

Abstract
Plin4 is a lipid droplet protein (LDP) found predominantly in white adipose tissue (WAT). The Plin4 gene is immediately downstream of the Plin5 gene; the two genes exhibit distinct though overlapping tissue expression patterns. Plin4 is absent in brown adipose tissue (BAT) and liver and expressed at low levels in heart and skeletal muscle, whereas Plin5 is highly expressed in these oxidative tissues but at a low level in WAT. The physiological role of Plin4 remains unclear. We have generated Plin4−/− mice by gene targeting. Loss of Plin4 has no effect on body weight or composition or on adipose mass or development. However, the triacylglycerol (TAG) content in heart, but not other oxidative tissues such as BAT, soleus muscle, and liver, is markedly reduced in Plin4−/− mice. The heart of Plin4−/− mice displays reduced Plin5 mRNA and protein levels (by ∼38 and 87%, respectively, vs. wild-type) but unchanged mRNA levels of other perilipin family genes (Plin2 and Plin3) or genes involved in glucose and lipid metabolism. Despite reduced cardiac TAG level, both young and aged Plin4−/− mice maintain normal heart function as wild-type mice, as measured by echocardiography. Interestingly, Plin4 deficiency prevents the lipid accumulation in the heart that normally occurs after a prolonged (48-h) fast. It also protects the heart from cardiac steatosis induced by high-fat diet or when Plin4−/− mice are bred into Lep−/− obese background. In conclusion, inactivation of Plin4 downregulates Plin5 and reduces cardiac lipid accumulation in mice.
Keywords: Plin4, Plin5, adipose tissue development, lipid droplet protein, cardiac lipid storage
eukaryotes from yeast to human share the capacity to synthesize and store neutral lipids in cytosolic lipid droplets (LDs). Lipid droplets are more than inert neutral lipid depots; they are functional subcellular organelles involved in multiple intracellular processes, including lipid metabolism, vesicle trafficking, and cell signaling (3, 12, 17). The cytosolic surfaces of lipid droplets are coated with one or more members of the perilipin (Plin) LDP family [originally called the PAT family, named after perilipin, adipocyte differentiation-related protein (ADRP), and tail-interacting protein of 47 kDa (TIP47)] that play critical roles in regulating neutral lipid distribution and metabolism (10, 16). PLIN family proteins share significant primary sequence similarities encompassing two conserved domains (the PAT1 and PAT2 domains) and the capability to associate with LDs (13, 16). Five members of the PLIN family have been identified to date: PLIN1, PLIN2 [also called ADRP, ADFP, or adipophillin (2)], PLIN3 [Tip47 (25)], PLIN4 [S3–12 (19)], and PLIN5 [Ox-PAT, MLDP, lsdp5, or PAT-1, (7, 24, 28)]; each PLIN family protein has unique tissue distribution, subcellular localization, and lipid binding characteristics.
A detailed sequence analysis of murine Plin family proteins reveals that Plin2, Plin3, and Plin5 share the highest degree of sequence similarity, whereas Plin1 and Plin4 display more divergent carboxyl and amino termini, respectively (16, 28). Like Plin1, Plin4 was first identified as an adipocyte-specific protein induced during adipocyte differentiation (19) and was shown to be a known PPARγ target (8, 18, 20). It is expressed predominantly in white adipose tissue (WAT) with a relatively low expression in heart and skeletal muscle and is completely absent in brown adipose tissue (BAT) and liver (24). In contrast, Plin5, the recently characterized Plin family protein, whose gene is immediately upstream of Plin4, is highly expressed in the oxidative tissues such as heart, skeletal muscle, BAT, and liver but is barely detectable in WAT. A unique feature of Plin4 is that approximately two-thirds of the Plin4 sequence is comprised of tandem repeats of an 11-residue motif shared by Plin2, Plin5, and Plin3 (8). These repeats may be involved in lipid binding, although their exact functions are not well characterized.
To date, little is known of the function of Plin4. Although both Plin4 and Plin1 are expressed in adipocytes, they reside in distinct LD populations (23, 26). In cultured mature adipocytes, like Plin3, Plin4 is normally found in cytosol in the absence of exogenous neutral lipid. Upon lipid loading, Plin4 coats nascent LDs to permit rapid packaging of newly synthesized triacylglycerol (TAG), an observation that suggests the participation of Plin4 in the early events of LD formation in adipocytes (23, 26). Plin4 seems to function as a regulated PAT protein that associates with LDs in a hormone- and substrate-dependent manner.
To determine the role of Plin4 in the control of LD biogenesis and maturation, we generated Plin4-deficient mice by gene targeting. Here, we report that Plin4−/− mice exhibit unperturbed adipocyte differentiation and development. Interestingly, however, they display markedly reduced amounts of TAG in the heart. Inactivation of Plin4 also leads to a concomitant reduction of Plin5, another perlipin family LDP, whose gene is located immediately upstream of the Plin4 gene, at both mRNA and protein levels. Our observations on the Plin4−/− mice suggest that Plin4, in association with Plin5, may control lipid accumulation in the heart.
MATERIALS AND METHODS
Chemicals, reagents, and antibodies.
We purchased tissue culture media from Invitrogen and lipid standards for thin-layer chromatography (TLC) analysis from Avanti Polar Lipids (Alabaster, AL). All other chemicals were purchased from Sigma Chemical (St. Louis, MO). Primary antibody against Plin4 (139.4 kDa) was a gift kindly provided by Dr. Perry E. Bickel (University of Texas Health Science Center, Houston TX). Primary antibodies against Plin2 (46.6 kDa), Plin3 (47.2 kDa), and Plin5 (50 kDa) were polyclonal antisera generated against recombinant His-tagged Plin2, Plin3, and Plin5, as described (4). The following antibodies were also used: Plin1 (56 kDa) (Progen, Heidelberg, Germany) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Millipore, Billerica, MA).
Animals.
Mice with Plin4 targeted deletion were generated at Regeneron Pharmaceuticals, using the strategy shown in Fig. 1A. A LacZ-polyA/neo selection cassette (pGK-em7-Neo) was inserted right after the ATG start codon, which replaced from half of exon 2 till the stop codon at exon 9, including the intronic regions of Plin4 (Fig. 1A). A thymidine kinase cassette was ligated to the 3′ end of the construct for selection against random insertion events. Homozygous mice were identified by genomic PCR genotyping (Fig. 1B). Complete deletion of Plin4 in homozygous mice was confirmed by Western blotting showing no Plin4 protein was detected in either the epididymal or subcutaneous WAT in the Plin4−/− mice compared with the wild-type counterparts (Fig. 1C). Plin4−/− mice were back-crossed to C57BL/6J for eight generations and maintained in a temperature-controlled facility with fixed 12:12-h light-dark cycles and free access to regular chow and water. Male animals of 8–30 wk old were used throughout this study unless otherwise indicated. Some experiments were done on animals fed a high-fat diet (HFD; 42% kcal fat; Harlan Teklad TD88137) from 6 wk old and kept for 10 wk. All animal experiments were done using protocols approved by the IACUC at Baylor College of Medicine.
Fig. 1.

Generation of (Plin4−/−) mice. A: targeting strategy. Exons 1-9 are represented by filled boxes. Replacement vector was used to replace the sequence encompassing exon 1 till part of exon 9 with the lacZ expression cassette and the neo gene driven by PGK promoter. B: PCR genotyping. A specific set of primers was used to amplify genomic tail DNAs extracted from WT, Plin4 heterozygous, and homozygous knockout littermates. The predicted 546 bp (p1&p2) and 454 bp (p1&p3) were successfully amplified from WT and knockout mice, respectively, with heterozygous mice containing both, suggesting successful deletion of Plin4 genomic sequence. C: protein expression of Plin4 in subcutaneous (SF) and gonadal fat (GF) of Plin4+/+ and Plin4−/− animals was analyzed by Western blot using specific antibody against NH2-terminal Plin4.
Plasma biochemistry and whole body fat content measurement.
Serum nonesterified fatty acid (NEFA; Wako), glycerol (Sigma), glucose (Therma Scientific), cholesterol, and total TAG levels (Therma Scientific) were measured by enzymatic assay kits for determination of their concentrations. Whole body fat and lean masses were measured using an EchoMRI Whole Body Composition Analyzer (Echo Medical Systems, Houston TX) according to the manufacturer's instructions and normalized to body weight.
Isolation of stromal vascular cells and adipocyte differentiation.
Murine primary preadipocytes from the subcutaneous fat stromal vascular fraction were prepared as described (9). Briefly, subcutaneous fat from 6- to 7-wk-old C57BL/6J mice were isolated, minced, and digested in 2 mg/ml collagenase IV (Sigma) with 20 mg/ml BSA at 37°C for 40 min. The digested mixtures were then filtered through a 100 μM cell strainer and spun at 250 g for 8 min. The pellets containing the stromal vascular fraction were then resuspended in preadipocyte growth medium (Cell applications) and cultured for induction of differentiation by using the standard 3T3-L1 differentiation protocol. The intracellular triglyceride levels in D8 differentiated stromal vascular cells (SVCs) were extracted by chloroform and methanol according to Bligh and Dyer (1). The amount of triglyceride was measured with an Infinity triglyceride assay kit (Thermo Scientific). The concentration of cellular protein was determined using a BCA protein assay (Bio-Rad). Intracellular triglyceride content was calculated as per milligram of protein.
Lipolysis in vitro and in vivo.
To determine lipolysis in vitro, D8 differentiated SVCs were first washed twice with DMEM and incubated in DMEM containing 2% fatty acid-free BSA in the presence or absence of 10 μM CL-316243 (a specific β3-adrenergic receptor agonist). A total of 100 μl of medium was collected at various time points for glycerol and NEFA measurements by enzymatic kit analyses (Wako). For in vivo lipolysis, mice were fasted for 4 h and treated with an intraperitoneal injection of the β3-adrenergic receptor agonist CL-316243 (0.1 mg/kg body wt). Blood was withdrawn from the tail before and 15 min after injection for determination of NEFA and glycerol levels.
Thin-layer chromatography and tissue lipid analysis.
Tissues were homogenized in standard PBS buffer. Lipids were extracted according to Bligh and Dyer (1). Total lipids were dissolved in chloroform, and the phospholipids (PL), TAG, free fatty acid, and cholesteryl ester fractions were separated by one-dimensional thin-layer chromatography (Silica Gel 60), using hexane-diethyl ether-glacial acetic acid (85:20:1). For quantitative analysis, total lipids were dissolved in 5% Triton X-100 in PBS. Triglyceride concentration was measured using a triglyceride assay kit (Thermo Scientific) and normalized to tissue weights.
Reverse transcription PCR and RNA quantitation.
Total RNA was isolated from tissues or cultured cells with TRIzol (Invitrogen) and reverse-transcribed with Superscript II reverse transcriptase using random primers (Invitrogen). Real-time quantitative RT-PCR was performed on the Strategene MX3000 real-time detection system using Perfecta SYBR Green PCR reagent kit (Quanta BioSciences).
Western blot analysis.
For Western blotting, equivalent amounts of protein homogenate were resolved by SDS-polyacrylamide gels and transferred to nitrocellulose membrane. The blots were probed with specific antibodies for visualization by enhanced chemiluminescence (SuperSignal kit, Pierce).
Echocardiography.
Transthoracis 2D and M-mode echocardiography analysis was used to assess heart function in conscious mice with a VisualSonics Vevo 770 echocardiography machine equipped with a 30-MHz probe (VisualSonics). At least seven animals from each group were measured, and the pooled data were analyzed for statistical significance.
Statistical analysis.
Student's t-test was used for statistical analysis. Differences were considered significant when P values were <0.05. Results are expressed as means ± SE.
RESULTS
Plin4 is dispensable for adipose tissue differentiation and lipolysis in vitro and in vivo.
Plin4−/− mice were born live with the expected Mendelian mode of inheritance and displayed no overtly abnormal phenotype. They were fertile and nursed their pups normally. Wild-type and Plin4−/− mice had similar body weights (Table 1). There were no differences in 4-h-fasted blood glucose, plasma NEFA, TAG, total cholesterol, or free glycerol between wild-type and Plin4−/− littermates (Table 1). The adiposity index, determined by echo magnetic resonance imaging, of mice on the regular chow diet showed similar lean body mass and total fat content (Table 1).
Table 1.
Body weight, organ weights, and plasma metabolites in male mice
| Parameters | Plin4+/+ | Plin4−/− | P Value |
|---|---|---|---|
| Body weight, g | 28.1 ± 2.1 | 28.3 ± 2.2 | 0.82 |
| Glucose, mg/dl | 168 ± 20 | 175 ± 26 | 0.50 |
| NEFA, mmol | 0.52 ± 0.21 | 0.50 ± 0.15 | 0.77 |
| Glycerol, mg/dl | 29.4 ± 5.5 | 29.9 ± 6.9 | 0.86 |
| TAG, mg/dl | 50.9 ± 6.4 | 47.4 ± 8.4 | 0.32 |
| TC, mg/dl | 105.7 ± 12.3 | 103.2 ± 21.2 | 0.76 |
| Perigonadal WAT, %body wt | 3.31 ± 0.52 | 2.37 ± 0.19 | 0.17 |
| BAT, %body wt | 0.28 ± 0.01 | 0.24 ± 0.01 | 0.10 |
| Fat mass, %body wt | 12.9 ± 5.7 | 9.14 ± 2.9 | 0.26 |
| Lean mass, %body wt | 77.7 ± 5.5 | 80.7 ± 2.4 | 0.33 |
Data are shown as means ± SD; n = 9 mice per group. Plasma concentrations of glucose, lipids, and metabolites were measured in 4-h-fasted male 12- to 13-wk-old mice maintained on a standard diet. Statistical significance was determined by 2-tailed Student's t-test. NEFA, nonesterified fatty acid; TAG, triacyglycerol; TC, total cholesterol; WAT, epididymal white adipose tissue; BAT, brown adipose tissue.
Plin4 is enriched in adipose tissue and highly induced during adipocyte differentiation (8). To determine whether Plin4 plays a role in fat cell differentiation, we performed in vitro differentiation using SVCs isolated from wild-type and Plin4−/− mice. Using a standard differentiation protocol (5), we found that wild-type and Plin4−/− cells behaved in an identical fashion in terms of the timing of upregulation of transcripts for the master adipogenesis transcription factors C/EBPα and PPARγ as well as mature adipocyte marker ap2 (Fig. 2A). The cellular TAG contents in the fully differentiated adipocytes at day 8 were also not different between wild-type and Plin4−/− cells (Fig. 2B). The identical findings in the two genotypes indicate that the absence of Plin4 does not affect mouse adipocyte differentiation in vitro.
Fig. 2.

Absence of Plin4 does not affect normal adipogenesis and lipolysis in vitro and in vivo. A: quantitative RT-PCR analyses of master adipocyte differentiation transcription factors (C/EBPα and PPARγ) as well as mature adipocyte marker ap2 during standard hormone-induced adipocyte differentiation of stromal vascular cells (SVC) isolated from subcutaneous fat of Plin4+/+ and Plin4−/− mice. Data were normalized to cyclophilin A and expressed as relative fold changes compared with WT at D0 (the day when the differentiation media was added). B: cellular triglyceride contents of fully differentiated cells isolated at day 8. C and D: basal and CL-316243 (CL)-induced in vitro lipolysis measured by NEFA (C) and glycerol (D) from fully differentiated SVC-derived adipocytes. E and F: 13-wk-old male Plin4+/+ and Plin4−/− mice (n = 5 each) were fasted for 4 h and treated with CL at 0.1 mg/kg ip for in vivo lipolysis. Basal and stimulated plasma NEFA (E) and glycerol (F) levels are presented as means ± SE. *P < 0.05 between −CL and +CL in the same genotype.
We observed no difference in the mass of WAT or BAT in wild-type and Plin4−/− mice (Table 1), indicating that the absence of Plin4 does not affect adipose differentiation and growth in vivo. The histological appearance of the fat cells and size of the lipid droplets in those two genotypes also appeared not to be different (data not shown). Therefore, loss of Plin4 has no effect on adipose tissue differentiation and growth in vivo.
The PAT domain protein Plin1 is known to regulate lipolysis. Plin4, like Plin1, is most highly expressed in WAT (8). We examined whether Plin4 shared lipolytic function like Plin1. We first measured lipolysis under basal conditions and after β-adrenergic stimulation in fully differentiated SVC adipocytes. Under basal conditions, there was lower but steady secretion of NEFA and glycerol from wild-type and Plin4−/− adipocytes (Fig. 2C). Treatment with CL-316243, a specific β3-adrenergic agonist, led to a higher release of both NEFA and glycerol at the 2- and 4-h time points. The release of NEFA plateaued at 2 h, whereas glycerol was still increasing till 4 h. However, there was no significant difference in either basal or CL-316243-stimulated lipolysis between wild-type and Plin4−/− adipocytes in vitro, both showing a ∼1.5-fold increase in the rate of NEFA release and a 2.5-fold increase in the rate of glycerol release following CL-316243 treatment (Fig. 2C).
We next measured the basal and stimulated in vivo lipolysis rates in Plin4−/− mice. As indicated in Fig. 2D, fasting basal plasma NEFA and glycerol levels were similar in Plin4−/− mice compared with their wild-type counterparts. Administration of CL-316243 led to a 1.5-fold stimulation of NEFA, and a 2-fold increase of glycerol, without any significant difference in response between wild-type and Plin4−/− mice. Overall, results from SVC-derived adipocytes in vitro and whole animals in vivo indicate that Plin4 does not play a significant role in the lipolysis of adipose tissue, nor is Plin4 required for the optimal lipolytic activity of Plin1.
Plin4-deficient mice have reduced cardiac TAG contents but maintain normal heart function.
Plin4 is expressed at relatively low levels in oxidative tissues such as skeletal muscle and heart (8, 24). We measured the lipid contents in these tissues in wild-type and Plin4−/− mice. TLC revealed a dramatic decrease of TAG content of the left ventricle of 4-h-fasted Plin4−/− mice (Fig. 3A). Enzymatic analysis of the total TAG content extracted from the heart of these animals confirmed that there was an ∼58% reduction in the intracardiac TAG content of male Plin4−/− mice (1.0 ± 0.12 μg/mg tissue) compared with their wild-type littermates (2.4 ± 0.16 μg/mg tissue) (Fig. 3B). There was no difference in the TAG contents of BAT and soleus muscle, whereas we consistently observed a tendency of lower, although nonsignificant, TAG content in the liver of Plin4−/− mice (Fig. 3B). Female Plin4−/− mice also showed a similar degree of reduction in the amount of intracardiac TAG (data not shown). Despite the reduced TAG content, the weights of Plin4−/− hearts as a percentage of body weight were similar in wild-type and Plin4−/− hearts (Fig. 3C). Furthermore, echocardiography revealed no difference in ventricular wall thicknesses or left ventricular inner diameters between the two genotypes in either young or aged mice (Table 2). The left ventricular mass tended to be lower in Plin4−/− than in wild-type mice in both young and aged animals, but the difference was statistically not significant (Table 2). Echocardiography analysis also showed that the percent fractional shortening and ejection fraction were similar in Plin4−/− and wild-type controls at the two age groups (Fig. 3D and Table 2), indicating that cardiac function remained normal despite reduced cardiac TAG contents in young and aged animals.
Fig. 3.

Plin4 absence reduces intracardiac triacylglycerol (TAG) content without affecting heart function. A: TLC analysis of total lipids extracted from ventricles isolated from 4-h-fasted 40-wk-old male Plin4+/+ and Plin4−/− mice (n = 5 each). Lipid standards were used to identify each lipid band. B: direct enzymatic analyses of triglyceride content of ventricle, soleus muscle (SM), brown adipose tissue (BAT), and liver of male Plin4+/+ and Plin4−/− mice (n = 7 in each group). **P < 0.005. C: ventricle wet weights as expressed as percentage of body weight in 13-wk-old male Plin4+/+ and Plin4−/− mice (n = 6–8 each). D: Percentage of fraction shortening in 13-wk-old male Plin4+/+ and Plin4−/− mice (n = 6–8 each).
Table 2.
Echocardiogram diameter and functional characteristics
| 18 wk old |
15 mo old |
|||
|---|---|---|---|---|
| Characteristics | Plin4+/+ (n = 6) | Plin4−/− (n = 8) | Plin4+/+ (n = 7) | Plin4−/− (n = 7) |
| Diastole | ||||
| IVST, mm | 0.51 ± 0.05 | 0.48 ± 0.04 | 0.55 ± 0.03 | 0.52 ± 0.02 |
| LVID, mm | 4.01 ± 0.08 | 3.94 ± 0.15 | 4.39 ± 0.1 | 4.09 ± 0.21 |
| LVPWT, mm | 0.9 ± 0.1 | 0.76 ± 0.03 | 0.78 ± 0.05 | 0.83 ± 0.08 |
| Systole | ||||
| IVST, mm | 0.72 ± 0.05 | 0.64 ± 0.03 | 0.79 ± 0.02 | 0.74 ± 0.02 |
| LVID, mm | 2.74 ± 0.05 | 2.79 ± 0.14 | 3.05 ± 0.08 | 2.83 ± 0.23 |
| LVPWT, mm | 1.26 ± 0.1 | 1.14 ± 0.04 | 1.1 ± 0.07 | 1.16 ± 0.12 |
| %EF | 61.9 ± 1.11 | 60.4 ± 2.8 | 57.9 ± 1.94 | 59.9 ± 2.98 |
| LV mass, mg | 103.5 ± 7.74 | 87.2 ± 2.2 | 109 ± 6.69 | 98 ± 7.1 |
Values are presented as means ± SE. Transthoracic echocardiogram imaging was performed on conscious 18-wk- and 15-mo-old male Plin4+/+ and Plin4−/− mice. M-mode images were analyzed. IVST, interventricular septum thickness; LVID, left ventricular inner diameter; LVPWT, left ventricular posterior wall thickness; %EF, percent ejection fraction; LV, left ventricle. No significant differences were found between the 2 genotypes at the same age.
Absence of Plin4 is associated with downregulated Plin5 expression with no effect on metabolic gene expression.
We examined the expression of LD protein genes in the heart of 13-wk-old male mice in response to the loss of Plin4 by real-time PCR. The mRNA level of Plin4 was reduced by 75% in Plin4−/− mice compared with wild-type mice when we used our real-time PCR primers that specifically quantified the 3′-noncoding region of Plin4. There was no change in the mRNA expression of Plin3 and Plin2, both highly expressed LDPs in the heart tissues. Unexpectedly, the mRNA level of Plin5, a recently identified LDP enriched in heart tissue, was decreased by 38% (Fig. 4A). Western blotting confirmed the complete absence of Plin4 protein, and no changes of Plin2 and Plin3 protein expressions in 13-wk-old Plin4−/− mice (Fig. 4, B and C). Interestingly, the protein level of Plin5 was reduced by 87% (Fig. 4, B and C), relatively more than expected from the moderately reduced mRNA level, suggesting that Plin5 expression may be reduced at both transcriptional and posttranscriptional levels in the absence of Plin4.
Fig. 4.

Plin4 inactivation blunted both mRNA and protein expressions of Plin5 in ventricles of Plin4−/− mice without affecting other genes involved in lipid and glucose metabolism. A: real-time PCR analyses of mRNA abundances of perlipin family LDPs (Plin4, -5, -2, and -3) in ventricles of 13-wk-old male Plin4+/+ and Plin4−/− (n = 5 each). We had to use primers targeting 3′-uncoding regions of Plin4 gene to achieve specificity, which resulted in 25% residual mRNA detected in ventricles of Plin4−/− mice. B: representative immunoblot (n = 4) of Plin4, Plin5, Plin2, and Plin3 in ventricles of 13-wk-old male Plin4+/+ and Plin4−/− mice. C: fold changes of Plin5, -2, and -3 protein expression by densitometry analyses using Image J. D: Quantitative RT-PCR analyses of mRNA expression of genes involved in lipid and glucose metabolism in Plin4+/+ and Plin4−/− ventricles (n = 5 each). Data were normalized to 4 housekeeping genes (cyclophilin A, Eelf1γ, Hmbs, and Gapdh) based on Genorm algorithm (medgen.ugent.be/genorm/) and expressed as fold changes relative to WT controls. **P < 0.005 between 2 genotypes.
To determine the effects of Plin4 gene ablation on cardiac substrate metabolism, we measured the expression of genes involved in glucose and fatty acid uptake, and peroxisomal and mitochondrial β-oxidation in the heart. The expression levels of fatty acid transport proteins (FATP1 and -4), CD36, Pparα, Cpt1α, Glut4, and pyruvate dehydrogenase kinase 4 (Pdk4) were similar in the two groups (Fig. 4C). There were also no differences in the mRNA expression of the two major cardiac intracellular TAG lipases (Atgl and Hsl) or the lipase responsible for fatty acid uptake from lipoprotein-TAG (Lpl) (Fig. 4C). These data suggest that those pathways or enzymes regulating cardiac substrate metabolism are not perturbed at the mRNA expression level, and they do not mediate the TAG reduction in the ventricles of Plin4−/− mice.
We next analyzed the mRNA expression of perilipin family LDPs in the WAT tissue and observed the same degree of Plin5 downregulation at the mRNA level in Plin4−/− mice as in the heart (Fig. 5A). No changes were observed in the mRNA expression of Plin1, Plin2, and Plin3. Meanwhile, the protein expression of Plin1 and Plin3 in the WAT was not different in the two genotypes (Fig. 5B). However, as previously reported, we failed to detect any Plin2 protein in mature WAT (27); likewise, the expression of Plin5 in mature WAT was also very low and below the detection limit of our antibody (data not shown; see also Ref. 24). In the liver, where Plin4 and Plin1 are normally not expressed at either mRNA or protein level (Ref. 24 and our own unpublished data), we observed similar degrees of Plin5 downregulation at both mRNA (47%) and protein levels (∼80%) in the Plin4−/− mice (Fig. 5, C and D). The expression of two major liver LDPs, Plin2 and Plin3, was not significantly changed at either mRNA or protein levels between wild-type or Plin4−/− mice. These findings indicate that deletion of Plin4 in our mice seems to cause downregulation of Plin5 at both mRNA and protein levels not only in heart but also in other tissues including WAT and liver.
Fig. 5.

Deletion of Plin4 leads to decreased Plin5 expression in other tissues including epididymal WAT and liver of Plin4−/− mice. A: mRNA abundances of perlipin family LDPs (Plin4, 5, 1, 2, and 3) in WAT of 13-wk-old male Plin4+/+ and Plin4−/− mice (n = 6 each). B: representative immunoblot (n = 4 each) of Plin1, -3, and -4 in the WAT of 13-wk-old male Plin4+/+ and Plin4−/− mice. C: mRNA abundances of Plin5, -2, and -3 (n = 6 each). D: representative immunoblot (n = 2) of Plin5, -2, and -3 in liver of 30-wk-old male Plin4+/+ and Plin4−/− mice. Real-time PCR data were normalized to 3 housekeeping genes (cyclophilin A, Eelf1γ, and Gapdh) based on Genorm algorithm (medgen.ugent.be/genorm/) and expressed as fold changes relative to WT controls. *P < 0.05 between 2 genotypes. β-Actin and Gapdh were used as loading controls for Western blot analyses in WAT and liver, respectively.
Plin4 inactivation protects the heart from induced cardiac steatosis associated with prolonged fasting, HFD, or genetic obesity.
Long-term fasting has been shown to induce TAG accumulation and Plin5 expression in heart (21). We studied how Plin4−/− mice respond to long-term fasting in terms of TAG accumulation and Plin5 protein expression. Analysis of cardiac TAG content demonstrated that there was a 2.27-fold increase in heart TAG after a 48-h fast relative to a 4-h fast in wild-type mice (Fig. 6A). We observed in 4-h-fasted Plin4−/− mice a 32% lower intracardiac TAG content compared with wild-type mice. A 48-h fast in Plin4−/− mice led to a 1.9-fold increase in cardiac TAG content compared with 4-h-fasted Plin4−/− mice. However, compared with wild-type mice fasted for 48 h, Plin4−/− mice fasted for the same time period displayed a total cardiac TAG content that was only 59% of that of wild-type (Fig. 6A, top). Immunoblot analysis revealed that both the Plin4 protein and mRNA levels of the left ventricle of wild-type mice were not much perturbed after a 48-h fast compared with 4 h, whereas that of Plin5 protein increased at least approximately twofold albeit no obvious upregulation at the mRNA level (Fig. 6, A, bottom, and B). Again, the Plin5 protein level in the heart of Plin4−/− mice was much lower than that in wild-type mice after a 4-h fast; the marked increase in cardiac Plin5 level after a 48-h fast in wild-type mice was also greatly curtailed in Plin4−/− animals, commensurate with changes in its mRNA level (Fig. 6, A, bottom, and B, middle). Consistent with what was reported in Plin5−/− heart (11), we also observed a lower protein expression of Plin2 in the hearts of Plin4−/− mice that were older than 24 wk at both 4-h and 48-h fasting conditions (Fig. 6A) despite no significant difference in mRNA level between wild-type and Plin4−/− mice (Fig. 6B, bottom). Long-term fasting increased cardiac mRNA and protein expressions of Plin2 in both wild-type and Plin4−/− mice. These data suggest that Plin5 and Plin2 levels increase to allow these two Plin proteins to coat the newly formed LD induced by long-term fasting.
Fig. 6.

Inactivation of Plin4 prevents long-term fast-induced cardiac steatosis accompanied by reduced Plin5 expression. A: TAG contents (n = 6–8 each) and representative immunoblot of cardiac Plin4, -5, and -2 expression in ventricles from 9-mo-old female Plin4+/+ and Plin4−/− under 4-h and 48-h fasts. B: real-time PCR analyses of Plin4, -5, and -2 in ventricles from 9-mo-old female Plin4+/+ and Plin4−/− under 4-h and 48-h fasts. **P < 0.005, *P < 0.05 between 2 genotypes; #P < 0.05 vs. 4-h fast among same genotype but no difference between the 2 genotypes at 48-h fast.
In wild-type (Plin4+/+) mice, both HFD feeding and the presence of leptin−/− genetic obesity led to an increase of cardiac TAG accumulation compared with normal chow diet (NCD)-fed wild-type mice (Fig. 7A). Analysis of LDP revealed that Plin2 was the major LDP that was modestly increased at mRNA and protein levels in the HFD-fed and genetically obese Lep−/− hearts. It is noteworthy that we found that cardiac Plin5 was upregulated only in the HFD model at both mRNA and protein levels, but there was no change in Plin4 expression at the protein or mRNA level in HFD and Lep−/− steatotic heart models (Fig. 7, B and C). Interestingly, in HFD-fed Plin4−/− mice, TAG accumulation in heart was only 61.6% that of wild-type (Fig. 8A, top). A similar degree of protection against left ventricular cardiac TAG accumulation was also observed in leptin-deficient mice with Plin4−/− background (2.94 ± 0.24 μg TAG/mg tissue) compared with leptin-deficient mice with wild-type background (Leptin−/− 5.21 ± 0.48 μg TAG/mg) (Fig. 8C, top). Immunoblot analysis confirmed that in both genotypes absence of Plin4 was associated with an ∼80% downregulation of Plin5 protein level in the left ventricles along with modestly lower expression of Plin2 (Fig. 8A and C bottom). Again, the mRNA levels of Plin5 in the heart were downregulated by no more than 40% in obese HFD or Lep−/− Plin4−/− mice, whereas Plin2 mRNA remained constant compared with wild-type (Fig. 8, B and D). These data highlight that loss of Plin4, accompanied by downregulation of Plin5, protects against TAG accumulation in the heart that occurs under different genetic and dietary manipulations. Furthermore, both Plin5 and Plin2 protein expressions are regulated posttranscriptionally in hearts.
Fig. 7.
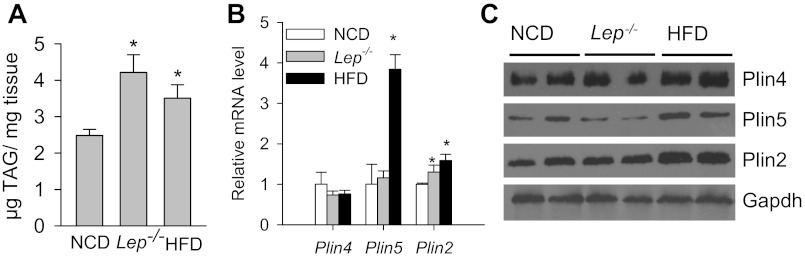
Perilipin family LDP expression in hearts with increased cardiac lipid accumulation. A: cardiac TAG accumulation in normal chow diet (NCD)-fed, obese lep−/−, and 10-wk high-fat diet (HFD)-fed WT (Plin4+/+) mice. Male mice 16–20 wk old were fasted for 4 h (n = 6–8 each). mRNA (B) and protein (C) analyses of major LDPs in ventricles from the aforesaid mice after 4-h fast. *P < 0.05 vs. NCD-fed mice.
Fig. 8.

Inactivation of Plin4 prevents HFD and geneticallty obese-induced cardiac steatosis accompanied by reduced Plin5 expression. A: TAG accumulation (n = 6–8 each, top) and representative immunoblot of cardiac Plin4, -5, and -2 expression (n = 2, bottom). B: real-time PCR analyses of LDPs in ventricles from 10-wk HFD-fed male Plin4+/+ and Plin4−/− mice after 4-h fast. C: TAG accumulation (n = 6–8 each, top) and representative immunoblot of cardiac Plin4, -5, and -2 expression (n = 2, bottom). D: real-time PCR analyses of LDPs in ventricles from 20-wk-old genetically obese female Lep−/−Plin4+/+ and Lep−/−Plin4−/− mice after 4-h fast. **P < 0.005, *P < 0.05 between 2 genotypes.
DISCUSSION
The two major findings of our study are that 1) as a relatively adipose-specific perilipin family LDP member, loss of Plin4 in mice does not affect adipose tissue development and maintenance; 2) loss of Plin4 concomitantly leads to reduction of Plin5 at both mRNA and protein levels, especially in heart, and decreases cardiac lipid accumulation under basal conditions while protecting it from dietary and genetically induced cardiac steatosis.
All four members of the perilipin family of lipid droplet proteins (Plin1, -2, -3,and -4) are expressed in adipose tissue. Plin1 is the most abundant PAT protein, which coats the mature lipid droplet in mature adipose tissue. Inactivation of Plin1 in mice leads to partial lipodystrophy due to unbridled basal lipolysis, suggesting a critical role of Plin1 in regulating adipose tissue function (14, 22). Plin2 was shown to coat newly formed small lipid droplets during the early stage of adipocyte differentiation but rapidly turns over via ubiquitination-mediated proteasome degradation in mature adipocytes (15, 27). Plin2 deficiency has no obvious effect on murine adipose tissue development (4). The two other LDPs, Plin3 and Plin4, are both expressed at basal levels in preadipocytes. During adipocyte differentiation, the expression of Plin4 is greatly induced, presumably through PPARγ activation, whereas the expression level of Plin3 stays relatively constant (8). Those two proteins have been shown only to coat the small nascent lipid droplets in 3T3-L1 adipocytes upon lipid loading. Little is known concerning the physiological function of Plin4 in adipose tissue development. In the Plin4−/− mice, there were no abnormalities observed on fat tissue development or distribution or lipolytic responses whether the mice were fed a normal chow (Table 1 and Fig. 2) or an HFD diet or when the mice were bred into a genetic obese (ob/ob) background (data not shown). Studies using Plin4−/− SVC cells further corroborated that adipocytes can mature normally and display unaltered basal and stimulated lipolysis in the absence of Plin4. No compensatory changes in Plin1, Plin2, or Plin3 at mRNA expression levels were detected during different stages of adipocyte differentiation in Plin4−/− SVC cells (data not shown). Therefore, Plin4 appears to be dispensable during fat cell development, which is not surprising considering the presence of other LDPs. These data suggest that Plin4 plays a redundant function in adipocyte development and homeostasis.
Unexpectedly, we found that deletion of Plin4 moderately downregulated Plin5 expression at the mRNA level. Interestingly, the level of Plin5 protein especially in tissues where it is highly expressed such as heart, was even more dramatically reduced, an effect likely to be mediated posttranscriptionally (Figs. 4 and 6). The exact reason why deletion of Plin4 affects mRNA expression of Plin5 remains unknown. In the murine genome, these two genes are next to each other, with the end of the Plin5 gene only 1.7 kb upstream of Plin4 in the same orientation, a genomic arrangement that is conserved in humans (Fig. 9). Plin5 has been shown to be activated by PPARα in liver and skeletal and cardiac muscles. It is also regulated by PPARγ in human and murine adipose tissues (24). However, promoter analysis of Plin5 failed to identify any cis-element in the 2.1-kb region upstream of the Plin5 initiation site that would confer transactivation by PPARs (28). Instead, functional peroxisome proliferator response elements (PPRE) are present downstream of Plin5 gene, which falls within the promoter region of Plin4 gene. Whether these PPREs are responsible for transactivation of Plin5 remains to be determined; nevertheless, these PPRE regions upstream of Plin4 gene are intact in Plin4−/− mice. Further analysis of the deleted 7.58-kb region immediately after the Plin4 ATG start codon to its stop codon by PPRE prediction algorithm (http://www.classicrus.com/PPRE/) revealed two potential PPRE regulatory elements at positions 2429 and 4035. Whether these predicted PPRE elements mediate the upstream transactivation of Plin5 awaits further detailed study (Fig. 9). It is noteworthy that the downregulation of Plin5 expression is much more drastic at the protein level (87%) vs. the mRNA level (∼38%), suggesting a significant posttranscriptional component to the regulation of Plin5. We speculate that, like Plin2, the protein expression of Plin5 may also be regulated posttranscriptionally depending on the presence and the amount of lipids in the tissue, a possibility that requires further investigation.
Fig. 9.

Genomic structure of Plin5 and Plin4 in the mouse genome. Relative distances between the genes and the characterized peroxisome proliferator response elements (PPREs) in the intact promoter region of Plin4 and the predicted PPREs in the deleted region of Plin4 gene were noted.
In Plin5−/− mice, the expression of Plin4 at the mRNA level was not perturbed by semiquantitative RT-PCR (11), although the protein level was not analyzed. This suggests that there is no reciprocal regulation between Plin4 and Plin5. We attempted to dissect whether in vitro manipulation of Plin4 by knockdown or overexpression affects Plin5 expression in cultured primary cardiac myocytes as well as in cardiac cell lines. Expression constructs of Plin4 cDNA fail to transduce primary cardiomyocytes or HL-1 cells [a contracting mouse cardiac muscle cell line that preserves much of the phenotypic characteristics of adult cardiomyocytes (6)], possibly partly because of the very large size of the cDNA (∼4.2 kb). Furthermore, neither Plin4 nor Plin5 protein is expressed in HL-1 cells (data not shown). We found that primary neonatal cardiomyocytes expressed Plin4, but they lost the expression as they were grown in culture, which makes it impossible to perform Plin4 knockdown experiments in these cells (data not shown). We further found that commercially available lentivirus that expressed shRNAs can knockdown Plin4 mRNA expression by no more than 30% in mature adipocytes, which abundantly express Plin4, but this degree of knockdown had no significant effect on Plin5 expression (data not shown). These experimental limitations have prevented us from further exploring the relationship between Plin4 and Plin5 expression in vitro.
The loss of Plin4 clearly protects against lipid accumulation in the heart. However, whether the absence of Plin4 is directly involved in the protection or via downregulation of Plin5 expression is unclear. Interestingly, Plin5−/− mice are also protected against cardiac TAG accumulation to about the same degree as Plin4−/− mice (11). Detailed studies revealed an essential role of Plin5 in maintaining LDs in the heart by antagonizing the hydrolysis of TAG by lipases (especially ATGL). Loss of Plin5 also subjects the heart to oxidative stress because of excess reactive oxygen species production, which further leads to impaired heart function in aged Plin5−/− mice (11). However, we failed to observe such deleterious consequences in Plin4−/− mice (Fig. 4, Table 2, and data not shown). It is likely that the persistence of ∼20% Plin5 protein in Plin4−/− mice may spare the mice the pathological effects of total absence of Plin5.
In summary, by creating Plin4−/− mice, we have identified that Plin4 is not essential for adipose development or lipolysis. Loss of Plin4 in mice leads to downregulated expression of Plin5 at both mRNA and protein levels. It also reduces cardiac TAG accumulation under basal conditions and after dietary or genetic manipulation.
GRANTS
This work was supported by grants from the US National Institutes of Health, HL-51586 (to L. Chan) and P30 DK-079638 for the Diabetes and Endocrinology Research Center at Baylor College of Medicine. L. Chan was also supported by the Betty Rutherford Chair in Diabetes Research.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: W.C., B.C., M.S., and L.C. conception and design of research; W.C., X.W., and L.L. performed experiments; W.C., X.W., and L.L. analyzed data; W.C., B.C., and L.C. interpreted results of experiments; W.C. prepared figures; W.C. drafted manuscript; W.C., M.S., and L.C. edited and revised manuscript; L.C. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank members of the Lawrence Chan laboratory for helpful discussions. We also thank Dr. Perry Bickel from The University of Texas for kindly providing the antibody against Plin4 and Dr. Nathen E. Wollins from Washington University in St. Louis for the Plin4 mammalian expression vector. We are grateful to Dr. William C. Claycomb from Louisiana State University Medical Center for providing the HL-1 cells.
W. Chen's current address: Dept. of Physiology, Medical College of Georgia at Georgia Regents University, 1120 15th St., Augusta GA 30912.
REFERENCES
- 1. Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37: 911–917, 1959 [DOI] [PubMed] [Google Scholar]
- 2. Brasaemle DL, Barber T, Wolins NE, Serrero G, Blanchette-Mackie EJ, Londos C. Adipose differentiation-related protein is an ubiquitously expressed lipid storage droplet-associated protein. J Lipid Res 38: 2249–2263, 1997 [PubMed] [Google Scholar]
- 3. Brown DA. Lipid droplets: proteins floating on a pool of fat. Curr Biol 11: R446–449, 2001 [DOI] [PubMed] [Google Scholar]
- 4. Chang BH, Li L, Paul A, Taniguchi S, Nannegari V, Heird WC, Chan L. Protection against fatty liver but normal adipogenesis in mice lacking adipose differentiation-related protein. Mol Cell Biol 26: 1063–1076, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen W, Chang B, Saha P, Hartig SM, Li L, Reddy VT, Yang Y, Yechoor V, Mancini MA, Chan L. Berardinelli-seip congenital lipodystrophy 2/seipin is a cell-autonomous regulator of lipolysis essential for adipocyte differentiation. Mol Cell Biol 32: 1099–1111, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Claycomb WC, Lanson NA, Jr, Stallworth BS, Egeland DB, Delcarpio JB, Bahinski A, Izzo NJ., Jr HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci USA 95: 2979–2984, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dalen KT, Dahl T, Holter E, Arntsen B, Londos C, Sztalryd C, Nebb HI. LSDP5 is a PAT protein specifically expressed in fatty acid oxidizing tissues. Biochim Biophys Acta 1771: 210–227, 2007 [DOI] [PubMed] [Google Scholar]
- 8. Dalen KT, Schoonjans K, Ulven SM, Weedon-Fekjaer MS, Bentzen TG, Koutnikova H, Auwerx J, Nebb HI. Adipose tissue expression of the lipid droplet-associating proteins S3–12 and perilipin is controlled by peroxisome proliferator-activated receptor-gamma. Diabetes 53: 1243–1252, 2004 [DOI] [PubMed] [Google Scholar]
- 9. Jimenez MA, Akerblad P, Sigvardsson M, Rosen ED. Critical role for Ebf1 and Ebf2 in the adipogenic transcriptional cascade. Mol Cell Biol 27: 743–757, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kimmel AR, Brasaemle DL, McAndrews-Hill M, Sztalryd C, Londos C. Adoption of PERILIPIN as a unifying nomenclature for the mammalian PAT-family of intracellular lipid storage droplet proteins. J Lipid Res 51: 468–471, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kuramoto K, Okamura T, Yamaguchi T, Nakamura TY, Wakabayashi S, Morinaga H, Nomura M, Yanase T, Otsu K, Usuda N, Matsumura S, Inoue K, Fushiki T, Kojima Y, Hashimoto T, Sakai F, Hirose F, Osumi T. Perilipin 5, a Lipid Droplet-binding Protein, Protects Heart from Oxidative Burden by Sequestering Fatty Acid from Excessive Oxidation. J Biol Chem 287: 23852–23863, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu P, Ying Y, Zhao Y, Mundy DI, Zhu M, Anderson RG. Chinese hamster ovary K2 cell lipid droplets appear to be metabolic organelles involved in membrane traffic. J Biol Chem 279: 3787–3792, 2004 [DOI] [PubMed] [Google Scholar]
- 13. Lu X, Gruia-Gray J, Copeland NG, Gilbert DJ, Jenkins NA, Londos C, Kimmel AR. The murine perilipin gene: the lipid droplet-associated perilipins derive from tissue-specific, mRNA splice variants and define a gene family of ancient origin. Mamm Genome 12: 741–749, 2001 [DOI] [PubMed] [Google Scholar]
- 14. Martinez-Botas J, Anderson JB, Tessier D, Lapillonne A, Chang BH, Quast MJ, Gorenstein D, Chen KH, Chan L. Absence of perilipin results in leanness and reverses obesity in Lepr(db/db) mice. Nat Genet 26: 474–479, 2000 [DOI] [PubMed] [Google Scholar]
- 15. Masuda Y, Itabe H, Odaki M, Hama K, Fujimoto Y, Mori M, Sasabe N, Aoki J, Arai H, Takano T. ADRP/adipophilin is degraded through the proteasome-dependent pathway during regression of lipid-storing cells. J Lipid Res 47: 87–98, 2006 [DOI] [PubMed] [Google Scholar]
- 16. Miura S, Gan JW, Brzostowski J, Parisi MJ, Schultz CJ, Londos C, Oliver B, Kimmel AR. Functional conservation for lipid storage droplet association among Perilipin, ADRP, and TIP47 (PAT)-related proteins in mammals, Drosophila, and Dictyostelium. J Biol Chem 277: 32253–32257, 2002 [DOI] [PubMed] [Google Scholar]
- 17. Murphy DJ. The biogenesis and functions of lipid bodies in animals, plants and microorganisms. Prog Lipid Res 40: 325–438, 2001 [DOI] [PubMed] [Google Scholar]
- 18. Nagai S, Shimizu C, Umetsu M, Taniguchi S, Endo M, Miyoshi H, Yoshioka N, Kubo M, Koike T. Identification of a functional peroxisome proliferator-activated receptor responsive element within the murine perilipin gene. Endocrinology 145: 2346–2356, 2004 [DOI] [PubMed] [Google Scholar]
- 19. Scherer PE, Bickel PE, Kotler M, Lodish HF. Cloning of cell-specific secreted and surface proteins by subtractive antibody screening. Nat Biotechnol 16: 581–586, 1998 [DOI] [PubMed] [Google Scholar]
- 20. Shimizu M, Yamashita D, Yamaguchi T, Hirose F, Osumi T. Aspects of the regulatory mechanisms of PPAR functions: analysis of a bidirectional response element and regulation by sumoylation. Mol Cell Biochem 286: 33–42, 2006 [DOI] [PubMed] [Google Scholar]
- 21. Suzuki J, Shen WJ, Nelson BD, Selwood SP, Murphy GM, Jr, Kanehara H, Takahashi S, Oida K, Miyamori I, Kraemer FB. Cardiac gene expression profile and lipid accumulation in response to starvation. Am J Physiol Endocrinol Metab 283: E94–E102, 2002 [DOI] [PubMed] [Google Scholar]
- 22. Tansey JT, Sztalryd C, Gruia-Gray J, Roush DL, Zee JV, Gavrilova O, Reitman ML, Deng CX, Li C, Kimmel AR, Londos C. Perilipin ablation results in a lean mouse with aberrant adipocyte lipolysis, enhanced leptin production, and resistance to diet-induced obesity. Proc Natl Acad Sci USA 98: 6494–6499, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wolins NE, Quaynor BK, Skinner JR, Schoenfish MJ, Tzekov A, Bickel PE. S3–12, Adipophilin, and TIP47 package lipid in adipocytes. J Biol Chem 280: 19146–19155, 2005 [DOI] [PubMed] [Google Scholar]
- 24. Wolins NE, Quaynor BK, Skinner JR, Tzekov A, Croce MA, Gropler MC, Varma V, Yao-Borengasser A, Rasouli N, Kern PA, Finck BN, Bickel PE. OXPAT/PAT-1 is a PPAR-induced lipid droplet protein that promotes fatty acid utilization. Diabetes 55: 3418–3428, 2006 [DOI] [PubMed] [Google Scholar]
- 25. Wolins NE, Rubin B, Brasaemle DL. TIP47 associates with lipid droplets. J Biol Chem 276: 5101–5108, 2001 [DOI] [PubMed] [Google Scholar]
- 26. Wolins NE, Skinner JR, Schoenfish MJ, Tzekov A, Bensch KG, Bickel PE. Adipocyte protein S3–12 coats nascent lipid droplets. J Biol Chem 278: 37713–37721, 2003 [DOI] [PubMed] [Google Scholar]
- 27. Xu G, Sztalryd C, Lu X, Tansey JT, Gan J, Dorward H, Kimmel AR, Londos C. Post-translational regulation of adipose differentiation-related protein by the ubiquitin/proteasome pathway. J Biol Chem 280: 42841–42847, 2005 [DOI] [PubMed] [Google Scholar]
- 28. Yamaguchi T, Matsushita S, Motojima K, Hirose F, Osumi T. MLDP, a novel PAT family protein localized to lipid droplets and enriched in the heart, is regulated by peroxisome proliferator-activated receptor alpha. J Biol Chem 281: 14232–14240, 2006 [DOI] [PubMed] [Google Scholar]