

Abstract
Acute kidney injury-induced organ fibrosis is recognized as a major risk factor for the development of chronic kidney disease, which remains one of the leading causes of death in the developed world. However, knowledge on molecules that may suppress the fibrogenic response after injury is lacking. In ischemic models of acute kidney injury, we demonstrate a new function of netrin-1 in regulating interstitial fibrosis. Acute injury was promptly followed by a rise in serum creatinine in both wild-type and netrin-1 transgenic animals. However, the wild-type showed a slow recovery of kidney function compared with netrin-1 transgenic animals and reached baseline by 3 wk. Histological examination showed increased infiltration of interstitial macrophages, extensive fibrosis, reduction of capillary density, and glomerulosclerosis. Collagen IV and α-smooth muscle actin expression was absent in sham-operated kidneys; however, their expression was significantly increased at 2 wk and peaked at 3 wk after reperfusion. These changes were reduced in the transgenic mouse kidney, which overexpresses netrin-1 in proximal tubular epithelial cells. Fibrosis was associated with increased expression of IL-6 and extensive and chronic activation of STAT3. Administration of IL-6 exacerbated fibrosis in vivo in wild-type, but not in netrin-1 transgenic mice kidney and increased collagen I expression and STAT3 activation in vitro in renal epithelial cells subjected to hypoxia-reoxygenation, which was suppressed by netrin-1. Our data suggest that proximal tubular epithelial cells may play a prominent role in interstitial fibrosis and that netrin-1 could be a useful therapeutic agent for treating kidney fibrosis.
Keywords: glomerular sclerosis, interstitial fibrosis, netrin-1, stat3, acute kidney injury
acute kidney injury (AKI) due to ischemia-reperfusion injury is a serious and frequent complication in clinical settings and frequently progresses to kidney failure. Despite advances in understanding the pathophysiology of AKI and improved supportive care, the prognosis of patients with AKI remains poor and is associated with increased cost, increased length of hospital stay, and adverse long-term outcomes with accelerated progression of chronic kidney disease (CKD) with a mortality rate of 40–80% in the intensive care setting (2, 10, 26). Similarly, preventive strategies have been unsuccessful, and the incidence of AKI is increasing and expected to double over the next decade (1). Therefore, there is an urgent need to develop an effective therapy for the prevention of AKI-associated CKD, but the molecular mechanisms linking AKI to CKD remain poorly understood. Animal studies show that ischemic kidney injury or toxic kidney injury damages multiple cell types, including endothelial and tubular epithelial cells. Recent studies suggest that proximal tubular epithelial injury alone induces interstitial fibrosis and glomerulosclerosis (8). Therefore, targeted suppression of tubular injury may prevent the development of interstitial fibrosis and glomerulosclerosis. We used cell type-specific overexpression of a molecule called netrin-1 to determine whether targeted suppression of tubular epithelial injury can prevent the development of ischemic AKI-induced interstitial fibrosis and glomerulosclerosis.
Netrin-1 is a laminin-related neuronal guidance cue widely expressed in many tissues, including kidney, and recent studies suggest that netrin-1 coordinates inflammation by regulating leukocyte migration across endothelial cells. Very little netrin-1 is expressed in kidney epithelial cells under normal conditions. However, netrin-1 protein expression is induced after injury. Studies from our laboratory had determined that netrin-1 induction after injury is regulated at the translational level (11). Administration of recombinant netrin-1 reduced ischemia-reperfusion injury (IRI) of the kidney, hypoxia-induced intestinal inflammation, and sepsis-induced lung inflammation (16, 18, 23, 28). Overexpression of netrin-1 is also known to suppress ischemia-reperfusion-induced apoptosis and the inflammatory response. However, its role in the development of IRI-associated fibrosis is unknown.
Here, we determined whether overexpression of netrin-1 in the proximal tubular epithelium can ameliorate AKI-induced interstitial fibrosis, glomerulosclerosis, and vascular dropout, features of the transition from acute to chronic injury.
MATERIALS AND METHODS
Renal ischemia-reperfusion.
Netrin-1 transgenic mice were characterized for transgene expression and phenotype as described previously (19, 27). Briefly, transgenic mice which overexpress chicken netrin-1 in the gut were developed to study the role of netrin-1 in colorectal carcinogenesis. The transgene expression was driven by a gut-specific l-fatty acid binding protein promoter. Characterization of transgene expression had shown that the promoter was also active in many tissues: colon, small intestine, spleen, lung, and kidney (17). The highest expression of the transgene was found in the kidney, and expression was localized in the proximal tubules. C57BL/6J mice (8–9 wk of age, Jackson Labs, Bar Harbor, ME) and age-matched netrin-1 transgenic mice were anesthetized with pentobarbital sodium (50 mg/kg body wt, intraperitoneally) and placed on a heating pad to maintain body temperature at 37°C. Both renal pedicles were identified through dorsal incisions and clamped for 26 min. Reperfusion was confirmed visually upon release of the clamps. As a control, sham-operated animals were subjected to the same surgical procedure except the renal pedicles were not clamped. Surgical wounds were closed, and mice were given 1 ml of warm saline, intraperitoneally. The mice were kept in a warm incubator until they regained consciousness. Animals were euthanized at 1, 2, and 3 wk after surgery, and kidney tissue was collected and processed for RNA isolation and protein extraction. Some animals received recombinant IL-6 (10 ng·animal−1·day−1, intraperitoneally) starting at day 3 after reperfusion to determine the effect of IL-6 on the fibrotic response, not on acute kidney injury. The Institutional Animal Care and Use Committee of Georgia Health Sciences University approved all of the protocols and procedures for using animals (approval no. BR10-10-369).
Cell culture.
Murine proximal tubule cells (TKPTS cells; kindly provided by Dr. E. Bello-Reuss, University of Texas Medical Branch, Galveston, TX) were cultured in advanced DMEM/F12 supplemented with glutamine, 5% FBS, and antibiotics. Serum-free medium was replaced before the start of the experiment. Cells were then treated with IL-6, netrin-1, or a combination of both IL-6 (10 ng/ml) and netrin-1 (250 ng/ml) for a period of 24 h. For hypoxia-reoxygenation of TKPTS cells, the medium was replaced at 80% confluence with HBSS and the culture plate was placed in a hypoxic bag (BD Biosciences) overnight. The plate was removed from the hypoxic bag, and HBSS was replaced with serum-free advanced DMEM/F12 medium. Cells were treated with IL-6, netrin-1, or a combination of both and incubated at 37°C in a CO2 incubator for an additional 24 h. Cells were harvested, and a lysate was prepared with RIPA buffer containing protease and phosphatase inhibitors for Western blot analysis. Some of the cells were used for RNA isolation and RT-PCR analysis.
Western blot analysis.
Protein extraction from kidneys and Western blot analysis were carried out as described before (22, 27). The membrane was probed with rabbit anti-α-smooth actin and antibodies against collagen IV, fibronectin (Abcam, Cambridge, MA), rabbit phospho-STAT3 (Phosphor tyrosine), and phospho JNK (Cell Signaling Technology, Danvers, MA). Proteins were detected using enhanced chemiluminescence detection reagents (Amersham Pharmacia Biotech). Protein loading was normalized to GAPDH expression using an anti-mouse GAPDH antibody (Abcam).
Quantification of mRNA by real-time RT-PCR.
Real-time RT-PCR was performed in an Applied Biosystems 7700 Sequence Detection System (Foster City, CA). Total RNA (1.5 μg) was reverse transcribed in a reaction volume of 20 μl using an Omniscript RT kit and random primers. The product was diluted to a volume of 150 μl, and 6-μl aliquots were used as templates for amplification using the SYBR Green PCR amplification reagent (Qiagen) and gene-specific primers or a PCR array for the fibrosis pathway (330231 PAMM-120ZA, Qiagen). The primer sets used were mouse collagen IV (forward: CAGATTCCGCAGTGCCCTA; reverse: GGAATAGCCGATCCACAGTGAG), collagen I (forward: GATGACGTGCAATGCAATGAA; reverse: CCCTCGACTCCTACATCTTCTGA), and transforming growth factor (TGF)-β1 (forward: TGACGTCACTGGAGTTGTACG; reverse: GGTTCATGTCATGGATGGTGC). The amount of DNA was normalized to the β-actin signal amplified in a separate reaction (forward: AGAGGGAAATCGTGCGTGAC; reverse: CAATAGTGATGACCTGGCCGT).
Renal function.
Renal function was assessed by measurements of serum creatinine (DZ072B, Diazyme Labs).
Kidney injury molecule-1 quantification in urine.
Kidney injury molecule-1 (KIM-1) was quantified in urine by ELISA (KT-634, Kamyia Biomedical, Seattle, WA).
Tissue preparation and histology.
Mice were euthanized under deep anesthesia. Kidneys were removed, cut sagitally, and fixed in 10% neutral-buffered formalin for paraffin embedding. Histological analysis was performed in paraffin-embedded and serially cut kidney sections (3 μM) stained with hematoxylin, periodic acid-Schiff (PAS), and Masson's trichrome. Trichrome staining was quantified by tracing the stained area in ×40 fields with cellSens Standard software (Olympus, Pittsburgh, PA). Five fields for each animal were traced, and the percent stained area was calculated. Semiquantitative analysis of chronic damage included the estimation of cortical tubular atrophy and interstitial fibrosis and was graded (1–4) as minimal (involving <5% of cortex), mild (5–24%), moderate (25–49%), or severe (50%). For assessment of glomerular involvement, an average of 50–70 glomeruli/section were examined on multiple levels. All scoring was performed in a blinded manner. To quantify leukocyte infiltration, sections were stained with a rat anti-mouse macrophage antibody (1:200 dilution, ab56297, Abcam) followed by a goat anti-rat biotin conjugate. Color was developed after incubation with ABC reagent (Vector Labs). Stained sections were photographed. To determine vascular density, sections were stained with a rabbit anti-CD36 antibody (1:100 dilution, Abcam) followed by a goat anti-rabbit biotin conjugate. IL-6 expression, the platelet-derived growth factor receptor-β (PDGFRβ), and STAT3 phosphorylation were localized using rabbit anti-IL-6 (Abcam), rabbit anti-PDGFRβ (Abcam), and rabbit anti-phospho STAT3 (tyrosine 705, Cell Signaling Technologies) antibodies. Color was developed after incubation with ABC reagent (Vector Labs). Five ×40 fields from stained sections were counted and photographed using an Olympus inverted microscope with a color CCD camera.
Statistics.
Data are given as means ± SE. Statistical analysis was performed using the unpaired Student's t-test to determine differences between two groups and ANOVA for multiple comparisons. P values of <0.05 were considered statistically significant.
RESULTS
Tubular-specific overexpression of netrin-1 enhanced recovery from acute ischemic injury.
Netrin-1 overexpression in proximal tubular epithelial cells of the kidney was confirmed by immunohistochemistry (Fig. 1, A and B). Both wild-type and netrin-1 transgenic mice were subjected to a similar period of ischemia, but no difference in peak serum creatinine levels was detected (Fig. 1). Consistent with our earlier studies (27), with less severe ischemia, netrin-1 transgenic mice are protected from AKI. Following peak serum creatinine on day 1, kidney function started to recover, as seen by declining serum creatinine levels, and reached near baseline on day 21. Compared with wild-type, netrin-1 transgenic animal kidney function recovered much more rapidly, as seen by a significant difference in serum creatinine level on days 3 and 7 after reperfusion. To determine netrin-1 overexpression on proximal tubular epithelial cell injury, KIM-1 excretion in urine was quantified by ELISA. As shown in Fig. 1D, 2 wk after reperfusion KIM-1 excretion was dramatically increased, which was significantly suppressed in netrin-1 transgenic mice, suggesting the protective effect of netrin-1 on the tubular epithelial cells.
Fig. 1.

Netrin-1 overexpression in proximal tubular epithelial cells accelerates recovery of kidney function in response to ischemia-reperfusion (IR) injury. A: a wild-type (WT) kidney does not show any staining for transgene chicken netrin-1. B: transgenic mouse kidney showing staining for chicken netrin-1 in proximal tubular epithelial cells. Some proximal tubules are not stained for chicken netrin-1. The reason for this is unclear. C: serum creatinine was measured at various time points after reperfusion in WT and netrin-1 transgenic mice subjected to sham operation or ischemia followed by reperfusion (IR). Values are means ± SE. *P < 0.01 vs. WT IR; n = 6 for each group. D: proximal tubular injury marker KIM-1 excretion was determined by ELISA at 2 wk after reperfusion. Kidney injury molecule-1 (KIM-1) excretion is increased after IR in WT mice, which was suppressed in netrin-1 transgenic animal subjected to IR. *P < 0.001 vs. sham. #P < 0.001 vs. WT IR; n = 6.
Fig. 2.

Netrin-1 overexpression in proximal tubular epithelial cells suppresses IR-induced tubular atrophy and glomerular sclerosis. Top: periodic acid-Schiff (PAS)-stained section (2 left panels) and trichrome-stained section (right most panel) showing tubular and glomerular injury in WT and netrin-1 transgenic animals after sham surgery or IR. Scale bar = 100 μM. Bottom: quantification of tubular and glomerular injury in PAS-stained section as described in materials and methods. Netrin-1 overexpression suppressed both tubular and glomerular injury significantly. Values are means ± SE; n = 6. *P < 0.001 vs. sham. #P < 0.05 vs. WT IR.
Tubular-specific overexpression of netrin-1 suppressed tubular atrophy and glomerular sclerosis.
To determine the long-term outcome of ischemia-reperfusion on the tubular epithelium and glomerulus, injury was scored as described in materials and methods. Atrophic tubules showed a flat and very simplified epithelial lining, with thickened and wrinkled tubular basement membranes that frequently contained hyaline casts (Fig. 2). Kidneys with advanced tubulointerstitial scarring additionally showed focal global and segmental glomerulosclerosis, generally involving <20–30% of all glomeruli. These changes were significantly suppressed in netrin-1 transgenic mice (Fig. 2).
Ischemia-reperfusion-induced interstitial fibrogenesis was suppressed by netrin-1 overexpression.
Ischemia-reperfusion of wild-type animal kidneys induced a large increase in the expression of several profibrotic [collagen, smooth muscle actin, platelet derived growth factor-B (PDGF-B), and TGF-β1] and proinflammatory genes (IL-1β, TNF-α, STAT1, and NF-κB) in the kidney compared with sham-operated animals (analyzed using a PCR array). Netrin-1 overexpression significantly suppressed the expression of both profibrotic and proinflammatory genes. These genes are known to mediate fibrosis in the kidney and other tissues. Consistent with the increased profibrotic gene expression and tubular epithelial injury, there was a large increase in interstitial fibrosis, as seen by an increase in trichrome staining in wild-type mice (Fig. 3). Netrin-1 overexpression suppressed the extent of interstitial fibrosis. Consistent with trichrome staining, Western blot analysis showed increased levels of α-smooth muscle actin and collagen IV expression in wild-type mouse kidneys, which were significantly suppressed in netrin-1 transgenic mice. Moreover, PCR with reverse transcription analyses demonstrated significantly increased kidney mRNA expression of the key profibrotic cytokine, TGF-β1, and major matrix components (collagen IVα1; Fig. 3C) in wild-type mice, which was significantly suppressed in netrin-1 transgenic mouse kidney, confirming PCR array results.
Fig. 3.

IR-induced fibrosis is suppressed in netrin-1 transgenic mice. Fibrotic gene expression was determined by real-time PCR, and fibrosis was determined using Masson's trichrome staining. Extent of fibrosis was calculated as percent stained area in the ×40 field. A: IR-induced profibrotic gene expression was suppressed with netrin-1 overexpression in the proximal tubular epithelial cells. *P < 0.05 vs. all other groups. B: Masson's trichrome staining shows fibrosis (blue color) in WT mice with IR, which was reduced in netrin-1 transgenic animals subjected to IR. C: quantification of fibrosis. Netrin-1 overexpression significantly reduced the extent of fibrosis. *P < 0.001 vs. sham. #P < 0.001 vs. WT IR. D: fibrotic gene expression after IR in WT and netrin-1 transgenic animals compared with sham surgery. *P < 0.05 vs. sham. #P < 0.05 vs. WT IR; n = 4–6. E: Western blot analysis of α-smooth muscle actin (α-SMA) and collage IV expression after IR in WT and netrin-1 transgenic mouse kidney; n = 4.
Reperfusion-induced capillary dropout was suppressed in netrin-1 transgenic mouse kidneys.
Capillary drop after reperfusion is known to contribute to chronic hypoxia and hamper recovery of kidney function. To determine whether improved kidney structure and function was associated with suppression of reperfusion-induced capillary dropout in transgenic mice, vascular capillary density was determined by staining with CD36 antibody. As shown Fig. 4, a larger network of peritubular capillaries was seen in the cortex and medulla in control kidneys. Reperfusion induced a large decrease in capillary density in wild-type mice, which was suppressed in netrin-1 transgenic mice.
Fig. 4.

Netrin-1 overexpression suppresses IR-induced drop in vascular density. Vascular density was determined by immunohistochemical staining of the vasculature using antibody against CD31 antigen. A dense network of vessels was seen in both cortex and outer medulla in sham-operated animals, which was drastically reduced after IR (2 wk) in WT animals. Netrin-1 overexpression suppressed IR-induced reduction in vascular density. Magnification ×660.
Ischemia-reperfusion-induced interstitial fibrogenesis associated with macrophage infiltration and PDGFRβ expression was suppressed by netrin-1 overexpression.
Enhanced macrophage infiltration after reperfusion injury was associated with increased interstitial fibrosis. To determine whether netrin-1 suppressed interstitial fibrosis through suppression of macrophage infiltration, sections were stained with a macrophage antibody, as described in materials and methods. Macrophage infiltration was extensive throughout the interstitium of wild-type mouse kidneys compared with sham controls. Netrin-1 overexpression suppressed macrophage infiltration (Fig. 5). PDGFRβ, which is known to mediate fibrosis, is predominantly expressed in fibrocytes and pericytes (1). As shown in Fig. 5, PDGFRβ expression was seen in the interstitium of sham-operated animals, which was increased tremendously after ischemia-reperfusion for 2 wk. However, netrin-1 overexpression suppressed the PDGFRβ expression increase in the interstitium. The increase in PDGFRβ expression was seen mostly in the damaged fibrotic area of the outer medulla. Moreover, increased macrophage filtration was associated with increased IL-6 expression in injured tubular epithelial cells, which was suppressed in netrin-1 transgenic animals (Fig. 6). IL-6 is an important cytokine and is known to induce macrophage infiltration in CKD. IL-6 is known to activate STAT3 phosphorylation, and increased STAT3 phosphorylation is known to mediate kidney fibrosis (21).
Fig. 5.

IR-induced infiltration of macrophages and PDGF receptor (PDGFR)-β expression is suppressed in netrin-1 transgenic animal kidney. Immunohistochemical localization of macrophages and PDGFRβ was carried out as described in materials and methods. A few interstitial macrophages and low levels of PDGFRβ expression are seen in sham-operated WT and netrin-1 transgenic animals. A large increase in the number of interstitial macrophages (arrows) and in PDGFRβ expression after IR in WT mouse kidney was seen, which was inhibited in netrin-1 transgenic animal kidney. Magnification ×400. Scale bar = 100 μM.
Fig. 6.

IR-induced IL-6 expression was suppressed by netrin-1 overexpression in tubular epithelial cells. A: immunohistochemical localization of IL-6 was performed as described in materials and methods. No staining was seen in sham-operated kidney. Intense immunostaining was seen in tubular epithelial cells after IR, which was suppressed in netrin-1 transgenic mouse kidney. Magnification ×660. B: Western blot analysis of IL-6 expression in kidney of WT and netrin-1 transgenic animals subjected to sham operation or IR. Densitometric quantification is shown at the bottom. IR-induced increase in IL-6 expression was significantly suppressed in netrin-1 transgenic animals. *P < 0.05 vs. sham. #P < 0.001 vs. IR WT; n = 3.
To determine whether netrin-1 suppresses STAT3 phosphorylation, thereby suppressing the fibrotic response of injured tubular epithelial cells, STAT3 phosphorylation was measured. As shown Fig. 7, no phosphor-STAT3 staining was detected in control kidneys. Two weeks after reperfusion, extensive phosphorylation was seen at tyrosine 705 of STAT3 in tubular epithelial cells and a few cells in the glomerulus as well. Interestingly, there was no increase in serine 727 phosphorylation after ischemia-reperfusion and netrin-1 overexpression did not alter its level. Netrin-1 overexpression suppressed STAT3 phosphorylation. Consistent with the immunostaining data, Western blot analysis showed increased STAT3 phosphorylation in wild-type kidneys, which was suppressed by netrin-1 expression.
Fig. 7.

IR-induced extensive and chronic activation of STAT3, which was suppressed in netrin-1 transgenic animals. A: phospho STAT3 immunostaining was carried out as described in materials and methods. No immunostaining for phospho STAT3 was seen in sham-operated kidney of both WT and netrin-1 transgenic animals. A larger increase in the number of STAT3-positive tubular epithelial cells, interstitial cells, and glomerular mesangial cells after IR was seen in WT mice. However, netrin-1 overexpression suppressed STAT3 activation. B: Western blot analysis of phospho STAT3 and phospho JNK in sham and IR-operated kidney. IR caused a large increase in phospho STAT3 and JNK phosphorylation, which was suppressed by netrin-1 overexpression. Protein loading was normalized to GAPDH. Magnification ×660. Scale bar = 100 μM. C: densitometric quantification of p-STAT3 and p-JNK. *P < 0.001 vs. N1 transgenic mice with corresponding reperfusion time and sham. #P < 0.05 vs. sham operated; n = 6.
Stress-activated MAPK JNK is known to mediate fibrosis after reperfusion injury in the kidney (31). To determine whether part of the netrin-1 effect on fibrosis is also mediated through suppression of JNK phosphorylation, we measured JNK phosphorylation by Western blot analysis. As shown in Fig. 7, 2 wk after reperfusion a large increase in JNK phosphorylation was seen, which was suppressed by netrin-1 overexpression in the tubules.
Consistent with in vivo results, IL-6 induced STAT3 phosphorylation in proximal tubular epithelial cells, which was partly suppressed by netrin-1 (Fig. 7). Neither IL-6 nor netrin-1 altered the total STAT3 expression in proximal tubular epithelial cells.
IL-6 enhanced the hypoxia-induced fibrotic response in mouse proximal tubular epithelial cells, which was suppressed by netrin-1.
To determine whether IL-6 increases the fibrotic response of the kidney by acting through tubular epithelial cells, TKPTS cells were subjected to normoxia or hypoxia and then treated with IL-6 or a combination of IL-6 and netrin-1 for 24 h. Consistent with our in vivo observation (Fig. 8), hypoxia-rexoygenation in vitro induced STAT3 activation, as seen by increased phosphorylation that was associated with increased collagen I production (Fig. 8). Hypoxia-reoxygenation-induced collagen I production was enhanced with IL-6 treatment. Both hypoxia-induced and IL-6 induced collagen I expression were suppressed by the addition of netrin-1, which was associated with suppression of STAT3 phosphorylation.
Fig. 8.
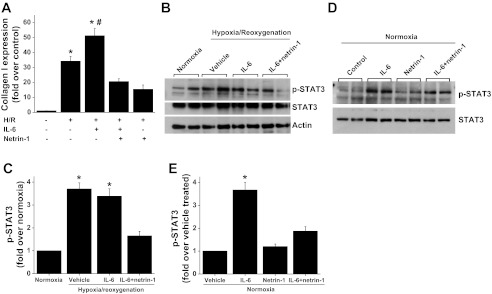
Hypoxia-reoxygenation-induced fibrotic response in mouse proximal tubular epithelial cells (TKPTS). A: TKPTS cells were subjected to hypoxia followed by 24 h of reoxygenation as described in materials and methods. Collagen I expression was determined by RT-PCR. Hypoxia-induced collagen I expression is enhanced with IL-6 treatment, which was suppressed by netrin-1 treatment. *P < 0.001 vs. all other groups. #P < 0.05 vs. hypoxia group. B: hypoxia- and IL-6-induced STAT3 activation (phosphorylation) is suppressed by netrin-1 treatment in TKPTS cells. C: densitometric quantification of p-STAT3. *P < 0.001 vs. all other groups. D: IL-6-induced STAT3 activation under normoxia was also suppressed by netrin-1 treatment in TKPTS cells. E: densitometric quantification of p-STAT3. *P < 0.01 vs. all other groups; n = 4.
Chronic administration of IL-6 enhanced ischemia-reperfusion-induced fibrosis.
Although a number of studies suggest that macrophages and their secreted components like IL-6 are known to mediate both acute and chronic kidney injury and fibrosis, opposing views have also been documented (3, 5, 6, 9, 12, 20, 30). To determine this directly, we administered recombinant IL-6 (10 ng/animal starting on day 3) for 2 wk. As shown in Fig. 9, administration of IL-6 to wild-type mice enhanced STAT3 phosphorylation, collagen I expression, and fibrosis of the kidney, suggesting that IL-6 does participate in renal fibrosis after ischemic injury, probably through activation of STAT3. Consistent with in vitro studies, chronic administration of IL-6 to netrin-1 transgenic mice did not exacerbate fibrosis (Fig. 10), suggesting that netrin-1 suppresses both IL-6 production as well as its activity.
Fig. 9.

Chronic administration of IL-6 enhanced fibrotic response after IR. WT mice were subjected to sham surgery or IR. Three days after IR, mice received either vehicle (0.1% BSA) or IL-6 (10 ng·animal−1·day−1) for 2 wk. Fibrosis was determined by Masson's trichrome staining, and fibrotic gene expression was quantified by RT-PCR. A: sham-operated WT kidney does not show any fibrosis. B: IR induced a large increase in interstitial fibrosis (blue staining). C: IL-6 administration enhanced fibrosis. D: quantification of collagen I, collagen IV, and transforming growth factor (TGF)-β1 gene expression by RT-PCR. IR induced a significant increase in collagen I, collagen IV, and TGF-β1 expression. IL-6 administration enhanced collagen I mRNA expression. *P < 0.05 vs. sham control. #P < 0.05 vs. vehicle-treated IR. E: kidney function was determined by measuring serum creatinine. IL-6 administration does not alter serum creatinine levels compared with vehicle-treated IR. *P < 0.001 vs. sham control. F: Western blot analysis of collagen IV, α-SMA, and phospho-STAT3 by Western blot analysis. IL-6 administration enhanced IR-induced collagen and SMA expression. G: densitometric quantification of Western blot analysis of collagen IV, α-SMA, and phospho-STAT3. *P < 0.001 vs. sham control. #P < 0.05 vs. IR+vehicle treated; n = 4–6.
Fig. 10.
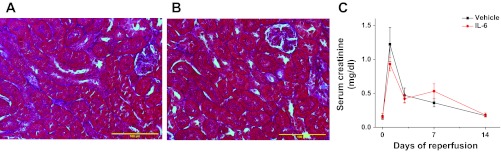
Chronic administration of IL-6 does not alter fibrotic response after IR in netrin-1 transgenic mice. Netrin-1 transgenic mice were subjected to sham surgery or IR. Three days after IR, mice received either vehicle (0.1% BSA) or IL-6 (10 10 ng·animal−1·day−1) for 2 wk. Fibrosis was determined by Masson's trichrome staining, and kidney function was determined by measuring serum creatinine. A: IR-induced interstitial fibrosis is largely suppressed as expected (blue staining) in vehicle-treated netrin-1 transgenic mice. B: IL-6 administration did not increase fibrosis. C: kidney function was determined by measuring serum creatinine. IL-6 administration does not alter serum creatinine levels compared with vehicle-treated IR; n = 6–8.
DISCUSSION
Kidney fibrosis commonly occurs after many forms of AKI and subsequently leads to development of CKD. Although the cell type that may initiate fibrotic response is not clear in humans, recent animal studies strongly suggest proximal tubular epithelial injury and aberrant recovery are major contributing factors to the fibrotic response that leads to interstitial fibrosis and glomerular sclerosis (8, 31). Although a few pathways were identified that may mediate tubular atrophy, interstitial fibrosis, the mechanism through which epithelial injury leads to interstitial fibrosis and glomerulosclerosis, is not completely understood. Moreover, currently there are no therapeutic approaches that limit this devastating fibrotic kidney disease.
We show here, for the first time, that tubular epithelial-specific overexpression of the guidance cue netrin-1 suppressed AKI-induced tubular atrophy, interstitial fibrosis, vascular dropout, and glomerular sclerosis. Moreover, we show that exacerbated chronic activation of the profibrotic STAT3 and JNK pathways was significantly suppressed by netrin-1 in tubular epithelial cells. Activation of STAT3 and JNK was associated with increased expression of IL-6, which was suppressed in netrin-1 transgenic animals. Administration of IL-6 increased the fibrotic response of the kidney. Our data suggest that netrin-1 may be useful as antifibrotic therapy in kidney diseases.
Mild to moderate ischemia followed by reperfusion protected the kidney against acute injury in netrin-1 transgenic mice (27). Therefore, in the current study, we compared the response of wild-type and netrin-1 transgenic mice exposed to ischemia with a similar degree of acute injury to determine the role of netrin-1 overexpression in recovery of the kidney. Although the peak creatinine level did not differ between the wild-type and netrin-1 transgenic mice, the subsequent recovery rate was hastened in transgenic mice compared with wild-type mice. The mechanism for enhanced recovery is not known. Netrin-1 is known to induce tubular epithelial cell proliferation and suppress apoptosis (27, 29). Since tubular epithelial cell proliferation and differentiation are critical for the rapid recovery, netrin-1 could enhance the recovery of kidney function through this mechanism.
Inflammation is a well-known feature of AKI (25) and the subsequent recovery phase (7, 13). Excessive infiltration of inflammatory cells may lead to aberrant recovery and interstitial fibrosis. Our data reveal that reperfusion of the kidney for 1–3 wk leads to a large increase in the number of interstitial macrophages, which was suppressed to a large extent by netrin-1 overexpression. The exact mechanisms that underlie the reduction in macrophage infiltration warrants further exploration. The current and recent data suggest that one of these mechanisms may include suppression of IL-6 and MCP-1 expression from injured epithelial cells, which may act as a chemoattractant for macrophages. This view was further supported by our IL-6 localization and chronic administration studies that show that netrin-1 suppressed IL-6 expression in the epithelial cells and that chronic administration of IL-6 enhanced the fibrotic response.
Despite the near normal return of serum creatinine levels, 3 wk of reperfusion led to maladaptive repair, which was manifested as tubulointerstitial fibrosis and tubular atrophy. Both interstitial fibrosis and tubular atrophy were significantly suppressed in netrin-1 transgenic animals. It is not clear whether the effect on fibrosis is direct or indirect, occurring through suppression of IL-6 and macrophage infiltration. Recent studies also show that STAT3 activation mediates interstitial fibrosis in the model of obstructive nephropathy (21). We see extensive and chronic activation of STAT3 in tubular epithelial cells of wild-type mice, which was suppressed in netrin-1 transgenic animals, suggesting that one of the possible mechanisms may include suppression STAT3 signaling, thereby suppressing fibrosis. The mechanism of STAT3 suppression by netrin-1 is unclear. NF-κB is known to interact with STAT3 and activate each other (24). Our recent studies had documented that netrin-1 suppresses NF-κB activation by suppression of IKBα degradation (Ranganathan PV, Jayakumar C, Mohamed R, Dong Z, Ramesh G, unpublished observations). Therefore, it is possible that either direct suppression of NF-κB-mediated STAT3 activation or suppression of NF-κB-induced IL-6 production by netrin-1 may mediate its inhibitory effects on STAT3. de Borst et al. (4) recently reported that JNK remained active in proximal tubule cells for weeks after an ischemic insult and was associated with tubulointerstitial inflammation and fibrosis. Moreover, JNK pathway activation and interstitial fibrosis have been directly linked in another study (31). Activation of JNK signaling was shown to upregulate profibrotic cytokines such as TGF-β1 and connective tissue growth factor in proximal tubule epithelium in ischemic, toxic, and obstructive models of kidney injury (31). Therefore, it is possible that inhibition of the JNK pathway could be another mechanism by which netrin-1 suppresses interstitial fibrosis. Finally, capillary loss, which was observed in wild-type mouse kidneys following 1–3 wk of reperfusion, was also suppressed. Injured tubular epithelial cells may communicate to peritubular capillary endothelial cells and pericytes through secretion of trophic factors such as PDGF-B, which may enhance proliferation and migration. This could disrupt the endothelium-pericyte cross talk, thereby contributing to capillary rarefaction, progressive ischemia, and aggravation of tubulointerstitial damage (14, 15). This was supported by suppression of PDGF-B in the kidney and PDGFRβ expression in the interstitium, suggesting that netrin-1 may regulate pericyte/fibrocyte proliferation or death either directly or indirectly.
Our findings also show that reperfusion-induced injury to renal epithelium also affects glomerular anatomy. Tubulointerstitial fibrosis/tubular atrophy was associated with a large increase in the number of globally or segmentally sclerosed glomeruli. Very few sclerosed glomeruli were detected in cases with mild tubulointerstitial fibrosis in netrin-1 transgenic animals. The mechanisms as to how tubular epithelial cell netrin-1 suppresses glomerular sclerosis is not clear. One reason for this may be the increasing loss of capillaries observed in tubulointerstitial fibrosis of wild-type kidneys, leading to a progressive reduction of glomerular blood flow. In addition, the proximity of early proximal tubular segments to the glomerular tuft and their per continuum connection via the urinary space could result in paracrine signaling from injured and regenerating/undifferentiated epithelium to directly impact the glomerulus. Alternatively, a progressive tubulointerstitial reaction originating around the atrophic and undifferentiated tubules may directly encroach upon the glomerular tuft. These processes may be suppressed in netrin-1 transgenic animals, thereby protecting the glomeruli from the damage.
In summary, we have demonstrated, for the first time, that targeted overexpression of netrin-1 in proximal tubular epithelial cells can suppress ischemia-reperfusion-induced tubular atrophy, interstitial fibrosis, and glomerular sclerosis. Part of the effects of netrin-1 may be mediated through suppression of STAT3 and JNK signaling and IL-6 expression in tubular epithelial cells. Netrin-1-based interventions aimed at proximal signaling events originating from the tubules may prevent pathological cascades that lead to CKD and warrant further examination for their potential use in the prevention of CKD.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases R01 Grant (1R01DK083379-01A3) to G. Ramesh.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: P.R. and C.J. performed experiments; P.R. and G.R. interpreted results of experiments; P.R. and G.R. prepared figures; P.R. and G.R. drafted manuscript; P.R., C.J., and G.R. approved final version of manuscript; C.J. and G.R. edited and revised manuscript; G.R. provided conception and design of research; G.R. analyzed data.
REFERENCES
- 1. Bonner JC. Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth Factor Rev 15: 255–273, 2004 [DOI] [PubMed] [Google Scholar]
- 2. Chertow GM, Soroko SH, Paganini EP, Cho KC, Himmelfarb J, Ikizler TA, Mehta RL. Mortality after acute renal failure: models for prognostic stratification and risk adjustment. Kidney Int 70: 1120–1126, 2006 [DOI] [PubMed] [Google Scholar]
- 3. Chow F, Ozols E, Nikolic-Paterson DJ, Atkins RC, Tesch GH. Macrophages in mouse type 2 diabetic nephropathy: correlation with diabetic state and progressive renal injury. Kidney Int 65: 116–128, 2004 [DOI] [PubMed] [Google Scholar]
- 4. de Borst MH, Prakash J, Sandovici M, Klok PA, Hamming I, Kok RJ, Navis G, van Goor H. c-Jun NH2-terminal kinase is crucially involved in renal tubulo-interstitial inflammation. J Pharmacol Exp Ther 331: 896–905, 2009 [DOI] [PubMed] [Google Scholar]
- 5. Furuichi K, Wada T, Iwata Y, Kitagawa K, Kobayashi K, Hashimoto H, Ishiwata Y, Asano M, Wang H, Matsushima K, Takeya M, Kuziel WA, Mukaida N, Yokoyama H. CCR2 signaling contributes to ischemia-reperfusion injury in kidney. J Am Soc Nephrol 14: 2503–2515, 2003 [DOI] [PubMed] [Google Scholar]
- 6. Galkina E, Ley K. Leukocyte recruitment and vascular injury in diabetic nephropathy. J Am Soc Nephrol 17: 368–377, 2006 [DOI] [PubMed] [Google Scholar]
- 7. Gandolfo MT, Jang HR, Bagnasco SM, Ko GJ, Agreda P, Satpute SR, Crow MT, King LS, Rabb H. Foxp3+ regulatory T cells participate in repair of ischemic acute kidney injury. Kidney Int 76: 717–729, 2009 [DOI] [PubMed] [Google Scholar]
- 8. Grgic I, Campanholle G, Bijol V, Wang C, Sabbisetti VS, Ichimura T, Humphreys BD, Bonventre JV. Targeted proximal tubule injury triggers interstitial fibrosis and glomerulosclerosis. Kidney Int 82: 172–183, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Harcourt BE, Forbes JM, Matthews VB. Obesity-induced renal impairment is exacerbated in interleukin-6-knockout mice. Nephrology 17: 257–262, 2012 [DOI] [PubMed] [Google Scholar]
- 10. Himmelfarb J, Joannidis M, Molitoris B, Schietz M, Okusa MD, Warnock D, Laghi F, Goldstein SL, Prielipp R, Parikh CR, Pannu N, Lobo SM, Shah S, D'Intini V, Kellum JA. Evaluation and initial management of acute kidney injury. Clin J Am Soc Nephrol 3: 962–967, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jayakumar C, Mohamed R, Ranganathan PV, Ramesh G. Intracellular kinases mediate increased translation and secretion of netrin-1 from renal tubular epithelial cells. PLoS ONE 6: e26776, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kielar ML, John R, Bennett M, Richardson JA, Shelton JM, Chen L, Jeyarajah DR, Zhou XJ, Zhou H, Chiquett B, Nagami GT, Lu CY. Maladaptive role of IL-6 in ischemic acute renal failure. J Am Soc Nephrol 16: 3315–3325, 2005 [DOI] [PubMed] [Google Scholar]
- 13. Lee S, Huen S, Nishio H, Nishio S, Lee HK, Choi BS, Ruhrberg C, Cantley LG. Distinct macrophage phenotypes contribute to kidney injury and repair. J Am Soc Nephrol 22: 317–326, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lin SL, Chang FC, Schrimpf C, Chen YT, Wu CF, Wu VC, Chiang WC, Kuhnert F, Kuo CJ, Chen YM, Wu KD, Tsai TJ, Duffield JS. Targeting endothelium-pericyte cross talk by inhibiting VEGF receptor signaling attenuates kidney microvascular rarefaction and fibrosis. Am J Pathol 178: 911–923, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lin SL, Kisseleva T, Brenner DA, Duffield JS. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am J Pathol 173: 1617–1627, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ly NP, Komatsuzaki K, Fraser IP, Tseng AA, Prodhan P, Moore KJ, Kinane TB. Netrin-1 inhibits leukocyte migration in vitro and in vivo. Proc Natl Acad Sci USA 102: 14729–14734, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mazelin L, Bernet A, Bonod-Bidaud C, Pays L, Arnaud S, Gespach C, Bredesen DE, Scoazec JY, Mehlen P. Netrin-1 controls colorectal tumorigenesis by regulating apoptosis. Nature 431: 80–84, 2004 [DOI] [PubMed] [Google Scholar]
- 18. Mirakaj V, Thix CA, Laucher S, Mielke C, Morote-Garcia JC, Schmit MA, Henes J, Unertl KE, Köhler D, Rosenberger P. Netrin-1 dampens pulmonary inflammation during acute lung injury. Am J Respir Crit Care Med 181: 815–824, 2010 [DOI] [PubMed] [Google Scholar]
- 19. Mohamed R, Jayakumar C, Ranganathan PV, Ganapathy V, Ramesh G. Kidney proximal tubular epithelial-specific overexpression of netrin-1 suppresses inflammation and albuminuria through suppression of COX-2-mediated PGE2 production in streptozotocin-induced diabetic mice. Am J Pathol 181: 1991–2002, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Navarro-Gonzalez JF, Mora-Fernandez C. The role of inflammatory cytokines in diabetic nephropathy. J Am Soc Nephrol 19: 433–442, 2008 [DOI] [PubMed] [Google Scholar]
- 21. Pang M, Ma L, Gong R, Tolbert E, Mao H, Ponnusamy M, Chin YE, Yan H, Dworkin LD, Zhuang S. A novel STAT3 inhibitor, S3I-201, attenuates renal interstitial fibroblast activation and interstitial fibrosis in obstructive nephropathy. Kidney Int 78: 257–268, 2010 [DOI] [PubMed] [Google Scholar]
- 22. Ramesh G, Reeves WB. p38 MAP kinase inhibition ameliorates cisplatin nephrotoxicity in mice. Am J Physiol Renal Physiol 289: F166–F174, 2005 [DOI] [PubMed] [Google Scholar]
- 23. Rosenberger P, Schwab JM, Mirakaj V, Masekowsky E, Mager A, Morote-Garcia JC, Unertl K, Eltzschig HK. Hypoxia-inducible factor-dependent induction of netrin-1 dampens inflammation caused by hypoxia. Nat Immunol 10: 195–202, 2009 [DOI] [PubMed] [Google Scholar]
- 24. Squarize CH, Castilho RM, Sriuranpong V, Pinto DS, Jr, Gutkind JS. Molecular cross-talk between the NFkappaB and STAT3 signaling pathways in head and neck squamous cell carcinoma. Neoplasia 8: 733–746, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tadagavadi RK, Wang W, Ramesh G. Netrin-1 regulates Th1/Th2/Th17 cytokine production and inflammation through UNC5B receptor and protects kidney against ischemia-reperfusion injury. J Immunol 185: 3750–3758, 2010 [DOI] [PubMed] [Google Scholar]
- 26. Waikar SS, Liu KD, Chertow GM. Diagnosis, epidemiology and outcomes of acute kidney injury. Clin J Am Soc Nephrol 3: 844–861, 2008 [DOI] [PubMed] [Google Scholar]
- 27. Wang W, Reeves WB, Pays L, Mehlen P, Ramesh G. Netrin-1 overexpression protects kidney from ischemia reperfusion injury by suppressing apoptosis. Am J Pathol 175: 1010–1018, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang W, Reeves WB, Ramesh G. Netrin-1 and kidney injury. I. Netrin-1 protects against ischemia-reperfusion injury of the kidney. Am J Physiol Renal Physiol 294: F739–F747, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang W, Reeves WB, Ramesh G. Netrin-1 increases proliferation and migration of renal proximal tubular epithelial cells via the UNC5B receptor. Am J Physiol Renal Physiol 296: F723–F729, 2009 [DOI] [PubMed] [Google Scholar]
- 30. Wang Y, Harris DCH. Macrophages in renal disease. J Am Soc Nephrol 22: 21–27, 2011 [DOI] [PubMed] [Google Scholar]
- 31. Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med 16: 535–543, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]