vivo, e.g. breast cancer treatment. This is consistent with our observation that all four of the mutations show normal plaque formation when expressed in HeLa cells. The detection of plaques indicates fairly normal trafficking of the Cx proteins to the cell membrane and subsequent organization into clusters of gap junctions, i.e., plaques. Of relevance, one of these four Cx47 mutations associated with secondary lymphedema, i.e., G146S, causes primary lymphedema inherited as an autosomal dominant mutation with reduced penetrance. As yet unknown modifying genetic or environmental factors must influence the expression of clinically detectable lymphedema. Variation in penetrance and expression has been demonstrated for other lymphedema genes such as FLT4 and FOXC2 (5, 28).
In two mutations, including G146S, we detected abnormal gap junction or Cx function using in vitro assays in HeLa cells. We documented autosomal dominant inheritance in two primary lymphedema families with Cx47 mutations and thus might expect a dominant negative effect of Cx47 mutations. However, these in vitro assays in HeLa cells are likely independent of such an effect since HeLa cells demonstrate little Cx expression (17) (and data not shown).
Functional data that supports the significance of these mutations in the development of secondary lymphedema is also demonstrated in human dermal LECs. We confirmed Cx47 expression in LECs (data not shown) along with other Cx species. Little is known about Cx expression and gap junction function in lymphatics, but gap junctions are important to the propagation of spontaneous contractions through mesenteric lymphatics in animal models (26, 27). It was recently reported that Cx47 had very limited expression in LECs in lymphatic valves in the thoracic duct and mesentery of developing mice. (29) We postulate that Cx47 mutations cause or contribute to the development of dermal lymphedema by impaired gap junction function causing impaired conduction of lymph from the periphery to more central lymphatic trunks. This is also supported through the identification of two Cx47 mutations in families with four limb lymphedema, one novel and one previously reported by us, where lymphoscintigraphy showed normal anatomy in distal lymphatics, but impaired uptake (24). The specific mechanisms through which Cx47 mutations result in increased susceptibility to secondary lymphedema remain to be determined. There is increasing evidence of the sensitivity of gap junction function in various connexin species to physiologic changes to ambient conditions such as pH and oxygenation (30–33). We suspect that these secondary mutations have pre-existing impairment of connexins’ ability to adapt to such changes in milieu that do not result in overt lymphedema until they are exacerbated by local changes in tissue post treatment. Further experimentation will be required before these mechanisms are clarified. Our findings of significant changes in gap junction function in LECs expressing the four mutations found in patients with secondary lymphedema: 1) confirms the significance of these mutations in patients with secondary lymphedema and 2) suggests impaired gap junction function as a novel mechanism for the development of lymphedema.
Identification of Cx47 mutations in secondary lymphedema, and previously in primary lymphedema, expands the clinical pathology of Cx47 in human disease. Until recently, Cx47 was only considered important for CNS myelination (34) because Cx47 mutations are causal for Pelizaeus-Merzbacher-like disease (11, 18, 35–38) (PMLD) and a milder phenotype of spastic paraplegia (39). These were all reported to be recessive mutations but recently dominant mutations in Cx47 were also identified as causing PMLD, among them a G146S mutation (identified as G149S) (12). In contrast to disease caused by recessive mutations, autosomal dominant mutations in Cxs are more likely to cause syndromes in a similar fashion to the Cx43 mutations causing oculodentodigital dysplasia and the Cx26 mutations causing hearing loss and a variety of skin diseases (21). Thus, the recent identification of dominant mutations in Cx47 causing PMLD coupled with our findings of Cx47 mutations causing and/or predisposing to lymphedema suggests some patients may manifest both neurologic and lymphatic deficits.
Our finding of four independent mutations in Cx47, including one shared mutation previously reported in primary lymphedema (16), not only supports these mutations as a genetic risk to the development of secondary lymphedema but raises the likelihood that other genes may contribute to such a genetic risk to secondary lymphedema as well. Gap junctions participate in a multi-protein complex and our observations implicate any of these proteins as potential candidates for risk mutations for secondary lymphedema and targets for novel drug therapy. A patient’s family history of lymphedema would be useful in identifying women at higher than normal risk of developing secondary lymphedema, and sequencing genes such as Cx47 and other genes known to cause primary lymphedema may prospectively identify a group of women who would benefit from early, aggressive surveillance and therapy prior to the clinical onset of lymphedema. Our findings challenge the commonly held view that secondary lymphedema is solely due to mechanical trauma (40, 41). Genetic susceptibility is an important risk factor which must be included with mechanical trauma, radiation, and/or chemical insult. A priori recognition of such a genetic susceptibility 1) raises the potential for early detection of a group at high risk, and 2) allows the possibility of altering surgical approach and/or chemotherapy radiation therapy or direct medical treatment of the lymphedema.
The prospect of preventive intervention or pharmacological treatment in secondary lymphedema is especially attractive given the estimated prevalence of up to 600,000 (2) women who suffer from secondary lymphedema following treatment for breast cancer. The limited treatment options currently available to these patients include early education and physiotherapy (42, 43). With regard to the Cx47 mutations specifically, there is potential for rapid translational progress given the pre-existing effort to develop Cx modifying drugs for application to cardiovascular disease (44). Our findings offer the possibility that early detection and intervention may be possible before breast cancer treatment is complete, and also offers the chance to ameliorate the severity of secondary lymphedema in a subset of breast cancer patients.
Supplementary Material
Acknowledgements
We acknowledge Dr. Atilla Soran for his encouragement and referrals, Dr. Geoffrey Murdoch for supplying CNS tissue for positive control samples, our genetic counseling students, the Clinical Translational Research Center nurses, and lastly, our patients without whom this work could not have been completed. We also acknowledge funding support for this work from the American Cancer Society through a generous donation provided by Longaberger, Inc, the Competitive Medical Research Fund of the UPMC Health System, Magee-Women’s Hospital Foundation, NIH grant HL092866 and grant UL1 RR024153 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research.
References
- 1.Hayes SC, Janda M, Cornish B, Battistutta D, Newman B. Lymphedema after breast cancer: incidence, risk factors, and effect on upper body function. J Clin Oncol. 2008;26:3536–3542. doi: 10.1200/JCO.2007.14.4899. [DOI] [PubMed] [Google Scholar]
- 2.Soran A, D'Angelo G, Begovic M, Ardic F, Harlak A, Samuel Wieand H, et al. Breast cancer-related lymphedema--what are the significant predictors and how they affect the severity of lymphedema? Breast J. 2006;12:536–543. doi: 10.1111/j.1524-4741.2006.00342.x. [DOI] [PubMed] [Google Scholar]
- 3.Poage E, Singer M, Armer J, Poundall M, Shellabarger MJ. Demystifying lymphedema: development of the lymphedema putting evidence into practice card. Clin J Oncol Nurs. 2008;12:951–964. doi: 10.1188/08.CJON.951-964. [DOI] [PubMed] [Google Scholar]
- 4.Fang J, Dagenais SL, Erickson RP, Arlt MF, Glynn MW, Gorski JL, et al. Mutations in FOXC2 (MFH-1), a forkhead family transcription factor, are responsible for the hereditary lymphedema-distichiasis syndrome. Am J Hum Genet. 2000;67:1382–1388. doi: 10.1086/316915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferrell RE, Levinson KL, Esman JH, Kimak MA, Lawrence EC, Barmada MM, et al. Hereditary lymphedema: evidence for linkage and genetic heterogeneity. Hum Mol Genet. 1998;7:2073–2078. doi: 10.1093/hmg/7.13.2073. [DOI] [PubMed] [Google Scholar]
- 6.Finegold DN, Schacht V, Kimak MA, Lawrence EC, Foeldi E, Karlsson JM, et al. HGF and MET mutations in primary and secondary lymphedema. Lymphat Res Biol. 2008;6:65–68. doi: 10.1089/lrb.2008.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Irrthum A, Devriendt K, Chitayat D, Matthijs G, Glade C, Steijlen PM, et al. Mutations in the transcription factor gene SOX18 underlie recessive and dominant forms of hypotrichosis-lymphedema-telangiectasia. Am J Hum Genet. 2003;72:1470–1478. doi: 10.1086/375614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stanton AW, Modi S, Bennett Britton TM, Purushotham AD, Peters AM, Levick JR, et al. Lymphatic drainage in the muscle and subcutis of the arm after breast cancer treatment. Breast Cancer Res Treat. 2009;117:549–557. doi: 10.1007/s10549-008-0259-z. [DOI] [PubMed] [Google Scholar]
- 9.Stout Gergich NL, Pfalzer LA, McGarvey C, Springer B, Gerber LH, Soballe P. Preoperative assessment enables the early diagnosis and successful treatment of lymphedema. Cancer. 2008;112:2809–2819. doi: 10.1002/cncr.23494. [DOI] [PubMed] [Google Scholar]
- 10.Ferrell RE, Kimak MA, Lawrence EC, Finegold DN. Candidate gene analysis in primary lymphedema. Lymphat Res Biol. 2008;6:69–76. doi: 10.1089/lrb.2007.1022. [DOI] [PubMed] [Google Scholar]
- 11.Uhlenberg B, Schuelke M, Ruschendorf F, Ruf N, Kaindl AM, Henneke M, et al. Mutations in the gene encoding gap junction protein alpha 12 (connexin 46.6) cause Pelizaeus-Merzbacher-like disease. Am J Hum Genet. 2004;75:251–260. doi: 10.1086/422763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diekmann S, Henneke M, Burckhardt BC, Gartner J. Pelizaeus-Merzbacher-like disease is caused not only by a loss of connexin47 function but also by a hemichannel dysfunction. Eur J Hum Genet. 2010;18:985–992. doi: 10.1038/ejhg.2010.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maeda S, Tsukihara T. Structure of the gap junction channel and its implications for its biological functions. Cell Mol Life Sci. 2011;68:1115–1129. doi: 10.1007/s00018-010-0551-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Orthmann-Murphy JL, Freidin M, Fischer E, Scherer SS, Abrams CK. Two distinct heterotypic channels mediate gap junction coupling between astrocyte and oligodendrocyte connexins. J Neurosci. 2007;27:13949–13957. doi: 10.1523/JNEUROSCI.3395-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruf N, Uhlenberg B. Analysis of human alternative first exons and copy number variation of the GJA12 gene in patients with Pelizaeus-Merzbacher-like disease. Am J Med Genet B Neuropsychiatr Genet. 2009;150B:226–232. doi: 10.1002/ajmg.b.30792. [DOI] [PubMed] [Google Scholar]
- 16.Ferrell RE, Baty CJ, Kimak MA, Karlsson JM, Lawrence EC, Franke-Snyder M, et al. GJC2 missense mutations cause human lymphedema. Am J Hum Genet. 2010;86:943–948. doi: 10.1016/j.ajhg.2010.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elfgang C, Eckert R, Lichtenberg-Frate H, Butterweck A, Traub O, Klein RA, et al. Specific permeability and selective formation of gap junction channels in connexin-transfected HeLa cells. J Cell Biol. 1995;129:805–817. doi: 10.1083/jcb.129.3.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Orthmann-Murphy JL, Enriquez AD, Abrams CK, Scherer SS. Loss-of-function GJA12/Connexin47 mutations cause Pelizaeus-Merzbacher-like disease. Mol Cell Neurosci. 2007;34:629–641. doi: 10.1016/j.mcn.2007.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Geback T, Schulz MM, Koumoutsakos P, Detmar M. TScratch: a novel and simple software tool for automated analysis of monolayer wound healing assays. Biotechniques. 2009;46:265–274. doi: 10.2144/000113083. [DOI] [PubMed] [Google Scholar]
- 20.Abbaci M, Barberi-Heyob M, Blondel W, Guillemin F, Didelon J. Advantages and limitations of commonly used methods to assay the molecular permeability of gap junctional intercellular communication. Biotechniques. 2008;45:33–52. doi: 10.2144/000112810. 6–62. [DOI] [PubMed] [Google Scholar]
- 21.Laird DW. Closing the gap on autosomal dominant connexin-26 and connexin-43 mutants linked to human disease. J Biol Chem. 2008;283:2997–3001. doi: 10.1074/jbc.R700041200. [DOI] [PubMed] [Google Scholar]
- 22.Wei CJ, Xu X, Lo CW. Connexins and cell signaling in development and disease. Annu Rev Cell Dev Biol. 2004;20:811–838. doi: 10.1146/annurev.cellbio.19.111301.144309. [DOI] [PubMed] [Google Scholar]
- 23.Xu X, Francis R, Wei CJ, Linask KL, Lo CW. Connexin 43-mediated modulation of polarized cell movement and the directional migration of cardiac neural crest cells. Development. 2006;133:3629–3639. doi: 10.1242/dev.02543. [DOI] [PubMed] [Google Scholar]
- 24.Ostergaard P, Simpson MA, Brice G, Mansour S, Connell FC, Onoufriadis A, et al. Rapid identification of mutations in GJC2 in primary lymphoedema using whole exome sequencing combined with linkage analysis with delineation of the phenotype. J Med Genet. 2011;48:251–255. doi: 10.1136/jmg.2010.085563. [DOI] [PubMed] [Google Scholar]
- 25.Laird DW. The gap junction proteome and its relationship to disease. Trends Cell Biol. 2010;20:92–101. doi: 10.1016/j.tcb.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 26.McHale NG, Meharg MK. Co-ordination of pumping in isolated bovine lymphatic vessels. J Physiol. 1992;450:503–512. doi: 10.1113/jphysiol.1992.sp019139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zawieja DC, Davis KL, Schuster R, Hinds WM, Granger HJ. Distribution, propagation, and coordination of contractile activity in lymphatics. Am J Physiol. 1993;264:H1283–H1291. doi: 10.1152/ajpheart.1993.264.4.H1283. [DOI] [PubMed] [Google Scholar]
- 28.Finegold DN, Kimak MA, Lawrence EC, Levinson KL, Cherniske EM, Pober BR, et al. Truncating mutations in FOXC2 cause multiple lymphedema syndromes. Hum Mol Genet. 2001;10:1185–1189. doi: 10.1093/hmg/10.11.1185. [DOI] [PubMed] [Google Scholar]
- 29.Kanady JD, Dellinger MT, Munger SJ, Witte MH, Simon AM. Connexin37 and Connexin43 deficiencies in mice disrupt lymphatic valve development and result in lymphatic disorders including lymphedema and chylothorax. Dev Biol. 2011;354:253–266. doi: 10.1016/j.ydbio.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gonzalez-Nieto D, Gomez-Hernandez JM, Larrosa B, Gutierrez C, Munoz MD, Fasciani I, et al. Regulation of neuronal connexin-36 channels by pH. Proc Natl Acad Sci U S A. 2008;105:17169–17174. doi: 10.1073/pnas.0804189105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Palacios-Prado N, Briggs SW, Skeberdis VA, Pranevicius M, Bennett MV, Bukauskas FF. pH-dependent modulation of voltage gating in connexin45 homotypic and connexin45/connexin43 heterotypic gap junctions. Proc Natl Acad Sci U S A. 2010;107:9897–9902. doi: 10.1073/pnas.1004552107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Palacios-Prado N, Sonntag S, Skeberdis VA, Willecke K, Bukauskas FF. Gating, permselectivity and pH-dependent modulation of channels formed by connexin57, a major connexin of horizontal cells in the mouse retina. J Physiol. 2009;587:3251–3269. doi: 10.1113/jphysiol.2009.171496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamaguchi DT, Ma D. Mechanism of pH regulation of connexin 43 expression in MC3T3-E1 cells. Biochem Biophys Res Commun. 2003;304:736–739. doi: 10.1016/s0006-291x(03)00633-8. [DOI] [PubMed] [Google Scholar]
- 34.Menichella DM, Goodenough DA, Sirkowski E, Scherer SS, Paul DL. Connexins are critical for normal myelination in the CNS. J Neurosci. 2003;23:5963–5973. doi: 10.1523/JNEUROSCI.23-13-05963.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bugiani M, Al Shahwan S, Lamantea E, Bizzi A, Bakhsh E, Moroni I, et al. GJA12 mutations in children with recessive hypomyelinating leukoencephalopathy. Neurology. 2006;67:273–279. doi: 10.1212/01.wnl.0000223832.66286.e4. [DOI] [PubMed] [Google Scholar]
- 36.Henneke M, Combes P, Diekmann S, Bertini E, Brockmann K, Burlina AP, et al. GJA12 mutations are a rare cause of Pelizaeus-Merzbacher-like disease. Neurology. 2008;70:748–754. doi: 10.1212/01.wnl.0000284828.84464.35. [DOI] [PubMed] [Google Scholar]
- 37.Salviati L, Trevisson E, Baldoin MC, Toldo I, Sartori S, Calderone M, et al. A novel deletion in the GJA12 gene causes Pelizaeus-Merzbacher-like disease. Neurogenetics. 2007;8:57–60. doi: 10.1007/s10048-006-0065-x. [DOI] [PubMed] [Google Scholar]
- 38.Wolf NI, Cundall M, Rutland P, Rosser E, Surtees R, Benton S, et al. Frameshift mutation in GJA12 leading to nystagmus, spastic ataxia and CNS dys-/demyelination. Neurogenetics. 2007;8:39–44. doi: 10.1007/s10048-006-0062-0. [DOI] [PubMed] [Google Scholar]
- 39.Orthmann-Murphy JL, Salsano E, Abrams CK, Bizzi A, Uziel G, Freidin MM, et al. Hereditary spastic paraplegia is a novel phenotype for GJA12/GJC2 mutations. Brain. 2009;132:426–438. doi: 10.1093/brain/awn328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moehler EaM, T E. In: Lymphedema: Etiology, clinical manifestations, and diagnosis. Basow DE, editor. Waltham, MA: 2010. UpToDate. [Google Scholar]
- 41.Schmitz KH, Ahmed RL, Troxel A, Cheville A, Smith R, Lewis-Grant L, et al. Weight lifting in women with breast-cancer-related lymphedema. N Engl J Med. 2009;361:664–673. doi: 10.1056/NEJMoa0810118. [DOI] [PubMed] [Google Scholar]
- 42.Schmitz KH, Ahmed RL, Troxel AB, Cheville A, Lewis-Grant L, Smith R, et al. Weight lifting for women at risk for breast cancer-related lymphedema: a randomized trial. JAMA. 2010;304:2699–2705. doi: 10.1001/jama.2010.1837. [DOI] [PubMed] [Google Scholar]
- 43.Torres Lacomba M, Yuste Sanchez MJ, Zapico Goni A, Prieto Merino D, Mayoral del Moral O, Cerezo Tellez E, et al. Effectiveness of early physiotherapy to prevent lymphoedema after surgery for breast cancer: randomised, single blinded, clinical trial. BMJ. 2010;340:b5396. doi: 10.1136/bmj.b5396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Clarke TC, Thomas D, Petersen JS, Evans WH, Martin PE. The antiarrhythmic peptide rotigaptide (ZP123) increases gap junction intercellular communication in cardiac myocytes and HeLa cells expressing connexin 43. Br J Pharmacol. 2006;147:486–495. doi: 10.1038/sj.bjp.0706631. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.