

Abstract
Immune cell infiltration of the kidney is a constant feature in salt-sensitive hypertension (SSHTN). We evaluated the relationship between the renal inflammation and pressure natriuresis in the model of SSHTN that results from transient oral administration of Nω-nitro-l-arginine methyl ester (l-NAME). Pressure natriuresis was determined in Wistar rats that received 4 wk of a high-salt (4% NaCl) diet, starting 1 wk after stopping l-NAME, which was administered alone (SSHTN group, n = 17) or in association with mycophenolate mofetil (MMF; MMF group, n = 15). The administration of MMF in association with l-NAME is known to prevent the subsequent development of SSHTN. Control groups received a high (n = 12)- and normal (0.4%)-salt diet (n = 20). Rats with SSHTN had increased expression of inflammatory cytokines and oxidative stress. The severity of hypertension correlated directly (P < 0.0001) with the number of tubulointerstitial immune cells and angiotensin II-expressing cells. Pressure natriuresis was studied at renal arterial pressures (RAPs) of 90, 110, 130, and 150 mmHg. Glomerular filtration rate was similar and stable in all groups, and renal blood flow was decreased in the SSHTN group. Significantly decreased natriuresis (P < 0.05) was found in the SSHTN group at RAPs of 130 and 150 mmHg, and there was an inverse correlation (P < 0.01) between the urinary sodium excretion and the number of tubulointerstitial inflammatory cells (lymphocytes and macrophages) and cells expressing angiotensin II. We conclude that tubulointerstitial inflammation plays a key role in the impairment of pressure natriuresis that results in salt-dependent hypertension in this experimental model.
Keywords: angiotensin II-positive cells, lymphocytes, macrophages, mycophenolate mofetil, urinary sodium excretion
arterial hypertension is a major cause of morbidity and mortality worldwide. Salt sensitivity, characterized by greater-than-expected blood-pressure changes in response to salt loading and restriction, is a feature of the majority of adult and elderly hypertensive patients (53) and is associated with renal tubulointerstitial inflammation in experimental models of hypertension (37, 38) and in primary hypertension in humans (11, 14, 49). The relevance of the renal inflammation in the pathogenesis of salt-sensitive hypertension (SSHTN) is underlined by the demonstration that treatments that suppress the renal inflammation result in amelioration or prevention of salt-driven hypertension (11, 38, 51).
It is assumed that the role played by renal inflammation is related to the impairment in the renal capacity to excrete sodium, since the central event in the pathogenesis of salt-induced hypertension is impairment in the pressure-natriuresis relationship (9). Pressure natriuresis refers to the increment in urinary sodium excretion (UNaV) that occurs when blood pressure rises and is the consequence of complex modifications in renal interstitial hydrostatic pressure, medullary blood flow, nitric oxide, reactive oxygen species, prostaglandins, and angiotensin II activity (8, 30, 36) that result in coordinated decreases in the surface distribution of apical sodium-hydrogen exchangers and basolateral Na-K ATPase activity (27). In SSHTN, the pressure-natriuresis relationship is less steep and shifted to the right so that higher blood-pressure levels are necessary to drive the natriuretic response required to maintain sodium balance (41).
Tubulointerstitial inflammation may impair UNaV via several mechanisms. Inflammation may lead to a loss of peritubular capillaries, tubular injury, and fibrotic interstitial changes. If these changes are severe and extensive, they may cause salt wasting, but milder changes that do not render inoperative the sodium reabsorption mechanisms are associated with SSHTN. This may be due to reduced oxygen delivery to regions of the kidney that function normally under relative hypoxia, causing a further reduction in oxygen partial pressure with the generation of oxidative stress and impaired pressure natriuresis (16). In addition, the infiltration of immune-competent cells in the renal interstitial space is associated with inflammation-induced generation of reactive oxygen species and increased local angiotensin II activity (7, 41, 42, 51) that suppress natriuresis.
The present investigations were directed to test the assumption that a consequence of tubulointerstitial inflammation is the blunting of the pressure-natriuresis response. In these studies, we selected an experimental model of SSHTN that follows recovery from nitric oxide synthase inhibition with Nω-nitro-l-arginine methyl ester (l-NAME) (34). In this model, 3 wk of l-NAME administration cause hypertension and significant renal injury and inflammation during the period l-NAME is given. Blood pressure returns to normal 1 wk after l-NAME is omitted, but if a high-salt diet is administered subsequently, hypertension develops in the following weeks. Treatment with mycophenolate mofetil (MMF) during l-NAME administration does not modify the blood-pressure elevation, due to inhibition of nitric oxide synthase, but ameliorates renal histological damage and inflammation caused by l-NAME and prevents the subsequent development of SSHTN (34). Important for the selection of this model in the present experiments are the findings that the histological damage that could, by itself, cause impairment in sodium excretion improves after l-NAME is discontinued, and several weeks later, the l-NAME-treated groups (with or without MMF treatment) present mild and comparable renal injury. At this time, the salient feature is that salt-induced hypertension is associated with tubulointerstitial infiltration of immune cells, a characteristic that is common to most, if not all, models of SSHTN (6, 34, 37–40, 42, 43).
MATERIALS AND METHODS
Male Wistar rats (250–320 g) were housed in institutional animal facilities with free access to food and water. All studies were performed in accordance with the Mexican Federal Regulation for Experimentation and Care (NOM-062-ZOO-2001), and the investigation was approved by Bioethics and Investigation Committees of Instituto Nacional de Cardiologia Ignacio Chavez. The following experimental groups were studied: 1) SSHTN group (n = 17), which received l-NAME (Sigma, St. Louis, MO) in the drinking water at a concentration of 70 mg/100 ml for 3 wk, followed (after a washout period of 1 wk and return to normal blood pressure) by the administration of a high-salt diet (4% NaCl Teklad diet; Harlan Laboratories, Madison, WI) for 4 wk; and 2) the MMF group (n = 15), which received l-NAME, as in the SSHTN group, but in addition, received MMF (CellCept; Roche Pharmaceutical, Nutley, NJ) by gastric gavage in daily doses of 20 mg/kg during the initial 3 wk of the study (nitric oxide synthesis inhibition) and then stopped; thus this drug was not given during the subsequent 5 wk that preceded the pressure-natriuresis studies (1 wk of washout and 4 wk of a high-salt diet). MMF is relatively insoluble in water and therefore, was suspended in water by vigorous agitation before administration, as in previous studies (38, 44). Control groups received a high (4% NaCl)-salt diet (C-HSD; n = 12) and normal (0.4% NaCl)-salt diet (C-NSD; n = 20).
Additional groups of rats were studied to assess the renal injury resulting from l-NAME administration. In these studies, rats that received l-NAME (n = 6) and l-NAME + MMF (n = 7) were killed at the end of 3 wk of l-NAME administration, and renal histology and immunohistology were examined. Control rats (n = 5) were studied at the same time. All of these rats were receiving a normal-salt diet.
Finally, whereas MMF was given only during the l-NAME administration and therefore, had been stopped 5 wk after the pressure-natriuresis studies (1 wk of washout and 4 wk of a high-salt diet), the possible lingering effects of MMF administration on the pressure natriuresis were evaluated in separate experiments done in normal rats studied 4 wk after they received MMF for 3 wk. These studies showed a normal pressure natriuresis response (data not shown).
Pressure-natriuresis experiments.
Pressure-natriuresis experiments were performed after 4 wk of a high-salt diet, as described by Wang et al. (52). Rats were anesthetized with pentobarbital sodium (30 mg/kg intraperitoneally) and placed on a surgical table with temperature control to maintain body temperature at 37°C. A patent airway was maintained with a PE-240 tube inserted in the trachea. A catheter in the left jugular vein was used for fluid administration, and a catheter was inserted in the left femoral artery to measure arterial pressure that was monitored continuously with a pressure transducer (model P23LX; Gould, Hato Rey, Puerto Rico) and recorded on a polygraph (Grass Instruments, Quincy, MA), as well as to take blood samples. The left kidney was exposed and placed in a lucite holder, covering the kidney surface with Ringer solution. A 2-mm flow probe was placed around the left renal artery for measuring renal blood flow (RBF) by an electromagnetic flow meter (model T10; Transonic Systems, Ithaca, NY) in seven to 10 rats of each experimental and control groups. An adjustable aortic clamp was placed above the left renal artery and used to control distal blood pressure. To obtain data at renal arterial pressure (RAP) of 150 mmHg in normotensive rats (seven rats from the C-NSD group, five rats from the C-HSD group, and six rats from the MMF group), the blood pressure was elevated by transient occlusion of carotid arteries (1), 20 min before beginning the first pressure-natriuresis evaluation, and was maintained for the rest of the experiment. Progressive reduction in RAP in steps of ∼20 mmHg was maintained at a stable level by means of the aortic clamp; urine and blood collections were obtained at 150, 130, 110, and 90 mmHg. The left ureter was catheterized for urinary collections. Rats were maintained euvolemic by infusion of 10 ml/kg body wt of isotonic rat plasma during surgery, followed by an infusion of 10% polyfructosan (Inutest; Laevosan-Gesellschafft, Austria) in 0.9% sodium saline solution at a 2.2-ml/h rate, and replacement of blood samples was obtained for analysis. The measurements were started after 1 h of stabilization. After 10 min equilibration at each blood-pressure level, 30-min urinary collections were obtained for analyses. Plasma samples were obtained at midpoint in urinary collections. At the end of the experiment, the kidneys were removed, weighted, and harvested; the animals were killed by a pentobarbital overdose; and the kidneys were weighed and harvested for histology and immunohistology studies.
Chemical analyses.
Blood and urine samples were collected for determination of glomerular filtration rate (GFR), UNaV, and fractional excretion of sodium (FNaE). Urine volume was determined gravimetrically. Polyfructosan concentrations were determined as described by Davidson and Sackner (5) and sodium concentrations by flame photometry.
Histology and immunohistology.
All histological evaluations were done without prior knowledge of the experimental group being studied. Histological studies were done in kidney sections of nine rats from the C-NSD group, eight rats from the C-HSD group, 10 rats from the SSHTN group, and 10 rats from the MMF group. Light microscopy was performed using formalin-fixed sections stained with periodic acid-Schiff and hematoxylin and eosin. Glomerulosclerosis index score and tubulointerstitial damage were evaluated as described in previous studies (34, 39, 40). The sclerosis in the glomeruli was graded from 0 to +4: grade 0 = normal, grade +1 = <25% involvement of the glomerular tuft, grade +2 = 25–50% involvement of the glomerular tuft, grade +3 = 50–75%, and grade +4 = sclerosis occupying >75% of the glomerular tuft. The glomerulosclerosis score was obtained as follows: [(1 × n glomeruli with +1) + (2 × n glomeruli with +2) + (3 × n glomeruli with +3) + (4 × n glomeruli with +4)] × 100/total number of glomeruli examined. The tubulointerstitium was evaluated in successive fields examined at 20× magnification in the entire cortical and juxtamedullary areas of each specimen using computer-assisted image analysis (Olympus BX51 system microscope and DP70 digital microscope camera with Sigma Scan Pro 5 software, San Jose, CA). Tubulointerstitial damage (tubular dilatation, tubular atrophy, and interstitial fibrosis) was graded on a 0–5 scale: 0 = no changes, grade 1 = <10%, grade 2 = 10–25%, grade 3 = 25–50%, grade 4 = 50–75%, grade 5 = 75–100.
Identification and quantification of tubulointerstitial immune cell infiltration.
Immunoperoxidase methodology was used to indentify lymphocytes and macrophages in the glomeruli (positive cells/glomerular cross-section) and tubulointerstitial regions (positive cells/mm2). Macrophages were identified with mouse CD68 anti-MAb and lymphocytes with mouse anti-rat CD3 (Biosource International, Camarillo, CA). CD4- and CD8-positive cells were identified with the corresponding MAb (anti-rat CD4, clone W3/25, mouse IgG1; and anti-rat CD8, no azide, clone OX-8, mouse IgG1), obtained from Cedarlane (Hornby, Canada). Angiotensin II-positive cells were identified with rabbit anti-ANG II-human IgG (Peninsula Laboratories, San Carlos, CA). Peroxidase-conjugated goat anti-mouse and IgG (Stressgen Bioreagents, Victoria, BC, Canada) and donkey anti-rabbit (Accurate Chemical & Scientific, Westbury, NY) were used as secondary antibodies. Infiltrating immune cells and angiotensin II-positive cells were rarely found in the glomeruli, and only the data in tubulointerstitial areas are given.
Inflammatory cytokines and oxidative stress.
Renal cortex from five animals of experimental (SSHTN and MMF) and control (C-NSD) groups were used for Western blots (IL-2, IL-6, and nitrotyrosine abundance) and malondialdehyde (MDA) determinations. For the Western blot analyses, 40 μg of protein was treated with 12% SDS-PAGE and transferred onto a nitrocellulose membrane, which was washed and probed with polyclonal antibody (1:1,000) against IL-2, IL-6, and nitrotyrosine (Santa Cruz Biotechnology, Santa Cruz, CA), and 1:1,000 goat horseradish peroxidase-labeled anti-rabbit IgG (Santa Cruz Biotechnology). Finally, enhanced chemiluminescence (ECL) detection solution was added, and hyperfilm ECL (GE Healthcare, Chalfont St. Giles, UK) was exposed to the membrane. Each membrane was stripped of bound antibody and reprobed with anti-β-actin on the same membrane for quantitative comparison. The bands were analyzed with a Kodak electrophoresis documentation and analysis system (EDAS 290).
MDA content was determined by the method of Ohkawa et al. (31). Briefly, kidney slices for MDA were placed in a cold mixure of 100 mM KCL and 0.003 EDTA, homogenized, and centrifuged at 600 g for 15 min. Supernatants of this preparation were used for MDA determination, as described previously (39, 45).
Statistical analyses.
GFR, RBF, UNaV, and the FNaE (percent of filtered) are expressed in relation to left-kidney weight. Multigroup ANOVA and Tukey Kramer post-tests were used to examine differences between groups. Serial changes were studied with paired t-tests. Correlations were analyzed with Pearson's correlation coefficient. Data are shown as means ± SE. Two-tailed P values <0.05 were considered significant. A commercially available statistical package (InStat; GraphPad Software, La Jolla, CA) was used for statistical calculations.
RESULTS
Blood pressure and histological findings induced by 3 wk of l-NAME administration are shown in Table 1. As in previous studies (34), MMF administration did not modify the hypertension induced by inhibition of nitric oxide synthase or the return to normal blood pressure, 1 wk after the discontinuation of l-NAME (l-NAME: basal = 120 ± 1.7 mmHg, 1 wk = 154 ± 3.1, 2 wk = 163 ± 3.2, 3 wk = 175.5 ± 3.05, 1 wk after discontinuation of l-NAME = 136 ± 3.0; l-NAME + MMF: basal = 117 ± 1.4, 1 wk = 151 ± 3.6, 2 wk = 151 ± 3.6, 3 wk = 176 ± 3.6, 1 wk after discontinuation of l-NAME = 139 ± 2.4). As shown in Table 1, l-NAME administration resulted in renal injury (glomerulosclerosis and tubulointerstitial damage) and renal infiltration of lymphocytes (CD3 + cells), macrophages (CD68 + cells), and angiotensin II + cells in tubulointerstitial areas. Treatment with MMF during the l-NAME resulted in a significant reduction (albeit not normalization) of renal injury and tubulointerstial inflammation.
Table 1.
Data after after 3 wk of l-NAME treatment
| Control NSD | l-NAME | l-NAME + MMF | |
|---|---|---|---|
| (n = 5) | (n = 6) | (n = 7) | |
| SBP, mmHg | 126.2 ± 4.27 | 175.5 ± 3.05a | 176 ± 3.6a |
| Plasma creatinine, mg/dl | 0.30 ± 0.03 | 0.40 ± 0.03 | 0.38 ± 0.03 |
| Proteinuria, mg/24 h | 2.8 ± 1.39 | 12.6 ± 4.80 | 11.5 ± 3.87 |
| GS index, 0–400 | 0 | 65.3 ± 7.00a,b | 45.7 ± 3.81a |
| TI damage score, 0–5 | 0.5 ± 0.21 | 1.96 ± 0.28a | 1.25 ± 0.154 |
| CD68 + cells/mm2 | 3.4 ± 1.12 | 88.8 ± 6.93a,c | 38.1 ± 5.28a |
| CD3 + cells/mm2 | 2.6 ± 1.07 | 75.3 ± 7.96a,d | 42.5 ± 5.17a |
| AII + cells/mm2 | 0.8 ± 0.58 | 28.5 ± 3.51a,c | 12.5 ± 1.23a |
Studies done in kidney sections harvested after 3 wk of Nω-nitro-l-arginine methyl ester (l-NAME) administration in the drinking water (l-NAME group) and l-NAME plus daily mycophenolate mofetil (MMF) treatment (MMF group). Control groups received no treatment. All rats were in a normal (0.4%)-salt diet (NSD). SBP, systolic blood pressure; GS, glomerulosclerosis; TI, tubulointerstitial; AII, angiotensin II-positive. Data are means ± SE.
Values higher than controls (P < 0.01 or lower). Differences between l-NAME and l-NAME + MMF groups are
P < 0.05,
P < 0.001 (multigroup variance analysis), and
P < 0.01.
General data obtained prior to the pressure-natriuresis experiments (5 wk after discontinuation of l-NAME) are shown in Table 2. As expected, systolic blood pressure was higher in the rats from the SSHTN group. As in previous studies (34), MMF treatment prevented the subsequent development of SSHTN, and the systolic blood pressure in the MMF group was not significantly different than in the control groups on a normal (C-NSD)- or high (C-HSD)-sodium diet (Table 2). Food intake, urine volume, and proteinuria were similar in all experimental groups (Table 2).
Table 2.
General results
| C-NSD (n = 20) | C-HSD (n = 12) | SSHTN (n = 17) | MMF (n = 15) | |
|---|---|---|---|---|
| Body weight, g | ||||
| Before l-NAME | 301.4 ± 4.92 | 308.0 ± 3.09 | 302.0 ± 1.35 | 307.5 ± 3.85 |
| Prior to pressure natriuresis | 438.9 ± 10.72 | 448.4 ± 16.44 | 450.0 ± 20.02 | 452.5 ± 8.69 |
| Left-kidney weight, g | 1.39 ± 0.025 | 1.45 ± 0.036 | 1.56 ± 0.040 | 1.56 ± 0.058 |
| Food ingestion, g/day | 30.2 ± 1.60 | 32.0 ± 0.88 | 30.0 ± 2.29 | 26.5 ± 1.41 |
| Urine volume, ml/24 h | 20.43 ± 4.51 | 20.0 ± 2.70 | 26.0 ± 5.38 | 22.0 ± 3.45 |
| Proteinuria, mg/24 h | 8.25 ± 1.03 | 8.62 ± 0.85 | 9.11 ± 1.20 | 8.77 ± 0.92 |
| MAP, mmHg | 116.0 ± 1.08 | 118.8 ± 1.54 | 157.9 ± 1.91a | 132.7 ± 1.64a |
Kidney weight was obtained at the end of pressure natriuresis experiments. Mean arterial pressure (MAP) was obtained by direct intra-arterial determinations prior to the pressure natriuresis experiments. Experimental [salt-sensitive hypertension (SSHTN) and MMF] and the control (C) high-salt diet (C-HSD) groups had been on a high-salt diet, whereas the C-NSD group was on a normal-sodium diet for 4 wk. Food ingestion means during the preceding 30 days; proteinuria and urine volume were measured the day before pressure natriuresis studies.
P < 0.01 vs. the rest.
Renal immunohistologic findings present in kidneys harvested immediately after the pressure-natriuresis experiments are shown in Table 3. During the 5 wk that elapsed after discontinuation of l-NAME (with and without MMF) and the pressure-natriuresis studies, the renal injury and tubulointerstitial inflammation were reduced with respect to the findings at the end of l-NAME treatment (Table 1). At the time when the pressure-natriuresis studies were performed (Table 3), the glomerulosclerosis and tubular damage, although higher in the SSHTN group, were not significantly different in any of the study groups and essentially comparable in the SSHTN group and the MMF group. In contrast, interstitial macrophages (CD68-positive cells), lymphocytes (CD3-positive cells, as well as CD4 and CD8 T cells), and angiotensin II-positive cells were several-fold higher in the group with SSHTN than in the control groups and the MMF group. Representative microphotographs are shown in Fig. 1.
Table 3.
Histology and immunohistology at the time of pressure natriuresis studies
| C-NSD (n = 7) | C-HSD (n = 7) | SSHTN (n = 10) | MMF (n = 10) | |
|---|---|---|---|---|
| GS index, 0–400 | 0.6 ± 0.24 | 0.6 ± 0.40 | 4.8 ± 2.63 | 3.4 ± 1.69 |
| TI damage score, 0–5 | 0.6 ± 0.24 | 0.8 ± 0.37 | 1.8 ± 0.58 | 1.2 ± 0.20 |
| CD68 + cells/mm2 | 7.82 ± 0.57 | 10.14 ± 0.66 | 35.65 ± 2.29a | 22.99 ± 1.49b |
| CD3 + cells/mm2 | 7.76 ± 0.63 | 11.66 ± 0.84 | 43.17 ± 2.76a | 26.77 ± 2.15b |
| CD4 + cells/mm2 | 6.78 ± 0.78 | 2.67 ± 1.09 | 29.3 ± 3.80c | 17.9 ± 2.29b |
| CD8 + cells/mm2 | 3.16 ± 1.08 | 6.28 ± 0.60 | 18.9 ± 2.12d | 9.81 ± 0.49e |
| AII + cells/mm2 | 8.09 ± 0.60 | 10.63 ± 0.64 | 32.06 ± 2.39c | 19.25 ± 0.42e |
Histology and immunohistology in the C-NSD and C-HSD groups and the experimental groups (SSHTN and MMF). Values are means ± SE.
P < 0.01 vs. the rest;
P < 0.01 vs. C-NSD and C-HSD;
P < 0.001 vs. C-NSD and C-HSD, and P < 0.05 vs. MMF;
P < 0.001 vs. the rest;
P < 0.05 vs. C-NSD.
Fig. 1.

Light microscopy and immunohistology of renal sections of rats from the control group, salt-sensitive hypertension (SSHTN) group, and mycophenolate mofetil (MMF) group. A: Light microscopy (periodic acid-Schiff staining), (B) macrophage (CD68-positive cells) infiltration, and (C) angiotensin II-positive cells. Tubulointerstitial inflammation and tubular cells and infiltrating cells staining positive for angiotensin II are evident in the SSHTN group. Scale marks correspond to the whole column. Staining details are in materials and methods.
Renal inflammatory cytokines and oxidative stress measurements are shown in Fig. 2. IL-2 (Fig. 2A), IL-6 (Fig. 2B), nitrotyrosine (Fig. 2C), and MDA (Fig. 2D) were increased in the SSHTN group and were suppressed in the group that had been administered MMF previously.
Fig. 2.

Western blots of renal content of IL-2 (A), IL-6 (B), nitrotyrosine (C), and malondialdehyde (D) in experimental (SSHTN and MMF) and control [normal salt diet (C-NSD)] groups. Western blot data are expressed as optical density (O.D.) relative to β-actin. Gel pictures are samples for 1 run. Data correspond to n = 5 in each group. Molecular weight shown in nitrotyrosine corresponds to nitrosylated tyrosine-containing proteins. *P < 0.05; **P < 0.01; ***P < 0.001.
RBF in the experimental groups at RAPs of 90, 110, 130, and 150 mmHg are shown in Fig. 3. The rats with SSHTN had lower values of RBF, and this was corrected by MMF treatment (Fig. 3A). Differences in GFR in the groups under study were not significantly different (Fig. 3B), and consequently, filtration fraction was increased in rats of the SSHTN group (Fig. 3C). The control groups (C-NSD and C-HSD) and the MMF group did not show significant modifications in the GFR. In contrast, rats with SSHTN increased the GFR from 110 to 150 mmHg RAP (P < 0.05; Fig. 3B).
Fig. 3.
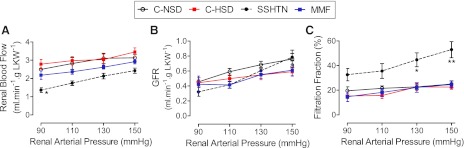
Renal blood flow (RBF) and glomerular filtration rate (GFR) at the studied renal arterial pressure (RAP) levels in the experimental (SSHTN and MMF) and control groups with C-NSD and a high-salt diet (C-HSD). A: no significant differences in RBF in C-NSD, C-HSD, and MMF groups. RBF in the SSHTN group was lower (*P < 0.01 vs. the rest). B: no significant differences among the GFRs at 90, 110, 130, and 150 mmHg RAPs in experimental and control groups. GFR in the SSHTN group increased progressively from 90 mmHg to 150 mmHg. C: filtration fraction in the SSHTN is significantly higher (*P < 0.05; **P < 0.01) than in the rest. Studies done with an induced (see text) RAP of 150 mmHg in normotensive rats from the C-NSD (n = 7), C-HSD (n = 5), and MMF (n = 6) groups, as indicated in materials and methods. Symbols and error bars are mean ± SE. LKW, left-kidney weight.
UNaV and FNaE at the corresponding RAPs are shown in Fig. 4, A and B, respectively. The essentially unchanged sodium-excretion rate in the SSHTN group contrasts with the values in the C-NSD and C-HSD that increase from 90 to 130 mmHg of RAP. Rats from the MMF group had values of UNaV and FNaE that were not significantly different from those in rats from the control groups.
Fig. 4.

Variations in (A) urinary sodium-excretion (UNaV) rate and (B) fractional sodium excretion (FNaE) at the studied RAPs. The progressive increment in UNaV and FNaE observed in the control and MMF groups was not present in the SSHTN group. Studies done with RAP of 150 mmHg in normotensive rats from the C-NSD (n = 5), C-HSD (n = 5), and MMF (n = 6) groups, as indicated in materials and methods. *P < 0.05 vs. the rest.
The relationship between the inflammatory infiltrate and UNaV and FNaE was examined at a RAP of 130 mmHg when a separation between experimental and control groups was present. The intensity of immune cell infiltration had a strong negative relationship with the UNaV rate (Fig. 5A) and with the FNaE (Fig. 5B). Similar negative relationships were found between the number of angiotensin II-positive cells and natriuresis (Fig. 6A) and FNaE (Fig. 6B).
Fig. 5.

Inverse relationship between interstitial immune cell (CD68- + CD3-positive cells) infiltration and (A) UNaV rate and (B) FNaE. Data correspond to values at RAP of 130 mmHg. Each symbol represents 1 animal of the corresponding groups.
Fig. 6.

Inverse relationship between the number of angiotensin II-positive cells and (A) UNaV rate and (B) FNaE. Data correspond to RAP of 130 mmHg. Symbols correspond to the experimental and control groups.
Not unexpectedly, a strong (P < 0.0001), positive relationship exists between the severity of hypertension (determined prior to the pressure-natriuresis experiments) and the intensity of tubulointerstial infiltration of immune-competent cells (Fig. 7A), as well as with cells expressing angiotensin II (Fig. 7B).
Fig. 7.

Direct relationship between the systolic blood pressure (SBP) and the number of infiltrating immune cells (CD68- + CD3-positive cells; A) and angiotensin II-positive cells (B). Symbols correspond to the experimental and control groups.
DISCUSSION
There is a strong association between renal inflammation and SSHTN (37, 38). The novel findings of this study are the demonstration of a direct association between the degree of impairment in pressure natriuresis and the severity of tubulointerstitial inflammation. These studies, therefore, provide compelling evidence that interstitial inflammation, in the absence of significant renal damage (Table 3), plays in the impairment of pressure natriuresis that drives salt-induced hypertension.
The RBF was lower in the group with SSHTN, consistent with renal vasoconstriction, likely as the result of intrarenal oxidative stress and angiotensin II overactivity generated by inflammation (41, 51). GFR did not differ significantly in experimental and control groups in the range of RAP studied. No significant changes in GFR between 90 and 150 mmHg RAP were found in control and MMF groups; in contrast, the rats in the SSHTN group had a progressive increment in GFR, as RAP was varied from 110 to 150 mmHg (paired t-test, P < 0.01). In pressure-natriuresis studies done in various laboratories, GFR in control groups has been found to be at steady levels or showing only a mild increment (15, 20, 26, 44, 52). In hypertension induced by angiotensin II, Mattson et al. (26) and Wang et al. (52) found a progressive increment in GFR with increasing RAP pressure, similar to what we observed in the SSHTN group. In addition, inflammatory cytokines, such as transforming growth factor β, impair the autoregulation of glomerular hemodynamics via the generation of reactive oxygen species (13, 47) and may cause afferent arteriolar remodeling (7, 46). Taken together, these findings suggest that the loss of GFR autoregulation in the SSHTN group may be related to renal angiotensin II overactivity and renal inflammation. Since GFR was essentially the same in the experimental and control groups, and the RBF was lower in the SSHTN group, the filtration fraction in the SSHTN group was increased, as has been reported in oxidative stress associated with a high-salt diet (25) and in the chronic, slow pressor responses induced by angiotensin II infusion (19).
The role played by oxidative stress in the medullary regions of the kidney, restricting nitric oxide availability and causing dysfunction of the pressure-natriuresis response, has been reviewed recently (8, 30, 36, 48). Reduction of oxidative stress by hypoxia-inducible factor 1α (23) can improve pressure natriuresis. The generation of reactive oxygen species is an obligatory consequence of inflammation (51), and reduction in nitric oxide, resulting from oxidative stress, is expected to restrict the vasodilatation capacity and reduce interstitial cGMP, which is critical for the natriuresis response (24). The increased renal nitrotyrosine and MDA content in the SSHTN group (Fig. 2, C and D) support this notion.
The pressure natriuresis in rats of the control group and rats with mild inflammatory infiltration of immune cells (MMF group) showed a progressive increment of natriuresis, and at 130 mmHg RAP and 150 mmHg RAP, the UNaV rate was more than three times and 13 times, respectively, higher than the values at 90 mmHg RAP pressure. In contrast, the SSHTN group showed a suppression of the pressure natriuresis that was strikingly evident at RAP values of 130 and 150 mmHg.
In agreement with previous studies (6, 40, 43), we found that severity of SSHTN is correlated directly (P < 0.001) with the intensity of the infiltration of immune-competent cells in the interstitial areas of the kidney. The association of immune cell infiltration and inflammatory activity is confirmed by the increment in renal expression of inflammatory cytokines (Fig. 2, A and B).
As reported by several investigators (2, 34, 35), l-NAME administration induces renal injury and inflammation, which are reduced after the discontinuation of l-NAME treatment. These findings were confirmed in the present studies, and at the time of the pressure-natriuresis experiments, the hypertensive rats of the SSHTN group and the normotensive rats of the other groups do not have significantly different histological damage. In contrast, the infiltration of immune cells and the number of angiotensin II-expressing cells in tubulointerstitial areas are significantly higher in the SSHTN group (Table 3). The causes responsible for the renal accumulation of lymphocytes and macrophages and its role in SSHTN are incompletely understood, but the role of immune reactivity in hypertension has been reviewed recently (10, 37, 42, 47), and recent work has identified the 70-kDa heat shock protein, overexpressed in the kidney, as a relevant autoantigen in SSHTN (33). In line with these emerging observations, elegant investigations have shown that mice lacking lymphocyte responses develop increased natriuresis and blunted angiotensin II-induced hypertension (4).
In the present experiments, we have documented that the suppression of the natriuresis induced by tubulointerstitial inflammation is evident at RAPs of 130 and 150 mmHg when there is a strong, negative correlation between the UNaV and FNaE and the number of inflammatory and angiotensin II-expressing cells, as described before (6, 34, 39, 40, 43).
The renal inflammation was associated with accumulation of angiotensin II-positive cells. The activation of a functional renin-angiotensin system in the immune-competent cells (12, 17) and tubular cells (21, 28) likely plays a major role in the blunting of pressure natriuresis induced by renal inflammation. In previous studies, we have shown that in experimental models of SSHTN, the plasma angiotensin is reduced as a result of sodium retention; in contrast, the renal angiotensin activity is increased (6). These findings underline the independence of intrarenal angiotensin from physiologic modulation of plasma angiotensin (29) and indicate a primary role of intrarenal angiotensin in the pathogenesis of SSHTN (6). Whereas in the present studies, we only identified angiotensin II-expressing cells by immunohistology, previous work from our group has established that the number of angiotensin II-positive cells, the intensity of renal angiotensin II activity, and the severity of tubulointerstitial inflammation are correlated directly with one another (6, 50). Antinatriuretic effects of angiotensin II result from reduction in sodium-filtered load and increased sodium reabsorption (26, 29) and are mediated by angiotensin II type 1 receptors (3, 18, 20–22, 28), whereas natriuresis is mediated by angiotensin III and type 2 receptors (32).
Oxidative stress and angiotensin II activity are key components of renal inflammation. The increased renal MDA content and inflammatory cytokines in the SSHTN group (Fig. 3) confirm this association, and it is reasonable to assume the participation of these elements in the impairment of pressure natriuresis resulting from renal inflammation.
In conclusion, this investigation found that interstitial inflammation is associated with suppression of the natriuresis resulting from increments in RAP pressure. At RAP levels of 130 mmHg, the UNaV is inversely correlated with the number of inflammatory cells and angiotensin II-positive cells in the kidney. These findings add insight to the pathogenesis of SSHTN and its relation to renal inflammation.
GRANTS
Financial support was provided by CONACYT Grant 79661, Mexico (to M. Franco), National Heart, Lung, and Blood Institute Grant HL-68607 (to R. J. Johnson), and grants from the National Science and Technology Fund (FONACIT), Venezuela, and the Asociación de Amigos del Riñón, Maracaibo, Venezuela (to B. Rodriguez-Iturbe and Y. Quiroz).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: M.F., R.J.J., and B.R-I. conception and design of research; M.F., E.T., R.B., U.P., and J.S. performed experiments; M.F., R.B., Y.Q., and B.R-I. analyzed data; M.F., R.B., Y.Q., and B.R-I. interpreted results of experiments; M.F., R.J.J., and B.R-I. prepared figures; M.F., R.J.J., and B.R-I. drafted manuscript; M.F. and B.R-I. edited and revised manuscript; M.F., R.J.J., and B.R-I. approved final version of manuscript.
ACKNOWLEDGMENTS
These data were presented in part as an oral abstract in the ASN 2011 meeting (TH-OR109), J Am Soc Nephrol 22: 27A, 2011.
REFERENCES
- 1.Arendshorst WJ. Autoregulation of renal flood flow in spontaneously hypertensive rats. Circ Res 44: 344–349, 1979 [DOI] [PubMed] [Google Scholar]
- 2.Bayliss C, Mitruka B, Deng A. Chronic blockade of nitric oxide synthesis in the rat produces systemic hypertension and glomerular damage. J Clin Invest 90: 278–281, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chugh G, Lokhandwala MF, Asghar M. Oxidative stress alters renal D1 and AT1 receptor functions and increases blood pressure in old rats. Am J Physiol Renal Physiol 300: F133–F138, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crowley SD, Song YS, Lin EE, Griffiths R, Kim HS, Ruiz P. Lymphocyte responses exacerbate angiotensin II-dependent hypertension. Am J Physiol Regul Integr Comp Physiol 298: R1089–R1097, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davidson WD, Sackner MA. Simplification of the anthrone method for the determination of inulin in clearance studies. J Lab Clin Med 62: 351–356, 1963 [PubMed] [Google Scholar]
- 6.Franco M, Martínez F, Quiroz Y, Galicia O, Bautista R, Johnson RJ, Rodríguez-Iturbe B. Renal angiotensin II concentration and interstitial infiltration of immune cells are correlated with blood pressure levels in salt-sensitive hypertension. Am J Physiol Regul Integr Comp Physiol 293: R251–R256, 2007 [DOI] [PubMed] [Google Scholar]
- 7.Franco M, Martínez F, Rodríguez-Iturbe B, Johnson RJ, Santamaría J, Montoya A, Nepomuceno T, Bautista R, Tapia E, Herrera-Acosta J. Angiotensin II, interstitial inflammation, and the pathogenesis of salt-sensitive hypertension. Am J Physiol Renal Physiol 291: F1281–F1287, 2006 [DOI] [PubMed] [Google Scholar]
- 8.Granger JP, Alexander BT, Llinas M. Mechanisms of pressure natriuresis. Curr Hypertens Rep 4: 152–159, 2002 [DOI] [PubMed] [Google Scholar]
- 9.Guyton AC. Blood pressure control: special role of the kidneys and body fluids. Science 252: 1813–1816, 1991 [DOI] [PubMed] [Google Scholar]
- 10.Harrison DG, TJ , Lob EH, Madur MS, Marvar PJ, Thabet SR, Vinh A, Weyand CM. Inflammation, immunity and hypertension. Hypertension 57: 132–140, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herrera J, Ferrebuz A, García Macgregor EG, Rodriguez-Iturbe B. Mycophenolate mofetil improves hypertension in patients with psoriasis and rheumatoid arthritis. J Am Soc Nephrol 17, Suppl 3: S218–S225, 2006 [DOI] [PubMed] [Google Scholar]
- 12.Hoch NE, Guzik TJ, Chen W, Deans T, Maalouf SA, Gratze P, Weyand C, Harrison DG. Regulation of T-cell function by endogenously produced angiotensin II. Am J Physiol Regul Integr Comp Physiol 296: R208–R216, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu T, RamachandraRao SP, Satish P, Siva S, Valancius C, Zhu Y, Mahadev K, Toh I, Goldstein BJ, Woolkalis M, Sharma K. Reactive oxygen species production via NADPH oxidase mediates TGF-beta-induced cytoskeletal alterations in endothelial cells. Am J Physiol Renal Physiol 289: F816–F825, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hughson MD, Gobe GC, Hoy WE, Manning RD, Jr, Douglas-Denton R, Bertram JF. Associations of glomerular number and birth weight with clinicopathological features of African Americans and whites. Am J Kidney Dis 52: 18–28, 2008 [DOI] [PubMed] [Google Scholar]
- 15.Jin XH, Mcgrath HE, Gildea JJ, Siragy HM, Felder RA, Carey RM. Renal interstitial guanosine cyclic 3, 5-monophosphate mediates pressure-natriuresis via protein kinase G. Hypertension 43: 1133–1139, 2004 [DOI] [PubMed] [Google Scholar]
- 16.Johnson RJ, Schreiner G. Hypothesis: the role of acquired tubulointerstitial disease in the pathogenesis of salt-dependent hypertension. Kidney Int 52: 1169–1179, 1997 [DOI] [PubMed] [Google Scholar]
- 17.Jurewicz M, Mcdermott DH, Sechler JM, Tinckam K, Takakura A, Carpenter CB, Milford E, Abdi R. Human T and natural killer cells posses a functional renin-angiotensin system: further mechanisms of angiotensin II-induced inflammation. J Am Soc Nephrol 18: 1093–1102, 2007 [DOI] [PubMed] [Google Scholar]
- 18.Kaschina E, Unger T. Angiotensin AT1/AT2 receptors: regulation, signalling and function. Blood Press 12: 70–88, 2003 [DOI] [PubMed] [Google Scholar]
- 19.Kawada N, Imai E, Karber A, Welch WJ, Wilcox CS. A mouse model of angiotensin II slow pressor response: role of oxidative stress. J Am Soc Nephrol 13: 2860–2868, 2002 [DOI] [PubMed] [Google Scholar]
- 20.Kline Liu F RL. Modification of pressure natriuresis by long-term losartan in spontaneously hypertensive rats. Hypertension 24: 467–473, 1994 [DOI] [PubMed] [Google Scholar]
- 21.Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev 59: 251–287, 2007 [DOI] [PubMed] [Google Scholar]
- 22.Lara LS, Mccormack M, Semprum-Prieto LS, Shenouda S, Majid DS, Kobori H, Navar LG, Prieto MC. AT1 receptor-mediated augmentation of angiotensinogen, oxidative stress, and inflammation in ANG II-salt hypertension. Am J Physiol Renal Physiol 302: F85–F94, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li N, Chen L, Yi F, Xia M, Li PL. Salt sensitive hypertension induced by decoy of transcription factor hypoxia-inducible factor-1 alpha in the renal medulla. Circ Res 102: 1101–1108, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lieb DC, Kemp BA, Howell NL, Gildea JJ, Carey RM. Reinforcing feedback loop of renal cyclic guanosine 3,5-monophosphate and interstitial hydrostatic pressure in pressure-natriuresis. Hypertension 54: 1278–1283, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Magalhaes JCG, Mota DL, Paixao ADO. Renal function in juvenile rats subjected to prenatal malnutrition and chronic salt overload. Exp Physiol 91: 611–619, 2006 [DOI] [PubMed] [Google Scholar]
- 26.Mattson DL, Raff H, Roman RJ. Influence of angiotensin II on pressure natriuresis in renal hemodynamics in volume expanded rats. Am J Physiol Regul Integr Comp Physiol 29: R1200–R1209, 1991 [DOI] [PubMed] [Google Scholar]
- 27.McDonough AA. Mechanisms of proximal tubule sodium transport regulation that link extracellular fluid volume and blood pressure. Am J Physiol Regul Integr Comp Physiol 298: R851–R861, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol 292: C82–C97, 2007 [DOI] [PubMed] [Google Scholar]
- 29.Navar LG, Prieto MC, Satou R, Kobori H. Intrarenal angiotensin II and its contribution to the genesis of chronic hypertension. Curr Opin Pharmacol 11: 180–186, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O'Connor PM, Cowley AW., Jr Modulation of pressure-natriuresis by renal medullary reactive oxygen species and nitric oxide. Curr Hypertens Rep 12: 86–92, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ohkawa H, Ohishi N, Yagi K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem 95: 351–359, 1979 [DOI] [PubMed] [Google Scholar]
- 32.Padia SH, Kemp BA, Howell NL, Fournie-Zaluski MC, Roques BP, Carey RM. Conversion of renal angiotensin II to angiotensin III is critical for AT2 receptor-mediated natriuresis in rats. Hypertension 51: 460–465, 2008 [DOI] [PubMed] [Google Scholar]
- 33.Pons H, Ferrebuz A, Quiroz Y, Romero-Vasquez F, Parra G, Johnson RJ, Rodriguez-Iturbe B. Immune reactivity to heat shock protein 70 expressed in the kidney is cause of salt sensitive hypertension. Am J Physiol Renal Physiol 304: F289–F299, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quiroz Y, Pons H, Gordon KL, Rincón J, Chávez M, Parra G, Herrera-Acosta J, Gómez-Garre D, Largo R, Egido J, Johnson RJ, Rodríguez-Iturbe B. Mycophenolate mofetil prevents the salt-sensitive hypertension resulting from short-term nitric oxide synthesis inhibition. Am J Physiol Renal Physiol 281: F38–F47, 2001 [DOI] [PubMed] [Google Scholar]
- 35.Ribeiro MO, Antunes E, de Nucci G, Lovisolo SM, Zatz R. Chronic inhibition of nitric oxide synthesis. A new model of arterial hypertension. Hypertension 20: 298–303, 1992 [DOI] [PubMed] [Google Scholar]
- 36.Rodriguez-Iturbe B, Franco M, Johnson Rj Impaired pressure natriuresis is associated with interstitial inflammation in salt-sensitive hypertension. Curr Opin Nephrol Hypertens 22: 37–44, 2013 [DOI] [PubMed] [Google Scholar]
- 37.Rodríguez-Iturbe B, Franco M, Tapia E, Quiroz Y, Johnson RJ. Renal inflammation, autoimmunity and salt-sensitive hypertension. Clin Exp Pharmacol Physiol 39: 96–103, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rodriguez-Iturbe B, Johnson RJ. The role of renal microvascular disease and interstitial inflammation in salt-sensitive hypertension. Hypertens Res 33: 975–980, 2010 [DOI] [PubMed] [Google Scholar]
- 39.Rodríguez-Iturbe B, Pons H, Quiroz Y, Gordon K, Rincón J, Chávez M, Parra G, Herrera-Acosta J, Gomez-Garre D, Largo R, Egido J, Johnson RJ. Mycophenolate mofetil prevents salt-sensitive hypertension resulting from angiotensin II exposure. Kidney Int 59: 2222–2232, 2001 [DOI] [PubMed] [Google Scholar]
- 40.Rodriguez-Iturbe B, Quiroz Y, Nava M, Bonet L, Chavez M, Herrera-Acosta J, Johnson RJ, Pons HA. Reduction of renal immune cell infiltration results in blood pressure control in genetically hypertensive rats. Am J Physiol Renal Physiol 282: F191–F201, 2002 [DOI] [PubMed] [Google Scholar]
- 41.Rodriguez-Iturbe B, Romero F, Johnson RJ. Pathophysiologic mechanisms of salt-dependent hypertension. Am J Kidney Dis 50: 655–672, 2007 [DOI] [PubMed] [Google Scholar]
- 42.Rodriguez-Iturbe B, Vaziri ND, Herrera-Acosta J, Johnson RJ. Oxidative stress, renal infiltration of immune cells and salt-sensitive hypertension: all for one and one for all. Am J Physiol Renal Physiol 286: F606–F616, 2004 [DOI] [PubMed] [Google Scholar]
- 43.Rodríguez-Iturbe B, Zhan Chang -DE, Quiroz Y, Sindhu RK, Vaziri ND. Antioxidant-rich diet improves hypertension and reduces renal immune infiltration in spontaneously hypertensive rats. Hypertension 41: 341–346, 2003 [DOI] [PubMed] [Google Scholar]
- 44.Roman RJ, Cowley AW. Abnormal pressure-diuresis-natriuresis response in spontaneously hypertensive rats. Am J Physiol 17: F199–F205, 1985 [DOI] [PubMed] [Google Scholar]
- 45.Romero F, Rodrıyguez-Iturbe B, Parra G, Gonzalez L, Herrera-Acosta J, Tapia E. Mycophenolate mofetil prevents the progression of renal failure induced by 5/6 renal ablation in rats. Kidney Int 55: 945–955, 1999 [DOI] [PubMed] [Google Scholar]
- 46.Sanchez-Lozada LG, Tapia E, Johnson RJ, Rodríguez-Iturbe Herrera-Acosta J B. Glomerular hemodynamic changes associated with arteriolar lesions and tubulointerstitial inflammation. Kidney Int Suppl Oct: S9–S14, 2003 [DOI] [PubMed] [Google Scholar]
- 47.Schiffrin EL. T lymphocytes: a role in hypertension? Curr Opin Nephrol Hypertens 19: 181–186, 2010 [DOI] [PubMed] [Google Scholar]
- 48.Sharma K, Cook A, Smith M, Valancius C, Inscho EW. TGF-beta impairs renal autoregulation via generation of ROS. Am J Physiol Renal Physiol 288: F1069–F1077, 2005 [DOI] [PubMed] [Google Scholar]
- 49.Sommers SC, Relman AS, Smithwick RH. Histologic studies of kidney biopsy specimens from patients with hypertension. Am J Pathol 34: 685–715, 1958 [PMC free article] [PubMed] [Google Scholar]
- 50.Vanegas V, Ferrebuz A, Quiroz Y, Rodriguez-Iturbe B. Hypertension in Page (cellophane wrapped) kidney is due to interstitial nephritis. Kidney Int 68: 1161–1170, 2005 [DOI] [PubMed] [Google Scholar]
- 51.Vaziri ND, Rodriguez-Iturbe B. Mechanisms of disease: oxidative stress and inflammation in the pathogenesis of hypertension. Nat Clin Pract Nephrol 2: 582–593, 2006 [DOI] [PubMed] [Google Scholar]
- 52.Wang CT, Chin SY, Navar LG. Impairment of pressure-natriuresis and renal autoregulation in ANG II-infused hypertensive rats. Am J Physiol Renal Physiol 279: F319–F325, 2000 [DOI] [PubMed] [Google Scholar]
- 53.Weinberg M, Fineberg N. Sodium and volume sensitivity of blood pressure. Age and pressure change over time. Hypertension 18: 67–71, 1991 [DOI] [PubMed] [Google Scholar]