

Abstract
Recent studies suggest that vascular endothelial growth factor (VEGF) can modulate smooth muscle phenotype and, consequently, the composition and function of arteries upstream from the microcirculation, where angiogenesis occurs. Given that hypoxia potently induces VEGF, the present study explores the hypothesis that, in fetal arteries, VEGF contributes to hypoxic vascular remodeling through changes in abundance, organization, and function of contractile proteins. Pregnant ewes were acclimatized at sea level or at altitude (3,820 m) for the final 110 days of gestation. Endothelium-denuded carotid arteries from full-term fetuses were used fresh or after 24 h of organ culture in a physiological concentration (3 ng/ml) of VEGF. After 110 days, hypoxia had no effect on VEGF abundance but markedly increased abundance of the Flk-1 (171%) and Flt-1 (786%) VEGF receptors. Hypoxia had no effect on smooth muscle α-actin (SMαA), decreased myosin light chain (MLC) kinase (MLCK), and increased 20-kDa regulatory MLC (MLC20) abundances. Hypoxia also increased MLCK-SMαA, MLC20-SMαA, and MLCK-MLC20 colocalization. Compared with hypoxia, organ culture with VEGF produced the same pattern of changes in contractile protein abundance and colocalization. Effects of VEGF on colocalization were blocked by the VEGF receptor antagonists vatalanib (240 nM) and dasatinib (6.3 nM). Thus, through increases in VEGF receptor density, hypoxia can recruit VEGF to help mediate remodeling of fetal arteries upstream from the microcirculation. The results support the hypothesis that VEGF contributes to hypoxic vascular remodeling through changes in abundance, organization, and function of contractile proteins.
Keywords: myosin light chain kinase, organ culture, regulatory myosin light chain, smooth muscle α-actin, vascular endothelial growth factor receptors
in utero hypoxia secondary to maternal diabetes (49), smoking (3), pulmonary insufficiency or placental malformation (36) is a frequent etiological factor in many complicated pregnancies (25). These hypoxic insults result in numerous perinatal and postnatal morbidities (30) that often include altered fetal cardiovascular function secondary to atypical patterns of vascular structure and contractility, collectively known as remodeling (16, 32). Whereas the exact molecular mechanisms governing hypoxic fetal vascular remodeling remain poorly understood, considerable evidence in adult vasculature demonstrates that chronic hypoxia orchestrates arterial wall thickening (17, 39), together with changes in contractile protein abundance (48) and organization (55). The functional consequences of these changes include altered arterial contractility (8, 51) due at least in part to modified reactivity of thick- and thin-filament contractile proteins (35).
Efforts to understand the mechanisms that drive hypoxic vascular remodeling have focused largely on the vasotrophic factors released by hypoxia, the most prominent of which is hypoxia-inducible factor (HIF) (46, 56). Upregulation, stabilization, and dimerization of HIF mediate transcription of multiple angiogenic genes, including vascular endothelial growth factor (VEGF; Fig. 1), which in turn is known traditionally to mediate capillary angiogenesis (18). In further support of angiogenesis, some studies have also suggested that hypoxia can modulate levels of VEGF receptors 1 and 2 (Flt-1 and Flk-1) in the vascular endothelium, as well as in certain tumors of rodent brain (43, 50) (Fig. 1). Whereas abundant previous work suggests that VEGF acts mainly on the vascular endothelium (18), recent findings further suggest that VEGF can exert potent trophic effects on multiple nonendothelial cell types (24), suggesting possible expression of VEGF receptors on such cell types. Of particular relevance to vascular remodeling is growing evidence that VEGF has potent trophic effects on smooth muscle cells (12) that result in alteration of contractile protein expression and organization (7).
Fig. 1.
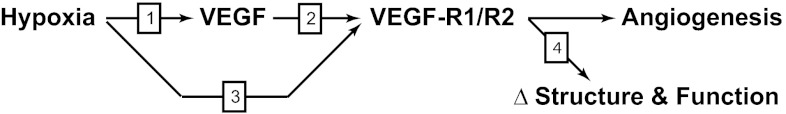
Summary of our approach to test the hypothesis that VEGF contributes to hypoxic vascular remodeling through changes in abundance, organization, and function of contractile proteins in fetal arteries. First, we propose that hypoxia induces short-term increases in VEGF (arrow 1) through upregulation of the transcription factor hypoxia-inducible factor. We further propose that these increases in VEGF act on VEGF receptors (VEGF R1/R2) expressed by smooth muscle cells (arrow 2). In addition, we propose that chronic hypoxia increases expression of smooth muscle VEGF receptors (arrow 3). Finally, we propose that activation of smooth muscle VEGF receptors leads to changes in contractile protein abundance and organization that result in changes in arterial structure and function (arrow 4). In this manner, we propose that hypoxic increases in VEGF mediate not only microcirculatory angiogenesis, but also arterial remodeling. Separate experiments were performed to test each of the numbered arrows in fetal arteries.
The current study explores the hypothesis that VEGF contributes to hypoxic fetal vascular remodeling through changes in abundance, organization, and function of contractile proteins. Through increases in VEGF and/or its receptors in smooth muscle, we propose that hypoxia could recruit VEGF to help mediate changes in ovine fetal vascular structure and function that are characteristic of chronic hypoxia. Given the central importance of the rate-limiting enzyme myosin light chain (MLC) kinase (MLCK) (41), its substrate 20-kDa regulatory MLC (MLC20) (20), and their contractile partner smooth muscle α-actin (SMαA) (15), our experimental design focused on mechanisms involved in hypoxia-induced changes in these contractile proteins. Comparisons between fetal arteries harvested from sheep maintained at sea level and from sheep maintained at high altitude (3,820 m) for 110 days served to define effects of chronic hypoxia, as previously described in detail (32). To assess the role of VEGF and its receptors in hypoxic remodeling (Fig. 1), we used organ cultures of whole carotid arteries, which preserved the spatial organization of all cell types in the arterial wall (24). To explain changes in smooth muscle phenotype orchestrated by hypoxia, which ultimately dictate contractile protein abundance and organization, we assessed contractile protein abundance via Western blot analysis and protein organization via a novel confocal colocalization imaging technique (9). Stress-strain measurements of myogenic reactivity normalized to arterial wall cross-sectional area (34) identified the functional consequences of arterial remodeling induced by hypoxia. Together, these studies provide a unique perspective of the direct contribution of VEGF and its receptors to hypoxic vascular remodeling in fetal ovine carotid arteries.
MATERIALS AND METHODS
All techniques, protocols, and procedures were approved by the Animal Research Committee of Loma Linda University and complied with all policies and codes of practice outlined in the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Procedures related to tissue harvesting, tissue preparation, and animal surgery are described in detail elsewhere (54).
Tissue harvest and preparation.
All experiments used common carotid arteries harvested from full-term (139–142 days gestation) fetal lambs delivered by cesarean section using strict sterile techniques. Pregnant ewes were anesthetized with pentobarbital sodium (30 mg/kg), intubated, and maintained on 1.5–2.0% halothane. The fetus was subsequently exteriorized via a midline vertical incision and killed by exsanguination via rapid removal of the heart. Normoxic arteries (FN) were harvested from animals maintained at sea level, whereas hypoxic arteries (FH) were harvested from animals acclimatized at altitude (3,820 m above sea level) for the final 110 days of gestation, as described in detail elsewhere (26, 32). This chronic hypoxia model yields 19 ± 1 Torr arterial Po2 in fetal sheep (26). Corresponding normoxic arterial Po2 averages 23 ± 1 Torr. Harvested common carotid arteries were kept in sterile HEPES buffer (122.1 mM NaCl, 25 mM HEPES, 5.16 mM KCl, 2.4 mM MgSO4, 11.1 mM dextrose, 1.6 mM CaCl2, and 50 μM EDTA). After thorough and careful removal of the loose connective tissues and blood, the arteries were denuded of endothelium via mechanical abrasion. Next, 3-mm lengths of artery were cut and distributed to the various experimental protocols. Medial thickness and exact arterial segment length were measured using an Olympus U-PMTVC optical microscope mounted with a Scion Visicapture Twain 1394 camera for image capture with ImagePro software (version 6.0, Media Cybernetics).
Contractility studies.
The 3-mm arterial segments were mounted on tungsten wires between isometric force transducers and micrometers used for precise positioning. Each artery was equilibrated for 1 h in Ca2+-replete Na+-Krebs buffer (pH 7.4) containing (in mM) 122 NaCl, 25.6 NaHCO3, 5.17 KCl, 2.56 dextrose, 2.49 MgSO4, 1.60 CaCl2, 0.114 ascorbic acid, and 0.027 EGTA. Artery viability was preserved by diffusional O2 supply enabled by continuous bubbling with 95% O2-5% CO2 at normal ovine core temperature (38°C) (40). Unstressed artery diameters were measured immediately following equilibration at a passive tension of 0.03 g. Relative to unstressed diameters (D0), working diameters (D) required to attain arterial strain ratios (D/D0) of 1.5, 1.8, 2.1, 2.3, 2.7, 3.0, and 3.3 were calculated. Under resting conditions and after addition of high-K+ buffer solutions, contractile stresses (dyn/cm2) were measured at each of the strain ratios to determine spontaneous myogenic tone and K+-induced active tone, respectively. The constituents of the high-K+ Krebs solution were (in mM) 122 KCl, 11.1 dextrose, 5.16 NaCl, 2.50 MgSO4, 2.15 NaHCO3, 1.60 CaCl2, 0.114 ascorbic acid, and 0.027 EDTA. Contractile responses to the high-K+ buffer were recorded until the arteries were stabilized; then they were returned to basal conditions using Na+-Krebs buffer and equilibrated at the next stretch ratio. After responses were recorded at the highest strain ratios, the arterial segments were frozen in liquid nitrogen to eliminate any active component of stress (7, 40). The segments were subsequently incubated in Ca2+-depleted Na+-Krebs solution containing 3 mM EGTA. Passive stresses produced at each strain ratio from the highest to the lowest were then recorded.
Differences between spontaneous tone measured before and after freezing with liquid nitrogen and EGTA at each level of strain were taken as spontaneous myogenic tone. Stiffness coefficients were determined by assessing the relations between strain ratios and passive stresses using nonlinear regression fits to a monotonic exponential model (Young's modulus) (13).
Fluorescence immunohistochemistry.
Segments of common carotid arteries were fixed overnight in 4% neutral buffered electron microscopy-grade formaldehyde (catalog no. 15713S, Electron Microscopy Sciences, Hatfield, PA), embedded in paraffin, and sectioned at 5 μm. Slides were deparaffinized in Histoclear solution (catalog no. HS-200, National Diagnostic, Atlanta, GA) and rehydrated in descending concentrations of alcohol. The samples were microwaved in citrate buffer (pH 6.03) to recover antigenicity; then the sections were permeabilized in 0.1% Triton X-100 (catalog no. T-8787, Sigma Aldrich, St. Louis, MO) and subsequently incubated in 1% bovine serum albumin (catalog no. SC-2323, Santa Cruz Biotechnology, Santa Cruz, CA) to block nonspecific binding. Incubations with primary antibodies were carried out overnight at 4°C. Antibody selectivity was confirmed with Western blots, and the titers used for immunohistochemistry were the lowest that optimized signal-to-noise ratio. The primary antibodies included monoclonal anti-SMαA (1:200 dilution; catalog no. A5228, Sigma Aldrich), polyclonal anti-MLCK (1:50 dilution; catalog no. SC-25428, Santa Cruz Biotechnology), and monoclonal anti-MLC20 (1:300 dilution; catalog no. M4401, Sigma-Aldrich). The slides were subjected to two 10-min wash cycles in PBS on the morning of the next day before the secondary antibody (DyLight 488-conjugated; catalog no. 35502, Pierce Chemical, Rockford, IL) was applied to the tissues on the slides for 2 h at room temperature. The slides were kept in darkness after incubation to preserve photosensitivity. The slides were subsequently covered and subjected to two 10-min wash cycles in PBS. SlowFade Gold antifade reagent with 4′,6-diaminido-2-phenylindole was added, coverslips were mounted on the slides, and the slides were stored in darkness until they were imaged. Tissue slides were imaged using a Zeiss Imager A1 on an AX10 fluorescence microscope with Spot software (version 4.6.4.5, Diagnostic Instruments).
Confocal microscopy.
Artery sections (5 μm thick) were double-stained with antibodies against MLCK and SMαA (see Fluorescence immunohistochemistry). After primary antibody incubation, the sections were washed in PBS and equilibrated in a dark room with two secondary antibodies labeled with DyLight 488 (SMαA) and DyLight 649 (for MLCK) for 2 h at room temperature. The sections were imaged with an Olympus FV1000 microscope at optical section thickness of 700 nm, lateral resolution of 200 nm, and numerical aperture of 18. The extent of colocalization between the two markers was determined using a custom-written nonparametric quadrant analysis that calculated the fraction of total pixels in the upper half of the intensity range for both markers relative to the number of pixels in the upper half of the intensity range for SMαA. We refer to this index as the percentage of pixels in the upper right quadrant (%UR). This method of quadrant analysis was adapted from flow-cytometry theory (1, 2) and is described elsewhere (9, 21). Sections (5 μm) on slides were also double-stained for MLC20 with SMαA and for MLCK with MLC20. Primary antibodies were monoclonal anti-MLC20 (1:300 dilution; catalog no. M4401, Sigma Aldrich) for MLC20, monoclonal anti-SMαA (1:200 dilution; catalog no. A5228, Sigma Aldrich) for SMαA, and polyclonal MYLK (1:50 dilution; catalog no. SC-25428, Santa Cruz Biotechnology) for MLCK. Secondary antibodies for MLC20-SMαA colocalization (DyLight 488 for SMαA and DyLight 633 for MLC20) and MLCK-MLC20 colocalization (DyLight 488 for MLC20 and DyLight 633 for MLCK, conjugated; catalog no. 35502, Pierce Chemical, Rockford, IL) were applied to the tissues; then the sections were imaged as described in Fluorescence immunohistochemistry.
Western blots.
Known weights of frozen arterial segments were processed using a glass-on-glass homogenizer in 8 M urea containing 500 mM NaCl, 20 mM Tris, 23 mM glycine, 10 mM EGTA, and 10% glycerol at pH 8.6 with protease inhibitor cocktail at 5 μl/ml buffer (catalog no. M1745, Sigma Aldrich). Centrifugation of the homogenates at 5,000 g for 20 min yielded supernatants, the protein concentrations of which were determined by the Bio-Rad Bradford assay. Protein homogenates were separated via SDS-PAGE, together with increasing concentrations of standards used to calibrate target protein abundance. The tissues for the standards were harvested from pooled adult ovine common carotid arteries. SDS-PAGE-separated proteins were transferred onto nitrocellulose membranes at 200 mA for 90 min in Towbin's buffer (25 mM Tris, 192 mM glycine, and 10% and 20% methanol) on ice. Continuous gentle shaking for 1 h at room temperature with 5% milk in Tris-buffered saline (TBS) at pH 7.5 (M-TBS) was used to block the membranes. After the initial blocking, subsequent washes and incubations were done in M-TBS containing 0.1% Tween 20. Incubations with primary antibody were performed for 3 h using the following dilutions: 1:3,000 for SMαA, 1:10,000 for MLCK, 1:200 for MLC20, and 1:750 for VEGF-A-165. All antibodies were obtained from the sources noted in Fluorescence immunohistochemistry, except anti-VEGF antibody, which was purchased from Abcam (catalog no. AB119). Visualization was performed after a 90-min secondary incubation using a secondary antibody conjugated to DyLight 800 (catalog no. 46422, Pierce Chemical). Imaging was completed on a LI-COR Bioscience Odyssey system.
For the VEGF receptor Western blot assay, artery segments were homogenized using a glass pestle and mortar containing buffer with (in mM) 500 NaCl, 50 Tris, and 5 EDTA at pH 7.4 with protease inhibitors, including (in μM) 500 2-(4-aminoethyl)benzenesulfonyl fluoride, 400 pepstatin-A, 20 bestatin, 10 E-64, 7.5 leupeptin, and 7 aprotinin (Sigma Aldrich) at 1:50 tissue-to-buffer ratio. Homogenates were centrifuged at 100,000 g for 1 h at 4°C. Pellets were resuspended at a 1:10 tissue-to-buffer ratio with 150 mM NaCl, 50 mM Tris, 10 mM DTT, 1% Triton X-100, 0.5% sodium deoxycholate, 0.2% SDS, and 10% glycerol with protease inhibitor cocktail as described above for 1 h with gentle shaking at room temperature. Samples were ultrasonicated six times for 5 s each at 20% amplitude to shear DNA and centrifuged at 10,000 g for 15 min, and the supernatants were collected. Protein concentrations were determined using Bradford's protein assay. Protein homogenates were separated on 5% SDS-PAGE with addition of 35 mM β-mercaptoethanol in the upper buffer reservoir, along with a pooled reference to normalize samples. Separated proteins were transferred onto 0.2-μm nitrocellulose (catalog no. BA83, Whatman) using 350 mA for 90 min in Towbin's buffer containing 25 mM Tris, 35 mM β-mercaptoethanol, 192 mM glycine, 0.01% SDS, and 20% methanol. After transfer, the membranes were blocked in M-TBS at pH 7.5 for 1 h at room temperature with continuous shaking. All subsequent washes and incubations were performed in M-TBS + 0.1% Tween 20. Primary antibodies were incubated for 3 h with a 1:200 dilution of Fms-like tyrosine kinase receptor (Flt-1; catalog no. SC-316, Santa Cruz Biotechnology) and a 1:200 dilution of fetal liver kinase 1 (Flk-1; catalog no. SC-504, Santa Cruz Biotechnology). Membranes were washed six times for 5 min each and then incubated for 90 min in secondary antibody conjugated to DyLight 800. Membranes were washed six times for 5 min each and then washed in TBS only. Membranes were imaged on a LI-COR Bioscience Odyssey system.
Organ culture.
As previously described (7), serial segments of individual arteries from both experimental groups (FN and FH) were maintained in DMEM fortified with 3.7 g/l Na2HCO3, 0.5% amino acid solution (catalog no. M5550, Sigma Aldrich), 1% nonessential amino acid solution (catalog no. M7145, Sigma Aldrich), 4 mM glutamine (catalog no. G7513, Sigma Aldrich), 2% antibiotic-antimycotic solution (catalog no. 15240-096, GIBCO, Carlsbad, CA), and 70 μg/ml gentamicin (catalog no. 15750-060, GIBCO) in an untreated 12-well plate and subsequently maintained in a humidified incubator with 5% CO2 in room air at 37°C. Matched sets of artery segments left in DMEM for 24 h served as control arteries, while those treated with a low dose (3 ng/ml) of VEGF were used to assess the effects of VEGF. Segments treated with vatalanib (240 nM) and dasatinib (6.3 nM) (42) were used to assess the effects of VEGF tyrosine kinase receptors; the concentrations were identified as the lowest effective concentrations in preliminary dose-finding experiments. In all organ culture experiments, the artery segments were first maintained in media without growth factors (DMEM) for 24 h. Control arteries remained in DMEM for another 24 h, and treated arteries were exposed to 3 ng/ml VEGF for 24 h. This low dose of VEGF was used to minimize nonspecific binding of VEGF to other non-VEGF receptors and represented physiological serum levels measured in gravid sheep (52).
Data analysis and statistics.
Contractile stresses generated from applied graded strains were calculated as force per cross-sectional area, where force = tension (g) × acceleration due to gravity and cross-sectional area = working wall thickness (μm) × segment length. Each animal contributed to the fresh, control, VEGF, and VEGF + vatalanib + dasatinib groups. Contractile stress, thickness, and stiffness values were compared using ANOVA. All data sets were confirmed to be normally distributed using the D'Agostino-Pearson analysis, and homogeneity of variance within ANOVA was verified using the Bartlett-Cochran test, as previously described (9). Statistical power was routinely ≥0.8.
All Western immunoblot measurements were calibrated against a standard curve generated from a pooled reference of common carotid tissues. Regional abundance values were compared using a two-way ANOVA with age and treatment as variables. Duncan's multiple range analysis was used to make post hoc comparisons between means within the ANOVA.
RESULTS
The study is based on a total of 124 segments from 17 normoxic fetal lambs and 93 segments from 12 hypoxic fetal lambs. In all cases, n indicates the number of animals used. Statistical significance implies P < 0.05. Values are means ± SE.
Effects of chronic hypoxia on carotid artery structure and stress-strain relations.
Medial thickness was significantly less in FN than FH arteries (341 ± 20 vs. 400 ± 28 μm; Fig. 2); chronic hypoxia significantly increased medial thickness by 17%. Similarly, arterial stiffness was significantly less in FN than FH arteries (8.02 ± 0.42 vs. 9.20 ± 0.69; Fig. 2); hypoxic increases in stiffness averaged 15%. Peak myogenic tone averaged 102.5 ± 21.8 dyn/cm2 in FN arteries but only 25.9 ± 6.3 dyn/cm2 in FH arteries. On average, chronic hypoxia significantly decreased peak myogenic tone by 75%.
Fig. 2.

Hypoxia remodels ovine carotid artery structure and function. Compared with normoxic arteries, arteries from chronically hypoxic animals exhibited an increased thickness (left) and stiffness (middle) of the medial layer. Determination of active stress-strain relations revealed that chronic hypoxia also significantly depressed myogenic tone but did not alter strain values at which contractile force was maximal (right). Values are means ± SE for arteries from normoxic (n = 17) and hypoxic (n = 12) fetuses. *P < 0.05.
Effects of chronic hypoxia on contractile protein abundance and colocalization.
The effects of chronic hypoxia on contractile protein abundances were highly protein-specific (Fig. 3). For SMαA, Western blot quantification revealed similar abundances (relative to standard) in FN (0.62 ± 0.07) and FH (0.68 ± 0.08) arteries. For MLCK, abundances were 90% less in FH than FN arteries. In contrast, the abundances of MLC20 were 61% greater in FH than FN arteries.
Fig. 3.
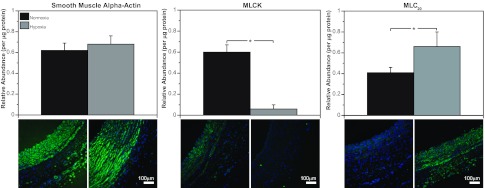
Chronic hypoxia alters smooth muscle contractile protein abundances. Western blot quantification of smooth muscle α-actin (SMαA) abundance yielded similar values in normoxic and hypoxic fetal carotid arteries (left). Myosin light chain (MLC) kinase (MLCK) abundance was markedly less in hypoxic than normoxic arteries (middle). Abundance of 20-kDa regulatory MLC (MLC20) was significantly greater in hypoxic than normoxic arteries (right). These results demonstrate that effects of chronic hypoxia on smooth muscle contractile protein abundances are highly protein-specific. Values are means ± SE; n ≥ 5 in all experimental groups. *P < 0.05 by ANOVA.
Chronic hypoxia significantly increased colocalization among all three pairs of contractile proteins (Fig. 4). For MLCK-SMαA, chronic hypoxia increased the coefficient of colocalization 42% relative to the normoxic group. Corresponding increases for MLC20-SMαA and MLCK-MLC20 colocalization averaged 123% and 237%, respectively.
Fig. 4.
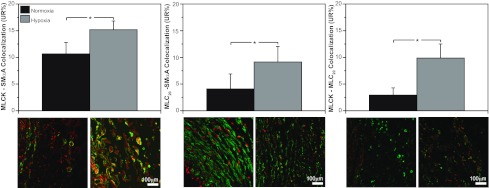
Chronic hypoxia increases colocalization among smooth muscle contractile proteins. As revealed by confocal microscopy, MLCK-SMαA colocalization was 42% greater in hypoxic than normoxic fetal arteries. Similarly, MLC20-SMαA colocalization was 42% greater and MLCK-MLC20 colocalization was 123% greater in hypoxic than normoxic fetal arteries. All these differences were statistically significant and suggest that the contractile proteins were becoming more compact and highly organized in response to chronic hypoxia. UR%, percentage of pixels in upper right quadrant. Values are means ± SE; n ≥ 5 in all experimental groups. *P < 0.05 by ANOVA.
Effects of long-term hypoxia on abundances of VEGF and VEGF receptors.
Western blot quantification of endogenous VEGF levels in FN (0.29 ± 0.07) and FH arteries (0.25 ± 0.06) revealed similar VEGF abundances (Fig. 5), suggesting that hypoxia had little effect on VEGF levels after 110 days of hypoxic acclimatization. In contrast, the abundances of the endogenous VEGF receptors 1 (Flt-1) and 2 (Flk-1) were 786% and 171% greater in FH than FN arteries (Fig. 6).
Fig. 5.

Effects of long-term hypoxia on VEGF abundance. Endogenous VEGF levels quantified via Western blot analysis exhibited similar abundances in normoxic and hypoxic fetal sheep. These results suggest that the well-documented increases in VEGF induced on exposure to hypoxia are transient and disappear after 110 days of hypoxic acclimatization. Values are means ± SE for normoxic (n = 7) and hypoxic (n = 5) fetal arteries.
Fig. 6.

Effects of long-term hypoxia on VEGF receptor expression. In homogenized endothelium-denuded fetal arteries, abundances of VEGF receptor 1 (Flt-1) and VEGF receptor 2 (Flk-1) were significantly increased by chronic hypoxia. Multiple bands on Western blots indicate different glycosylation states of the receptors. Values represent total of all glycosylation states for Flk-1. Because of the absence of the 230-kDa form of Flt-1 in hypoxic arteries, abundances of the 250-kDa form are compared. Standards on Western blots were prepared from mixed samples of normoxic adult arteries. Values are means ± SE; n = 6 in all experimental groups. *P < 0.05 by ANOVA.
VEGF receptors mediate VEGF-induced changes in contractile protein organization.
To confirm that VEGF altered contractile protein organization through a VEGF receptor-dependent mechanism, MLC20-SMαA colocalization was used as a reporter assay (Fig. 7). In organ culture, a low physiological concentration (3 ng/ml) of VEGF increased MLC20-SMαA colocalization by 237%. This effect of VEGF was blocked completely in the presence of the VEGF receptor blockers vatalanib (240 nM) and dasatinib (6.3 nM).
Fig. 7.

VEGF activation of its tyrosine kinase receptors mediates increased contractile protein colocalization. Organ culture of endothelium-denuded normoxic fetal arteries with VEGF (3 ng/ml) significantly increased MLC20-SMαA colocalization. Addition of the VEGF receptor antagonists vatalanib (240 nM) and dasatinib (6.3 nM) completely blocked this effect of VEGF. Values are means ± SE; n = 5 in all experimental groups. *P < 0.05 by ANOVA.
Effects of VEGF on contractile protein abundance and colocalization.
In organ culture, treatment with 3 ng/ml VEGF had no significant effect on SMαA abundance in FN (0.59 ± 0.05 vs. 0.64 ± 0.07) or FH (0.66 ± 0.07 vs. 0.64 ± 0.09) arteries (Fig. 8). However, VEGF significantly increased MLCK abundance by 100% in FN arteries but decreased MLCK abundance by 33% in FH arteries; after VEGF treatment, MLCK abundance was significantly greater in FN than FH arteries. For MLC20, VEGF had no individually significant effect in FN (0.23 ± 0.04 vs. 0.27 ± 0.05) or FH (0.38 ± 0.03 vs.0.44 ± 0.08) arteries, but after treatment with VEGF, MLC20 abundance was significantly less in FN than FH arteries. As indicated by comparison of Fig. 3 with Fig. 8, chronic hypoxia and VEGF had highly similar effects on the abundance of all three contractile proteins.
Fig. 8.
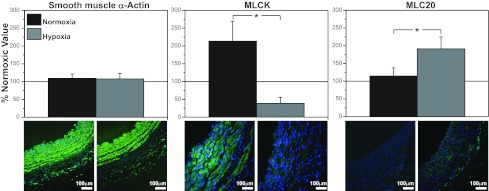
Effects of VEGF on contractile protein abundances are highly protein-specific. As revealed by Western blot quantification, organ culture with 3 ng/ml VEGF had no significant effect on SMαA abundance in endothelium-denuded arteries from normoxic or hypoxic fetuses. In contrast, organ culture with 3 ng/ml VEGF significantly increased MLCK abundance in endothelium-denuded arteries from normoxic fetuses but decreased it in arteries from hypoxic fetuses. Conversely, organ culture with 3 ng/ml VEGF had no significant effect on MLC20 abundance in arteries from normoxic fetuses but significantly increased MLC20 in arteries from hypoxic fetuses. This pattern of effects emphasizes that effects of VEGF on contractile protein abundance are highly protein-specific, markedly influenced by hypoxic acclimatization, and closely similar to effects of chronic hypoxia (Fig. 3). Values are means ± SE; n = 5 in all experimental groups. *P < 0.05 by ANOVA.
The effects of VEGF and chronic hypoxia on patterns of contractile protein colocalization were also highly similar. In parallel with the effects of chronic hypoxia (Fig. 4), organ culture with 3 ng/ml VEGF increased MLCK-SMαA colocalization by 55%, increased MLC20-SMαA colocalization by 237%, and increased MLCK-MLC20 colocalization by 75% (Fig. 9). All these effects of VEGF on contractile protein colocalization were statistically significant.
Fig. 9.
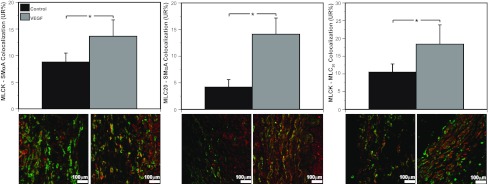
VEGF increases colocalization among smooth muscle contractile proteins. In endothelium-denuded arteries from normoxic fetuses, organ culture with 3 ng/ml VEGF increased MLCK-SMαA colocalization by 55%, MLC20-SMαA colocalization by 237%, and MLCK-MLC20 colocalization by 75% compared with corresponding untreated controls. These results demonstrate that VEGF can act directly on arterial smooth muscle to enhance contractile protein colocalization. This pattern of effects was also closely similar to effects of chronic hypoxia on contractile protein colocalization (Fig. 4). Values are means ± SE; n ≥ 5 in all experimental groups. *P < 0.05 by ANOVA.
DISCUSSION
This study offers three original findings suggesting a nonangiogenic role of VEGF in hypoxic remodeling of fetal ovine carotid arteries. 1) In homogenates of endothelium-denuded fetal carotid arteries, 110 days of hypoxic acclimatization had no effect on VEGF levels but significantly increased abundances of both main VEGF receptors (Flt-1 and Flk-1) relative to normoxic controls. 2) Chronic hypoxia and organ culture with VEGF were without effect on SMαA abundance, decreased MLCK abundance, and increased MLC20 abundance in endothelium-denuded fetal carotid arteries. 3) Chronic hypoxia and organ culture with VEGF increased MLCK-SMαA, MLC20-SMαA, and MLCK-MLC20 colocalization. Together, these findings support the general hypothesis that VEGF contributes to hypoxic fetal vascular remodeling through changes in the abundance, organization, and function of contractile proteins.
Effect of hypoxia on structure and contractility.
Previous studies of hypoxic vascular remodeling reported hypoxic increases in total arterial wall thickness involving the medial and adventitial layers (17, 37, 47). In the present study, chronic hypoxia increased medial thickness (Fig. 2), reinforcing the view that hypoxia promotes expansion of arterial smooth muscle (11, 47), an important determinant of overall arterial stiffness. Correspondingly, hypoxia also increased arterial stiffness, suggesting altered smooth muscle composition and structure (31) and, possibly, increased collagen cross-linking and collagen-to-elastin ratios (53). The associated hypoxic decreases in contractile stress, measured in dynes per square centimeter (Fig. 2), suggest that hypoxia increased the ratio of noncontractile to contractile proteins in the artery wall in these fetal ovine carotid arteries (7, 17). Such changes were probably mediated by a unique, but unknown, combination of numerous possible mechanisms (31).
Consistent with previous studies (17), hypoxic structural changes in the fetal arteries were associated with decreased myogenic reactivity (Fig. 2). Hypoxic depression of myogenic tone must involve changes in regulation of cytosolic Ca2+ or myofilament Ca2+ sensitivity (34, 41). Whereas effects of chronic hypoxia on Ca2+ signaling in nonpulmonary smooth muscle remain largely unreported, evidence from our group suggests that chronic hypoxia significantly alters myofilament Ca2+ sensitivity in ovine cranial arteries (35). Correspondingly, the present study explored the hypothesis that hypoxia may alter myofilament Ca2+ sensitivity and contractility through changes in contractile protein abundance and organization.
Effects of hypoxia on contractile protein abundances.
The most abundant contractile protein in smooth muscle is SMαA, a ∼42-kDa monomer that polymerizes to form the thin filaments essential for contraction (38). Expression of SMαA did not differ significantly from normoxic basal levels after 110 days of hypoxic acclimatization (Fig. 3, left), suggesting that the signaling pathways that govern expression of this contractile protein were not altered by hypoxia.
Expression of MLCK, another contractile protein, varies with smooth muscle phenotype (4, 7, 22); MLCK is the rate-limiting enzyme that phosphorylates and activates regulatory MLC20 (19). Hypoxia potently decreased MLCK (Fig. 2, middle), possibly due to phenotypic transformation or an effect at the level of transcription, translation, or turnover. For transcription, hypoxia might act through HIF-1α to activate a hypoxia response element (HRE) that decreases MLCK gene transcription. No evidence for a repressor element in the MLCK gene has been published, although Qi et al. (44) reported that HIF can upregulate MLCK expression in cultured pulmonary vein endothelial cells, suggesting the presence of a HRE in the MLCK promoter. Alternatively, hypoxia might enhance MLCK degradation; MLCK abundance dropped precipitously in arteries organ-cultured under serum-starved conditions (7). This drop in MLCK would require rapid degradation, which might be stimulated by hypoxia (28). Hypoxia might also drive phenotypic transformation toward a less contractile, more synthetic, phenotype that expresses less MLCK (19). Hypoxia's ability to promote such transformation has been demonstrated in cultured rat pulmonary artery smooth muscle (23, 57) but not previously in whole arteries adapted to chronic hypoxia.
In sharp contrast to the pattern of change observed for MLCK, MLC20 abundances significantly increased in hypoxic compared with normoxic arteries (Fig. 3, right). Although hypoxic increases in MLC20 abundance could result from increased transcription due to a HRE in the promoter for MLC20, literature searches yielded no evidence of a HRE in the promoters for MLC20 or myosin heavy chain, with which MLC20 should be coexpressed (14). Hypoxic increases in MLC20 abundance also might result from increases in translation efficiency, but available evidence suggests only inhibition of translation by hypoxia (33). Hypoxic increases in MLC20 might be explained by phenotypic transformation toward a synthetic phenotype, which is characteristic of hypoxia (21). Such a transformation would increase MLC20 as a component of increased nonmuscle myosin heavy chain expression, which is induced by chronic hypoxia in fetal carotid arteries (21). In light of evidence that hypoxia can increase protein degradation (45), this mechanism might also contribute to hypoxic increases in MLC20 abundance. Equally important, the diverse effects of hypoxia on SMαA, MLCK, and MLC20 abundances emphasize that the effects of hypoxia on contractile protein expression are unique and highly specific for each protein.
Effects of hypoxia on contractile protein organization.
Vascular contractility is determined not only by contractile protein abundances, but also by protein organization within smooth muscle cells (6). Chronic hypoxia increased colocalization of MLCK with SMαA (Fig. 4, left), suggesting that hypoxia depressed release of an inhibitory factor or enhanced release of a stimulatory factor from the endothelium or adventitia to enhance MLCK-SMαA colocalization. Although the molecular mechanisms responsible remain unknown, this result is consistent with evidence that MLCK binds strongly with SMαA (29) through mechanisms that are physiologically regulated (6).
As for MLCK-SMαA colocalization, hypoxia also enhanced MLC20-SMαA (Fig. 4, middle) and MLCK-MLC20 (Fig. 4, right) colocalization, demonstrating that hypoxia orchestrated processes that enhanced interaction among multiple contractile proteins. Importantly, this pattern of increased colocalization occurred concurrently with markedly decreased MLCK abundance (Fig. 3), which raises the possibility that increased colocalization may help preserve hypoxic contractility (Fig. 2). Together, these results emphasize that hypoxia exerts coordinated, but separate, influences on contractile protein abundance, organization, and function through mechanisms that remain largely unidentified.
Effects of chronic hypoxia on VEGF and VEGF receptors.
To better understand the mechanisms that mediate the effects of hypoxia on contractile protein abundance, organization, and function, we studied established mediators of hypoxic effects. The most prominent of these is HIF (46, 56), which is well known to increase capillary angiogenesis through induction of VEGF (18). In light of recent evidence from our laboratory that VEGF can act directly on arterial smooth muscle to alter phenotype, contractile protein expression, and contractility (7, 21), we formulated the hypothesis that VEGF contributes to hypoxic fetal vascular remodeling through changes in the abundance, organization, and function of contractile proteins (Fig. 1). Our first test of this hypothesis (Fig. 1, arrow 1) was to examine the effects of 110 days of hypoxia on VEGF levels in endothelium-denuded fetal carotid homogenates. VEGF levels were unchanged by chronic hypoxia (Fig. 5), in agreement with previous reports that VEGF rises and then returns to baseline within 21 days of chronic hypoxia (10). In addition, this finding also suggested that elevated VEGF could not explain the sustained changes in contractile protein abundance and colocalization observed in hypoxic arteries.
In light of evidence that hypoxia can modulate levels of VEGF receptors in the vascular endothelium and certain tumors of rodent brain (43, 50), we also tested the possibility that chronic hypoxia increased VEGF receptor levels in fetal arterial smooth muscle (Fig. 1, arrow 3). These measurements yielded the novel finding that both main VEGF receptors (Flk-1 and Flt-1) were expressed in fetal ovine carotid arteries, as suggested by previous measurements of VEGF receptor mRNA (7). More importantly, these measurements revealed that chronic hypoxia significantly enhanced VEGF receptor abundances (Fig. 6). Together with our VEGF measurements (Fig. 5), these findings support the novel view that hypoxia promotes VEGF-mediated effects in the short term through increases in VEGF abundance (10) but in the long term through increases in the abundances of VEGF receptors.
Effects of VEGF on contractile protein abundance and organization.
To confirm that our previously reported effects of VEGF on contractile proteins in fetal lamb carotid arteries (7, 21) were mediated by action on smooth muscle VEGF receptors (Fig. 1, arrow 2), we examined the effects of VEGF on MLC20-SMαA colocalization in the presence and absence of vatalanib and dasatinib, two well-characterized VEGF receptor antagonists (27, 42). Organ culture of endothelium-denuded fetal carotid arteries with a low physiological concentration (3 ng/ml) of VEGF (7, 52) significantly increased MLC20-SMαA colocalization (Fig. 7). At concentrations found to be optimal in previous dose-finding experiments (21), 240 nM vatalanib with 6.3 nM dasatinib in coculture with VEGF completely blocked the effects of VEGF on MLC20-SMαA colocalization. These results thus verified that VEGF can act through VEGF receptors to alter contractile protein organization in fetal carotid arteries.
As a further test of the hypothesis that VEGF contributes to hypoxic vascular remodeling, our next series of experiments examined the effects of VEGF on contractile protein abundances (Fig. 1, arrow 4). Organ culture with a physiological concentration (3 ng/ml) of VEGF (52) had no effect on SMαA abundance, significantly depressed MLCK abundance, and significantly increased MLC20 abundance in hypoxic relative to normoxic arteries (Fig. 8). Most importantly, this pattern of effects was closely similar to the effects of chronic hypoxia on the same contractile proteins in fresh arteries. When we examined the effects of organ culture with VEGF on contractile protein colocalization, VEGF increased MLCK-SMαA, MLC20-SMαA, and MLCK-MLC20 colocalization. Again this pattern of effects was quite similar to the effects of chronic hypoxia on contractile protein colocalization. Together, these abundance and colocalization measurements support the hypothesis that VEGF contributes to hypoxic fetal vascular remodeling through changes in the abundance, organization, and function of contractile proteins.
Overview.
The present study explores the mechanisms that couple chronic hypoxia to changes in the structure and function of large systemic fetal arteries (Fig. 1). Consistent with numerous previous studies (21, 32), our findings demonstrate that chronic hypoxia alters fetal artery structure and contractility (Fig. 2). A diverse and abundant literature also supports the idea that hypoxia potently increases VEGF in the short term (10, 46). The present study expands this concept by demonstrating that, in the long term, chronic hypoxia has little effect on VEGF (Fig. 5) but potently increases the VEGF receptor abundances (Fig. 6). Our results also augment previous findings that VEGF can modulate the expression, organization, and function of smooth muscle contractile proteins (7, 21) by demonstrating that these effects are dependent on activation of smooth muscle VEGF receptors (Fig. 7). Finally, the present study demonstrates that the qualitative effects of chronic hypoxia on the abundance (Fig. 3) and colocalization (Fig. 4) of SMαA, MLCK, and MLC20 are closely similar to the corresponding effects of VEGF (Figs. 8 and 9). Together with the other evidence obtained, the findings as a whole are highly consistent with the general hypothesis that VEGF contributes to hypoxic fetal vascular remodeling through changes in the abundance, organization, and function of contractile proteins. Without doubt, VEGF is not the only factor contributing to hypoxic vascular remodeling (5), and in vivo studies that involve selective interruption of VEGF signaling in large systemic arteries are required to quantitatively assess the role of VEGF in hypoxic vascular remodeling. The present findings suggest that such future studies are warranted and advance the idea that hypoxic increases in VEGF act simultaneously in the microcirculation to increase capillary density, while also acting in the large upstream arteries to alter the abundance, organization, and function of contractile proteins.
GRANTS
This work was supported by National Institutes of Health Grants HL-54120, HD-31266, HL-64867, and NS-076945 and the Loma Linda University School of Medicine.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
O.O.A., S.M.B., and J.M.W. performed the experiments; O.O.A., M.C.H., A.J.S., J.M.W., and W.J.P. analyzed the data; O.O.A., J.M.W., and W.J.P. interpreted the results of the experiments; O.O.A. prepared the figures; O.O.A. drafted the manuscript; O.O.A. and W.J.P. edited and revised the manuscript; S.M.B. and W.J.P. are responsible for conception and design of the research; W.J.P. approved the final version of the manuscript.
REFERENCES
- 1.Agnati LF, Fuxe K, Torvinen M, Genedani S, Franco R, Watson S, Nussdorfer GG, Leo G, Guidolin D. New methods to evaluate colocalization of fluorophores in immunocytochemical preparations as exemplified by a study on A2A and D2 receptors in Chinese hamster ovary cells. J Histochem Cytochem 53: 941–953, 2005 [DOI] [PubMed] [Google Scholar]
- 2.Bagwell CB, Hudson JL, Irvin GL., 3rd Nonparametric flow cytometry analysis. J Histochem Cytochem 27: 293–296, 1979 [DOI] [PubMed] [Google Scholar]
- 3.Bakker H, Jaddoe VW. Cardiovascular and metabolic influences of fetal smoke exposure. Eur J Epidemiol 26: 763–770, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belik J, Kerc E, Pato MD. Rat pulmonary arterial smooth muscle myosin light chain kinase and phosphatase activities decrease with age. Am J Physiol Lung Cell Mol Physiol 290: L509–L516, 2006 [DOI] [PubMed] [Google Scholar]
- 5.Bir SC, Kolluru GK, Fang K, Kevil CG. Redox balance dynamically regulates vascular growth and remodeling. Semin Cell Dev Biol 23: 745–757, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blue EK, Goeckeler ZM, Jin Y, Hou L, Dixon SA, Herring BP, Wysolmerski RB, Gallagher PJ. 220- and 130-kDa MLCKs have distinct tissue distributions and intracellular localization patterns. Am J Physiol Cell Physiol 282: C451–C460, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Butler SM, Abrassart JM, Hubbell MC, Adeoye O, Semotiuk A, Williams JM, Mata-Greenwood E, Khorram O, Pearce WJ. Contributions of VEGF to age-dependent transmural gradients in contractile protein expression in ovine carotid arteries. Am J Physiol Cell Physiol 301: C653–C666, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chakrabarty K, Fahim M. Modulation of the contractile responses of guinea pig isolated tracheal rings after chronic intermittent hypobaric hypoxia with and without cold exposure. J Appl Physiol 99: 1006–1011, 2005 [DOI] [PubMed] [Google Scholar]
- 9.Charles SM, Zhang L, Cipolla MJ, Buchholz JN, Pearce WJ. Roles of cytosolic Ca2+ concentration and myofilament Ca2+ sensitization in age-dependent cerebrovascular myogenic tone. Am J Physiol Heart Circ Physiol 299: H1034–H1044, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chavez JC, Agani F, Pichiule P, LaManna JC. Expression of hypoxia-inducible factor-1α in the brain of rats during chronic hypoxia. J Appl Physiol 89: 1937–1942, 2000 [DOI] [PubMed] [Google Scholar]
- 11.Christou H, Yoshida A, Arthur V, Morita T, Kourembanas S. Increased vascular endothelial growth factor production in the lungs of rats with hypoxia-induced pulmonary hypertension. Am J Respir Cell Mol Biol 18: 768–776, 1998 [DOI] [PubMed] [Google Scholar]
- 12.Clyman RI, Seidner SR, Kajino H, Roman C, Koch CJ, Ferrara N, Waleh N, Mauray F, Chen YQ, Perkett EA, Quinn T. VEGF regulates remodeling during permanent anatomic closure of the ductus arteriosus. Am J Physiol Regul Integr Comp Physiol 282: R199–R206, 2002 [DOI] [PubMed] [Google Scholar]
- 13.Cox RH. Three-dimensional mechanics of arterial segments in vitro: methods. J Appl Physiol 36: 381–384, 1974 [DOI] [PubMed] [Google Scholar]
- 14.Eddinger TJ, Meer DP. Myosin II isoforms in smooth muscle: heterogeneity and function. Am J Physiol Cell Physiol 293: C493–C508, 2007 [DOI] [PubMed] [Google Scholar]
- 15.Frid MG, Moiseeva EP, Stenmark KR. Multiple phenotypically distinct smooth muscle cell populations exist in the adult and developing bovine pulmonary arterial media in vivo. Circ Res 75: 669–681, 1994 [DOI] [PubMed] [Google Scholar]
- 16.Giussani DA, Camm EJ, Niu Y, Richter HG, Blanco CE, Gottschalk R, Blake EZ, Horder KA, Thakor AS, Hansell JA, Kane AD, Wooding FB, Cross CM, Herrera EA. Developmental programming of cardiovascular dysfunction by prenatal hypoxia and oxidative stress. PLos One 7: e31017, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Griffith SL, Rhoades RA, Packer CS. Pulmonary arterial smooth muscle contractility in hypoxia-induced pulmonary hypertension. J Appl Physiol 77: 406–414, 1994 [DOI] [PubMed] [Google Scholar]
- 18.Hoeben A, Landuyt B, Highley MS, Wildiers H, Van Oosterom AT, De Bruijn EA. Vascular endothelial growth factor and angiogenesis. Pharmacol Rev 56: 549–580, 2004 [DOI] [PubMed] [Google Scholar]
- 19.Hong F, Haldeman BD, Jackson D, Carter M, Baker JE, Cremo CR. Biochemistry of smooth muscle myosin light chain kinase. Arch Biochem Biophys 510: 135–146, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horowitz A, Menice CB, Laporte R, Morgan KG. Mechanisms of smooth muscle contraction. Physiol Rev 76: 967–1003, 1996 [DOI] [PubMed] [Google Scholar]
- 21.Hubbell MC, Semotiuk AJ, Thorpe RB, Adeoye OO, Butler SM, Williams JM, Khorram O, Pearce WJ. Chronic hypoxia and VEGF differentially modulate abundance and organization of myosin heavy chain isoforms in fetal and adult ovine arteries. Am J Physiol Cell Physiol 303: C1090–C1103, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Injeti ER, Sandoval RJ, Williams JM, Smolensky AV, Ford LE, Pearce WJ. Maximal stimulation-induced in situ myosin light chain kinase activity is upregulated in fetal compared with adult ovine carotid arteries. Am J Physiol Heart Circ Physiol 295: H2289–H2298, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jie W, Guo J, Shen Z, Wang X, Zheng S, Wang G, Ao Q. Contribution of myocardin in the hypoxia-induced phenotypic switching of rat pulmonary arterial smooth muscle cells. Exp Mol Pathol 89: 301–306, 2010 [DOI] [PubMed] [Google Scholar]
- 24.Jin KL, Mao XO, Greenberg DA. Vascular endothelial growth factor: direct neuroprotective effect in in vitro ischemia. Proc Natl Acad Sci USA 97: 10242–10247, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Julian CG. High altitude during pregnancy. Clin Chest Med 32: 21–31, 2011 [DOI] [PubMed] [Google Scholar]
- 26.Kamitomo M, Longo LD, Gilbert RD. Right and left ventricular function in fetal sheep exposed to long-term high-altitude hypoxemia. Am J Physiol Heart Circ Physiol 262: H399–H405, 1992 [DOI] [PubMed] [Google Scholar]
- 27.Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, Faraoni R, Floyd M, Hunt JP, Lockhart DJ, Milanov ZV, Morrison MJ, Pallares G, Patel HK, Pritchard S, Wodicka LM, Zarrinkar PP. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol 26: 127–132, 2008 [DOI] [PubMed] [Google Scholar]
- 28.Kim HA, Lim S, Moon HH, Kim SW, Hwang KC, Lee M, Kim SH, Choi D. Hypoxia-inducible vascular endothelial growth factor gene therapy using the oxygen-dependent degradation domain in myocardial ischemia. Pharm Res 27: 2075–2084, 2010 [DOI] [PubMed] [Google Scholar]
- 29.Kohama K, Ye LH, Hayakawa K, Okagaki T. Myosin light chain kinase: an actin-binding protein that regulates an ATP-dependent interaction with myosin. Trends Pharmacol Sci 17: 284–287, 1996 [DOI] [PubMed] [Google Scholar]
- 30.Kurinczuk JJ, White-Koning M, Badawi N. Epidemiology of neonatal encephalopathy and hypoxic-ischaemic encephalopathy. Early Hum Dev 86: 329–338, 2010 [DOI] [PubMed] [Google Scholar]
- 31.Lacolley P, Challande P, Osborne-Pellegrin M, Regnault V. Genetics and pathophysiology of arterial stiffness. Cardiovasc Res 81: 637–648, 2009 [DOI] [PubMed] [Google Scholar]
- 32.Longo LD, Hull AD, Long DM, Pearce WJ. Cerebrovascular adaptations to high-altitude hypoxemia in fetal and adult sheep. Am J Physiol Regul Integr Comp Physiol 264: R65–R72, 1993 [DOI] [PubMed] [Google Scholar]
- 33.Magagnin MG, van den Beucken T, Sergeant K, Lambin P, Koritzinsky M, Devreese B, Wouters BG. The mTOR target 4E-BP1 contributes to differential protein expression during normoxia and hypoxia through changes in mRNA translation efficiency. Proteomics 8: 1019–1028, 2008 [DOI] [PubMed] [Google Scholar]
- 34.Meininger GA, Davis MJ. Cellular mechanisms involved in the vascular myogenic response. Am J Physiol Heart Circ Physiol 263: H647–H659, 1992 [DOI] [PubMed] [Google Scholar]
- 35.Nauli SM, Williams JM, Gerthoffer WT, Pearce WJ. Chronic hypoxia modulates relations among calcium, myosin light chain phosphorylation, and force differently in fetal and adult ovine basilar arteries. J Appl Physiol 99: 120–127, 2005 [DOI] [PubMed] [Google Scholar]
- 36.Neerhof MG, Thaete LG. The fetal response to chronic placental insufficiency. Semin Perinatol 32: 201–205, 2008 [DOI] [PubMed] [Google Scholar]
- 37.Ooi CY, Wang Z, Tabima DM, Eickhoff JC, Chesler NC. The role of collagen in extralobar pulmonary artery stiffening in response to hypoxia-induced pulmonary hypertension. Am J Physiol Heart Circ Physiol 299: H1823–H1831, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Owens GK, Loeb A, Gordon D, Thompson MM. Expression of smooth muscle-specific α-isoactin in cultured vascular smooth muscle cells: relationship between growth and cytodifferentiation. J Cell Biol 102: 343–352, 1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Packer CS, Roepke JE, Oberlies NH, Rhoades RA. Myosin isoform shifts and decreased reactivity in hypoxia-induced hypertensive pulmonary arterial muscle. Am J Physiol Lung Cell Mol Physiol 274: L775–L785, 1998 [DOI] [PubMed] [Google Scholar]
- 40.Pearce WJ, Hull AD, Long DM, Longo LD. Developmental changes in ovine cerebral artery composition and reactivity. Am J Physiol Regul Integr Comp Physiol 261: R458–R465, 1991 [DOI] [PubMed] [Google Scholar]
- 41.Pfitzer G. Regulation of myosin phosphorylation in smooth muscle. J Appl Physiol 91: 497–503, 2001 [DOI] [PubMed] [Google Scholar]
- 42.Pichot CS, Hartig SM, Xia L, Arvanitis C, Monisvais D, Lee FY, Frost JA, Corey SJ. Dasatinib synergizes with doxorubicin to block growth, migration, and invasion of breast cancer cells. Br J Cancer 101: 38–47, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Plate KH, Breier G, Millauer B, Ullrich A, Risau W. Up-regulation of vascular endothelial growth factor and its cognate receptors in a rat glioma model of tumor angiogenesis. Cancer Res 53: 5822–5827, 1993 [PubMed] [Google Scholar]
- 44.Qi H, Wang P, Liu C, Li M, Wang S, Huang Y, Wang F. Involvement of HIF-1α in MLCK-dependent endothelial barrier dysfunction in hypoxia. Cell Physiol Biochem 27: 251–262, 2011 [DOI] [PubMed] [Google Scholar]
- 45.Ramchandran R, Pilipenko E, Bach L, Raghavan A, Reddy SP, Raj JU. Hypoxic regulation of pulmonary vascular smooth muscle cyclic guanosine monophosphate-dependent kinase by the ubiquitin conjugating system. Am J Respir Cell Mol Biol 46: 323–330, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Semenza GL. Expression of hypoxia-inducible factor 1: mechanisms and consequences. Biochem Pharmacol 59: 47–53, 2000 [DOI] [PubMed] [Google Scholar]
- 47.Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res 99: 675–691, 2006 [DOI] [PubMed] [Google Scholar]
- 48.Taggart M, Wray S. Hypoxia and smooth muscle function: key regulatory events during metabolic stress. J Physiol 509: 315–325, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Teramo KA. Obstetric problems in diabetic pregnancy: the role of fetal hypoxia. Best Pract Res Clin Endocrinol Metab 24: 663–671, 2010 [DOI] [PubMed] [Google Scholar]
- 50.Tipoe GL, Fung ML. Expression of HIF-1α, VEGF and VEGF receptors in the carotid body of chronically hypoxic rat. Respir Physiol Neurobiol 138: 143–154, 2003 [DOI] [PubMed] [Google Scholar]
- 51.Villamor E, Kessels CG, Ruijtenbeek K, van Suylen RJ, Belik J, de Mey JG, Blanco CE. Chronic in ovo hypoxia decreases pulmonary arterial contractile reactivity and induces biventricular cardiac enlargement in the chicken embryo. Am J Physiol Regul Integr Comp Physiol 287: R642–R651, 2004 [DOI] [PubMed] [Google Scholar]
- 52.Vonnahme KA, Wilson ME, Li Y, Rupnow HL, Phernetton TM, Ford SP, Magness RR. Circulating levels of nitric oxide and vascular endothelial growth factor throughout ovine pregnancy. J Physiol 565: 101–109, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang Z, Chesler NC. Role of collagen content and cross-linking in large pulmonary arterial stiffening after chronic hypoxia. Biomech Model Mechanobiol 11: 279–289, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Williams JM, White CR, Chang MM, Injeti ER, Zhang L, Pearce WJ. Chronic hypoxic decreases in soluble guanylate cyclase protein and enzyme activity are age dependent in fetal and adult ovine carotid arteries. J Appl Physiol 100: 1857–1866, 2006 [DOI] [PubMed] [Google Scholar]
- 55.Yablonka-Reuveni Z, Christ B, Benson JM. Transitions in cell organization and in expression of contractile and extracellular matrix proteins during development of chicken aortic smooth muscle: evidence for a complex spatial and temporal differentiation program. Anat Embryol (Berl) 197: 421–437, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yamakawa M, Liu LX, Date T, Belanger AJ, Vincent KA, Akita GY, Kuriyama T, Cheng SH, Gregory RJ, Jiang C. Hypoxia-inducible factor-1 mediates activation of cultured vascular endothelial cells by inducing multiple angiogenic factors. Circ Res 93: 664–673, 2003 [DOI] [PubMed] [Google Scholar]
- 57.Yin H, Li Q, Qian G, Wang Y, Li Y, Wu G, Wang G. Rab1 GTPase regulates phenotypic modulation of pulmonary artery smooth muscle cells by mediating the transport of angiotensin II type 1 receptor under hypoxia. Int J Biochem Cell Biol 43: 401–408, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]