

Abstract
Among the over 300 members of the solute carrier (SLC) group of integral plasma membrane transport proteins are the nine electroneutral cation-chloride cotransporters belonging to the SLC12 gene family. Seven of these transporters have been functionally described as coupling the electrically silent movement of chloride with sodium and/or potassium. Although in silico analysis has identified two additional SLC12 family members, no physiological role has been ascribed to the proteins encoded by either the SLC12A8 or the SLC12A9 genes. Evolutionary conservation of this gene family from protists to humans confirms their importance. A wealth of physiological, immunohistochemical, and biochemical studies have revealed a great deal of information regarding the importance of this gene family to human health and disease. The sequencing of the human genome has provided investigators with the capability to link several human diseases with mutations in the genes encoding these plasma membrane proteins. The availability of bacterial artificial chromosomes, recombination engineering techniques, and the mouse genome sequence has simplified the creation of targeting constructs to manipulate the expression/function of these cation-chloride cotransporters in the mouse in an attempt to recapitulate some of these human pathologies. This review will summarize the three human disorders that have been linked to the mutation/dysfunction of the Na-Cl, Na-K-2Cl, and K-Cl cotransporters (i.e., Bartter's, Gitleman's, and Andermann's syndromes), examine some additional pathologies arising from genetically modified mouse models of these cotransporters including deafness, blood pressure, hyperexcitability, and epithelial transport deficit phenotypes.
Keywords: Bartter's syndrome, Na-Cl cotransport, Na-K-2Cl cotransport, K-Cl cotransport, Gitelman's syndrome, Bartter's syndrome, Andermann's syndrome, brain excitability, inner ear, mouse knockouts
the solute carrier 12 (SLC12) gene family encodes electroneutral inorganic cation-chloride cotransporters (CCCs) that are plasma membrane proteins mediating the movement of inorganic sodium (Na+) and/or potassium (K+) cations, tightly coupled to the movement of chloride (Cl−) anions. These transporters play several important roles in human physiology, many of which we will thoroughly examine in this review. Most of our current knowledge is based on basic comparative studies involving many diverse animal species. In this review, we will focus on the increasing use of genetically modified mice to model human pathologies and diseases.
The SLC12 family can be divided into two major branches, defined not only by their transport properties but also by their amino acid sequences which point to an ancient separation of their respective genes (Fig. 1A). SLC12A1–A3 form the Na+-dependent branch of CCCs with two Na-K-2Cl cotransporters (NKCCs, members A1 and A2) and one Na-Cl cotransporter (NCC, member A3), whereas genes SLC12A4–A7 encode four Na+-independent K-Cl cotransporters (KCCs). As with many other SLC transporters, the SLC12 family of proteins includes two orphan members, SLC12A8 and SLC12A9, which encode proteins for which no function has yet been ascribed. The topology of cation-chloride cotransporters resembles that of many other SLC families, with an intracellular amino terminal domain, a core segment consisting of 12 transmembrane domains (TMDs), and a large intracellular carboxyl terminal domain. Interestingly, primary sequence and structural homology of the SLC12 transmembrane core segment with amino acid permeases and bacterial transporters place the NCCs, NKCCs, and KCCs within the amino acid-polyamine-organocation (APC) superfamily of transmembrane transporter proteins (155).
Fig. 1.

Evolution of solute carrier 12 (SLC12) proteins. A: cluster dendogram of human SLC12A cotransporters. The two major branches of cation-chloride cotransporters (CCC) separated early during evolution. Functionally uncharacterized CCC9 separated early from the Na+-dependent branch of cation-chloride cotransporters. In a similar fashion, CCC8 separated early from the Na+-independent K-Cl cotransporters. The human Na+-independent cotransporters are more closely related to one another than are the Na+-dependent cotransporters. NKCC2, Na-K-2Cl cotransporter 2. B: cluster dendogram illustrating separation of the Na+-independent cation-chloride cotransporters during vertebrate evolution. Although multiple “K-Cl like” cotransporters are found in roundworms and arthropods, the modern K-Cl cotransporters diverged early during vertebrate evolution. The amino acid sequences used to create the dendograms in both A and B were aligned using Vector Nti Suite software (version 6.0; Invitrogen/Life Technologies), saved as a text file, and then reformatted for use with Promlk, a software component of the Phylogeny Inference Package (PHYLIP; http://evolution.gs.washington.edu/phylip.html). Length of tree branches can be compared with the reference bar, which represents 0.1 amino acid substitutions per site. Dm, Drosophila melanogaster; Ce, Caenorhabditis elegans.
Proteomic analysis of bacterial CCCs reveals a variety of proteins with similarity to mammalian NKCC1. A key characteristic of Na+-dependent CCCs in general, and NKCC1 in particular, is the presence of a large extracellular loop between transmembrane domains TMD7 and TMD8. However, this loop is missing in bacterial CCC ancestors. In fact, one bacterial CCC member actually displays a large extracellular loop between TMD3 and TMD4. This observation when added to the large number of prokaryotic NKCC sequences indicates that there is as much, if not more, diversity among prokaryote NKCC sequences than among eukaryote NKCCs. NCC and NKCC2 are first found in fish, indicating a relatively late evolutionary branching of these two Na+-dependent CCC-related proteins. As there are more differences between NCC and NKCC1 than there are between NKCC2 and NKCC1, phylogenetic analysis places the branching of NCC before that of NKCC2 (see Fig. 1A). Whether this is the actual order of gene duplication is currently unknown.
Analysis of a protozoan genome (Capsaspora) reveals the presence of one NKCC and two KCC genes, indicating an ancient separation between the NKCC and KCC branches. Instead of a large extracellular loop between transmembrane domains TMD5 and TMD6 (a key characteristic of higher organism K-Cl cotransporters) one Capsaspora KCC sequence displays an unusual loop between TMD9 and TMD10. Although the C. elegans and Drosophila genomes also contain two KCC-like genes (CE-Kcc-1, CE-Kcc-2, DM-Kcc-1, and DM-Kcc-2), it is only later in vertebrate evolution that the modern SLC12A4–A7 genes first appeared (Fig. 1B).
Since only one KCC gene is found in echinoderms and tunicates, the first major duplication of the KCC branch likely occurred during early vertebrate evolution, giving rise to the SLC12A4 and A6 (KCC1 and KCC3) branch and the SLC12A5 and A7 (KCC2 and KCC4) branch. As all four K-Cl cotransporters are found in the modern teleost fish, it is clear that the two branches divided once again during fish evolution. Note that there are some intriguing absences of KCC genes in vertebrate genomes. For instance, SLC12A7 (KCC4) is found in a variety of fish genomes such as zebrafish (Danio rerio), puffer fish (Tetraodon nigroviridis), and tilapia (Oreochromis niloticus), but it is absent from amphibian (Xenopus laevis) and reptile (Anolis carolinensis) genomes. Whether it truly constitutes a gene loss in modern amphibians and reptiles or absence due to the paucity of amphibian and reptile sequenced genomes is unknown. Similarly, SLC12A6 (KCC3) is found in amphibians, reptiles, and mammals but intriguingly not in birds. With the annotation of the chicken and zebra finch genomes nearly complete, it is quite likely that KCC3 is essentially absent in birds. Although only one of the two orphan cotransporters (SLC12A8) is found in protists, phylogenetic analysis (based on amino acid sequences) suggests that SLC12A9 is actually evolutionarily older than SLC12A8. In this review, we will focus on human diseases linked to the mutation/dysfunction of the Na-Cl, Na-K-2Cl, and K-Cl cotransporters and examine and discuss the phenotypes associated with targeted disruption of these transporters in the mouse.
ADVANCES In GENE TARGETING
Homologous recombination in cultured embryonic stem cells has been used to create novel mouse strains which knock out gene expression or knock in mutations that either silence or activate the translated protein, often to mimic a human disease. Before the sequencing of the human (76, 149) and mouse (96) genomes, a map of the exons, introns, and rare restriction sites had to be created to design a targeting vector. This was a very time-consuming and labor-intensive project, often taking months to years to complete. First, genomic DNA isolated from mouse tissue was digested with a panel of restriction enzymes and Southern blot analysis was performed with 32P-labeled cDNA fragments to localize the exons. Next, more genomic DNA was digested into 10- to 20-kb fragments to create a phage genomic DNA library, which was then screened by Southern blot analysis using the radiolabeled cDNA fragments of the target gene as probes. After rescue of smaller genomic DNA fragments into plasmids, the DNA/plasmid was sequenced to differentiate exons from introns and identify unique restriction enzyme sites for construction of a targeting vector with an antibiotic-resistance gene cassette (Fig. 2, steps 1–7). There were so many potential pitfalls that it is still amazing that these multi month/year projects yielded successful outcomes. Today, steps 1–6 have been replaced by the mouse genome sequencing project. The entire design of a targeting construct can be made in a few hours and the creation of the construct can be made in a few weeks with the availability of the entire mouse genome sequence, bacterial artificial chromosomes (BACs), and genetic recombineering techniques (101). The first genetically modified mouse models of SLC12 cotransporters were generated in the late 1990s and involved the Na+-dependent cation-chloride cotransporters (25, 124, 138).
Fig. 2.
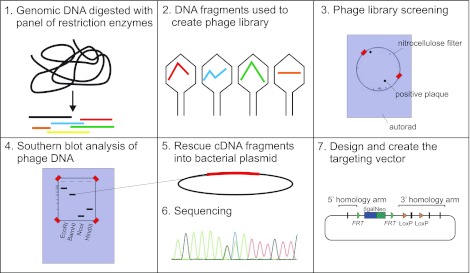
Storyboard of gene-targeted manipulation. Before the complete sequencing of animal genomes, investigators wishing to target specific genes for knockout in animal models were required to initially map genomic DNA to identify the exons, introns, and unique restriction sites within the gene of interest. Steps involved include 1) endonuclease digestion of genomic DNA; 2) creation of a phage DNA library; 3) screening of phage library; 4) Southern blot analysis with phage DNA probes; 5) rescue of cDNA fragments into bacterial plasmids; 6) sequencing; and 7) design and construction of a targeting vector with upstream and downstream arms of DNA to promote homologous recombination.
Na-Cl Cotransporter (SLC12A3 Gene)
The distal nephron is a major site of parallel reabsorption of urinary Na+ and Ca2+. The observation that administration of thiazide diuretics simultaneously decreased Na+ reabsorption while increasing Ca2+ reabsorption led Dr. Mackenzie Walser to postulate that the diuretic dissociates cation reabsorption in the distal nephron by inhibiting net sodium transport and secondarily enhancing net calcium transport (153). As thiazides did not affect the transepithelial potential difference, it was later postulated that Na+ and Cl− “conductances” were inhibited to the same extent (20). Microperfusion experiments placed the site of Na+ and Ca2+ reabsorption to the distal convoluted tubule (DCT) (19). It was while studying the flounder urinary bladder, which is anatomically and functionally an extension of the kidney, that Dr. John Stokes demonstrated the presence of a thiazide-sensitive, electroneutral Na-Cl cotransporter (NCC) at the mucosal membrane (133). This finding indicated that the mechanism for Na+ reabsorption in the mammalian DCT was likely not an ion channel but a membrane carrier. It was from the urinary bladder of the flounder, which constituted a rich source of NCC RNA, that Dr. Steven Hebert and coworkers initially isolated (by expression cloning) the cDNA encoding NCC (43). This was followed a year later by the cloning of the mammalian NCC through homology screening of a rat kidney library (42). As indicated in Fig. 3, the Na-Cl cotransporter is expressed in the DCT, where it recovers 10% of the overall urinary Na+ (see Fig. 4B).
Fig. 3.

SLC12A cotransporters in the kidney nephron. KCC3 is localized to the basolateral membrane of proximal tubule (PT), NKCC2 on the apical membrane of the thick ascending limb (TAL), and Na-Cl cotransporter (NCC) expression on the apical membrane of the distal convoluted tubule (DCT); KCC4 is found in the DCT and cortical connecting tubule (CNT); and NKCC1 is expressed in the inner medullar collecting duct (IMCD), the afferent arteriole of the glomerulus, and the intra- and extraglomerular mesangium.
Fig. 4.

Major SLC12 transporters in mammalian kidneys. A: thick ascending limb epithelial cell of the Loop of Henle. NKCC2 participates in the reabsorption of Na+ and recycles the K+ that exits through the ATP-dependent renal outer medullary potassium channel (ROMK). Additional Na+ ions follow the paracellular pathway driven by an electric gradient generated by ROMK on the apical membrane and CLCK (CLCKA and CLCKB) on the basolateral membrane. B: epithelial cell of the distal convoluted tubule (DCT). NCC expressed on the apical membrane contributes to Na+ reabsorption. The segment also reabsorbs Ca2+ through apical Ca2+ channel (transient receptor potential vanniloid type 5, TRPV5) and basolateral Na+/Ca2+ exchanger (NCX1) and Ca2+ pump. In the DCT cell, Ca2+ is buffered and transported by parvalbumin (Pvlb; DCT1) or calbindin-2 (Calb; DCT2). PMCA1b, plasma membrane Ca2+-ATPase 1b.
Many insights into the function of NCC were gained from naturally occurring mutations found in the human population. A rather large number of mutations in the human SLC12A3 gene have been identified in patients suffering from a salt-wasting disorder called Gitelman's syndrome. The first report (129) listed 14 single amino acid substitutions or missense mutations, one nonsense mutation, and two splice site mutations. Interestingly, only three individuals out of 17 were homozygous for a single mutation, whereas the remaining individuals were heterozygous for multiple mutations. Another study in an Italian population reported two previously described and 10 additional new mutations in the SLC12A3 gene (86). In a recent study, Glaudemans and coworkers identified 114 SLC12A3 mutations in 163 patients with Gitelman's syndrome, 31 of which were novel mutations (49). Again, the number of patients homozygous for a single mutation (24%) was nearly threefold less than patients with compound heterozygous mutations (61%). This latest study closely examined the effect of these mutations on cotransporter expression and function. While several mutations led to the complete absence of NCC expression, others led to trafficking defects and targeting of the glycoprotein for degradation. Interestingly, there was a large subset of mutations that neither affected protein expression nor trafficking of the cotransporter to the cell surface but still decreased Na+ uptake when expressed in Xenopus laevis oocytes (49).
Genetic data demonstrate that in the Caucasian population, mutations in this transporter are found with a frequency of 1:100 people (88). However, they also indicate that compound heterozygous mutations are less deleterious in combination than mutations found in homozygous individuals who feature the classical symptoms of Gitelman's disease. The major clinical manifestations of Gitleman's syndrome are metabolic alkalosis, hypokalemia, hypomagnesemia, hypocalciuria, and high renin-aldostenone levels (129). As NCC function is absent or significantly reduced in Gitleman patients, there is a significant decrease in Na+ and water reabsorption, resulting in volume contraction. The decreased Na+ reabsorption in the DCT results in increased delivery of Na+ to the collecting duct, where its reabsorption via epithelial sodium channel (ENaC) stimulates both K+ and H+ secretion and leads to hypokalemia and metabolic alkalosis. Furthermore, the reduced blood volume activates the renin-angiotensin-aldosterone (RAA) system: angiotensin II promotes proximal H+ secretion via the Na+/H+ exchanger and aldosterone activates ENaC activity, increasing the driving force for secretion of K+ and H+ and further hypokalemia and metabolic alkalosis. The loss of K+ can lead to associated symptoms such as fatigue, tetany, paralysis, and increased risk of ventricular arrhythmia. Additional features of the disease are hypomagnesemia, which is likely due to loss of the transient receptor potential cation channel (TRPM6) expression in the DCT, and hypocalciuria due to increased proximal tubule passive calcium reabsorption secondary to extracellular volume contraction.
In 1998, Shull and coworkers generated a NCC-null mouse by disrupting exon 12 of the SLC12A3 gene (124). Despite the absence of the NaCl cotransporter, these mice appeared healthy, grew normally, and were fertile. Similar to Gitelman's syndrome patients, mice deficient in NCC exhibited reduced levels of urinary Ca+2 and serum Mg+2. When maintained under standard diet conditions, blood pH, serum K+ levels, and plasma electrolytes from NCC-null mice were indistinguishable from wild-type mice. However, when placed on a low-Na+ diet, both genotypes demonstrated a significant reduction in urinary Na+ excretion, possibly indicating increased Na+ reabsorption regardless of the presence/absence of the Na-Cl cotransporter. However, a significant decrease in arterial blood pressure (72.3 ± 5 vs. 86.1 ± 3.9 mmHg) was observed in the NCC-null mice (compared with wild-type mice) maintained on a low-Na+ diet, similar to patients with Gitelman's syndrome (124). NCC-null mice also displayed a twofold increase in renin mRNA levels, whereas no significant increase in serum aldosterone was measured. The absence of aldosterone stimulation is consistent with absence of volume contraction, which typically activates the RAA axis, leaving the increased renin levels to be due to the intrarenal renin-angiotensin system. NCC-null mice maintained on a low-K+ diet exhibited a significant reduction in plasma K+ concentration, whereas wild-type mice, placed on the same diet conditions, were able to maintain their plasma K+ concentration for a period of 7 days (93). These data suggest that under normal diet conditions, NCC-null mice are somehow able to compensate for the cotransporter-mediated reduction in Na+ transport.
One possible compensatory mechanism to maintain sodium balance in these NCC-null mice is the increased expression of the γ-subunit of the epithelial sodium channel, ENaC (14). Indeed, the collecting ducts, which are distal to the DCT, are also involved in reclaiming a portion (∼5%) of filtered urinary Na+ through the combined actions of ENaC and the Cl−/HCO3− exchanger, Pendrin. Individually, the NCC and Pendrin knockout mice only develop hypotension under dietary salt restriction, but when combined into a double knockout, the animals demonstrate severe volume depletion, salt and fluid wasting, as well as a significant reduction in arterial blood pressure under basal conditions (131). The variable phenotypes observed within Gitleman's syndrome patients and NCC-null mice can therefore be attributed to the degree of compensation by other salt transporters, which in turn can be influenced by a variety of factors. In fact, a similar variability in phenotypes is observed in various studies of the SPAK-null mouse, knockout of a kinase which regulates NCC activity (41, 53, 87). Similar to human kidneys treated with thiazide diuretics, the number of DCT epithelial cells decreases in the NCC-null mice, indicating atrophy of the segment. Conversely, hypertrophy of the DCT is observed in a mouse model of pseudohypoaldosteronism type II, where NCC function is increased (75). These morphological changes are thought to be directly due to alterations in Na+ transport activity.
Although the NCC-null mouse phenotypically recapitulates the hypocalciuria and hypomagnesemia observed in patients with Gitleman's syndrome, the knockout mouse does not exhibit hypokalemic alkalosis. As a result, future studies with this mouse model may provide valuable insight into the pathology of milder forms of Gitleman's syndrome.
Na-K-2Cl Cotransporter-1 (SLC12A2 Gene)
In 1971, Floyd Kregenow demonstrated that duck erythrocytes recover their volume after hypertonic shrinkage by activating an ouabain-insensitive, Na+-dependent, “active” accumulation of K+. Two years later at the 2nd Annual Meeting of the Red Blood Cell Club at Yale University (New Haven, CT), he showed that the shrinkage-induced cation uptake was mediated by a furosemide-sensitive Na+ plus K+ cotransport system (referenced in Ref. 69). It was seven years later that a more complete description of an electrically silent Na-K-2Cl cotransport mechanism was proposed (44), and another fourteen years before the identification at the molecular level of two distinct Na-K-2Cl cotransporters (26, 42, 108, 158).
Design and engineering of the Delpire NKCC1 knockout construct began in 1996 at Brigham and Women's Hospital (Boston, MA). Relatively large (8–18 kb) gene fragments were first isolated by screening lambda-phage genomic libraries using small cDNA fragments of NKCC1 as probes. Precise maps of these genomic clones were obtained by a panel of restriction enzyme digests, and smaller fragments were then subcloned into plasmids for sequencing to identify the exons. Extreme care was necessary to select unique or rare restriction sites with which to move fragments of these lambda-phage genomic clones into a targeting vector containing a neomycin-resistance gene cassette. This labor-intensive work provided the first partial map of the SLC12A2 gene (116) and a targeting construct for homologous recombination that could produce a straight knockout. The decision to proceed to the production of the mouse took many months because of fears that the mouse would be embryonically lethal since the cotransporter is expressed in so many tissues. The NKCC1 knockout mouse did turn out to be viable and the first animals were born in late 1998. Interestingly, two months after our 1999 Nature Genetics report (26), Dixon (28) reported that NKCC1 was responsible for the inner ear defects previously described (27) in a mouse mutant line existing since the early 1940s (55). Shortly after, two additional groups reported the generation of their own NKCC1 knockout mice (38, 105). Over the past 12 years, multiple studies have been conducted with these three independently created lines of NKCC1 knockout mice (see Table 1). In 2010, Ten Hagen and coworkers confirmed that SLC12A2 was not essential for survival, as knockout of Ncc69 (the fruit fly ortholog of NKCC1) in Drosophila melanogaster did not affect animal viability (134). However, it is puzzling that no mutations to date have been found in the human population, suggesting that NKCC1 function is essential to the development and survival of the human fetus.
Table 1.
Phenotypes of NKCC1 knockout mice
| Phenotype | Mouse | Reference |
|---|---|---|
| Inner ear dysfunction | Delpire, Shull, Koller, Dixon | (25, 28, 38, 106) |
| Small size | Delpire | (25) |
| Male infertility | Delpire, Koller | (25) |
| Postnatal hyperexcitability | Delpire | (162) |
| Postnatal anti-convulsant | Delpire | (33) |
| Pain perception | Delpire | (74, 136) |
| Intestinal transit | Shull | (38) |
| Intestinal transit (ICC) | Delpire | (157) |
| Normal intestinal secretion | Shull | (151) |
| Olfactory neurons | Delpire | (117) |
| Normal olfaction | Shull | (98, 130) |
| Abnormal Cl− currents (DRG) | Delpire | (136) |
| Decreased saliva production | Shull | (36) |
| Normal lung function | Koller | (47) |
| Protection against lung sepsis | Koller | (97) |
| Low blood pressure | Delpire | (Unpublished observations) |
| Low blood pressure | Shull | (38, 92) |
| Normal blood pressure | Shull | (68) |
The table recapitulates the phenotypes reported with each NKCC1 knockout mouse. ICC, interstitial cells of Cajal; DRG, dorsal root ganglion.
Sensorineural deafness.
The most striking phenotype of the NKCC1 knockout mouse is the shaker/waltzer phenotype or head bobbing and circling behavior characteristic of inner ear dysfunction (25, 38). Hearing and balance are affected in these mice, pointing to both cochlear and vestibular defects. Histology of the cochlea revealed absence of endolymphatic fluid as well as loss of hair cells and supporting outer phalangeal cells in the organ of Corti (25, 38). These data were confirmed and extended by ultrastructural studies of the inner ear of NKCC1 knockout mice (106). Placed on the basolateral membrane of stria vascularis cells, the Na-K-2Cl cotransporter together with the Na+-K+-ATPase provide an entry pathway for K+ into the cells. Na+ that enters through the cotransporter can be recycled by the Na+/K+ pump, and K+ channels located on the apical membrane (Kv7.1/KvLQT1) provide the exit pathway for K+ out of the stratified epithelial cells (see Fig. 10 in section on KCC3 knockout and age-related deafness). The structural changes and behavioral phenotype of the NKCC1 knockout mouse confirms the critical role that NKCC1 plays in the production/secretion of the K+-rich endolymphatic fluid. Consistent with this observation, knockout of Kv7.1 or KCNE1/minK (a Kv7.1 modifier protein) leads to inner ear deficits similar to the NKCC1 knockout phenotype. In fact, both NKCC1 and Kv7.1 expression has been shown to decrease in the inner ear with age-related deafness (79). Note that immunohistochemical analysis in the inner ear revealed high expression levels of NKCC1 not only in the stria vascularis but also in spiral and vestibular ganglia (25). The physiology of NKCC1 has not yet been studied in these particular neurons, but it is tempting to associate this observation to the high level of NKCC1 expression detected in primary afferent dorsal root ganglion neurons (see below). The similarity between the anomalies of the zebrafish little ear (lte) phenotype and the NKCC1-null mouse prompted Geisler and coworkers to map the lte phenotype to the chromosomal location of the zebrafish Nkcc1 gene (45). Subsequent developmental studies demonstrated a loss of endolymph fluid and collapse of the endolymphatic space in the zebrafish lte phenotype at 75 h postfertilization (1). These studies indicate that the function of NKCC1 in the inner ear is conserved throughout vertebrate evolution.
CNS excitability: is NKCC1 pro- or anticonvulsant?
Central nervous system (CNS) neurons born in the periventricular zone migrate, differentiate, and acquire their final characteristics by forming synapses and communicating with neighboring neurons during embryonic development. This neuronal migration and differentiation utilizes the neurotransmitter GABA, which in immature neurons produces excitatory postsynaptic potentials. Kriegstein and coworkers assessed the developmental changes in the GABA receptor reversal potential and found that the intracellular Cl− concentration in neurons is highest when the neurons are born and then slowly decreases during neuronal maturation (104). It is this high intracellular Cl− that facilitates GABA-receptor mediated depolarization of the neuronal membrane. For instance, in immature hippocampus, excitatory GABA facilitates the development of giant depolarizing potentials that originate in the network spontaneously (9, 10). One of the mechanisms potentially involved in maintaining this high intracellular Cl− is NKCC1, which uses the favorable energy of the Na+ and Cl− chemical potential differences to maintain intracellular Cl− above its electrochemical potential equilibrium. In support of this hypothesis, expression of NKCC1 was shown to be highest in young neurons and decreases during maturation (114). Despite the developmental decrease in NKCC1 expression, only minimal changes in Cl− reversal potential have been measured with bumetanide in young CNS neurons (33, 162) or using the NKCC1-null mouse (7, 159), challenging the idea that the cotransporter is primarily responsible for accumulating Cl− above its electrochemical potential equilibrium in immature neurons. In fact, it is possible that the primary role of NKCC1 is not to “set” the Cl− concentration to values above its electrochemical potential equilibrium, but rather to provide a pathway for fast reaccumulation of Cl− in conditions that lower its concentration.
Experiments performed in rat hippocampal slices showed that bumetanide, a potent inhibitor of NKCC1, suppresses synchronous bursts of action potentials (33). The spontaneous synchronous network activity returned shortly after washout of bumetanide. Furthermore, the loop diuretic was also shown to suppress inter-ictal and ictal-like epileptiform activity induced by high K+ in hippocampal slices in an age-dependent manner (33). As expected, the NKCC1-null mice confirmed the specificity of the bumetanide effect. In a recent study, the authors demonstrated that bumetanide enhanced phenobarbital efficacy in preventing seizures induced by exposure to low Mg2+ (32). Thus, NKCC1 in these studies was proconvulsant. In contrast, we found a higher frequency of action potential firing in CA3 pyramidal neurons from homozygous NKCC1-null mice, compared with wild-type mice. These data were confirmed by applying bumetanide to slices isolated from wild-type mice (162). Furthermore, when we used 4-aminopyridine to induce hyperexcitability, we found synchronized ictal-like epileptiform activities arising in NKCC1-null hippocampal slices but only interictal-like activities appearing in wild-type NKCC1 hippocampal slices. These results indicated that instead of facilitating seizure activity, NKCC1 actually inhibited the generation of seizures (162). Thus, there is still a controversy to the role of NKCC1 in CNS excitability. In the Zhu et al. (162) study, we discussed in some detail possible reasons for both the pro- and anticonvulsant results of the two studies. One possibility is that NKCC1 is a substitute mechanism for clearance of external K+. Another possible explanation comes from a study performed in C57BL/6 mice that demonstrated that based on the protocol used to induce hyperexcitability, bumetanide exerts differential effects on network excitability (67). Furthermore, a recent study demonstrated a significant increase in neuronal Cl− during the preparation of brain slices, questioning the validity of the recordings performed in freshly obtained tissue preparations (31).
Sensory neurons.
The role of NKCC1 in accumulating Cl− in primary afferent neurons is much less controversial than that of central neurons. Early work performed in amphibian dorsal root ganglion (DRG) neurons demonstrated a concentration of Cl− that was 2.6 times higher than predicted from electrochemical potential equilibrium conditions, dependent on external Na+ and K+ ions, and sensitive to the loop diuretic furosemide (3). All of these properties are characteristics of the Na-K-2Cl cotransporter. Cloning of the mammalian NKCC1 (26) and subsequent development of polyclonal antibodies (65) allowed for the identification of this isoform in DRG neurons (4, 113). A recent study confirmed the expression of NKCC1 in both large and small DRG neurons and also demonstrated a progressive developmental decrease in intracellular Cl−, although still much higher than that of mature central neurons (85). Gramicidin-perforated patch-clamp measurements demonstrated a return of the intracellular Cl− to equilibrium distribution in DRG neurons isolated from NKCC1 knockout mice (136). High intracellular Cl− concentration in the terminals of sensory afferent fibers facilitates GABA depolarization and presynaptic inhibition. Thus, once the fiber terminal is depolarized or shunted, it will prevent the transmission of an incoming action potential (Fig. 5A). This might be a mechanism set up to filter sensory noise, only allowing strong sensory signals to go through, when the fiber has recovered from GABA depolarization.
Fig. 5.

Nociception phenotype in NKCC1 knockout mice. A: model of sensory circuitry in the spinal cord. Nociceptive signals are carried by unmyelinated (C) and thinly myelinated (Aδ) afferent fibers that synapse onto spinal cord lamina I and II neurons. Interneurons (orange), which are activated by projection neurons in deeper spinal cord layers, release GABA at the terminals of the C and Aδ fibers. Because the Cl− concentration is high in these afferent neurons, GABA produces a depolarization of the nerve terminal and inhibition of incoming pain signals from the periphery. B: withdrawal latencies for hot plate and tail flick assays in wild-type, heterozygous, and homozygous NKCC1 knockout mice. Both tests were performed at 52°C. Data represent means ± SE. *Significant difference with P < 0.05. Hot plate and tail flick data were taken from references 136 and 74, respectively.
The SLC12A2 knockout mouse has also been used to confirm the role of NKCC1 in peripheral nerve degeneration (112). Following axonotomy of the sciatic nerve, it was observed that the reversal potential of muscimol-induced GABAA currents shifted toward depolarized potentials in wild-type but not NKCC1-null animals. Confirmation that this shift was a result of NKCC1 activity was demonstrated by 1) extracellular Cl−-dependence, 2) increased phosphorylation of existing NKCC1 protein, and 3) bumetanide sensitivity. Pieraut and coworkers (112) used time-lapse video microscopy to show that initiation and growth of neurites was significantly greater in axotomized nerves, compared with control neurons. Furthermore, depletion of intracellular Cl− significantly decreased neurite growth velocity, indicating that regenerative growth is dependent on the regulation of intracellular Cl− concentration. The use of intrathecal injection of bumetanide, small interfering RNA targeting NKCC1, and NKCC1-null mice all prevented neurite growth (following axotomy), clearly indicating that the cotransporter is involved.
As NKCC1-null mice exhibit a severe inner ear deficit, it is not unexpected that they would demonstrate a severe locomotor deficit, as demonstrated by their inability to perform on the accelerated rotarod assay (136). Unfortunately, the inner ear-driven locomotor phenotype prevented us from making any conclusions regarding a possible sensory afferent proprioceptive deficit. However, these mice do exhibit a significant nociception phenotype (Fig. 5B), as demonstrated by the hot plate test (136), as well as the tail flick and intradermal capsaicin injection assays (74). This decreased sensitivity (or increased threshold) to pain might be explained by the absence of presynaptic inhibition and an inability of the mice to differentiate nociceptive signals from sensory noise. Because developmental compensation is known to occur in knockout animal models, it was important to confirm the role of NKCC1 in pain perception using approaches independent of genetically modified animals. This was done in a 2005 study that found that the flinching behavior produced by an injection of 1% formalin in the mouse paw could be decreased by an intrathecal injection of NKCC1 inhibitors (50).
Olfactory receptor neurons.
When odorants bind to G-coupled receptors of the ciliary membrane, they trigger an increase in cAMP that, in turn, activates Ca2+ channels. As Ca2+ enters into the cell, it stimulates a Ca2+-dependent Cl− channel that depolarizes and excites the neuronal membrane (40). NKCC1 expression is high in the olfactory epithelium but absent in the adjacent respiratory epithelium (117). The cotransporter appears to be localized to the proximal dendrite and soma of the olfactory receptor neuron (ORN) and is required for the generation of the inward Cl− current. Indeed, using heptanal as an odorant, Reisert and coworkers (117) demonstrated in ORNs from wild-type mice the appearance of a large receptor current blocked by niflumic acid, a Cl− channel blocker. This current was substantially reduced after incubation of the tissue with 50 μM bumetanide for 30 min. Importantly, in ORNs from NKCC1-null mice, the receptor current was seven times smaller than the current in wild-type ORNs and unaffected by niflumic acid. These electrophysiological data showing that NKCC1 accumulates Cl− were obtained in isolated olfactory neurons and were consistent with previous data obtained with Cl−-sensitive fluorescent dyes (64). However, electro-olfactogram data obtained in intact olfactory epithelium tissue showed that NKCC1-null mice still generate odor-induced field potentials, although reduced by 39% compared with wild-type mice (98). Furthermore, NKCC1-null mice were shown to have normal olfactory sensitivity, as demonstrated by a two-sample discrimination task performed in an olfactometer operant chamber (130). Thus, in this case as well, there might be a significant difference between data obtained from freshly isolated neurons versus data obtained with intact tissue.
Interstitial cells of Cajal.
Specialized cells in the interstitium of the gastrointestinal tract produce a slow wave electrical pacemaker activity that controls intestinal motility. These cells exist in close association with smooth muscle cells and neurons. In the small intestine, interstitial cells of Cajal (ICCs) surrounding the myenteric plexus generate slow waves that propagate to adjacent smooth muscle cells. Using suppression subtractive hybridization between RNA isolated from wild-type mice and RNA isolated from two ICC-deficient mouse strains, Wouters and collaborators (157) identified SLC12A2 as a gene downregulated in the mutant strains, indicating a possible role for the Na-K-2Cl cotransporter in the function of these cells. To confirm this, they measured slow waves in the mouse jejunum of wild-type mice. These waves had amplitudes ranging from 400 to 600 μV and frequencies of 36.1 ± 2.1 min−1. Upon addition of 40 μM bumetanide, the amplitude and frequency decreased to 50–100 μV and 27.4 ± 1.3 min−1, respectively. The effect of the loop diuretic on shape, amplitude, and frequency of the slow waves was reversible. The decreased amplitude and frequency of these slow waves in the presence of bumetanide was similar to the decreased wave amplitude and frequency measured in the isolated jejunum of NKCC1 knockout mice. The observed decreased intestinal motility, whether induced pharmacologically or genetically, might explain the morbidity related to hemorrhage, intussusception, and fecal impaction that have been reported around the weaning period in NKCC1 knockout animals (38).
Spermatogenesis.
Another striking phenotype of the NKCC1 knockout mouse is the male infertility (25, 105). Gross examination demonstrates smaller testis size in the NKCC1 knockout mouse (105). Figure 6A reports both a smaller body weight and testis size (as measured by weight). Histological analysis of the testis from NKCC1 knockout mice revealed a smaller diameter of the seminiferous tubules and complete absence of mature spermatids near the lumen of the tubule (105). Additionally, larger numbers of spermatogonia were observed in the tubules of NKCC1 knockout mice, with a fraction of them demonstrating apoptotic cell death (Fig. 6, C and D). This deficiency might be related to a CNS deficit as we demonstrated reduced circulating levels of testosterone and luteinizing hormone (LH) in NKCC1 knockout animals (Fig. 6B). Note that intratesticular testosterone which is also reduced in male NKCC1 knockout mice is produced by Leidig cells under the control of anterior pituitary LH, which itself is controlled by secretion of gonadotropin-releasing hormone (GnRH) from the hypothalamus (Fig. 6A, inset). Interestingly, suppression of fertility in physiological and pathological situations often involves inhibition of GnRH neurons, and these neurons, in contrast to most CNS neurons, exhibit GABAA-receptor mediated membrane depolarization due to NKCC1-facilitated Cl− accumulation (23). Indeed, the intracellular Cl− in untreated GnRH neurons is 25.6 ± 0.3 mM, compared with 11.5 ± 0.9 mM after addition of 50 μM bumetanide. It is therefore possible that the infertility phenotype observed in male NKCC1 knockout mice results from a deficit in hypothalamic neurons involved in the secretion of hormones important for the development of the male reproductive organ.
Fig. 6.

NKCC1 expression/function alters reproduction and sterility. A: relationship between body weight and testicle weight in heterozygous (red squares) and homozygous (blue circles) NKCC1 knockout mice. Distribution of weight is indicated by the 6 data points per genotype. Inset: control of testosterone secretion. Hypothalamic neurons release gonadotropin releasing hormone (GnRH), which stimulates the pituitary to release luteinizing hormone (LH) and follicle-stimulating hormone (FSH). The two anterior pituitary hormones then stimulate the testis to produce testosterone. B: plasma levels of testosterone and LH determined in homozygous (blue bars) and heterozygous (red bars) NKCC1 knockout mice. C: TUNEL staining in control testis. Note the single layer of large dark cells (spermatogonia) at the base of the seminiferous tubule and large number of spermatids filling the lumen. D: TUNEL staining in homozygous NKCC1-null testis. Note the accumulation and disorganization of spermatogonia. Also note the number of TUNEL-positive (red stained) cells, indicating apoptotic cell death. Sections were counterstained with thionin (unpublished observations from the Delpire laboratory).
Epithelia-salivary gland.
One of the tissues that express the highest levels of NKCC1 is the salivary gland. Located on the basolateral membrane of the acinar cells, the cotransporter participates in the secretion of fluid that accompanies secreted proteins, such as α-amylase for the serous secretion of the parotid gland, or mucin for seromucous secretion of the sublingual and submandibular glands. Disruption of NKCC1 causes significant reduction in the volume of saliva secreted in response to pilocarpine, a muscarinic agonist (36). Consistent with the loss of NKCC1 function, a decrease in bumetanide-sensitive Cl− influx into acinar cells was also observed. However, it was also shown that salivary gland tissue was able to somewhat compensate for the loss of NKCC1 by enhancing Cl− movement through the Cl−/HCO3− exchanger.
Epithelia-lung.
NKCC1 is expressed on the basolateral membrane of airway epithelium which secretes Cl− and the fluid responsible for the hydration/fluidity of the overlying mucus layer (Fig. 7A). The key mechanism for Cl− transport is the cystic fibrosis transmembrane conductance regulator (CFTR), a Cl− channel that when mutated causes cystic fibrosis. NKCC1 transports Cl− from the blood side into the epithelial cells, so it can then be secreted by CFTR, osmotically driving water across the epithelium. Analysis of the lung revealed high NKCC1 expression in the trachea of neonatal mice, but much lower expression in adult mice (54). In the fetal lung, the cotransporter is also involved in the production of the fluid, which fills the lung and seems important for its maturation. Consistent with this hypothesis are electrical measurements across the tracheal epithelium, which showed sizable bumetanide-sensitive short-circuit currents (SCCs) in neonatal tissue but no significant bumetanide-sensitive SCCs in adult trachea. This seems, however, to be species specific, as adult rabbit and human trachea demonstrate NKCC1-mediated Cl− uptake. Note that in neonatal trachea, in addition to the bumetanide-sensitive component of the SCC, Cl− also enters the basolateral membrane through a DIDS-sensitive Cl−/HCO3− transport mechanism. The fact that adult NKCC1-null mice do not exhibit an obvious lung phenotype suggests that they are able to adequately compensate for the absence of the cotransporter. Alternative anion transport mechanisms can also sustain liquid production at near normal levels in the fetal lung of NKCC-null mice (47). These data indicate that there is redundancy in the mechanisms of Cl− transport, indicating its importance to lung physiology. Remarkably, NKCC1-null mice are also protected against hypothermic sepsis and invasion of bacteria in the blood due to lung infection, indicating that NKCC1 may also play a role in the acute inflammatory response of the lung (97).
Fig. 7.

Illustration of differential NKCC1 expression and function. A: prototypical Cl− secreting epithelial cells (e.g., salivary, lung, intestine) express NKCC1 on the basolateral membrane. The Na+-K+-ATPase (yellow), coexpressed with NKCC1, recycles Na+ ions thus maintaining the driving force for NKCC1. Chloride ions exit the apical membrane of epithelial through a Cl− channel, the cystic fibrosis transmembrane conductance regulator. Lines represent the mucus layer adjacent to the apical membrane. B: apical localization of NKCC1 and the Na+-K+-ATPase in choroid plexus epithelial cells uncouples the reabsorption of K+ from Na+ secretion. Potassium ions exit the basolateral membrane through K-Cl cotransporters and K+ channels.
Epithelia-intestinal tract.
Because of the incidence of intestinal obstruction in NKCC1-null mice (16% greater than that in wild-type mice; 38), the secretory properties of the intestine were closely examined. Similar to airway epithelium, Cl− secretion in the NKCC1-null mouse duodenum involves the concerted movement of the anion through basolateral Na-K-2Cl cotransporters and apical CFTR Cl− channels (Fig. 7A). Similar to the compensatory mechanisms in the lung, Cl− entry into the intestine of NKCC1-null mice is facilitated by the activity of a Cl−/HCO3− exchanger (151). Comparative experiments performed with bumetanide-treated wild-type and knockout tissue, as well as analysis of Cl−/HCO3− exchanger mRNA expression levels, indicate that the compensation is at the activity level of the transporters and not at the expression level (i.e., genetic compensation). These data indicate that some ion transporters function at submaximal activity levels. Thus, by increasing the activity of existing transporters, they can compensate for the loss of other exchangers and cotransporters.
Epithelia-kidney.
Although a kidney-specific Na-K-2Cl cotransporter isoform exists (SLC12A1, see next section), NKCC1 expression has been demonstrated in several locations in the mouse kidney including the glomerular afferent arteriole, glomerular and extraglomerular mesangium, and the inner medullary collecting duct (65). In rat kidney, expression was similar in glomerular afferent arteriole, but type A intercalated cells of the outer medullary collecting duct expressed the cotransporter on the basolateral membrane (34). Presence of NKCC1 in renin-expressing smooth muscle cells of the juxtaglomerular afferent arteriole indicates a possible role of the cotransporter in the renin-angiotensin system and control of glomerular filtration. Detailed studies have shown that NKCC1-null mice have increased levels of plasma renin concentration associated with increased granulation of afferent arterioles and intra-renal renin mRNA (17, 152). As renin production by the afferent arteriole smooth muscle cells depends on the delivery of Na+ to the macula densa, Castrop and coworkers (17) tested the effect of furosemide injection on plasma renin levels in wild-type and NKCC1-null mice. They observed increases in plasma renin levels in both genotypes, consistent with furosemide inhibition of NKCC2 function and increased salt delivery to macula densa cells. When they isolated juxtaglomerular cells from wild-type and knockout animals, they observed that furosemide increased renin release in wild-type cells, but not in NKCC1-null cells. This observation indicates that NKCC1 tonically suppresses renin synthesis and renin release. However, this interpretation is confounded by the low blood pressure measured in the NKCC1-null animals [e.g., 114.5 ± 2.2 mmHg in-null vs. 131.8 ± 2.5 mmHg in wild-type mice; (92)]. In fact, as blood pressure decreases, the rate of glomerular filtration and salt delivery to the macula densa is lower. This signal by itself would lead to an increased renin secretion; however, it has been argued that the blood pressure decrease in NKCC1-null mice is not severe enough to explain the extent of the renin increase observed in these animals. Note that another study examining the same mouse line observed no significant difference in blood pressure between wild-type and knockout mice, unless the mice were fed with a high-salt diet, and then NKCC1-null mice exhibited an increase in mean arterial blood pressure that was absent in wild-type animals. Differences between the studies might be related to the methodologies used for blood pressure measurement: tail cuffs (92, 152) versus telemetry (68). The underlying cause of the reduced blood pressure has been closely examined by Susan Wall (152). Hypotension could be due to impaired cardiac output, vasodilation of blood vessels, or extracellular volume depletion. It was also shown that NKCC1-null mice had similar cardiac function as their wild-type counterpart, eliminating the heart as a factor in the observed hypotension. Furthermore, as plasma aldosterone levels were unchanged, extracellular volume depletion was also eliminated as the cause of the hypotension. Although no significant differences were observed in aortic contractility between the two genotypes, there was a significant decrease in contractility of the portal vein in NKCC1-null mice compared with wild-type, indicating that the cotransporter plays a significant role in vascular tone. Aside the increased renin release, there are several other kidney-related abnormalities in NKCC1-null mice. Analysis of Na+ transport in the knockout versus wild-type mice revealed increased NCC expression under both high-Na+ and low-Na+ diets and increased sodium–hydrogen exchanger 3 (NHE3), NKCC2 and Na+-K+-ATPase expression under a low-Na+ diet condition. Increased expression of these transporters suggests increased Na+ transport, which is also inconsistent with a decreased blood pressure. In each of these specific epithelial examples, the NKCC1-null mouse demonstrates both the physiological importance of Cl− transport and the various mechanisms that exist to compensate for the loss of NKCC1 activity.
Na-K-2Cl Cotransporter-2 (SLC12A1 Gene)
Functional identification of a Na+ reabsorption mechanism in the thick ascending limb (TAL) of the Loop of Henle came with the discovery in the early 1960s of furosemide and its effect on the renal medullary sodium gradient (39, 58). The functional characteristics of this mechanism came shortly after the description by Geck and coworkers (44) of a furosemide-sensitive Na-K-2Cl cotransporter mechanism in Ehrlich Ascites. Indeed, Reiner Greger and colleagues (51, 52) showed that the TAL carrier was electrically silent, yet apparently transported 2Cl− for 1 Na+, and that this cotransport of Na+ and Cl− was K+ dependent. It was not until 1994 with the cloning of Na-K-2Cl cotransporter cDNAs from rat and rabbit kidney medulla (42, 108) and the cloning of separate Na-K-2Cl cotransporter cDNAs from mouse inner medullary collecting duct (26) and shark rectal gland (158) that the “widely expressed” NKCC1 and “kidney-specific expression” of NKCC2 were identified as two distinct isoforms encoded by two separate genes.
Mutations in human SLC12A1 results in Bartter's syndrome type 1, an autosomal recessive salt-wasting disorder characterized by polyuria, renal tubular hypokalemic alkalosis, and hypercalciuria. Disruption of the NKCC2 gene can be due to missense mutations (Asp648Asn, Val272Phe) (128) or (Gly193Arg, Arg199Gly, Gly224Asp, Ala267Ser, Gly243Glu, Arg302Gln, Gly319Arg, Cys436Ser, GlyAla478, AlaThr508, Ala510Asp, Ala578Thr) (148), premature stop codons (Trp625X) (72) (Tyr998X) (148), frameshift stops at positions 706 and 1318 (148), or splicing defects (100). All of these mutations result in the lack of NKCC2 function in the TAL, providing a window into the significance of the cotransporter in fluid and salt homeostasis in the kidney. Defective NaCl reabsorption in the TAL results in salt and fluid wasting, accompanied by blood volume reduction. As in Gitelman's syndrome, the decreased Na+ reabsorption in the TAL results in increased delivery of Na+ to the collecting duct where its reabsorption via ENaC stimulates both K+ and H+ secretion and leads to hypokalemia and metabolic alkalosis. Furthermore, the reduced blood volume activates the RAA system through intrarenal baroreceptors (renin cells) and elevated sympathetic nerve activity. Angiotensin II promotes proximal secretion of protons via the Na+/H+ exchanger, and aldosterone activates ENaC activity, increasing the driving force for secretion of K+ and H+ and further hypokalemia and metabolic alkalosis.
Less than a year after the publication of several NKCC1-knockout animals (see above), Takahashi and coworkers (138) produced a homozygous NKCC2 knockout mouse that exhibited severe dehydration, high plasma renin and low potassium concentrations, and metabolic acidosis. Unfortunately, the severe extracellular volume depletion resulted in a failure of the pups to thrive and none survived past weaning. As a result, a follow-up study was performed using mice heterozygous with one allele of NKCC2 knocked down. Various physiological parameters (e.g., levels of blood creatinine, daily urine volume, blood pressure, urea nitrogen, osmolality) were indistinguishable between wild-type and heterozygous NKCC2 mice (137). Quantitative real-time-PCR determined that although the heterozygous NKCC2 mice had approximately one-half the mRNA of wild-type NKCC2 mice, no significant difference in protein levels of NKCC2 could be observed. The authors hypothesized that the lack of any physiological difference between the two genotypes was a result of alterations in the rate of protein turnover. To localize the site of posttranslational compensation, they isolated portions of the kidney nephron and measured the transepithelial voltage. No discernible difference in NaCl reabsorption between the two genotypes was observed in the isolated TAL, strongly indicating that phenotypic compensation for the single allelic copy of the SLC12A1 gene occurred in that portion of the kidney nephron (137).
There are actually three splice variants of NKCC2 (A, B, and F) derived from alternative splicing of exon 4. Along with different levels of overall expression, each isoform also has localized expression in the different portions of the TAL (48, 62, 108). The most abundant NKCC2 isoform, NKCC2F, is expressed in the medullary TAL. NKCC2B is expressed mainly in the cortical TAL, and NKCC2A is expressed in both the cortical and the medullary TAL (see Fig. 3). Each of the three NKCC2 variants exhibit different affinities for Cl−; thus their expression in different regions of the TAL allows for a wide range of ion transport activity (103).
Macula densa epithelial sensor cells detect tubular salt concentration through the transport activity of NKCC2 located on their apical (luminal) membrane (111, 123). Macula densa cells were found to exclusively express the high Cl− affinity NKCC2B isoform, and mice genetically modified to be deficient for NKCC2B had no gross anatomic, behavioral, or fertility abnormalities (102). Interestingly, the targeting of NKCC2B did not result in a compensatory change in the expression levels of either NKCC2A or NKCC2F. Even more surprising, the urine-concentrating ability of the NKCC2B-null mice was similar to that of wild-type mice following 48 h of water deprivation. Although plasma renin concentrations (PRC) were similar between both genotypes under basal conditions, and when all isoforms of NKCC2 were inhibited by acute administration of furosemide, NKCC2B-null mice significantly lowered their PRC when maintained on a chronic high-salt diet. Conversely, a chronic low-salt diet stimulated PRC in both genotypes. Blood pressure and heart rate were similar in both wild-type and NKCC2B-null mice, whereas plasma aldosterone levels changed in parallel to changes in PRC. Distal tubular fluid chloride concentrations in NKCC2B-null mice were greater than in wild-type mice, whereas absolute Cl− absorption along the Loop of Henle was significantly higher in wild-type versus NKCC2B-null mice (but only at low microperfusion rates) (102). Measurement of fractional chloride and water absorption were also higher in wild-type versus NKCC2B-null mice, but again only at low microperfusion rates. Isoform-specific in situ hybridization experiments found that macula densa cells express both NKCC2B and NKCC2A, but not NKCC2F. As a result, macula densa cells deficient for NKCC2B still possessed some NKCC2 protein and explained why much of the macula densa function was preserved in the NKCC2B-null mice (102).
Similar to the NKCC2B-null mice, genetic disruption of the low Cl− affinity NKCC2A isoform produced viable mice without any salt-wasting phenotype. Although knockout of NKCC2A did not affect mRNA levels of NKCCF, the level of NKCC2B mRNA was actually greater than that observed in wild-type mice, indicating compensation. However, it is interesting that total NKCC2 protein levels, determined by Western blot analysis, were slightly reduced in the NKCC2A-null mouse (103). Whereas wild-type mice given a single intravenous injection of isotonic saline significantly suppressed plasma renin concentration (PRC), NKCC2A-null mice given the same treatment did not exhibit the same significant decrease in the PRC response. Microperfusion of superficial nephrons demonstrated that distal Cl− concentrations, absolute Cl− absorption, and water absorption between the two genotypes were similar at low flow rates but significantly different at high perfusion rates (103). Conversely, measurement of tubuloglomerular feedback responsiveness in NKCC2A-null mice increased at low perfusion rates, but decreased at saturating perfusion rates. Altogether, the overlapping expression of NKCC2A and NKCC2B in the TAL and macula densa appears to significantly increase the efficiency of salt absorption over a wide range of Cl− concentrations (103). Thus, the NKCC2A and NKCC2B knockout mouse models have demonstrated that renal salt absorption is dynamic along the nephron and that the expression/affinity of these different isoforms serves to meet these varying salt absorption concentrations.
Swelling-Induced Cl−-Dependent K+ Efflux
Earlier, we described how Floyd Kregenow demonstrated a hypertonic shrinkage induced Na+-dependent accumulation of K+ in duck erythrocytes (71). In an accompanying manuscript, Kregenow suggested that the pump and leak concept of Tosteson and Hoffman (144) being responsible for cell volume regulation could not explain the observed monovalent cation transport across duck erythrocyte membranes in nonhemolytic hypotonic solutions (70). He observed that after an initial swelling phase, duck erythrocytes returned to their original volume by a loss of cellular K+ and Cl− and cell water, with little or no change in Na+. As the loss of osmotic particles is controlled and even stop once cells have returned to their original volume, Kregenow suggested that an intrinsic intracellular regulatory mechanism must be responsible (70). It was not until 1980 that Phillip Dunham and Clive Ellory in England and Peter Lauf in the U.S. characterized a Na+-independent, Cl−-dependent ouabain-insensitive K+ efflux activated by hypotonicity (29) or pretreatment with N-ethylmaleimide (77), an efflux now known to be mediated by electroneutral K-Cl cotransport. In fact, there are four isoforms of the Na+-independent K-Cl cotransporters encoded by separate genes (46, 56, 57, 95, 109, 115). With the exception of the relationship between KCC3 and the human peripheral neuropathy, Andermann's syndrome, no overt human pathologies have been directly linked to mutation/dysfunction of the other K-Cl cotransporters. For the purpose of this review, we will focus on how genetic knockdown of each KCC isoform in the mouse has produced multiple physiological phenotypes (e.g., erythrocyte sickling; seizure susceptibility; peripheral neuropathy; and sensorineural deafness).
K-Cl Cotransporter-1 (SLC12A4 Gene)
Despite the ubiquitous expression of KCC1, neither specific roles of this K-Cl cotransporter isoform nor any KCC1-associated human diseases have been identified. Generation of a KCC1-null mouse by deletion of exons 4 and 5 resulted in a shift in the open reading frame and premature termination of the encoded protein. Homozygous KCC1-null mice were indistinguishable from their wild-type littermates with similar body weight, physical appearance, response to auditory and visual stimuli, and susceptibility to seizures. Furthermore, histological examination found no obvious abnormalities in any of their major organ systems (121). Crossing of the KCC1 and KCC3-null mice to produce a double knockout mouse resulted in litters at the expected Mendelian ratios; however, matings were rarer and litters much smaller. In addition, the postnatal development and growth of the double knockout animals was less than that of wild-type mice, and they exhibited an increased mortality rate when compared with the KCC3-null mice, indicating an additive effect by incorporating disruption of the KCC1 gene. Red blood cell (RBC) counts of the double knockout animals were reduced when compared with wild-type littermates; however, hemoglobin values were within the normal range and thus the animals were not anemic. Small differences were observed in the mean corpuscular volume of RBCs and reticulocytes, which was elevated, and in the osmotic resistance, which was decreased, in the double knockout mice compared with wild-type animals (121).
Human sickle cell disease is an inherited condition caused by a mutated hemoglobin gene that results in RBCs assuming a rigid and abnormal “sickle” shape (99). Transgenic expression of a modified hemoglobin gene in the SAD mouse produces polymerized hemoglobin and erythrocyte sickling (145). Elevated K-Cl cotransporter activity is believed to be a major factor in the dehydration of RBCs and accelerates the polymerization of hemoglobin in sickle cells (78). Interesting results, however, were obtained when crossing the double knockout with sickle cell mice. Hubner and coworkers thus crossed the KCC1/KCC3-null mouse with the SAD mouse and produced offspring with RBC mean corpuscular volume and osmotic resistance that were similar to wild-type mice (121). This experiment clearly indicates that K-Cl cotransport is a mechanism that when activated dehydrates the RBCs. Whereas transient dehydration of normal red cells has no long-term consequences, dehydration of RBCs containing sickle hemoglobin greatly affects the morphology of the diseased cells. Absence of K-Cl cotransport has therefore a protective effect against sickling. The required addition of the genetic disruption of KCC3 to demonstrate a KCC1 knockout phenotype is consistent with evidence of multiple isoforms being expressed in RBCs and other cell types. Furthermore, we have learned that two separate K-Cl cotransporter isoforms contribute to the dehydration of RBCs, and targeting of one isoform can be compensated for by the other isoform. It should be noted that this result could not have been uncovered with pharmacological agents, as isoform-specific inhibitors are not yet available.
K-Cl Cotransporter-2 (SLC12A5 Gene)
Originally identified and cloned from a rat brain cDNA library, KCC2 has been shown to be expressed exclusively in brain and spinal cord neurons (109). Based on this selective expression, KCC2 was immediately postulated to be the so-called “Cl− pump” mechanism that drives Cl− out of the neuron against its electrochemical driving force, thus confirming a 10-year-old hypothesis that a K-Cl cotransport mechanism was responsible for the outward movement of Cl− in mammalian cortical neurons (141). The gradual increase in cotransporter expression during rodent postnatal development (18, 82) was consistent with a gradual decrease in intracellular Cl− and consistent with the strengthening of GABA hyperpolarizing responses during maturation of the inhibitory system. Because the driving force for K-Cl cotransport is very close to thermodynamic equilibrium, small changes in either intracellular Cl− or external K+ can easily change the direction of transport (107). This is significant as repeated neuronal firing is likely to result in transient increase in extracellular K+, thus affecting excitability. Also note that KCC2 is the only one of the four K-Cl cotransporter isoforms that exhibits any activity under isotonic conditions. The three other K-Cl cotransporters need to be activated by hypotonicity to detect transport activity, the significance of which is still to be determined. The isotonic activity of KCC2 is known to be encoded by a small portion of the cytoplasmic COOH-terminal tail of the cotransporter (89). A direct relationship between KCC2 and the development of GABA hyperpolarizing responses during neuronal maturation was first demonstrated using KCC2-specific antisense oligonucleotides (118). Confirmation of this role came with the development of two knockout mouse models (60, 156). The release of our knockout data in poster form at the Society for Neuroscience (SFN) in 2001 produced a bit of a stir, as our animals survived for about two weeks after birth, whereas Thomas Jentsch's mice died shortly after birth. At the time of the SFN meeting, both studies had yet to be published. We now know that the difference between the two models can be explained by the targeting strategy. The first published knockout mouse, from Thomas Jentsch's laboratory, targeted exon 5, which encodes for part of the highly conserved second transmembrane domain and first intracellular loop between TM2 and TM3. Although these knockout animals had normal diaphragms and normal heart rates, they died at birth due to respiratory failure (60). The pre-Botzinger complex is a cluster of interneurons in the brainstem essential to the generation of respiratory rhythm. Examination of the respiratory-related motor output of E18.5 isolated brainstems from wild-type animals showed a regular and stable rhythm of 14 bursts per minute; however, preparations from Jentschs' KCC2 knockout animals failed to show any rhythmic activity. Our SLC12A5-knockout model, published one year later, targeted exon 1 and produced viable mice that still expressed 5–10% of KCC2 protein as detected by Western blot analysis (156). Although at the time we did not understand the reason for this result, the remaining KCC2 expression in the brain and/or spinal cord was enough to allow our mice to bypass the early postnatal lethality observed in the Jentsch mice. In 2007, a novel NH2-terminal isoform of the neuronal K-Cl cotransporter (KCC2a) was identified that involved an alternative promoter and first exon (146). It is now clear that by targeting exon 1b, we had eliminated only one isoform (KCC2b), leaving untouched the expression of the KCC2a isoform that is initiated from a distinct promoter on the SLC12A5 gene. Elimination of exon 1b disrupted expression of the major isoform (KCC2b), resulting in pups that demonstrated frequent tonic/clonic seizures that ultimately led to their deaths between postnatal day 12 and 17 (P12–P17). These seizures could be prevented by injecting the animals with 2.5–5 mg/kg phenytoin (also known as dilantin) before P12–P13; however, owing to developmental factors that are still unknown, the compound became ineffective in older pups.
Because of the sporadic seizures we observed in aging heterozygous mice, we decided to determine their seizure threshold versus wild-type mice using repeated injections of a sub-dose (60 mg/kg) of the proconvulsant agent pentylenetetrazole. As seen in Fig. 8A, heterozygous mice demonstrated a twofold increase in seizure susceptibility when compared with wild-type mice. To demonstrate that this seizure activity was directly related to the amount of KCC2 expression and not to some other developmental factors, we created an inducible line of total KCC2 knockout mice. Mice carrying two KCC2 alleles with exon 5 flanked by loxP sites, a CAMKIIα-tTA transgene (84), and a phCMV-tetO-CRE transgene (122) were mated in the presence of doxycycline to prevent expression of the recombinase. When the mice reached P40, doxycycline was removed from the diet and Kaplan-Meier survival curves show that all mice died between P90 and P110 (see Fig. 8B). This data demonstrated that there was no developmental component to the occurrence of the seizures, as these events could be triggered later in life by decreasing expression of the cotransporter. The KCC2b knockout animals have also been used to study the role of KCC2 in isolated cortical pyramidal neurons. It was shown that KCC2 expression increases in cultured neurons and, as anticipated, the cotransporter mediates a progressive decrease in the intracellular Cl− concentration (161). Experiments performed with long-term GABA application or with membrane depolarization combined with GABA application demonstrated that wild-type neurons were able to prevent large Cl− changes and recover quickly from small changes in the intracellular Cl− concentration. In contrast, younger neurons (P2–P6), or older neurons (P13–P17) isolated from KCC2b knockout pups were much slower responding to Cl− changes. These data clearly indicate that KCC2 mediates the developmental decrease in intracellular Cl− and is active in mature neurons to maintain the low intracellular Cl− necessary for GABA hyperpolarization (161). In agreement with this role in maintaining Cl− and preventing the development of epileptiform activity, field recordings performed in the CA1 region of the hippocampus showed a significant increase in amplitude and frequency of spontaneous spikes in the KCC2 heterozygous mice when compared with wild-type mice (162). Furthermore, addition of the convulsive agent 4-aminopyridine to the brain slices induced significantly more seizure-like events in the brain slices isolated from KCC2 heterozygous mice than in the brain slices from their wild-type counterparts (162).
Fig. 8.

Decrease in KCC2 expression leads to seizures and lethality. A: pentylenetetrazole (PTZ)-induced seizures in 6-wk-old wild-type and heterozygote KCC2-null mice. Two groups of mice (21 wild-type and 21 heterozygotes) were injected daily with 60 mg/kg PTZ for a total of 4 days. The number of mice exhibiting seizures scoring >3 within 1 h after the injection are represented. [Modified from Ref. 156, with permission.] B: Kaplan-Meier plots of KCC2b knockouts and CAMKIIα-driven inducible KCC2 knockouts [where mice were fed with doxycycline chow (200 mg/kg, BioServ, Frenchtown, NJ) since conception until doxycycline withdrawal at postnatal day 40 (P40)]. Lethality occurred between P110 and P130, indicating both slow elimination of the drug and a slow decrease in KCC2 expression. C: Western blot analysis of KCC2 expression at P60 in hindbrain and forebrain of a control and an inducible KCC2 knockout mouse after 2 wk doxycycline withdrawal.
Maintaining a low neuronal intracellular Cl− concentration is an evolutionary old function of the K-Cl cotransporter as it is observed in C. elegans. Indeed, in the roundworm, the GABAA receptor (UNC-49) inhibits GABAergic motor neurons enabling coordinated body bends (8). Treatment with muscimol (a GABAA agonist) results in an uncoordinated contraction/relaxation response known as the “rubber band” phenotype (22). However, muscimol treatment of C. elegans kcc-2 mutants did not exhibit this rubber band phenotype, demonstrating that the cotransporter is involved in establishing the necessary chloride gradient for the inhibitory action of UNC-49 in body-wall muscle cells (139).
KCC2's role in locomotion and spasticity was also examined using wild-type and heterozygous KCC2b knockout mice (13). It was shown that KCC2 expression is significantly affected following spinal cord injury and that decreased KCC2 expression led to a reduction in network inhibition. Furthermore, the decreased KCC2 expression in the KCC2b heterozygous mice, or decreased KCC2 function with the use of DIOA as a KCC2 inhibitor, resulted in decreased rate-dependent depression of the Hoffmann reflex. This reflex is commonly used to assess primary type Ia afferents-mediated motoneuronal excitability in individuals suffering from spasticity (13). A follow-up study demonstrated that reduction in KCC2 expression resulted in a depolarized equilibrium potential for Cl− in lumbar motoneurons, associated with increased spontaneous motor activity and a faster locomotor-like activity (132).
Another line of KCC2 hypomorph mice was generated by Vilen and coworkers (150) by inserting a neomycin resistance gene cassette floxed by loxP sites in exon 4 through Mu transposon. Analysis of these mice, which express ∼20% KCC2, showed that they are viable and fertile but exhibit a growth deficit leading to adult mice having a 20% reduction in body weight. As the KCC2b knockout mice express only 5–10% KCC2, this suggests that the amount of KCC2 necessary for survival revolves between 10 and 20%. Cotransporter expression below that level affects animal viability. This also suggests that in our KCC2 inducible knockout mouse (Fig. 8B), the level of KCC2 expression upon doxycycline removal must have dropped well below 10–20%. Behavioral analysis of the Vilen mice revealed normal locomotor activity and motor coordination but increased anxiety-like behavior, as measured by the elevated plus maze assay (143). Mice were also tested for sensory perception, including response to heat-evoked nociception and tactile sensitivity. In all tests, the mutant mice demonstrated a decreased response to the sensory stimulation. This behavior is somewhat contradictory to studies that have shown increased nociception associated with reduction of KCC2 expression (for examples see Refs. 21 and 160) and to a report showing that injection of a KCC2-specific inhibitor in the intrathecal space of a wild-type mouse decreased the latency to response to heat-evoked pain stimuli (6). As for the KCC2b knockout mice, these hypomorphic mice also demonstrated hypersensitivity to pentylenetetrazole (PTZ)-induced seizures, and greater reversal of these PTZ-induced seizures to the general anesthetic propofol. In a subsequent study, the mice also tested for compound-induced locomotion effects (142). The study showed decreased sensitivity to diazepam-induced but normal responses to alcohol-induced motor impairment. The mice also showed normal responses to gaboxadol-induced sedation and neurosteroid-induced hypnosis. These data were consistent with KCC2 involvement in fast hyperpolarizing inhibition rather than tonic GABAergic inhibition. In retrospect, without the differential phenotypes of the Jentsch and Delpire KCC2 knockout mice, the alternatively spliced exon 1 may not have been identified. It raises the question of whether genetic disruption of other cotransporters (and in fact any other gene) at different exons would yield novel information.
Considering the key role of KCC2 in regulating neuronal Cl− and preventing CNS hyperexcitability, it is curious that no mutation or polymorphism in the human KCC2 gene has been identified in relationship to epilepsy or other hyperexcitability disorders. Note, however, that significant changes in KCC2 expression were observed in the postmortem brains from patients with schizophrenia (61, 140). In a recent study, Wolfgang Liedtke's group showed that carbon nanotubes, when exposed to cultured neurons, accelerate the developmental up-regulation of KCC2 (80). These data were verified in situ using organotypic cultures of cortical slices isolated from a novel mouse model. In these experiments, cortical slices from neonatal mice expressing luciferase under the KCC2 promoter were prepared and cultured in the absence or presence of carbon nanotubes for 3 days. Bioluminescence was measured after luciferin addition, and slices exposed to carbon nanotube demonstrated significantly higher luminescence, demonstrating accelerated KCC2 transcription.
K-Cl Cotransporter-3 (SLC12A6 Gene)
KCC3 identification and cloning were reported in 1999 by three independent groups (56, 95, 115). The cotransporter is expressed in multiple tissues as demonstrated by Northern blot analysis—muscle, lung, placenta, liver, heart, kidney, and brain—with highest mRNA levels in muscle, heart, kidney, and brain (95). Expression of KCC3 has been examined in some detail in the nervous system of the mouse using immunofluorescence (110) and in situ hybridization (126). Using an antibody directed against the NH2-terminal tail, the cotransporter was localized to the basolateral membrane of choroid plexus (Fig. 7B), in white matter tracts, large pyramidal neurons in cortex, and Purkinje cells in the cerebellum. In the spinal cord, the signal was intense in dorsal and lateroventral columns and weaker in the central cord. Expression of KCC3 was low at birth and increased significantly during postnatal development (110). Using in situ hybridization, Shekarabi and coworkers (126) demonstrated expression of KCC3 in parvalbumin- and calbindin-positive interneurons as well as in radial or radial-like glia.
Human peripheral neuropathy.
Andermann's syndrome, also named ACCPN (Agenesis Corpus Callosum with Peripheral Neuropathy) or HSMN/ACC (Hereditary Sensory Motor neuropathy/Agenesis Corpus Callosum) is an early onset peripheral neuropathy associated with various degrees of agenesis of the corpus callosum. The syndrome is prevalent in the Charlevoix and Saguenay-Lac-Saint-Jean regions of Quebec and was first described in the medical literature by Andermann et al. (5). Briefly, patients exhibit various degrees of hypotonia during their first year of life with delayed motor milestones. They often require braces for walking, then later a wheelchair, and eventually are bedridden as the neuropathy worsens. The affected individuals also suffer from mental retardation, psychotic symptoms, and bone deformities (30, 37, 73). Dr. Guy Rouleau, a neurologist at McGill University (Montreal, Canada), mapped this neurodegenerative disorder to chromosome 15q13–15q15 (16). The cloning and mapping of KCC3 to human chromosome 15q14 prompted Dr. David Mount (at the time working at Vanderbilt University) to contact Dr. Rouleau, who sequenced all SLC12A6 exons in ACCPN patients, while we worked on creating a KCC3 knockout mouse. Both studies appeared in a consolidated paper in Nature Genetics in 2002 (59). Complete sequencing of exons from Quebec patients revealed deletion of a glycine residue codon in exon 18, resulting in a frame shift (T813fsX) termination of the open reading frame. Interestingly, one affected individual was found to be only heterozygous for the T813fsX mutation. Sequencing of his SLC12A6 exons revealed a separate mutation in the second allele Phe529fsX532. Two mutations were also found in Turkish and Italian families: Arg1011X and Arg675X, respectively (59). Following the original report, other mutations have been reported from isolated families (119, 147). In Table 2 we summarize how all of these mutations (with the exception of R207C and G539D) lead to a codon frame shift and premature protein truncation. Note, however, that these two amino acids are predicted to be located in transmembrane domains 1 and 7, respectively, and their mutation into cysteine and aspartic acid might result into misfolded proteins.
Table 2.
Point mutations in HSN/ACC (KCC3) patients
| Mutation | Exon | Location | Remark | Origin | Reference |
|---|---|---|---|---|---|
| Thr813fsX | Exon 18 | COOH terminus | Most prevalent | Quebec | (59) |
| Phe529fsX532 | Exon 11 | TM7 | Compound heterozygote | Quebec | (59) |
| Arg1011X | Exon 22 | COOH terminus | Turkish | (59) | |
| Arg675X | Exon 15 | ICL5 | Italian | (59) | |
| Tyr678LeufsX41 | Exon 15 | TM11 | Compound heterozygote | German | (147) |
| Phe493CysfsX48 | Exon 10 | TM6 | Compound heterozygote | (147) | |
| Ile301SerfsX15 | Exon 8 | ECL2 | German | (147) | |
| Arg207Cys | Exon 5 | TM1 | Turkish | (147) | |
| Gly539Asp | Exon 12 | TM7 | Compound heterozygote | German | (119) |
| Pro373SplicX42 | Exon 8 | ECL3 | Compound heterozygote | (119) | |
| Tyr192SerfsX12 | Exon 5 | TM1 | Brazilian | (81) |
In the mutation column, three-letter abbreviations represent the amino acids that are mutated into other amino acids or stop codons (X). HSN/ACC, hereditary sensory neuropathy/agenesis corpus callosum; KCC, K-Cl cotransporter; TM, transmembrane domain; ICL and ECL, intra- and extracellular loops, respectively.
We created our KCC3 knockout mouse by targeting exon 3 of the SLC12A6 gene. This mouse has an intact corpus callosum but exhibited severe locomotor deficits when challenged by rotarod, beam task, and wire hang assays (59). The absence of a corpus callosum phenotype is not that surprising since the phenotype in ACCPN patients is highly variable and therefore likely to be influenced by additional factors. Thus, the peripheral neuropathy is clearly the common denominator between ACCPN patients and the mouse knockout. A separate KCC3 knockout mouse was generated in Thomas Jentsch's laboratory by fusing the first conserved KCC3 exon with a β-galactosidase and a stop codon (12). A third knockout mouse model harboring a KCC3 mutation that spontaneously arose at the Jackson Laboratory facility (the gaxp mouse) has also been reported (63). These two additional models exhibit severe locomotor deficits consistent with our knockout mouse and the neuropathy of Andermann's syndrome.
KCC3 knockout and neurodegeneration.
Neurodegeneration has been described in both the CNS and the peripheral nervous system (PNS) of KCC3 knockout mice. In the CNS, degeneration was observed in the outer molecular layer of the dentate gyrus (hippocampus), as well as in both dorsal and ventral roots of the spinal cord (12). As pathology of human peripheral nerves demonstrated a moderate to severe reduction of myelinated nerve fibers and the presence of “onion bulb” formations (24, 147), we conducted a detailed ultramicroscopic analysis of the peripheral nerves of KCC3 knockout mice (see Fig. 9). It revealed axonal and periaxonal swelling, indicating a possible involvement of both neurons and Schwann cells (15). Cross sections of sciatic nerves showed that only a fraction of the fibers were affected, whereas the axon and myelin compaction was perfectly normal in the majority of fibers. This observation could indicate that only a subpopulation of fibers is affected by the absence of KCC3. Alternatively, there is the possibility that the pathology could be limited to “random” sections along a fiber's length and thus the number of affected fibers could be underestimated. Associated with the peripheral nerve pathology, both Andermann patients as well as KCC3 knockout animals display nerve conduction velocity deficits (15, 135, 147).
Fig. 9.

Axonal defects in KCC3-null mice. A and B: electron micrographs of P8 distal sciatic nerves isolated from wild-type (A) and KCC3-null (B) mice. Scale bar in A, 2 μm. Myelin compaction in mutant fibers (B, inset) is indistinguishable from that of wild-type (A, inset, scale bar, 500 nm). Fibers with periaxonal fluid accumulation appear only in KCC3-null nerves (asterisk, B). C–F: varying degrees of the severity of the abnormal fluid accumulation in distal KCC3-null sciatic nerve fibers. Scale bars, 500 nm. Myelin debris (arrows) is observed in the periaxonal space (C and E). a, Axon; pn, paranodes; bv, blood vessel. [Modified from Ref. 15, with permission.]
Neuronal origin of the locomotor phenotype was recently demonstrated by using a synapsin-driven CRE × KCC3flox strategy (125). In this study, the authors demonstrated a severe locomotor deficit in mice where KCC3 knockout was confined to neurons reminiscent of the global KCC3 knockout phenotype. The authors also reported mouse hyperactivity, which is likely the result of CNS dysfunction. As we wrap up this section, we will note that birds (e.g., chicken and finches whose genomes have been sequenced) lack a SLC12A6 gene (see introduction). This raises a series of questions related to differences between bird and mammalian nerve physiology: Is it that another K-Cl cotransporter fulfills the role of KCC3 in the peripheral nerves of birds? Or is it possible that the K-Cl cotransporter function in nerves is specific to mammalian species? A better understanding of the role of KCC3 in mammalian nerve physiology and a better understanding of why deletion of KCC3 in humans and mice results in a nerve degeneration disorder will help in answering these questions.
KCC3 function in CNS and PNS.
Function of KCC3 has been assessed in central and peripheral neurons using the gramicidin-perforated patch method (12, 83). Boettger and coworkers (12) reported that KCC3 knockout mice demonstrated a reduced threshold to flurothyl-induced convulsions or seizures. This observation indicates a role for KCC3 in neuronal Cl− regulation, maybe similar to the role of KCC2 (see above). Consequently, the authors examined the Cl− reversal potential in cerebellar Purkinje cells, which abundantly express KCC3. They observed a small but significant shift in EGABA in neurons isolated from KCC3 knockout mice versus wild-type mice. While the shift is much smaller than the shift observed when KCC2 is disrupted, these data confirm a role of KCC3 in neuronal Cl− homeostasis. This observation also supports immunocytochemical data that both KCC2 and KCC3 can be coexpressed in neurons. Thus, it would be interesting if future studies were to determine whether the transporters act independently of each other or affect each other's function.
The KCC3 knockout mouse has also been used to assess the role of KCC3 in peripheral (DRG) neurons (83). The Na-K-2Cl cotransporter (described above) accumulates Cl− in sensory DRG neurons, and recent data using MQAE-fluorescence showed that DRG neurons from younger mice have slightly higher Cl− concentrations than DRG neurons isolated from adult mice (85). This observation was also made using the gramicidin-perforated patch technique (83). This latest method revealed that while a decrease in NKCC1 expression is likely involved in the postnatal decrease in Cl−, a subpopulation of DRG neurons (∼30%) acquire a DIOA-mediated depolarization during the course of development from young to adult DRG neurons, concomitant with an increase in KCC3a transcript (83). Remarkably, 30% of adult DRG neurons isolated from KCC3 knockout mice maintained a relatively higher Cl− concentration versus 0% of adult wild-type DRG neurons. It is tempting to speculate that these are the same 30% that responded to DIOA. The role of KCC3 and the lower Cl− concentration in this subpopulation of DRG neurons is still unknown. Note that KCC3 also plays a role in cell volume homeostasis and it was shown that hippocampal neurons and proximal tubule epithelial cells isolated from KCC3 knockout mice elicit an abnormal regulatory volume decrease response compared with wild-type cells when exposed to hypotonic media (12). Whether or not the inability of the KCC3-deficient cells to volume regulate contributes to the pathology of the neurons has yet to be determined.
KCC3 knockout and psychiatric disorders.
As Andermann patients demonstrate psychotic behaviors, we tested KCC3 knockout mice for prepulse inhibition, a known neurological phenomenon in which a weak prepulse stimulus delivered shortly before a stronger pulse stimulus inhibits the response to this strong stimulus. Abnormal prepulse inhibition indicates an inability of the tested circuitry to filter out background or unnecessary information. Such deficits are observed in individuals suffering from schizophrenia and other psychotic disorders. In this case, we measured the startle response following auditory stimuli. Whereas heterozygous KCC3 mice demonstrated a prepulse inhibition phenotype similar to wild-type mice, homozygous KCC3 knockout mice displayed a significant reduction in this sensorimotor gating behavior (59). It is worth mentioning that a study also demonstrated an association between rare SLC12A6 variants and bipolar disorder (91), and these variants (due to methylation) likely result in decreased KCC3 expression (94).
KCC3 knockout and age-related deafness.
Auditory brainstem response analysis showed that KCC3 knockout mice slowly develop hearing loss during their first year (12). This progressive loss of hearing was also observed in KCC4-null mice (11), although in those animals the hearing deficit developed much sooner (see KCC4 knockout section below). KCC3 localization was examined in the inner ear and shown to be expressed in the inner and outer hair supporting cells, as well as in other epithelial cells of the organ of Corti and in type I and type III fibrocytes (see Fig. 10). Over time, cochlear degeneration was observed in the knockout animals together with a significant reduction in the passive steady-state potential, which depends on the difference in ionic composition of the endolymph versus perilymph and the permeability of the cell layers enclosing the scala media (12).
Fig. 10.

Expression of SLC12 cotransporters in the inner ear. Cartoon depicts the scala vestibule, scala tympani, and scala media of the mammalian inner ear. Illustrated are the organ of Corti, inner and outer hair cells, phalangeal cells, Deiter's cells, and stria vascularis cells. Note the basolateral expression of NKCC1 on the stria vascularis cells that line the scala media. Inner and outer hair cells express KCC3, and supporting phalangeal and Deiter's cells express KCC4.
KCC3 knockout mice and high blood pressure.
Two studies have demonstrated elevated blood pressure in KCC3 knockout mice (2, 12). In the first study, arterial blood pressure was measured in awake, 3- to 5-mo-old male mice. Although the mean pressure of KCC3 knockout animals (118 ± 2 mmHg) was significantly higher than the arterial pressure of wild-type littermates (100 ± 2 mmHg), there was no difference in heart rates (12). In the second study, arterial blood pressure was measured continuously by telemetry for a 24-h period to capture the pressure during both the light (inactive) and the dark (active) periods in 5- to 6-mo-old mice. KCC3 knockout animals demonstrated a greater increase (more than 30 mmHg) in mean arterial blood pressure during both day and night periods, when compared with controls. Again, no difference between genotypes was observed for heart rate as well as for other parameters, such as body weight, plasma osmolarity and hematocrit; however, increased water consumption was noticed in the KCC3 knockout mice. Drinking volume, measured as milliliters of water per 24 h, increased by 22%, and drinking activity, measured by the number of licks per 24 h, increased markedly by 48% (2). The increase in blood pressure was later shown to not be due to vascular smooth muscle tone but related to an elevated sympathetic tone (120). Indeed, α1-adrenergic blockade or inhibition of ganglionic transmission was shown to restore the arterial blood pressure in KCC3 knockout animals.
KCC3 knockout and kidney phenotype?
The SLC12A6 cotransporter is expressed on the basolateral membrane of proximal tubule cells from segments S1 to S3 (see Fig. 3) (12, 90). Beyond one study, presented as a poster at the 2003 Experimental Biology meeting (San Diego, CA), which indicated impaired fluid and bicarbonate absorption in the proximal tubule of KCC3-null mice (154), little is known about the role of KCC3 in kidney. Despite the phylogenetic similarity of KCC1 and KCC3, knockout of KCC3 clearly produces multiple phenotypes involving multiple systems which are not observed with the genetic disruption of KCC1. Although the expression of KCC1 is widespread and the principle function is considered to be: housekeeping,“, the phenotypic discrepancies suggest that KCC3 (and possibly KCC4) performs additional key functions for which KCC1 cannot serve as “redundant” backup isoform.
K-Cl Cotransporter-4 (SLC12A7 Gene)
The fourth isoform of the electroneutral K-Cl cotransporters, KCC4, was cloned in 1999 and found predominantly expressed in kidney, heart, lung, and liver tissue (95). In 2002, the Jentsch Laboratory generated a KCC4-null mouse by disrupting the SLC12A7 gene with loxP sites inserted in front of the exon coding for the first and part of the second transmembrane domain and the following exon, which encodes for the remainder of the second transmembrane domain. Southern and Western blot analyses demonstrated disrupted genomic DNA and absence of protein expression, respectively (11). KCC4-null mice were both viable and fertile, producing offspring in the expected Mendelian ratios. Behavioral experiments did not initially indicate any overt phenotypic abnormalities; however, between 14 and 21 days after birth, the mice developed severe hearing loss. Histological analysis of the inner ear demonstrated total loss of the outer hair cells (OHCs) that mediate high-frequency hearing. A similar loss of outer and inner hair cells was observed in the NKCC1 null mouse (25, 38, 106) and the KCC3 null mouse (12). Histological localization of these cotransporters in the inner ear is illustrated in Fig. 10. The K+ ions that cycle through these hair cells must be removed by uptake into Deiter's cells and subsequent diffusion through gap junctions to adjoining epithelial cells. As these Deiter's cells do not express significant amounts of Na+-K+-ATPase, the use of KCC4-mediated uptake of K+ would not require metabolic energy and occur near electrochemical equilibrium (11). A similar model of ionic buffering of extracellular K+ by neuronal K-Cl cotransporters was proposed in 1997 (107). Loss of this K+ “recycling” in the KCC4-null mouse would alter the ionic composition around these OHCs and possibly lead to their degeneration. Unlike the cochlea of the NKCC1 null mouse (25, 38) with no endolymphatic fluid, there was no collapse of Reissner's membrane in either the KCC4 null or KCC3 null mice, indicating that the two cotransporters are not involved in K+ secretion or the production of endolymph. The generation of cotransporter knockout animal models has clearly revealed a major role for NKCC1, KCC3, and KCC4 in inner ear physiology.
Interestingly, along with the severe hearing loss, disruption of KCC4 expression in portions of the kidney nephron resulted in mice with renal tubular acidosis (11). Blood gas analysis that demonstrated a compensatory metabolic acidosis was consistent with the increased urine alkalinity observed in the KCC4 null mice. The α-intercalated cells of the distal nephron utilize a combination of apical H+-ATPase with basolateral anion exchanger to secrete H+ into the lumen. The lack of basolateral KCC4 expression in the α-intercalated cells of the knockout mouse resulted in an increase in both the intracellular Cl− and the electrochemical gradient, thereby inhibiting apical H+ secretion. In this way, KCC4 becomes the third transport protein in α-intercalated cells whose inactivation results in renal tubular acidosis (11). Mutations in several other membrane transport proteins have also resulted in hearing loss and kidney dysfunction. Autosomal recessive mutations in the apical H+-ATPase were found to be the common link between improper pH balance in collecting duct α-intercalated cells and epithelial cells of the cochlear scala media (66). Two variants of Bartter's syndrome with sensorineural deafness have been linked to mutations in CLCKB chloride channel and Barttin, an essential β subunit of CLCK (35, 127). Interestingly, despite significant expression of KCC4 in multiple tissues, to date only the inner ear and kidney appear affected. This again suggests a “redundancy” of cotransporter isoforms with expression/function of KCC3 having a more important physiological role than KCC4 and KCC1.
SUMMARY
Although only seven of the nine members of this small protein family have been well characterized, the electroneutral cation-chloride cotransporters clearly serve many important physiological roles. The sequencing of the mouse genome has provided a unique opportunity to genetically manipulate the expression of these cotransporters at the cellular, tissue, and systemic level. Knockout mice of all three Na+-dependent cotransporters were generated in the late 1990s. The NCC-null mouse exhibited phenotypic symptoms similar to Gitleman's syndrome. Inner ear dysfunction, nervous system deficits, and multiple epithelial-related disorders are some of the most striking defects exhibited by the NKCC1-null mouse. The NKCC2-null mouse exhibited Bartter-like symptoms including severe dehydration, high plasma renin and potassium concentrations, and metabolic acidosis. Although genetic knockout of KCC1 did not produce any overt phenotype, when combined with the knockout of KCC3, the double knockout mouse restored the volume and osmotic resistance to the red blood cells of another mouse model for human sickle cell disease. Identification of a novel KCC2 NH2-terminal isoform with an alternative promoter and first exon ultimately explained why different targeting strategies for the knockout of KCC2 in the mouse produced both animals that died immediately after birth and animals that survived for more than two weeks. The partial knockout of KCC2 altered intracellular chloride concentrations in neurons increasing seizure-susceptibility and decreasing nociception and tactile sensitivity. Generation of a KCC3-null mouse exhibited severe locomotor deficits and a peripheral neuropathy seen in humans afflicted with Andermann's syndrome. Additionally, knockout of KCC3 resulted in elevated blood pressure, age-related deafness, and neuronal prepulse inhibition. Finally, disruption of KCC4 resulted in mice with severe hearing loss and renal tubular acidosis.
Clearly, the genetic manipulation of the murine SLC12 gene family has produced mammalian model organisms that exhibit phenotypic syndromes seen in several human physiological disorders. Continued use of these animal models to test novel pharmacological and therapeutic interventions will undoubtedly be beneficial to human health and disease prevention.
GRANTS
The work in the authors' laboratory was funded by the National Institute of Neurological Disease and Stroke Grant NS36758 and the National Institute of General Medical Sciences Grant GM74771.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
K.B.G. and E.D. prepared the figures; K.B.G. and E.D. drafted the manuscript; K.B.G. and E.D. edited and revised the manuscript; K.B.G. and E.D. approved the final version of the manuscript.
REFERENCES
- 1. Abbas L, Whitfield TT. Nkcc1 (SLC12A2) is required for the regulation of endolymph volume in the otic vesicle and swim bladder volume in the zebrafish larva. Development 136: 2837–2848, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Adragna NC, Chen Y, Delpire E, Lauf PK, Morris M. Hypertension in K-Cl cotransporter-3 knockout mice. Adv Exp Med Biol 559: 379–385, 2004 [DOI] [PubMed] [Google Scholar]
- 3. Alvarez-Leefmans FJ, Gamiño SM, Giraldez F, Nogueron I. Intracellular chloride regulation in amphibian dorsal root ganglion neurons studied with ion-selective microelectrodes. J Physiol 406: 225–246, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Alvarez-Leefmans FJ, Leon-Olea M, Mendoza-Sotelo J, Alvarez FJ, Anton B, Garduno R. Immunolocalization of the Na(+)-K(+)-2Cl(-) cotransporter in peripheral nervous tissue of vertebrates. Neuroscience 104: 569–582, 2002 [DOI] [PubMed] [Google Scholar]
- 5. Andermann F, Andermann E, Joubert M, Karpati G, Carpenter S, Melancon D. Familial agenesis of the corpus callosum with anterior horn cell disease: a syndrome of mental retardation, areflexia and paraparesis. Trans Am Neurol Assoc 97: 242–244, 1972 [Google Scholar]
- 6. Austin TM, Delpire E. Inhibition of KCC2 in mouse spinal cord neurons leads to hyper-sensitivity to thermal stimulation. Anesth Analg 113: 1509–1515, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Balakrishnan V, Becker M, Lohrke S, Nothwang HG, Guresir E, Friauf E. Expression and function of chloride transporters during development of inhibitory neurotransmission in the auditory brainstem. J Neurosci 23: 4134–4145, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bamber BA, Beg AA, Twyman RE, Jorgensen EM. The Caenorhabditis elegans unc-49 locus encodes multiple subunits of a heteromultimeric GABA receptor. J Neurosci 19: 5348–5359, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ben-Ari Y. Excitatory actions of gaba during development: the nature of the nurture. Nat Rev Neurosci 3: 728–739, 2002 [DOI] [PubMed] [Google Scholar]
- 10. Ben-Ari Y, Cherubini E, Corradetti R, Gaiarsa JL. Giant synaptic potentials in immature rat CA3 hippocampal neurones. J Physiol 416: 303–325, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Boettger T, Hubner CA, Maier H, Rust MB, Beck FX, Jentsch TJ. Deafness and renal tubular acidosis in mice lacking the K-Cl co-transporter Kcc4. Nature 416: 874–878, 2002 [DOI] [PubMed] [Google Scholar]
- 12. Boettger T, Rust MB, Maier H, Seidenbecher T, Schweizer M, Keating DJ, Faulhaber J, Ehmke H, Pfeffer C, Scheel O, Lemcke B, Horst J, Leuwer R, Pape HC, Volkl H, Hubner CA, Jentsch TJ. Loss of K-Cl co-transporter KCC3 causes deafness, neurodegeneration and reduced seizure threshold. EMBO J 22: 5422–5434, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Boulenguez P, Liabeuf S, Bos R, Bras H, Jean-Xavier C, Brocard C, Stil A, Darbon P, Cattaert D, Delpire E, Marsala M, Vinay L. Down-regulation of the potassium-chloride cotransporter KCC2 contributes to spasticity after spinal cord injury. Nat Med 16: 302–307, 2010 [DOI] [PubMed] [Google Scholar]
- 14. Brooks HL, Sorensen AM, Terris J, Schultheis PJ, Lorenz JN, Shull GE, Knepper MA. Profiling of renal tubule Na+ transporter abundances in NHE3 and NCC-null mice using targeted proteomics. J Physiol 530: 359–366, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Byun N, Delpire E. Axonal and periaxonal swelling precede peripheral neurodegeneration in KCC3 knockout mice. Neurobiol Dis 28: 39–51, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Casaubon LK, Melanson M, Lopes-Cendes I, Marineau C, Andermann E, Andermann F, Weissenbach J, Prevost C, Bouchard JP, Mathieu J, Rouleau GA. The gene responsible for a severe form of peripheral neuropathy and agenesis of the corpus callosum maps to chromosome 15q. Am J Hum Genet 58: 28–34, 1996 [PMC free article] [PubMed] [Google Scholar]
- 17. Castrop H, Lorenz JN, Hansen PB, Friis U, Mizel D, Oppermann M, Jensen BL, Briggs J, Skøtt O, Schnermann J. Contribution of the basolateral isoform of the Na-K-2Cl− cotransporter (NKCC1/BSC2) to renin secretion. Am J Physiol Renal Physiol 289: F1185–F1192, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Clayton GH, Owens GC, Wolf JS, Smith RL. Ontogeny of cation-Cl− cotransporter expression in rat neocortex. Brain Res Dev Brain Res 109: 281–292, 1998 [DOI] [PubMed] [Google Scholar]
- 19. Costanzo LS. Localization of diuretic action in microperfused rat distal tubules: Ca and Na transport. Am J Physiol Renal Fluid Electrolyte Physiol 248: F527–F535, 1985 [DOI] [PubMed] [Google Scholar]
- 20. Costanzo LS, Windhager EE. Calcium and sodium transport by the distal convoluted tubule of the rat. Am J Physiol Renal Fluid Electrolyte Physiol 235: F492–F506, 1978 [DOI] [PubMed] [Google Scholar]
- 21. Coull JA, Boudreau D, Bachand K, Prescott SA, Nault F, Sik A, De Koninck P, De Koninck Y. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature 424: 938–942, 2003 [DOI] [PubMed] [Google Scholar]
- 22. de la Cruz IP, Levin JZ, Cummins C, Anderson P, Horvitz HR. sup-9, sup-10, and unc-93 may encode components of a two-pore K+ channel that coordinates muscle contraction in Caenorhabditis elegans. J Neurosci 27: 9133–9145, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. DeFazio RA, Heger S, Ojeda SR, Moenter SM. Activation of A-type gamma-aminobutyric acid receptors excites gonadotropin-releasing hormone neurons. Mol Endocrinol 16: 2872–2891, 2002 [DOI] [PubMed] [Google Scholar]
- 24. Deleu D, Bamanikar SA, Muirhead D, Louon A. Familial progressive sensorimotor neuropathy with agenesis of the corpus callosum (Andermann syndrome): a clinical, neuroradiological and histopathological study. Eur Neurol 37: 104–109, 1997 [DOI] [PubMed] [Google Scholar]
- 25. Delpire E, Lu J, England R, Dull C, Thorne T. Deafness and imbalance associated with inactivation of the secretory Na-K-2Cl co-transporter. Nat Genet 22: 192–195, 1999 [DOI] [PubMed] [Google Scholar]
- 26. Delpire E, Rauchman MI, Beier DR, Hebert SC, Gullans SR. Molecular cloning and chromosome localization of a putative basolateral Na-K-2Cl cotransporter from mouse inner medullary collecting duct (mIMCD-3) cells. J Biol Chem 269: 25677–25683, 1994 [PubMed] [Google Scholar]
- 27. Deol MS. The development of the inner ear in mice homozygous for shaker-with-syndictalism. J Embryol Exp Morphol 11: 493–512, 1963 [PubMed] [Google Scholar]
- 28. Dixon MJ, Gazzard J, Chaudhry SS, Sampson N, Schulte BA, Steel KP. Mutation of the Na-K-Cl co-transporter gene SLC12A2 results in deafness in mice. Hum Mol Genet 8: 1579–1584, 1999 [DOI] [PubMed] [Google Scholar]
- 29. Dunham PB, Steward GW, Ellory JC. Chloride-activated passive potassium transport in human erythrocytes. Proc Natl Acad Sci USA 77: 1711–1715, 1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dupré N, Howard HC, Mathieu J, Karpati G, Vanasse M, Bouchard JP, Carpenter S, Rouleau GA. Hereditary motor and sensory neuropathy with agenesis of the corpus callosum. Ann Neurol 54: 9–18, 2003 [DOI] [PubMed] [Google Scholar]
- 31. Dzhala V, Valeeva G, Glykys J, Khazipov R, Staley K. Traumatic alterations in GABA signaling disrupt hippocampal network activity in the developing brain. J Neurosci 32: 4017–4031, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dzhala VI, Brumback AC, Staley K. Bumetanide enhances phenobarbital efficacy in a neonatal seizure model. Ann Neurol 63: 222–235, 2008 [DOI] [PubMed] [Google Scholar]
- 33. Dzhala VI, Talos DM, Sdrulla DA, Brumback AC, Mathews GC, Benke TA, Delpire E, Jensen FE, Staley KJ. NKCC1 transporter facilitates seizures in the developing brain. Nat Med 11: 1205–1213, 2005 [DOI] [PubMed] [Google Scholar]
- 34. Ecelbarger CA, Terris J, Hoyer JR, Nielsen S, Wade JB, Knepper MA. Localization and regulation of the rat renal Na+-K+-2Cl− cotransporter, BSC-1. Am J Physiol Renal Fluid Electrolyte Physiol 271: F619–F628, 1996 [DOI] [PubMed] [Google Scholar]
- 35. Estevez R, Boettger T, Stein V, Birkenhager R, Otto E, Hildebrandt F, Jentsch TJ. Barttin is a Cl− channel beta-subunit crucial for renal Cl− reabsorption and inner ear K+ secretion. Nature 414: 558–561, 2001 [DOI] [PubMed] [Google Scholar]
- 36. Evans RL, Park K, turner RJ, Watson GE, Nguyen HV, Dennett MR, Hand AR, Flagella M, Shull GE, Melvin JE. Severe impairment of salivation in Na+/K+/2Cl− cotransporter (NKCC1)-deficient mice. J Biol Chem 275: 26720–26726, 2000 [DOI] [PubMed] [Google Scholar]
- 37. Filteau MJ, Pourcher E, Bouchard RH, Baruch P, Mathieu J, Bédart F, Simard N, Vincent P. Corpus callosum agenesis and psychosis in Andermann syndrome. Arch Neurol 48: 1275–1280, 1991 [DOI] [PubMed] [Google Scholar]
- 38. Flagella M, Clarke LL, Miller ML, Erway LC, Giannella RA, Andringa A, Gawenis LR, Kramer J, Duffy JJ, Doetschman T, Lorenz JN, Yamoah EN, Cardell EL, Shull GE. Mice lacking the basolateral Na-K-2Cl cotransporter have impaired epithelial chloride secretion and are profoundly deaf. J Biol Chem 274: 26946–26955, 1999 [DOI] [PubMed] [Google Scholar]
- 39. Fraser AG, Cowie JF, Lambie AT, Robson JS. The effects of furosemide on the osmolality of the urine and the composition of renal tissue. J Pharmacol Exp Ther 158: 475–486, 1967 [PubMed] [Google Scholar]
- 40. Frings S. Chloride-based signal amplification if olfactory sensory neurons. In: Physiology and Pathology of Chloride Transporter and Channels in the Nervous System: From Molecules to Diseases, edited by Alvarez-Leefmans FJ, Delpire E. London: Academic, 2009, p. 413–424 [Google Scholar]
- 41. Gagnon KB, Delpire E. Molecular physiology of SPAK and OSR1: two Ste20-related protein kinases regulating ion transport. Physiol Rev 92: 1577–1617, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gamba G, Miyanoshita A, Lombardi M, Lytton J, Lee WS, Hediger M, Hebert SC. Molecular cloning, primary structure, and characterization of two members of the mammalian electroneutral sodium-(potassium)-chloride cotransporter family expressed in kidney. J Biol Chem 269: 17713–17722, 1994 [PubMed] [Google Scholar]
- 43. Gamba G, Saltzberg SN, Lombardi M, Miyanoshita A, Lytton J, Hediger MA, Brenner BM, Hebert SC. Primary structure and functional expression of a cDNA encoding the thiazide-sensitive, electroneutral sodium-chloride cotransporter. Proc Natl Acad Sci USA 90: 2749–2753, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Geck P, Pietrzyk C, Burckhardt BC, Pfeiffer B, Heinz E. Electrically silent cotransport of Na+, K+ and Cl− in Ehrlich cells. Biochim Biophys Acta 600: 432–447, 1980 [DOI] [PubMed] [Google Scholar]
- 45. Geisler R, Rauch GJ, Geiger-Rudolph S, Albrecht A, van Bebber F, Berger A, Busch-Nentwich E, Dahm R, Dekens MP, Dooley C, Elli AF, Gehring I, Geiger H, Geisler M, Glaser S, Holley S, Huber M, Kerr A, Kirn A, Knirsch M, Konantz M, Küchler AM, Maderspacher F, Neuhauss SC, Nicolson T, Ober EA, Praeg E, Ray R, Rentzsch B, Rick JM, Rief E, Schauerte HE, Schepp CP, Schönberger U, Schonthaler HB, Seiler C, Sidi S, Söllner C, Wehner A, Weiler C, Nüsslein-Volhard C. Large-scale mapping of mutations affecting zebrafish development. BMC Genomics 8: 11, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gillen CM, Brill S, Payne JA, Forbush BI. Molecular cloning and functional expression of the K-Cl cotransporter from rabbit, rat, and human. A new member of the cation-chloride cotransporter family. J Biol Chem 271: 16237–16244, 1996 [DOI] [PubMed] [Google Scholar]
- 47. Gillie DJ, Pace AJ, Coakley RJ, Koller BH, Barker PM. Liquid and ion transport by fetal airway and lung epithelia of mice deficient in sodium-potassium-2-chloride transporter. Am J Respir Cell Mol Biol 25: 14–20, 2001 [DOI] [PubMed] [Google Scholar]
- 48. Giménez I, Isenring P, Forbush B. Spatially distributed alternative splice variants of the renal Na-K-Cl cotransporter exhibit dramatically different affinities for the transported ions. J Biol Chem 277: 8767–8770, 2002 [DOI] [PubMed] [Google Scholar]
- 49. Glaudemans B, Yntema HG, San-Cristobal P, Schoots J, Pfundt R, Kamsteeg EJ, Bindels RJ, Knoers NVAM, Hoenderop JG, Hoefsloot LH. Novel NCC mutants and functional analysis in a new cohort of patients with Gitelman syndrome. Eur J Hum Genet 20: 263–270, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Granados-Soto V, Arguelles CF, Alvarez-Leefmans FJ. Peripheral and central antinociceptive action of Na–K–2Cl cotransporter blockers on formalin-induced nociception in rats. Pain 114: 231–238, 2005 [DOI] [PubMed] [Google Scholar]
- 51. Greger R, Schlatter E. Presence of luminal K+, a prerequisite for active NaCl transport in the cortical thick ascending limb of Henle's loop of rabbit kidney. Pflügers Arch 392: 92–94, 1981 [DOI] [PubMed] [Google Scholar]
- 52. Greger R, Schlatter E, Lang F. Evidence for electroneutral sodium chloride cotransport in the cortical thick ascending limb of Henle's loop of rabbit kidney. Pflügers Arch 386: 308–314, 1983 [DOI] [PubMed] [Google Scholar]
- 53. Grimm PR, Taneja TK, Liu J, Coleman R, Chen YY, Delpire E, Wade JB, Welling PA. SPAK isoforms and OSR1 regulate sodium-chloride co-transporters in a nephron-specific manner. J Biol Chem 287: 37673–37690, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Grubb BR, Pace AJ, Lee E, Koller BH, Boucher RC. Alterations in airway ion transport in NKCC1-deficient mice. Am J Physiol Cell Physiol 281: C615–C623, 2001 [DOI] [PubMed] [Google Scholar]
- 55. Hertwig P. Neue Mutationen und Koppelungsgruppen bei der Hausmaus. Z Indukt Abstamm Vererbungsl 80: 220–226, 1942 [Google Scholar]
- 56. Hiki K, D'Andrea RJ, Furze J, Crawford J, Woollatt E, Sutherland GR, Vadas MA, Gamble JR. Cloning, characterization, and chromosomal location of a novel human K+-Cl− cotransporter. J Biol Chem 274: 10661–10667, 1999 [DOI] [PubMed] [Google Scholar]
- 57. Holtzman EJ, Kumar S, Faaland CA, Warner F, Logue PJ, Erickson SJ, Ricken G, Waldman J, Kumar S, Dunham PB. Cloning, characterization, and gene organization of K-Cl cotransporter from pig and human kidney and C. elegans. Am J Physiol Renal Physiol 275: F550–F564, 1998 [DOI] [PubMed] [Google Scholar]
- 58. Hook JB, Williamson HE. Effect of furosemide on renal medullary sodium gradient. Proc Soc Exp Biol Med 118: 372–374, 1965 [DOI] [PubMed] [Google Scholar]
- 59. Howard HC, Mount DB, Rochefort D, Byun N, Dupré N, Lu J, Fan X, Song L, Rivière JB, Prévost C, Welch R, England R, Zhan FQ, Mercado A, Siesser WB, George AL, Horst J, Simonati A, McDonald MP, Bouchard JP, Mathieu J, Delpire E, Rouleau GA. Mutations in the K-Cl cotransporter KCC3 cause a severe peripheral neuropathy associated with agenesis of the corpus callosum. Nat Genet 32: 384–392, 2002 [DOI] [PubMed] [Google Scholar]
- 60. Hubner CA, Stein V, Hermans-Borgmeyer I, Meyer T, Ballanyi K, Jentsch TJ. Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition. Neuron 30: 515–524, 2001 [DOI] [PubMed] [Google Scholar]
- 61. Hyde TM, Lipska BK, Ali T, Mathew SV, Law AJ, Metitiri OE, Straub RE, Ye T, Colantuoni C, Herman MM, Bigelow LB, Weinberger DR, Kleinman JE. Expression of GABA signaling molecules KCC2, NKCC1, and GAD1 in cortical development and schizophrenia. J Neurosci 31: 11088–11095, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Igarashi P, Vanden Heuvel GB, Payne JA, Forbush BI. Cloning, embryonic expression and alternative splicing of a murine kidney specific Na-K-Cl cotransporter. Am J Physiol Renal Fluid Electrolyte Physiol 269: F405–F418, 1995 [DOI] [PubMed] [Google Scholar]
- 63. Jiao Y, Jin X, Yan J, Zhang C, Jiao F, Li X, Roe BA, Mount DB, Gu W. A deletion mutation in Slc12a6 is associated with neuromuscular disease in gaxp mice. Genomics 91: 407–414, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kaneko H, Putzier I, Frings S, Kaupp UB, Gensch T. Chloride accumulation in mammalian olfactory sensory neurons. J Neurosci 24: 7931–7938, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kaplan MR, Plotkin MD, Brown D, Hebert SC, Delpire E. Expression of the mouse Na-K-2Cl cotransporter, mBSC2, in the terminal IMCD, the glomerular and extraglomerular mesangium and the glomerular afferent arteriole. J Clin Invest 98: 723–730, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Karet FE, Kinberg KE, Nelson RD, Nayir A, Mocan H, Sanjad SA, Rodriguez-Soriano J, Santos F, Cremers CW, Di Pietro A, Hoffbrand BI, Winiarski J, Bakkaloglu A, Ozen S, Dusunsel R, Goodyer P, Hulton SA, Wu DK, Skvorak AB, Morton CC, Cunningham MJ, Jha V, Lifton RP. Mutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nat Genet 21: 84–90, 1999 [DOI] [PubMed] [Google Scholar]
- 67. Kilb W, Sinning A, Luhmann HJ. Model-specific effects of bumetanide on epileptiform activity in the in-vitro intact hippocampus of the newborn mouse. Neuropharmacology 53: 524–533, 2007 [DOI] [PubMed] [Google Scholar]
- 68. Kim SM, Eisner C, Faulhaber-Walter R, Mizel D, Wall SM, Briggs JP, Schnermann J. Salt sensitivity of blood pressure in NKCC1-deficient mice. Am J Physiol Renal Physiol 295: F1230–F1238, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kregenow FM. Osmoregulatory salt transport mechanisms: control of cell volume in anisotonic media. Annu Rev Physiol 43: 493–505, 1981 [DOI] [PubMed] [Google Scholar]
- 70. Kregenow FM. The response of duck erythrocytes to nonhemolytic hypotonic media. Evidence for a volume-controlling mechanism. J Gen Physiol 58: 372–395, 1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kregenow FM. The respose of duck erythrocytes to hypertonic media. Further evidence for a volume-controlling mechanism. J Gen Physiol 58: 396–411, 1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kurtz CL, Karolyi L, Seyberth HW, Koch MC, Vargas R, Feldmann D, Vollmer M, Knoers NVAM, Madrigal G, Guay-Woodford LM. A common NKCC2 mutation in Costa Rican Bartter's syndrome patients: evidence for a founder effect. J Am Soc Nephrol 8: 1706–1711, 1997 [DOI] [PubMed] [Google Scholar]
- 73. Labrisseau A, Vanasse M, Brochu P, Jasmin G. The Andermann syndrome: agenesis of the corpus callosum associated with mental retardation and progressive sesorimotor neuronopathy. Can J Neurol Sci 11: 257–261, 1984 [DOI] [PubMed] [Google Scholar]
- 74. Laird JM, Garcia-Nicas E, Delpire EJ, Cervero F. Presynaptic inhibition and spinal pain processing in mice: a possible role of the NKCC1 cation-chloride co-transporter in hyperalgesia. Neurosci Lett 361: 200–203, 2004 [DOI] [PubMed] [Google Scholar]
- 75. Lalioti MD, Zhang J, Volkman HM, Kahle KT, Hoffmann KE, Toka HR, Nelson-Williams C, Ellison DH, Flavell R, Booth CJ, Lu Y, Geller DS, Lifton RP. Wnk4 controls blood pressure and potassium homeostasis via regulation of mass and activity of the distal convoluted tubule. Nat Genet 38: 1124–1132, 2006 [DOI] [PubMed] [Google Scholar]
- 76. Lander ES, Linton LMBB, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann N, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, Mullikin JC, Mungall A, Plumb R, Ross M, Shownkeen R, Sims S, Waterston RH, Wilson RK, Hillier LW, McPherson JD, Marra MA, Mardis ER, Fulton LA, Chinwalla AT, Pepin KH, Gish WR, Chissoe SL, Wendl MC, Delehaunty KD, Miner TL, Delehaunty A, Kramer JB, Cook LL, Fulton RS, Johnson DL, Minx PJ, Clifton SW, Hawkins T, Branscomb E, Predki P, Richardson P, Wenning S, Slezak T, Doggett N, Cheng JF, Olsen A, Lucas S, Elkin C, Uberbacher E, Frazier M, Gibbs RA, Muzny DM, Scherer SE, Bouck JB, Sodergren EJ, Worley KC, Rives CM, Gorrell JH, Metzker ML, Naylor SL, Kucherlapati RS, Nelson DL, Weinstock GM, Sakaki Y, Fujiyama A, Hattori M, Yada T, Toyoda A, Itoh T, Kawagoe C, Watanabe H, Totoki Y, Taylor T, Weissenbach J, Heilig R, Saurin W, Artiguenave F, Brottier P, Bruls T, Pelletier E, Robert C, Wincker P, Smith DR, Doucette-Stamm L, Rubenfield M, Weinstock K, Lee HM, Dubois J, Rosenthal A, Platzer M, Nyakatura G, Taudien S, Rump A, Yang H, Yu J, Wang J, Huang G, Gu J, Hood L, Rowen L, Madan A, Qin S, Davis RW, Federspiel NA, Abola AP, Proctor MJ, Myers RM, Schmutz J, Dickson M, Grimwood J, Cox DR, Olson MV, Kaul R, Raymond C, Shimizu N, Kawasaki K, Minoshima S, Evans GA, Athanasiou M, Schultz R, Roe BA, Chen F, Pan H, Ramser J, Lehrach H, Reinhardt R, McCombie WR, de la Bastide M, Dedhia N, Blöcker H, Hornischer K, Nordsiek G, Agarwala R, Aravind L, Bailey JA, Bateman A, Batzoglou S, Birney E, Bork P, Brown DG, Burge CB, Cerutti L, Chen HC, Church D, Clamp M, Copley RR, Doerks T, Eddy SR, Eichler EE, Furey TS, Galagan J, Gilbert JG, Harmon C, Hayashizaki Y, Haussler D, Hermjakob H, Hokamp K, Jang W, Johnson LS, Jones TA, Kasif S, Kaspryzk A, Kennedy S, Kent WJ, Kitts P, Koonin EV, Korf I, Kulp D, Lancet D, Lowe TM, McLysaght A, Mikkelsen T, Moran JV, Mulder N, Pollara VJ, Ponting CP, Schuler G, Schultz J, Slater G, Smit AF, Stupka E, Szustakowski J, Thierry-Mieg D, Thierry-Mieg J, Wagner L, Wallis J, Wheeler R, Williams A, Wolf YI, Wolfe KH, Yang SP, Yeh RF, Collins F, Guyer MS, Peterson J, Felsenfeld A, Wetterstrand KA, Patrinos A, Morgan MJ, de Jong P, Catanese JJ, Osoegawa K, Shizuya H, Choi S, Chen YJ. International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature 409: 860–921, 2001 [DOI] [PubMed] [Google Scholar]
- 77. Lauf PK, Theg BE. A chloride dependent K+ flux induced by N-ethylmaleimide in genetically low K+ sheep and goat erythrocytes. Biochem Biophys Res Commun 70: 221–242, 1980 [DOI] [PubMed] [Google Scholar]
- 78. Lew VL, Bookchin RM. Ion transport pathology in the mechanism of sickle cell dehydration. Physiol Rev 85: 179–200, 2005 [DOI] [PubMed] [Google Scholar]
- 79. Li JL, Chu HQ, Zhou LQ, Xiong H, Wang Y, Chen QG, Chen J, Li ZY, Liu Y, Cui YH. Association of age-related hearing loss with ion transporter KCNQ1 and NKCC1 in cochlea of C57BL/6J mice. Zhonghua Er Bi Yan Hou Tou Jing Wai Ke Za Zhi 46: 139–143, 2011 [PubMed] [Google Scholar]
- 80. Liedtke W, Yeo M, Zhang H, Wang Y, Gignac M, Miller S, Berglund K, Liu J. Highly conductive carbon nanotube matrix accelerates developmental chloride extrusion in central nervous system neurons by increased expression of chloride transporter KCC2. Small. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lourenço CM, Dupré N, Rivière JB, Rouleau GA, Marques VD, Genari AB, Santos AC, Barreira AA, Marques WJ. Expanding the differential diagnosis of inherited neuropathies with non-uniform conduction: Andermann syndrome. J Peripher Nerv Syst 17: 123–127, 2012 [DOI] [PubMed] [Google Scholar]
- 82. Lu J, Karadsheh M, Delpire E. Developmental regulation of the neuronal-specific isoform of K-Cl cotransporter KCC2 in postnatal rat brains. J Neurobiol 39: 558–568, 1999 [PubMed] [Google Scholar]
- 83. Lucas O, Hilaire C, Delpire E, Scamps F. KCC3-dependent chloride extrusion in adult sensory neurons. Mol Cell Neurosci 50: 211–220, 2012 [DOI] [PubMed] [Google Scholar]
- 84. Mansuy IM, Winder DG, Moallem TM, Osman M, Mayford M, Hawkins RD, Kandel ER. Inducible and reversible gene expression with the rtTA system for the study of memory. Neuron 21: 257–265, 1998 [DOI] [PubMed] [Google Scholar]
- 85. Mao S, Garzon-Muvdi T, Difulvio M, Chen Y, Delpire E, Alvarez FJ, Alvarez-Leefmans FJ. Molecular and functional expression of cation-chloride cotransporters in doersal root ganglion neurons during postnatal maturation. J Neurophysiol 108: 834–852, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Mastroianni N, Bettinelli A, Bianchetti M, Colussi G, De Fusco M, Sereni F, Ballabio A, Casari G. Novel molecular variants of the Na-Cl cotransporter gene are responsible for Gitelman syndrome. Am J Hum Genet 59: 1019–1026, 1996 [PMC free article] [PubMed] [Google Scholar]
- 87. McCormick JA, Mutig K, Nelson JH, Saritas T, Hoorn EJ, Yang CL, Rogers S, Curry J, Delpire E, Bachmann S, Ellison DH. A SPAK isoform switch modulates renal salt transport and blood pressure. Cell Metab 14: 352–364, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Melander O, Orho-Melander M, Bengtsson K, Lindblad U, Râstam L, Groop L, Hulthén UL. Genetic variants of thiazide-sensitive NaCl-cotransporter in Gitelman's syndrome and primary hypertension. Hypertension 36: 389–394, 2000 [DOI] [PubMed] [Google Scholar]
- 89. Mercado A, Broumand V, Zandi-Nejad K, Enck AH, Mount DB. A C-terminal domain in KCC2 confers constitutive K+-Cl− cotransport. J Biol Chem 281: 1016–1026, 2006 [DOI] [PubMed] [Google Scholar]
- 90. Mercado A, Vazquez N, Song L, Cortes R, Enck AH, Welch R, Delpire E, Gamba G, Mount DB. NH2-terminal heterogeneity in the KCC3 K+-Cl− cotransporter. Am J Physiol Renal Physiol 289: F1246–F1261, 2005 [DOI] [PubMed] [Google Scholar]
- 91. Meyer J, Johannssen K, Freitag CM, Schraut K, Teuber I, Hahner A, Mainhardt C, Mössner R, Volz HP, Wienker TF, McKeane D, Stephan DA, Rouleau G, Reif A, Lesch KP. Rare variants of the gene encoding the potassium chloride co-transporter 3 are associated with bipolar disorder. Int J Neuropsychopharmacol 8: 495–504, 2005 [DOI] [PubMed] [Google Scholar]
- 92. Meyer JW, Flagella M, Sutliff RL, Lorenz JN, Nieman ML, Weber CS, Paul RJ, Shull GE. Decreased blood pressure and vascular smooth muscle tone in mice lacking basolateral Na+-K+-2Cl− cotransporter. Am J Physiol Heart Circ Physiol 283: H1846–H1855, 2002 [DOI] [PubMed] [Google Scholar]
- 93. Morris RG, Hoorn EJ, Knepper MA. Hypokalemia in a mouse model of Gitelman's syndrome. Am J Physiol Renal Physiol 290: F1416–F1420, 2006 [DOI] [PubMed] [Google Scholar]
- 94. Moser DES, Kumsta R, Palmason H, Bock C, Athanassiadou Z, Lesch KP, Meyer J. Functional analysis of a potassium-chloride co-transporter 3 (SLC12A6) promoter polymorphism leading to an additional DNA methylation site. Neuropsychopharmacology 34: 458–467, 2009 [DOI] [PubMed] [Google Scholar]
- 95. Mount DB, Mercado A, Song L, Xu J, George JAL, Delpire E, Gamba G. Cloning and Characterization of KCC3 and KCC4, new members of the cation-chloride cotransporter gene family. J Biol Chem 274: 16355–16362, 1999 [DOI] [PubMed] [Google Scholar]
- 96. Mouse Genome Sequencing Consortium, Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, Antonarakis SE, Attwood J, Baertsch R, Bailey J, Barlow K, Beck S, Berry E, Birren B, Bloom T, Bork P, Botcherby M, Bray N, Brent MR, Brown DG, Brown SD, Bult C, Burton J, Butler J, Campbell RD, Carninci P, Cawley S, Chiaromonte F, Chinwalla AT, Church DM, Clamp M, Clee C, Collins FS, Cook LL, Copley RR, Coulson A, Couronne O, Cuff J, Curwen V, Cutts T, Daly M, David R, Davies J, Delehaunty KD, Deri J, Dermitzakis ET, Dewey C, Dickens NJ, Diekhans M, Dodge S, Dubchak I, Dunn DM, Eddy SR, Elnitski L, Emes RD, Eswara P, Eyras E, Felsenfeld A, Fewell GA, Flicek P, Foley K, Frankel WN, Fulton LA, Fulton RS, Furey TS, Gage D, Gibbs RA, Glusman G, Gnerre S, Goldman N, Goodstadt L, Grafham D, Graves TA, Green ED, Gregory S, Guigó R, Guyer M, Hardison RC, Haussler D, Hayashizaki Y, Hillier LW, Hinrichs A, Hlavina W, Holzer T, Hsu F, Hua A, Hubbard T, Hunt A, Jackson I, Jaffe DB, Johnson LS, Jones M, Jones TA, Joy A, Kamal M, Karlsson EK, Karolchik D, Kasprzyk A, Kawai J, Keibler E, Kells C, Kent WJ, Kirby A, Kolbe DL, Korf I, Kucherlapati RS, Kulbokas EJ, Kulp D, Landers T, Leger JP, Leonard S, Letunic I, Levine R, Li J, Li M, Lloyd C, Lucas S, Ma B, Maglott DR, Mardis ER, Matthews L, Mauceli E, Mayer JH, McCarthy M, McCombie WR, McLaren S, McLay K, McPherson JD, Meldrim J, Meredith B, Mesirov JP, Miller W, Miner TL, Mongin E, Montgomery KT, Morgan M, Mott R, Mullikin JC, Muzny DM, Nash WE, Nelson JO, Nhan MN, Nicol R, Ning Z, Nusbaum C, O'Connor MJ, Okazaki Y, Oliver K, Overton-Larty E, Pachter L, Parra G, Pepin KH, Peterson J, Pevzner P, Plumb R, Pohl CS, Poliakov A, Ponce TC, Ponting CP, Potter S, Quail M, Reymond A, Roe BA, Roskin KM, Rubin EM, Rust AG, Santos R, Sapojnikov V, Schultz B, Schultz J, Schwartz MS, Schwartz S, Scott C, Seaman S, Searle S, Sharpe T, Sheridan A, Shownkeen R, Sims S, Singer JB, Slater G, Smit A, Smith DR, Spencer B, Stabenau A, Stange-Thomann N, Sugnet C, Suyama M, Tesler G, Thompson J, Torrents D, Trevaskis E, Tromp J, Ucla C, Ureta-Vidal A, Vinson JP, Von Niederhausern AC, Wade CM, Wall M, Weber RJ, Weiss RB, Wendl MC, West AP, Wetterstrand K, Wheeler R, Whelan S, Wierzbowski J, Willey D, Williams S, Wilson RK, Winter E, Worley KC, Wyman D, Yang S, Yang SP, Zdobnov EM, Zody MC, Lander ES. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520–562, 2002 [DOI] [PubMed] [Google Scholar]
- 97. Nguyen M, Pace AJ, Koller BH. Mice lacking NKCC1 are protected from development of bacteremia and hypothermic sepsis secondary to bacterial pneumonia. J Exp Med 204: 1383–1393, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Nickell WT, Kleene NK, Gesteland RC, Kleene SJ. Neuronal chloride accumulation in olfactory epithelium of mice lacking NKCC1. J Neurophysiol 95: 2003–2006, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Norman BJ, Miller SD. Human genome project and sickle cell disease. Soc Work Public Health 26: 405–416, 2011 [DOI] [PubMed] [Google Scholar]
- 100. Nozu K, Iijima K, Kawai K, Nozu Y, Nishida A, Takeshima Y, Fu XJ, Hashimura Y, Kaito H, Nakanishi K, Yoshikawa N, Matsuo M. In vivo and in vitro splicing assay of SLC12A1 in an antenatal salt-losing tubulopathy patient with an intronic mutation. Hum Genet 126: 533–538, 2009 [DOI] [PubMed] [Google Scholar]
- 101. Ohtsuka M, Kimura M, Tanaka M, Inoko H. Recombinant DNA technologies for construction of precisely designed transgene constructs. Curr Pharm Biotechnol 10: 244–251, 2009 [DOI] [PubMed] [Google Scholar]
- 102. Oppermann M, Mizel D, Huang G, Li C, Deng C, Theilig F, Bachmann S, Briggs J, Schnermann J, Castrop H. Macula densa control of renin secretion and preglomerular resistance in mice with selective deletion of the B isoform of the Na,K,2Cl co-transporter. J Am Soc Nephrol 17: 2143–2152, 2006 [DOI] [PubMed] [Google Scholar]
- 103. Oppermann M, Mizel D, Kim SM, Chen L, Faulhaber-Walter R, Huang Y, Li C, Deng C, Briggs J, Schnermann J, Castrop H. Renal function in mice with targeted disruption of the A isoform of the Na-K-2Cl co-transporter. J Am Soc Nephrol 18: 440–448, 2007 [DOI] [PubMed] [Google Scholar]
- 104. Owens DF, Boyce LH, Davis MBE, Kriegstein AR. Excitatory GABA responses in embryonic and neonatal cortical slices demonstrated by gramicidin perforated-patch recordings and calcium imaging. J Neurosci 16: 6414–6423, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Pace AJ, Lee E, Athirakul K, Coffman TM, O'Brien DA, Koller BH. Failure of spermatogenesis in mouse lines deficient in the Na+-K+-2Cl− cotransporter. J Clin Invest 105: 441–450, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Pace AJ, Madden VJ, Henson OWJ, Koller BH, Henson MM. Ultrastructure of the inner ear of NKCC1-deficient mice. Hear Res 156: 17–30, 2001 [DOI] [PubMed] [Google Scholar]
- 107. Payne JA. Functional characterization of the neuronal-specific K-Cl cotransporter: implications for [K+]o regulation. Am J Physiol Cell Physiol 273: C1516–C1525, 1997 [DOI] [PubMed] [Google Scholar]
- 108. Payne JA, Forbush BI. Alternatively spliced isoforms of the putative renal Na-K-Cl cotransporter are differentially distributed within the rabbit kidney. Proc Natl Acad Sci USA 91: 4544–4548, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Payne JA, Stevenson TJ, Donaldson LF. Molecular characterization of a putative K-Cl cotransporter in rat brain. A neuronal-specific isoform. J Biol Chem 271: 16245–16252, 1996 [DOI] [PubMed] [Google Scholar]
- 110. Pearson M, Lu J, Mount DB, Delpire E. Localization of the K-Cl cotransporter, KCC3, in the central and peripheral nervous systems: expression in choroid plexus, large neurons, and white matter tracts. Neuroscience 103: 483–493, 2001 [DOI] [PubMed] [Google Scholar]
- 111. Persson AE, Salomonsson M, Westerlund P, Greger R, Schlatter E, Gonzalez E. Macula densa cell function. Kidney Int Suppl 32: S39–S44, 1991 [PubMed] [Google Scholar]
- 112. Pieraut S, Matha V, Sar C, Hubert T, Méchaly I, Hilaire C, Mersel M, Delpire E, Valmier J, Scamps F. NKCC1 phosphorylation stimulates neurite growth of injured adult sensory neurons. J Neurosci 27: 6751–6759, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Plotkin MD, Kaplan MR, Peterson LN, Gullans SR, Hebert SC, Delpire E. Expression of the Na+-K+-2Cl− cotransporter BSC2 in the nervous system. Am J Physiol Cell Physiol 272: C173–C183, 1997 [DOI] [PubMed] [Google Scholar]
- 114. Plotkin MD, Snyder EY, Hebert SC, Delpire E. Expression of the Na-K-2Cl cotransporter is developmentally regulated in postnatal rat brains: a possible mechanism underlying GABA's excitatory role in immature brain. J Neurobiol 33: 781–795, 1997 [DOI] [PubMed] [Google Scholar]
- 115. Race JE, Makhlouf FN, Logue PJ, Wilson FH, Dunham PB, Holtzman EJ. Molecular cloning and functional characterization of KCC3, a new K-Cl cotransporter. Am J Physiol Cell Physiol 277: C1210–C1219, 1999 [DOI] [PubMed] [Google Scholar]
- 116. Randall J, Thorne T, Delpire E. Partial cloning and characterization of Slc12a2: the gene encoding the secretory Na+-K+-2Cl− cotransporter. Am J Physiol Cell Physiol 273: C1267–C1277, 1997 [DOI] [PubMed] [Google Scholar]
- 117. Reisert J, Lai J, Yau KW, Bradley J. Mechanism of the excitatory Cl− response in mouse olfactory receptor neurons. Neuron 45: 553–561, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397: 251–255, 1999 [DOI] [PubMed] [Google Scholar]
- 119. Rudnik-Schöneborn S, Hehr U, von Kalle T, Bornemann A, Winkler J, Zerres K. Andermann syndrome can be a phenocopy of hereditary motor and sensory neuropathy–report of a discordant sibship with a compound heterozygous mutation of the KCC3 gene. Neuropediatrics 40: 129–133, 2009 [DOI] [PubMed] [Google Scholar]
- 120. Rust MB, Faulhaber J, Budack MK, Pfeffer C, Maritzen T, Didie M, Beck FX, Boettger T, Schubert R, Ehmke H, Jentsch TJ, Hubner CA. Neurogenic mechanisms contribute to hypertension in mice with disruption of the K-Cl cotransporter KCC3. Circ Res 98: 549–556, 2006 [DOI] [PubMed] [Google Scholar]
- 121. Rust MB, Alper SL, Rudhard Y, Shmukler BE, Vicente R, Brugnara C, Trudel M, Jentsch TJ, Hübner CA. Disruption of erythroid K-Cl cotransporters alters erythrocyte volume and partially rescues erythrocyte dehydration in SAD mice. J Clin Invest 117: 1708–1717, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Saam JR, Gordon JI. Inducible gene knockouts in the small intestinal and colonic epithelium. J Biol Chem 274: 38071–38082, 1999 [DOI] [PubMed] [Google Scholar]
- 123. Schlatter E, Salomonsson M, Persson AE, Greger R. Macula densa cells sense luminal NaCl concentration via furosemide sensitive Na+2Cl−K+ cotransport. Pflügers Arch 414: 286–290, 1989 [DOI] [PubMed] [Google Scholar]
- 124. Schultheis PJ, Lorenz JN, Meneton P, Nieman ML, Riddle TM, Flagella M, Duffy JJ, Doetschman T, Miller ML, Shull GE. Phenotype resembling Gitelman's syndrome in mice lacking the apical Na+-Cl− cotransporter of the distal convoluted tubule. J Biol Chem 273: 29150–29155, 1998 [DOI] [PubMed] [Google Scholar]
- 125. Shekarabi M, Moldrich RX, Rasheed S, Salin-Cantegrel A, Laganière J, Rochefort D, Hince P, Huot K, Gaudet R, Kurniawan N, Sotocinal SG, Ritchie J, Dion PA, Mogil JS, Richards LJ, Rouleau GA. Loss of neuronal potassium/chloride cotransporter 3 (KCC3) is responsible for the degenerative phenotype in a conditional mouse model of hereditary motor and sensory neuropathy associated with agenesis of the corpus callosum. J Neurosci 32: 3865–3876, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Shekarabi M, Salin-Cantegrel A, Laganiere J, Gaudet R, Dion P, Rouleau GA. Cellular expression of the K(+)-Cl(−) cotransporter KCC3 in the central nervous system of mouse. Brain Res 1374: 15–26, 2011 [DOI] [PubMed] [Google Scholar]
- 127. Simon DB, Bindra RS, Mansfield TA, Nelson-Williams C, Mendonca E, Stone R, Schurman S, Nayir A, Alpay H, Bakkaloglu A, Rodriguez-Soriano J, Morales JM, Sanjad SA, Taylor CM, Pilz D, Brem A, Trachtman H, Griswold W, Richard GA, John E, Lifton RP. Mutations in the chloride channel gene, CLCNKB, cause Bartter's syndrome type III. Nat Genet 17: 171–178, 1997 [DOI] [PubMed] [Google Scholar]
- 128. Simon DB, Karet FE, Hamdan JM, Di Pietro A, Sanjad SA, Lifton RP. Bartter's syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na-K-2Cl cotransporter NKCC2. Nat Genet 13: 183–188, 1996 [DOI] [PubMed] [Google Scholar]
- 129. Simon DB, Nelson-Williams C, Johnson Bia M, Ellison D, Karet FE, Morey Molina A, Vaara I, Iwata F, Cushner HM, Koolen M, Gainza FJ, Gitelman HJ, Lifton RP. Gitelman's variant of Bartter's syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat Genet 12: 24–30, 1996 [DOI] [PubMed] [Google Scholar]
- 130. Smith DW, Thach S, Marshall EL, Mendoza MG, Kleene SJ. Mice lacking NKCC1 have normal olfactory sensitivity. Physiol Behav 93: 44–49, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Soleimani M, Barone S, Xu J, Shull GE, Siddiqui F, Zahedi K, Amlal H. Double knockout of pendrin and Na-Cl cotransporter (NCC) causes severe salt wasting, volume depletion, and renal failure. Proc Natl Acad Sci USA 109: 13368–13373, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Stil A, Jean-Xavier C, Liabeuf S, Brocard C, Delpire E, Vinay L, Viemari JC. Contribution of the potassium-chloride co-transporter KCC2 to the modulation of lumbar spinal networks in mice. Eur J Neurosci 33: 1212–1222, 2011 [DOI] [PubMed] [Google Scholar]
- 133. Stokes JB, Lee I, D'Amico M. Sodium chloride absorption by the urinary bladder of the winter flounder: A thiazide-sensitive, electrically neutral transport system. J Clin Invest 74: 7–16, 1984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Sun Q, Tian E, Turner RJ, Ten Hagen KG. Developmental and functional studies of the SLC12 gene family members from Drosophila melanogaster. Am J Physiol Cell Physiol 298: C26–C37, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Sun YT, Lin TS, Tzeng SF, Delpire E, Shen MR. Deficiency of electroneutral K+-Cl− cotransporter 3 causes a disruption in impulse propagation along peripheral nerves. Glia 58: 1544–1452, 2010 [DOI] [PubMed] [Google Scholar]
- 136. Sung KW, Kirby M, McDonald MP, Lovinger DM, Delpire E. Abnormal GABAA-receptor mediated currents in dorsal root ganglion neurons isolated from Na-K-2Cl cotransporter-null mice. J Neurosci 20: 7531–7538, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Takahashi N, Brooks HL, Wade JB, Liu W, Kondo Y, Ito S, Knepper MA, Smithies O. Posttranscriptional compensation for heterozygous disruption of the kidney-specific NaK2Cl cotransporter gene. J Am Soc Nephrol 13: 604–610, 2002 [DOI] [PubMed] [Google Scholar]
- 138. Takahashi N, Chernavvsky DR, Gomez RA, Igarashi P, Gitelman HJ, Smithies O. Uncompensated polyuria in a mouse model of Bartter's syndrome. Proc Natl Acad Sci USA 97: 5434–5439, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Tanis JE, Bellemer A, Moresco JJ, Forbush B, Koelle MR. The potassium chloride cotransporter KCC-2 coordinates development of inhibitory neurotransmission and synapse structure in Caenorhabditis elegans. J Neurosci 29: 9943–9954, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Tao R, Li C, Newburn EN, Ye T, Lipska BK, Herman MM, Weinberger DR, Kleinman JE, Hyde TM. Transcript-specific associations of SLC12A5 (KCC2) in human prefrontal cortex with development, schizophrenia, and affective disorders. J Neurosci 32: 5216–5222, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Thompson SM, Deisz RA, Prince DA. Outward chloride/cation co-transport in mammalian cortical neurons. Neurosci Lett 89: 49–54, 1988 [DOI] [PubMed] [Google Scholar]
- 142. Tornberg J, Segerstrale M, Kulesskaya N, Voikar V, Taira T, Airaksinen MS. KCC2-deficient mice show reduced sensitivity to diazepam, but normal alcohol-induced motor impairment, gaboxadol-induced sedation, and neurosteroid-induced hypnosis. Neuropsychopharmacology 32: 911–918, 2006 [DOI] [PubMed] [Google Scholar]
- 143. Tornberg J, Voikar V, Savilahti H, Rauvala H, Airaksinen MS. Behavioural phenotypes of hypomorphic KCC2-deficient mice. Eur J Neurosci 21: 1327–1337, 2005 [DOI] [PubMed] [Google Scholar]
- 144. Tosteson DC, Hoffman JF. Regulation of cell volume by active cation transport in high and low potassium sheep red cells. J Gen Physiol 44: 169–194, 1960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Trudel M, De Paepe ME, Chrétien N, Saadane N, Jacmain J, Sorette M, Hoang T, Beuzard Y. Sickle cell disease of transgenic SAD mice. Blood 84: 3189–3197, 1994 [PubMed] [Google Scholar]
- 146. Uvarov P, Ludwig A, Markkanen M, Pruunsild P, Kaila K, Delpire E, Timmusk T, Rivera C, Airaksinen MS. A novel N-terminal isoform of the neuron-specific K-Cl cotransporter KCC2. J Biol Chem 282: 30570–30576, 2007 [DOI] [PubMed] [Google Scholar]
- 147. Uyanik G, Elcioglu N, Penzien J, Gross C, Yilmaz Y, Olmez A, Demir E, Wahl D, Scheglmann K, Winner B, Bogdahn U, Topaloglu H, Hehr U, Winkler J. Novel truncating and missense mutations of the KCC3 gene associated with Andermann syndrome. Neurology 66: 1044–1048, 2006 [DOI] [PubMed] [Google Scholar]
- 148. Vargas-Poussou R, Feldmann D, Vollmer M, Konrad M, Kelly L, van den Heuvel LP, Tebourbi L, Brandis M, Karolyi L, Hebert SC, Lemmink HH, Deschenes G, Hildebrandt F, Seyberth HW, Guay-Woodford LM, Knoers NV, Antignac C. Novel molecular variants of the Na-K-2Cl cotransporter gene are responsible for antenatal Bartter syndrome. Am J Hum Genet 62: 1332–1340, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Venter JC, Adams MDME, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, Gocayne JD, Amanatides P, Ballew RM, Huson DH, Wortman JR, Zhang Q, Kodira CD, Zheng XH, Chen L, Skupski M, Subramanian G, Thomas PD, Zhang J, Gabor Miklos GL, Nelson C, Broder S, Clark AG, Nadeau J, McKusick VA, Zinder N, Levine AJ, Roberts RJ, Simon M, Slayman C, Hunkapiller M, Bolanos R, Delcher A, Dew I, Fasulo D, Flanigan M, Florea L, Halpern A, Hannenhalli S, Kravitz S, Levy S, Mobarry C, Reinert K, Remington K, Abu-Threideh J, Beasley E, Biddick K, Bonazzi V, Brandon R, Cargill M, Chandramouliswaran I, Charlab R, Chaturvedi K, Deng Z, Di Francesco V, Dunn P, Eilbeck K, Evangelista C, Gabrielian AE, Gan W, Ge W, Gong F, Gu Z, Guan P, Heiman TJ, Higgins ME, Ji RR, Ke Z, Ketchum KA, Lai Z, Lei Y, Li Z, Li J, Liang Y, Lin X, Lu F, Merkulov GV, Milshina N, Moore HM, Naik AK, Narayan VA, Neelam B, Nusskern D, Rusch DB, Salzberg S, Shao W, Shue B, Sun J, Wang Z, Wang A, Wang X, Wang J, Wei M, Wides R, Xiao C, Yan C, Yao A, Ye J, Zhan M, Zhang W, Zhang H, Zhao Q, Zheng L, Zhong F, Zhong W, Zhu S, Zhao S, Gilbert D, Baumhueter S, Spier G, Carter C, Cravchik A, Woodage T, Ali F, An H, Awe A, Baldwin D, Baden H, Barnstead M, Barrow I, Beeson K, Busam D, Carver A, Center A, Cheng ML, Curry L, Danaher S, Davenport L, Desilets R, Dietz S, Dodson K, Doup L, Ferriera S, Garg N, Gluecksmann A, Hart B, Haynes J, Haynes C, Heiner C, Hladun S, Hostin D, Houck J, Howland T, Ibegwam C, Johnson J, Kalush F, Kline L, Koduru S, Love A, Mann F, May D, McCawley S, McIntosh T, McMullen I, Moy M, Moy L, Murphy B, Nelson K, Pfannkoch C, Pratts E, Puri V, Qureshi H, Reardon M, Rodriguez R, Rogers YH, Romblad D, Ruhfel B, Scott R, Sitter C, Smallwood M, Stewart E, Strong R, Suh E, Thomas R, Tint NN, Tse S, Vech C, Wang G, Wetter J, Williams S, Williams M, Windsor S, Winn-Deen E, Wolfe K, Zaveri J, Zaveri K, Abril JF, Guigó R, Campbell MJ, Sjolander KV, Karlak B, Kejariwal A, Mi H, Lazareva B, Hatton T, Narechania A, Diemer K, Muruganujan A, Guo N, Sato S, Bafna V, Istrail S, Lippert R, Schwartz R, Walenz B, Yooseph S, Allen D, Basu A, Baxendale J, Blick L, Caminha M, Carnes-Stine J, Caulk P, Chiang YH, Coyne M, Dahlke C, Mays A, Dombroski M, Donnelly M, Ely D, Esparham S, Fosler C, Gire H, Glanowski S, Glasser K, Glodek A, Gorokhov M, Graham K, Gropman B, Harris M, Heil J, Henderson S, Hoover J, Jennings D, Jordan C, Jordan J, Kasha J, Kagan L, Kraft C, Levitsky A, Lewis M, Liu X, Lopez J, Ma D, Majoros W, McDaniel J, Murphy S, Newman M, Nguyen T, Nguyen N, Nodell M, Pan S, Peck J, Peterson M, Rowe W, Sanders R, Scott J, Simpson M, Smith T, Sprague A, Stockwell T, Turner R, Venter E, Wang M, Wen M, Wu D, Wu M, Xia A, Zandieh A, Zhu X. The sequence of the human genome. Science 291: 1304–1351, 2001 [DOI] [PubMed] [Google Scholar]
- 150. Vilen H, Eerikäinen S, Tornberg J, Airaksinen MS, Savilahti H. Construction of gene-targeting vectors: a rapid Mu in vitro DNA transposition-based strategy generating-null, potentially hypomorphic, and conditional alleles. Transgenic Res 10: 69–80, 2001 [DOI] [PubMed] [Google Scholar]
- 151. Walker NM, Flagella M, Gawenis LR, Shull GE, Clarke LL. An alternate pathway of cAMP-stimulated Cl secretion across the NKCC1-null murine duodenum. Gastroenterology 123: 531–541, 2002 [DOI] [PubMed] [Google Scholar]
- 152. Wall SM, Knepper MA, Hassell KA, Fischer MP, Shodeinde A, Shin W, Pham TD, Meyer JW, Lorenz JN, Beierwaltes WH, Dietz JR, Shull GE, Kim YH. Hypotension in NKCC1-null mice: role of the kidneys. Am J Physiol Renal Physiol 290: F409–F416, 2006 [DOI] [PubMed] [Google Scholar]
- 153. Walser M. Calcium-sodium interdependence in renal transport. In: Renal Pharmacology, edited by Fisher JW. New York: Appleton-Century-Crofts, 1971, p. 21–41 [Google Scholar]
- 154. Wang T, Delpire E, Giebisch G, Hebert SC, Mount DB. Impaired fluid and bicarbonate absorption in proximal tubules (PT) of KCC3 knockout mice. FASEB J 17: A464, 2003 [Google Scholar]
- 155. Wong FH, Chen JS, Reddy V, Day JL, Shlykov MA, Wakabayashi ST, Saier MH., Jr The amino acid-polyamine-organocation superfamily. J Mol Microbiol Biotechnol 22: 105–113, 2012 [DOI] [PubMed] [Google Scholar]
- 156. Woo NS, Lu J, England R, McClellan R, Dufour S, Mount DB, Deutch AY, Lovinger DM, Delpire E. Hyper-excitability and epilepsy associated with disruption of the mouse neuronal-specific K-Cl cotransporter gene. Hippocampus 12: 258–268, 2002 [DOI] [PubMed] [Google Scholar]
- 157. Wouters M, De Laet A, Ver Donck L, Delpire E, van Bogaert PP, Timmermans JP, de Kerchove d'Exaerde A, Smans K, Vanderwinden JM. Subtractive hybridization unravels a role for the ion co-transporter NKCC1 in the murine intestinal pacemaker. Am J Physiol Gastrointest Liver Physiol 290: G1219–G1227, 2006 [DOI] [PubMed] [Google Scholar]
- 158. Xu JC, Lytle C, Zhu TT, Payne JA, Benz EJ, Forbush BI. Molecular cloning and functional expression of the bumetanide-sensitive Na-K-2Cl cotransporter. Proc Natl Acad Sci USA 91: 2201–2205, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Zhang LL, Delpire E, Vardi N. NKCC1 does not accumulate chloride in developing retinal neurons. J Neurophysiol 98: 266–177, 2007 [DOI] [PubMed] [Google Scholar]
- 160. Zhang W, Liu LY, Xu TL. Reduced potassium-chloride co-transporter expression in spinal cord dorsal horn neurons contributes to inflammatory pain hypersensitivity in rats. Neuroscience 152: 502–510, 2008 [DOI] [PubMed] [Google Scholar]
- 161. Zhu L, Lovinger D, Delpire E. Cortical neurons lacking KCC2 expression show impaired regulation of intracellular chloride. J Neurophysiol 93: 1557–1568, 2005 [DOI] [PubMed] [Google Scholar]
- 162. Zhu L, Polley N, Mathews GC, Delpire E. NKCC1 and KCC2 prevent hyperexcitability in the mouse hippocampus. Epilepsy Res 79: 201–212, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]