

Abstract
Cerebral hyperperfusion syndrome (CHS) may occur as a severe complication following surgical treatment of carotid stenosis. However, the mechanism inducing neurological symptoms in CHS remains unknown. We describe a patient with CHS presenting with seizures 24 h following carotid endarterectomy. Imaging demonstrated early ipsilateral blood–brain barrier (BBB) breakdown with electroencephalographic evidence of cortical dysfunction preceding brain edema. Using in vitro experiments on rat cortical tissue, we show that direct exposure of isolated brain slices to a serum-like medium induces spontaneous epileptiform activity, and that neuronal dysfunction is triggered by albumin. We propose BBB breakdown and subsequent albumin extravasation as a novel pathogenic mechanism underlying CHS and a potential target for therapy.
Keywords: Albumin, Blood–brain barrier, Cerebral hyperperfusion syndrome, Magnetic resonance imaging, Seizure, Astrocytes, TGF-βR
Introduction
Cerebral hyperperfusion syndrome (CHS) is a complication observed after both carotid endarterectomy (CEA) and carotid angioplasty with stenting [11, 25]. The syndrome is characterized by headache, seizures, focal neurological deficits and cognitive impairment, appearing within hours and up to 1 month after surgery [11, 15, 16, 25]. Failure of autoregulation of cerebral blood flow was identified as an important factor initiating excess perfusion. This in turn may result in severe brain edema and intracranial hemorrhage. However, neurological symptoms may develop prior to, or without the occurrence of such gross structural alterations. The mechanisms underlying the progression from vascular hyperperfusion to neuronal dysfunction during CHS remain unknown.
Breakdown of the BBB was observed in an animal model of cerebral hyperperfusion [19]. Recent animal experiments have shown that increased permeability of brain blood vessels induces neuronal hyperexcitability [2–4, 7, 21], delayed neuronal degeneration and impairment of cortical function [4, 22]. In this study we demonstrate a clinical case of CHS presenting BBB opening and neurological symptoms prior to edema formation. The experimental results indicate serum components as a potent trigger of neuronal dysfunction. Our data suggests BBB breakdown as a pathogenic mechanism underlying the development of CHS.
Methods
Brain magnetic resonance imaging (MRI)
Fluid-attenuated inversion-recovery (FLAIR) sequences were performed before, as well as 1, 2 and 5 days following CEA with standard protocols using a 1.5 Tesla device (Intera, Philips Medical Systems, Best, the Netherlands).
Electroencephalography
Electroencephalography was carried out 1 and 5 days after CEA using a clinical 128 channel digital EEG acquisition unit (CEEGRAPH IV, Bio-logic Systems Corp., Mundelein, Illinois), with a digitization rate of 256 Hz, as previously reported [24]. Twenty-seven conventional AgCl surface electrodes were placed according to the international 10–20 electrode system. Scalp electrode impedances were kept below 10 kΩ. Fast Fourier Transform was applied to the EEG waveforms recorded from each electrode. Source localization was achieved, as reported, using standardized low resolution electrotomography (sLORETA see [17, 27].
Electrophysiological recordings in vitro
All experimental procedures were approved by the Berlin and Beer-Sheva animal ethics committees and principles of laboratory care were followed. Standard techniques [7] were employed for the electrophysiological recordings. In brief, rats were deeply anaesthetized and decapitated. Brains were quickly removed and 6–8 transverse slices (400 μm thick) were prepared using a vibratome (Campden Instruments, Loughborough, UK). Slices were divided in 3 groups for incubation in (1) artificial cerebrospinal fluid (ACSF, composition in mM: 129 NaCl, 21 NaHCO3, 1.25 NaH2PO4, 1.8 MgSO4, 1.6 CaCl2, 3 KCl, 10 glucose; n = 9 slices from 3 animals), (2) artificial serum (ASERUM, composition based on ACSF and modified to fit serum composition [21]: 0.8 MgSO4, 1.3 CaCl2, 5.7 KCl, 1 L-glutamine, 0.1 serum albumin, according to 25% serum concentration; n = 9 slices from 3 animals), or (3) ASERUM without the addition of serum albumin (n = 8 slices from 3 animals) in a standard interface chamber providing humidified, carbogenated (5% CO2, 95% O2) gas atmosphere at 36 ± 1°C. Incubation time ranged from 6 to 10 h. Field potentials were recorded during this period using extra cellular glass microelectrodes (~3 MΩ) filled with 154 mM NaCl and positioned in cortical layer IV. Slices were stimulated with brief (100 μs) pulses using bipolar stimulation electrodes placed at the border between white and gray matter. Signals were amplified (SEC-10L, NPI, Tamm, Germany), digitized on-line (CED-1401, Cambridge, England) and stored for off-line analysis. Statistical analysis was performed using SPSS 12.0.1 for Windows. Differences in the rate of occurrence of epileptiform activity between the groups were determined by the Pearson’s chi-square test. P<0.05 was taken as the level of statistical significance.
Results
Clinical case
A 67-year-old patient with a 90% occlusion of the left carotid artery underwent CEA. The patient’s history included mild hypertension. Pre-operative neurological examination and brain imaging were normal. One day after surgery, she developed confusion and partial seizures limited to her right arm and leg. FLAIR weighted MRI images after gadolinium administration displayed high signals from the left subarachnoidal space, indicating acute breakdown of the blood–CSF barrier (Fig. 1a). MRI of head and neck showed patency of the operated carotid, T2* perfusion imaging was symmetrically normal. 24 h later, prominent cortical edema and gray matter enhancement developed over the left hemisphere (Fig. 1b). On day 5, clinical improvement was associated with normalization of the MRI (Fig. 1c). The clinical and radiological signs were associated with pathological slowing (delta-theta range, 2–5 Hz) in the electroencephalography (EEG) over her left hemisphere (Fig. 1d), which normalized on day 5 (Fig. 1e).
Fig. 1.

MRI and EEG findings in a patient suffering from CHS. a Axial FLAIR MRI image 4 h after clinical presentation: signal enhancement in the subarachnoid space over the left hemisphere is observed, indicating breakdown of the Blood–CSF barrier. b Axial MR image on day 2 after surgery, showing diffuse cortical edema with gray matter enhancement. c Normalization of the MRI findings on day 5. d EEG on day 1, 6 h after clinical presentation: note the diffuse slowing in electrodes over the left hemisphere. Below: Spectral analysis showing abnormal slowing in the theta range and presumed localization of the abnormal theta rhythm (2–5 Hz) using sLORETA. e EEG traces on day 5 after surgery demonstrating EEG normalization
Animal experiments
Recent animal experiments suggested that BBB breakdown played a role in the pathogenesis of seizures [10], epilepsy [2–4, 21, 26], and neuronal dysfunction and damage [4, 7, 22]. We thus challenged the hypothesis that direct exposure of brain tissue to serum components is sufficient to induce neuronal dysfunction and hypersynchrony. Electrophysiological recordings were simultaneously performed at different locations within the neocortical in vitro slice preparation (see Fig. 2a). 6–10 h after incubation with an artificial serum-like solution (ASERUM), spontaneous epileptiform interictal-like activity was recorded in 66.7% of the slices (n = 9 slices, 3 animals). Epileptiform activity was recorded simultaneously in large regions of the neocortex and propagated within the neo- and perirhinal cortices (Fig. 2b). Such abnormal activity was never recorded in slices incubated in artificial CSF-like solution (ACSF, n = 9 slices, 3 animals). Importantly, abnormal activity was never recorded in albumin-free ASERUM solution (n = 8 slices, 3 animals, Chi Square: p<0.01). Abnormal neuronal activity was also observed in response to brief afferent electrical stimulation at the gray-white matter border of the cortex in 77.8% of slices from the group exposed to ASERUM (Fig. 2c). This was in contrast to the normal, short duration, non-propagating field potential observed in albumin-free medium (Fig. 2d).
Fig. 2.
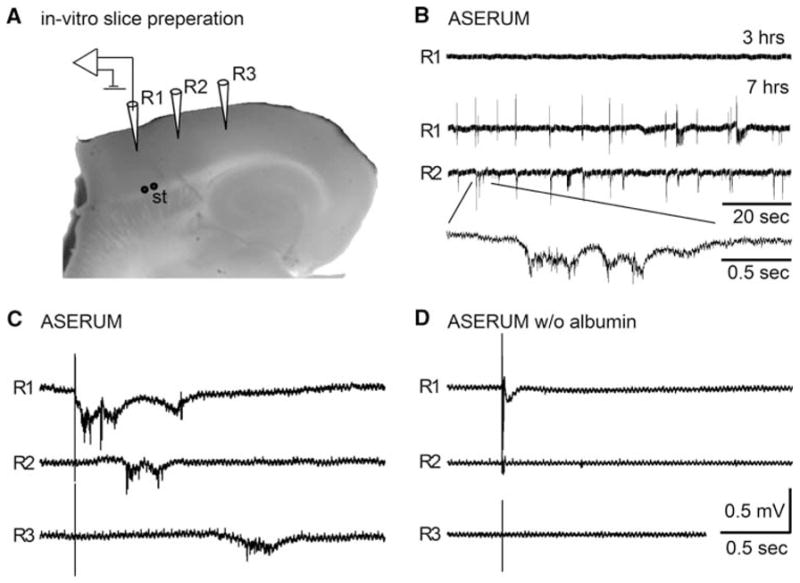
Spontaneous epileptiform activity after cortical exposure to serum components. a Photograph of the transverse brain slice preparation demonstrating the positioning of extracellular recording (R1–3) and stimulation (St) electrodes. b Representative traces of spontaneous activity recorded 3 and 7 h after exposure of the slice to artificial serum solution (ASERUM). Note the appearance of spontaneous hyper-synchronization of electrical activity within the cortex, as shown in simultaneous recordings from R1 and R2. c, d Representative electrophysiological traces demonstrating the response to brief electrical stimulation of afferent fibers in the white matter. Abnormally propagating epileptiform activity was evoked 5–7 h after incubation in ASERUM (c), but did not appear in slices incubated in albumin-free ASERUM (d) or ACSF (data not shown)
Discussion
In the present study we describe a patient showing a robust blood–CSF barrier breakdown as the earliest imaging sign of CHS, associated with diffuse EEG slowing and followed by brain edema (Fig. 1). This sequence of events raised the hypothesis that exposure of brain tissue to serum components directly induces abnormal neuronal activity and associated clinical symptoms. To challenge this hypothesis, we exposed rat brain slices to artificial serum and compared neuronal network activity to that observed under artificial CSF. We found that several hours after brain exposure to serum-imitating medium, neuronal activity is significantly altered and spontaneous abnormal hypersynchronous epileptiform activity is induced, similar to that previously published in animal models of epilepsy (e.g., [2–5, 18, 21]).
Our experimental data support the conclusion that direct exposure of the cerebral cortex to a serum-like medium leads to a robust change in the electrophysiological properties of the neuronal network. Breakdown of the BBB is expected to impair the maintenance of a unique extracellular environment of the brain tissue, and to change the concentrations of electrolytes (e.g., increase K+, decrease Mg2+ and Ca2+), amino acids (e.g., glutamine— a precursor for glutamate and GABA), and proteins (e.g., the most abundant serum protein, albumin, is not produced by brain cells in any significant amount). In line with our previous studies, we show here that serum-derived albumin was sufficient to induce stimulus-evoked abnormal network responses [7, 21], while serum concentrations of electrolytes (and glutamine) without albumin did not induce abnormal activity (Fig. 2c, d). However, alteration of electrolytes facilitated the appearance of albumin-induced spontaneous epileptiform activity. The time window between the onset of hyperperfusion and the appearance of clinical symptoms is reflected in our experimental model by the period of time between exposure of the cortex to albumin and neuronal dysfunction. Vasogenic edema formation, which occurs in parallel to albumin extravasation under in vivo conditions, may contribute to the observed symptoms and EEG changes. However, it is noteworthy that in the presented case, significant brain edema was found in the MRI 1 day after BBB breakdown and seizure generation. A more direct evidence for a direct role of BBB breakdown and albumin extravasation comes from the animal experiments showing that activation of the transforming growth factor beta (TGFβ) signaling pathway by albumin or by low concentrations of TGFβ1 induce seizure-like activity with no prior edema [2]. The delayed appearance of hyperexcitability suggests the involvement of complex network changes, probably associated with altered gene expression and synaptic plasticity [2–4]. The involvement of specific signaling pathways and cell populations in albumin-mediated epileptogenesis may offer novel targets for the prevention and treatment of CHS.
BBB breakdown is observed under numerous neurological conditions [1, 14], but its role in the pathogenesis of a clinical disease has only recently been suggested in animal [2–4, 7, 10, 22, 26] and human studies [9, 24]. The role of BBB breakdown in CHS has never been described. Production of free radicals and nitric oxide during carotid clamping may promote endothelial damage [25]. In fact, vasogenic edema has frequently been reported in CHS patients [18, 23], suggesting that following carotid surgery, BBB breakdown, as we presented in our case study, is a likely event. This is more likely to occur in patients with impaired autoregulation [20]. It should be noted that due to the lack of standardized methods for quantification of BBB breakdown, such an early event—if mild—may be missed in conventional imaging studies [23, 24]. While failure of autoregulation is a risk factor for cerebral hyperperfusion following CEA, seizure activity induced by BBB breakdown during CHS ([10, 26], and see Fig. 2) may additionally disturb vessel regulation and modify the course of CHS. Both hyper- and hypoperfusion were reported in the human epileptic cortex [8, 13]. Increased cerebral blood flow (CBF) in the ictal phase [8] may promote BBB breakdown and generation of seizure activity. This sequence of events may be self-enhancing and increase brain edema, as observed in the presented case (Fig. 2). Reduction of CBF [13] as a result of worsening edema, in the presence of increased energy consumption and free radical production during seizure activity, may compromise vascular and cellular integrity and lead to delayed neuronal and functional damage [22, 24]. Vascular damage and BBB breakdown may further increase the risk for intracerebral hemorrhage as suggested following stroke [6]. However, when hyperperfusion is reduced and the BBB is repaired, parallel electrophysiological and clinical improvements are expected (Fig. 1). Since consequences of CHS may be severe and difficult to treat, prevention is a central element in current management strategies and is based on identification of vulnerable patients. Early postoperative diagnosis involves perfusion monitoring and detection of complications such as cerebral edema, hemorrhage, infarction and seizures. Intensive control of systemic blood pressure plays a crucial role in prevention, as well as treatment of the syndrome. Additionally, prompt handling of the individual complications is required (for review see [11, 25]). Characterization of extent and time course of BBB leakage may facilitate identification of patients at risk for delayed cognitive impairment [15] or for development of epilepsy [12]. Since BBB disruption may occur at an early stage of CHS, interference with astrocytic albumin uptake mechanisms (e.g., by pharmacological blockade of TGF-βR) could provide a valuable preventive tool to ameliorate the progression of CHS.
We propose that BBB breakdown is an important initiating factor in the early and delayed neurological deficits following carotid procedures. Animal experiments suggest that activation of TGF-β signaling by serum albumin is involved as a key signaling element. Clinical studies are awaited in order to furnish prospective measurements of BBB permeability in patients following carotid procedures. Coupled with further research on animals, these studies will help target BBB breakdown for the prevention and treatment of CHS.
Acknowledgments
The authors would like to thank Asaf Krah for technical assistance. This study was supported by the Israeli Science Foundation (566/07), the Israel-USA Binational Science foundation and the Sonderforschungsbereich TR3 (C8).
Contributor Information
Sebastian Ivens, Department of Psychiatry and Psychotherapy, Charité Universitätsmedizin, Berlin, Germany. Institute of Neurophysiology, Neurocure Research Center, Charité Universitätsmedizin, Berlin, Germany.
Szendro Gabriel, Department of Vascular Surgery, Soroka University Medical Center, Ben-Gurion University of the Negev, Beersheba, Israel.
George Greenberg, Department of Vascular Surgery, Soroka University Medical Center, Ben-Gurion University of the Negev, Beersheba, Israel.
Alon Friedman, Email: alonf@bgu.ac.il, Institute of Neurophysiology, Neurocure Research Center, Charité Universitätsmedizin, Berlin, Germany. Department of Physiology and Neurosurgery, Soroka University Medical Center, Ben-Gurion University of the Negev, Beersheba, Israel. Department of Physiology, Faculty of Health Sciences, Ben-Gurion University of the Negev, 84105 Beersheba, Israel.
Ilan Shelef, Department of Neuroradiology, Soroka University Medical Center, Ben-Gurion University of the Negev, Beersheba, Israel.
References
- 1.Abbott NJ, Ronnback L, Hansson E. Astrocyte–endothelial interactions at the blood–brain barrier. Nat Rev Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- 2.Cacheaux LP, Ivens S, David Y, Lakhter AJ, Bar-Klein G, Shapira M, Heinemann U, Friedman A, Kaufer D. Transcriptome profiling reveals TGF-beta signaling involvement in epileptogenesis. J Neurosci. 2009;29:8927–8935. doi: 10.1523/JNEUROSCI.0430-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.David Y, Flores LP, Ivens S, Heinemann U, Kaufer D, Friedman A. Astrocytic dysfunction in epileptogenesis: consequences of altered potassium and glutamate buffering? J Neurosci. 2009;29:10588–10599. doi: 10.1523/JNEUROSCI.2323-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Friedman A, Kaufer D, Heinemann U. Blood–brain barrier breakdown-inducing astrocytic transformation: novel targets for the prevention of epilepsy. Epilepsy Res. 2009;85:142–149. doi: 10.1016/j.eplepsyres.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gutnick MJ, Connors BW, Prince DA. Mechanisms of neocortical epileptogenesis in vitro. J Neurophysiol. 1982;48:1321–1335. doi: 10.1152/jn.1982.48.6.1321. [DOI] [PubMed] [Google Scholar]
- 6.Hjort N, Wu O, Ashkanian M, Solling C, Mouridsen K, Christensen S, Gyldensted C, Andersen G, Ostergaard L. MRI detection of early blood–brain barrier disruption: parenchymal enhancement predicts focal hemorrhagic transformation after thrombolysis. Stroke. 2008;39:1025–1028. doi: 10.1161/STROKEAHA.107.497719. [DOI] [PubMed] [Google Scholar]
- 7.Ivens S, Kaufer D, Flores LP, Bechmann I, Zumsteg D, Tomkins O, Seiffert E, Heinemann U, Friedman A. TGF-beta receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain. 2007;130:535–547. doi: 10.1093/brain/awl317. [DOI] [PubMed] [Google Scholar]
- 8.Kim JH, Im KC, Kim JS, Lee SA, Lee JK, Khang SK, Kang JK. Ictal hyperperfusion patterns in relation to ictal scalp EEG patterns in patients with unilateral hippocampal sclerosis: a SPECT study. Epilepsia. 2007;48:270–277. doi: 10.1111/j.1528-1167.2006.00847.x. [DOI] [PubMed] [Google Scholar]
- 9.Korn A, Golan H, Melamed I, Pascual-Marqui R, Friedman A. Focal cortical dysfunction and blood–brain barrier disruption in patients with Postconcussion syndrome. J Clin Neurophysiol. 2005;22:1–9. doi: 10.1097/01.wnp.0000150973.24324.a7. [DOI] [PubMed] [Google Scholar]
- 10.Marchi N, Angelov L, Masaryk T, Fazio V, Granata T, Hernandez N, Hallene K, Diglaw T, Franic L, Najm I, Janigro D. Seizure-promoting effect of blood–brain barrier disruption. Epilepsia. 2007;48:732–742. doi: 10.1111/j.1528-1167.2007.00988.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moulakakis KG, Mylonas SN, Sfyroeras GS, Andrikopoulos V. Hyperperfusion syndrome after carotid revascularization. J Vasc Surg. 2009;49(4):1060–1068. doi: 10.1016/j.jvs.2008.11.026. [DOI] [PubMed] [Google Scholar]
- 12.Naylor AR, Evans J, Thompson MM, London NJ, Abbott RJ, Cherryman G, Bell PR. Seizures after carotid endarterectomy: hyperperfusion, dysautoregulation or hypertensive encephalopathy? Eur J Vasc Endovasc Surg. 2003;26:39–44. doi: 10.1053/ejvs.2002.1925. [DOI] [PubMed] [Google Scholar]
- 13.Nelissen N, Van PW, Baete K, Van LK, Palmini A, Van BH, Dupont P. Correlations of interictal FDG-PET metabolism and ictal SPECT perfusion changes in human temporal lobe epilepsy with hippocampal sclerosis. Neuroimage. 2006;32:684–695. doi: 10.1016/j.neuroimage.2006.04.185. [DOI] [PubMed] [Google Scholar]
- 14.Neuwelt EA. Mechanisms of disease: the blood–brain barrier. Neurosurgery. 2004;54:131–140. doi: 10.1227/01.neu.0000097715.11966.8e. [DOI] [PubMed] [Google Scholar]
- 15.Ogasawara K, Inoue T, Kobayashi M, Endo H, Yoshida K, Fukuda T, Terasaki K, Ogawa A. Cerebral hyperperfusion following carotid endarterectomy: diagnostic utility of intraoperative transcranial Doppler ultrasonography compared with single-photon emission computed tomography study. AJNR Am J Neuroradiol. 2005;26:252–257. [PMC free article] [PubMed] [Google Scholar]
- 16.Ogasawara K, Mikami C, Inoue T, Ogawa A. Delayed cerebral hyperperfusion syndrome caused by prolonged impairment of cerebrovascular autoregulation after carotid endarterectomy: case report. Neurosurgery. 2004;54:1258–1261. doi: 10.1227/01.neu.0000120064.55339.f9. [DOI] [PubMed] [Google Scholar]
- 17.Pascual-Marqui RD. Standardized low-resolution brain electromagnetic tomography (sLORETA): technical details. Methods Find Exp Clin Pharmacol. 2002;24(Suppl D):5–12. [PubMed] [Google Scholar]
- 18.Prince DA, Futamachi KJ. Intracellular recordings in chronic focal epilepsy. Brain Res. 1968;11:681–684. doi: 10.1016/0006-8993(68)90156-x. [DOI] [PubMed] [Google Scholar]
- 19.Sakaki T, Tsujimoto S, Nishitani M, Ishida Y, Morimoto T. Perfusion pressure breakthrough threshold of cerebral autoregulation in the chronically ischemic brain: an experimental study in cats. J Neurosurg. 1992;76:478–485. doi: 10.3171/jns.1992.76.3.0478. [DOI] [PubMed] [Google Scholar]
- 20.Schwartz RB. Hyperperfusion encephalopathies: hypertensive encephalopathy and related conditions. Neurologist. 2002;8:22–34. doi: 10.1097/00127893-200201000-00003. [DOI] [PubMed] [Google Scholar]
- 21.Seiffert E, Dreier JP, Ivens S, Bechmann I, Tomkins O, Heinemann U, Friedman A. Lasting blood–brain barrier disruption induces epileptic focus in the rat somatosensory cortex. J Neurosci. 2004;24:7829–7836. doi: 10.1523/JNEUROSCI.1751-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tomkins O, Friedman O, Ivens S, Reiffurth C, Major S, Dreier JP, Heinemann U, Friedman A. Blood–brain barrier disruption results in delayed functional and structural alterations in the rat neocortex. Neurobiol Dis. 2007;25:367–377. doi: 10.1016/j.nbd.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 23.Tomkins O, Kaufer D, Korn A, Shelef I, Golan H, Reichenthal E, Soreq H, Friedman A. Frequent blood–brain barrier disruption in the human cerebral cortex. Cell Mol Neurobiol. 2001;21:675–691. doi: 10.1023/a:1015147920283. [DOI] [PubMed] [Google Scholar]
- 24.Tomkins O, Shelef I, Kaizerman I, Eliushin A, Afawi Z, Misk A, Gidon M, Cohen A, Zumsteg D, Friedman A. Blood–brain barrier disruption in post-traumatic epilepsy. J Neurol Neurosurg Psychiatry. 2008;79:774–777. doi: 10.1136/jnnp.2007.126425. [DOI] [PubMed] [Google Scholar]
- 25.van Mook WN, Rennenberg RJ, Schurink GW, van Oostenbrugge RJ, Mess WH, Hofman PA, de Leeuw PW. Cerebral hyperperfusion syndrome. Lancet Neurol. 2005;4:877–888. doi: 10.1016/S1474-4422(05)70251-9. [DOI] [PubMed] [Google Scholar]
- 26.van Vliet EA, da Costa AS, Redeker S, van Schaik R, Aronica E, Gorter JA. Blood–brain barrier leakage may lead to progression of temporal lobe epilepsy. Brain. 2007;130:521–534. doi: 10.1093/brain/awl318. [DOI] [PubMed] [Google Scholar]
- 27.Zumsteg D, Friedman A, Wennberg RA, Wieser HG. Source localization of mesial temporal interictal epileptiform discharges: correlation with intracranial foramen ovale electrode recordings. Clin Neurophysiol. 2005;116:2810–2818. doi: 10.1016/j.clinph.2005.08.009. [DOI] [PubMed] [Google Scholar]