

Summary
Histone H3 phosphorylation is the hallmark of mitosis deposited by aurora kinase B. Benzo[e]pyridoindoles are a family of potent, broad, ATP-competitive aurora kinase inhibitors. However, benzo[e]pyridoindole C4 only inhibits histone H3 phosphorylation in prophase but not in metaphase. Under the C4 treatment, the cells enter into mitosis with dephosphorylated histone H3, assemble chromosomes normally and progress to metaphase, and then to anaphase. C4 also induces lagging chromosome in anaphase but we demonstrated that these chromosome compaction defects are not related to the absence of H3 phosphorylation in prophase. As a result of C4 action, mitosis lasts longer and the cell cycle is slowed down.
We reproduced the mitotic defects with reduced concentrations of potent pan aurora kinase as well as with a specific aurora B ATP-competitive inhibitor; we therefore propose that histone H3 phosphorylation and anaphase chromosome compaction involve the basal activity of aurora kinase B. Our data suggest that aurora kinase B is progressively activated at mitosis entry and at anaphase onset. The full activation of aurora kinase B by its partners, in prometaphase, induces a shift in the catalytic domain of aurora B that modifies its affinity for ATP. These waves of activation/deactivation of aurora B correspond to different conformations of the chromosomal complex revealed by FRAP.
The presence of lagging chromosomes may have deleterious consequences on the daughter cells and, unfortunately, the situation may be encountered in patients receiving treatment with aurora kinase inhibitors.
Key words: Cancer, Chromosome compaction, Aurora kinase, Kinase inhibitors, Survivin, Chromosomal passenger complex, Histone H3 phosphorylation
Introduction
Histone phosphorylation plays a direct role in major cell events like mitosis, cell death, repair, replication and recombination (Baek, 2011). The same residue phosphorylation may have dual role. For example, histone H3 phosphorylations on Ser10 and Ser 28 were reported both in interphase where chromatin needs to be decondensed for transcriptional activation of genes, and in mitosis where chromatin condensation is necessary (Hans and Dimitrov, 2001). Their definitive roles in these processes have not yet been elucidated.
In late G2 phase, the phosphorylation of histone H3 only occurs on pericentromeric chromatin. Then it spreads along the chromosomes and is complete in prophase. At the end of mitosis, histone H3 is dephosphorylated. Aurora kinase B is a well-known primary mitotic kinase responsible for histone H3 phosphorylation on Serine 10 and 28 (Baek, 2011).
Aurora kinases are a family of serine/threonine protein kinases that play a key role in mitosis progression (Carmena et al., 2009; Vader and Lens, 2008). In human, three aurora kinases have been identified: A, B and C. Aurora A is first associated with centrosomes and then with spindle microtubules whereas aurora B is a chromosomal passenger protein travelling from centromeres to microtubules. Aurora A is required for centrosome duplication, entry into mitosis, formation of bipolar spindle and mitotic checkpoint (Hannak et al., 2001; Sardon et al., 2008; Seki et al., 2008). Aurora B is essential for chromosome condensation, kinetochore functions, spindle checkpoint activation and cytokinesis completion (Carmena et al., 2009; Lens and Medema, 2003; Mendoza et al., 2009; Saurin et al., 2011). Aurora C is mostly involved in spermatogenesis (Vader and Lens, 2008).
Both aurora A and B kinases are activated through interactions with their substrates or partners and priming by other kinases. Activation of aurora B occurs through a two-step mechanism: first, aurora B binds to the C-terminal IN-box sequence of INCENP and is then autophosphorylated at threonine 232 (Thr232) within its activation loop (Honda et al., 2003; Yasui et al., 2004). In the second step, the kinase phosphorylates INCENP and is subsequently fully activated. Aurora kinase B activity is enhanced through its phosphorylation by Chk1 and priming of its substrates by Plk1 and Mps1 (Jelluma et al., 2008; Petsalaki et al., 2011; Rosasco-Nitcher et al., 2008). Despite this description of the aurora kinase B activation, the physicochemical properties of the enzyme are poorly documented since aurora B itself has not been crystallized in the absence of inhibitors (Girdler et al., 2008; Sessa et al., 2005). Most of the available data derive from the study of the ATP-binding pocket of aurora A (Dodson et al., 2010; Martin et al., 2012). The catalytic domains of human aurora-A and aurora-B are 76% identical at the primary sequence level, and just differ by three amino acids in their ATP-binding sites. With the aim to derive specific kinase inhibitors, efforts are devoted to the description of the different conformations adopted by the ATP-binding sites.
Aurora kinases A and B, exclusively expressed in mitosis, are over-represented in many cancers, including primary colon and breast cancers (Sen et al., 1997; Vader and Lens, 2008). In the light of these observations, aurora kinases have emerged as potential promising targets in the development of anti-cancer therapy and small molecule inhibitors of aurora kinase have been identified (Garuti et al., 2009; Girdler et al., 2006; Harrington et al., 2004; Hoang et al., 2009; Soncini et al., 2006). Several of these ATP-competitive inhibitors are currently in clinical development. Preliminary data from clinical trials generally indicate disease stabilization, the best response being achieved in solid tumours or in refractory chronic myeloid leukemia (Cheetham et al., 2007; Kollareddy et al., 2012; Vu et al., 2010).
Besides their possible efficiency in therapy, aurora kinase inhibitors are tools for describing these enzymes. In a previous study, we found that benzo[e]pyridoindoles are potent ATP-competitive inhibitors of aurora kinases (Hoang et al., 2009). In vitro, they inhibited aurora kinases at nanomolar range; in cells, they prevented mitosis progression. In this study, playing with these aurora kinase inhibitors, we showed that the phosphorylation of histone H3 (Ser10 and 28) is not a prerequisite for chromosome assembly and is accounted for by basal aurora kinase B activity. Moreover, at anaphase onset, the compaction of DNA was also perturbed by low concentrations of ATP competitive inhibitors. These data revealed modifications of the ATP-binding site of aurora kinase B during mitosis and their consequences are discussed.
Results
By high throughput screening, we found that benzo[e]pyridoindoles are potent aurora kinase inhibitors (Hoang et al., 2009). In vitro, they prevented histone H3 phosphorylation on Ser10. C1 was the first hit and C4 was the fourth inhibitor of this series. The efficiency of molecules C1 and C4, at two different concentrations, is recalled in Fig. 1A. C1, the more potent of the two molecules, was already shown to be an ATP competitive inhibitor of aurora kinase B that prevents mitosis progression (Hoang et al., 2009). By immunofluorescence, we showed that histone H3 phosphorylation was fully inhibited in all mitotic cells by C1 (1 µM) but only in prophase by C4 (3 µM) (Fig. 1A).
Fig. 1. Delay of histone H3 phosphorylation at mitotic entry upon C4 treatment.

(A) C1 and C4 are two benzo[e]pyridole inhibitors of the catalytic domain of aurora kinase A (Hoang et al., 2009). Molecules and their efficiency in vitro towards aurora A catalytic domain are recalled (Hoang et al., 2009). These data derived from the high throughput screening performed under non-saturating conditions. In vitro, IC50 of C1 towards aurora A, B are 61 nM and 31 nM respectively (Hoang et al., 2009). Immunofluorescences of histone H3 (Ser10) phosphorylation on HeLa cells treated by either C1 (1 µM) or C4 (3 µM) are represented; Histone H3 (Ser10) phosphorylation is shown in green whereas DNA is in red. Both prophase and pro-metaphase are imaged and compared to control cells. (B) Immunofluorescence of Histone H3 (Ser10 and Ser 28) phosphorylations on MPM2 positive HeLa cells. (C) Analysis of the percentage of positive histone H3-phospho Ser 10 at mitotic entry. Two independent experiments were conducted and 100 cells in G2/prophase scored in each. (D) Western blots were realized on cells synchronized by a MG-132 block and then released for 3 hours. Histone H3 (Ser10 and Ser 28) phosphorylations were analyzed. The same membrane was also revealed using an antibody against aurora kinase B for estimation of the amount of mitotic cells.
MPM2 recognizing phospho-mitotic epitopes allowed the detection of early mitotic cells. The normal MPM2 signal in the presence of C4 demonstrated that C4 did not induce an overall inhibition of phosphorylation in prophase, but only inhibited histone H3 phosphorylation on both Ser 10 and 28 (Fig. 1B). At the concentration of 3 µM, C4 completely abolished histone H3 phosphorylation since 94±4% of the cells in G2/prophase were scored negative with this marker (Fig. 1C). This inhibition was further confirmed by immunoblotting with the reduction of Ser 10 and Ser 28 phospho-histone H3 signals in C4 treated cells (Fig. 1D).
By immunoblotting, we followed the appearance of histone H3 phosphorylation on Ser 10 and Ser 28 in synchronized cells (Fig. 2A). These signals were reduced in C4 treated cells, at mitotic entry, but they were quite similar to control cells, 8 hours later (supplementary material Table S1), showing that C4 only inhibited the histone H3 phosphorylation at G2/prophase. Meanwhile, we did not observe any differences in activity of aurora kinases A and B. They were respectively evaluated through the autophosphorylation of aurora A and the phosphorylation of CenP-A on Ser 7, these two phosphorylations occurring upon priming by the partners (Fig. 2A; supplementary material Fig. S1). By immunofluorescence, we noticed that, in prometaphase, under C4 treatment, although chromosomes were well-condensed, histone H3 was only partly phosphorylated (Fig. 2B). The centromeres, detected through the survivin signal, were found positive for phospho-H3 whereas the chromosome arms were still mostly negative. This pattern is consistent with the enrichment of aurora kinase B on centromeres (Fig. 2C). In the C4 treated cells, aurora kinase B was also found on the whole chromosomes in prometaphase (Fig. 2C), co-localizing with survivin. In the presence of C4, the level of aurora kinase B was found similar to the control (supplementary material Table S1).
Fig. 2. Histone H3 phosphorylation appears at prometaphase upon C4 treatment.
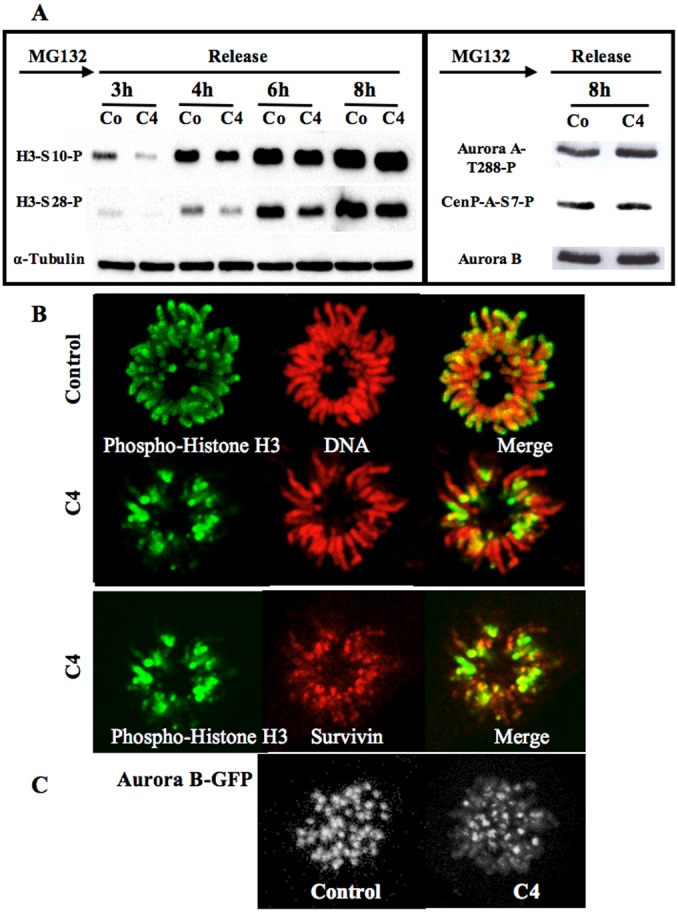
(A) Western blots were realized on cells synchronized at mitotic entry in the presence of MG-132 and then released for varying times. Histone H3 (Ser10 and Ser 28) phosphorylations were analyzed in control (Co) and C4 treated cells (C4). The same membrane was also revealed using an antibody against α-tubulin for estimation of the amount of cells. The signals were quantified and the results are in supplementary material Table S1. The phosphorylations of Cenp-A (Ser 7) and aurora kinase A (Thr 288) are also shown in control (Co) and C4 treated cells (C4). Aurora kinase B detection is used for estimation of the amount of mitotic cells. Its level was quantified using α-tubulin as internal standard (supplementary material Table S1; Fig. S2). (B) Detection of histone H3 (Ser10) phosphorylation on control and C4 treated cells. Phospho-histone H3 is in green and DNA in red. On the same C4-treated cells, the histone H3 (Ser10) phosphorylation signal (in green) colocalized with survivin (represented in red). (C) Detection of aurora kinase B on prometaphasic cells. Z stacks were imaged and 3D-projections are shown. The settings were kept identical for both conditions.
Time-lapse experiments on cells expressing fluorescent histone H2A suggested a delay in mitosis progression. The anaphase chromosome compaction was perturbed and lagging chromosomes were observed (Fig. 3). Around 60% of cells in anaphase were scored abnormal under the treatment and only few in the control (Fig. 6A). The screening was stringent; abnormal meant at least two chromosomes from one pole connected to one opposite chromosome. This segregation defects are thus a major consequence of the C4 treatment. Knowing that both aurora kinases A and B are key enzymes in anaphase (Mora-Bermúdez et al., 2007; Hégarat et al., 2011), we looked to their localization. In late anaphase, we did not observe any modification of aurora kinase A localization whereas aurora kinase B was partly found in the midzone and on the chromatin (Fig. 4A). At anaphase onset, passenger proteins like aurora kinase B and survivin are instantly transferred on microtubules when chromosomes start to segregate. Therefore we followed survivin and aurora kinase B localizations by time lapse (Fig. 4B,C) during mitosis.
Fig. 3. Time lapse on mitotic cells.
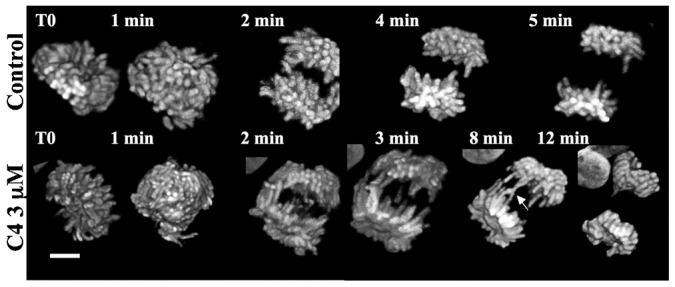
Mitotic Hek stably expressing histone H2A-GFP were continuously imaged. Z stacks were acquired and 3D-projections are shown. The settings were kept identical for all conditions. Representative control and C4 treated cells are shown and elapse times indicated on each image. Scale bar: 5 µm.
Fig. 6. Quantification of the mitotic defect induced by C4.
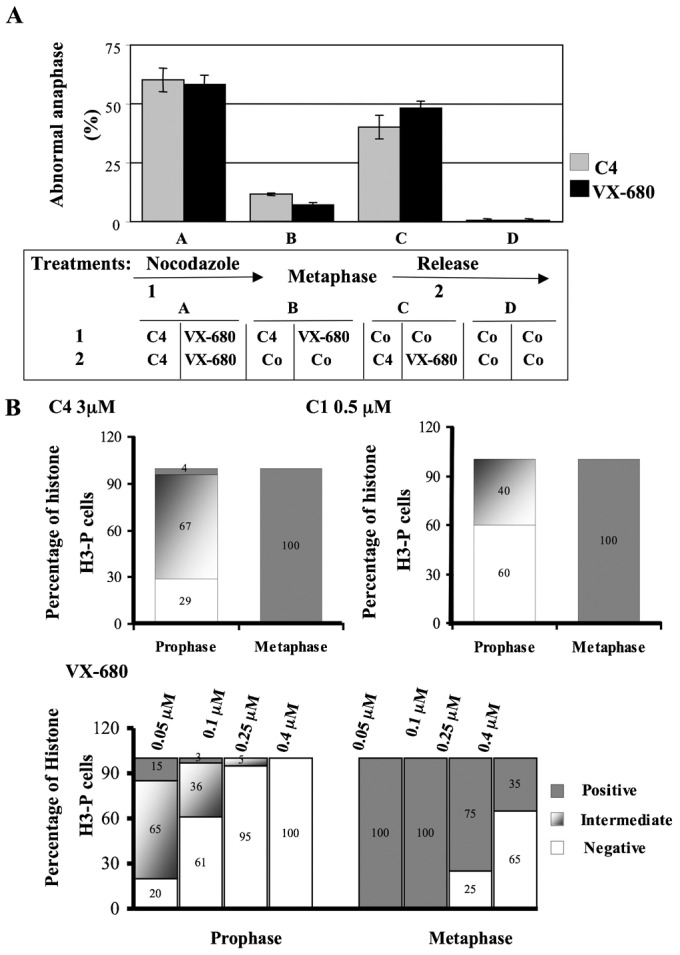
(A) Abnormal anaphases presenting connecting chromosomes were scored by immunofluorescence. DNA was labelled by Hoechst. Cells were synchronized in prometaphase by nocodazole (0.33 µM) for 16 hours and anaphase was induced upon release (post-anaphase onset, 3 hours). Compounds C4 (3 µM) and VX-680 (0.1 µM) were present in different steps: either in (pro)metaphase (1) and (or) post-metaphase (2). In assays A, compounds were added in both steps (1) and (2). In assays B, compounds were only present in prometaphase and were then withdrawn in (2); conversely in assays C, compounds were added after metaphase (step 2). Co means control experiments without compound. Control assays in the absence of compounds are in D. A view of the course of the experiment is shown. Around 200 mitotic cells were analysed in each condition and two independent experiments were conducted. (B) The percentage of phospho-histone H3 (Ser 10) was defined by immunofluorescence, in both prophasic and metaphasic HeLa cells treated either by C4 (3 µM), C1 (0.5 µM) or VX-680 (the concentration of VX-680 varied from 0.05 µM to 0.4 µM). Cells were scored as positive, negative or intermediate. Intermediate signal means a significant reduction of the phosphorylation compared to the control. Around 100 cells were scored in each assay and two independent experiments were conducted.
Fig. 4. Consequences of chromosome compaction defects.
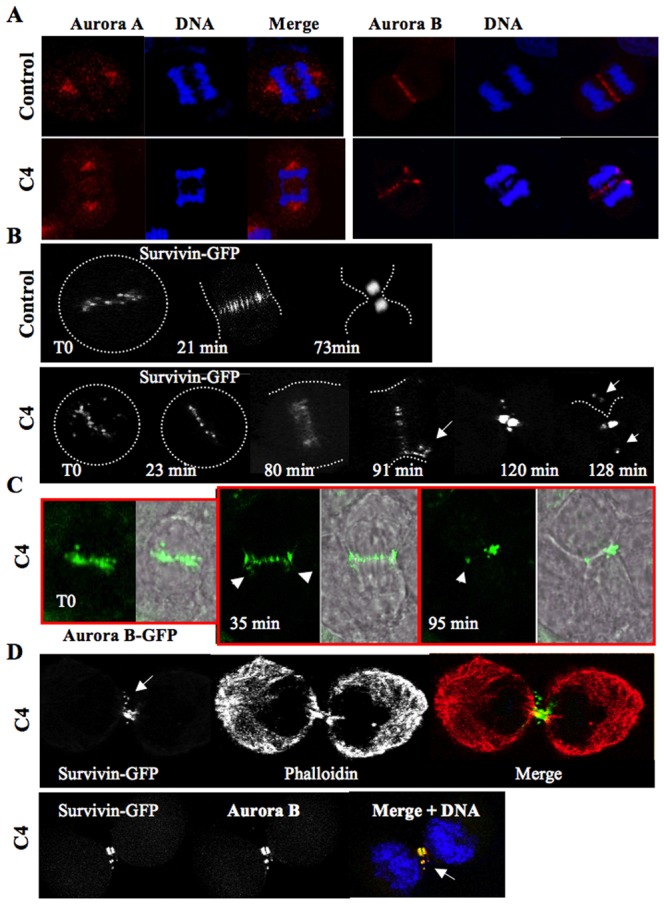
(A) Localization of aurora kinases A and B by immunofluorescence in control and C4 treated cells. A late anaphase is imaged. (B) Time lapse on mitotic HeLa cells stably expressing survivin-GFP. Time-lapse microscopy of mitotic HeLa cells expressing the survivin-GFP fusion was performed in the absence (Control) or in the presence of C4 (at 3 µM). The compound was added to the cell culture and then mitotic cells were continuously imaged. Representative photos, made at the times indicated, are presented. For a better understatement, the shape of the cells is recalled by a white dotted line on each fluorescent image. Two independent experiments were conducted and at least 15 cells were followed in each. (C) Time lapse on mitotic HeLa cells stably expressing aurora B-GFP. The experiment described in B was reproduced with HeLa cells stably expressing aurora B-GFP. A representative cell is shown and the fluorescent signal is merged with the wide field image. Elapsed times are indicated. (D) Immunofluorescence was realized on HeLa cells expressing survivin-GFP. Cells in cytokinesis were imaged. The cell was labelled with phalloidin-TRITC in order to visualize its shape meanwhile aurora kinase B was revealed with antibodies.
In control cells, survivin-GFP was transferred to microtubules and fully concentrated in the midbody (Fig. 4B). Conversely in the C4 treated cells, survivin-GFP was present mostly on microtubules but fluorescence was detected outside the midzone (Fig. 4B, indicated by arrows). Finally the survivin signal was also detected outside the midbody in cytokinesis. The localization of aurora B-GFP was found very similar to survivin-GFP and again the kinase was partly mis-localized in late anaphase and telophase (Fig. 4C). The partial mislocalization of passenger proteins was confirmed by immunofluorescence (Fig. 4D). Survivin and aurora kinase were mostly concentrated in the midbody but the passenger complex was also detected on the plasma membrane. The shape of the cell was highlighted by phaloidin labelling (Fig. 4D). Other major actors of cytokinesis like MKPL1 were also partly delocalized in the vicinity of the passengers (data not shown). We found that tubulin was disorganized in the midzone (supplementary material Fig. S2), it may be a consequence of the presence of lagging chromosomes in this area.
The next question was whether cells could complete mitosis and progress in the cell cycle upon C4 treatment. We determined by FACS the repartition of the cells, at different times of treatment (Fig. 5; supplementary material Table S2). The C4 treatment did not significantly modify the timing of mitotic entry (Fig. 5; 4 hours) but delayed mitosis exit (Fig. 5; 8 and 10 hours). In the presence of C4, we did not observe any increase of cell ploidy and the cell viability was also not affected (Fig. 5; supplementary material Table S1). Thus, benzo[e]pyridoindole C4 targeted mostly mitotic events.
Fig. 5. Cell cycle progression upon C4 treatment.
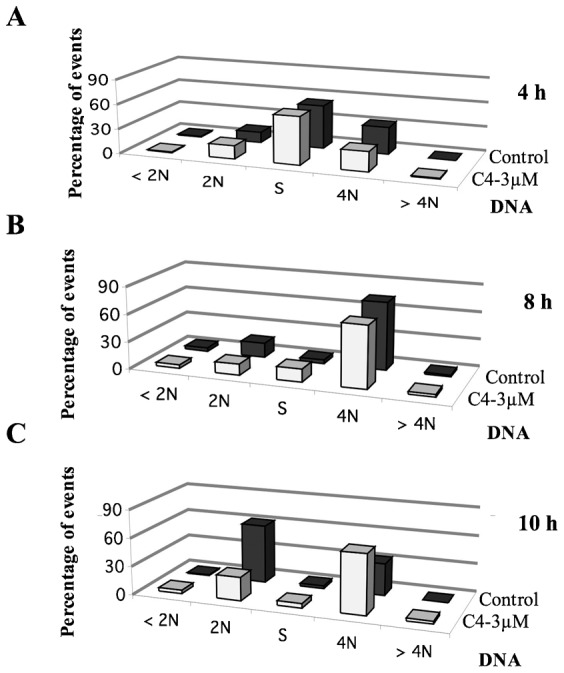
FACS analyses show the repartition of synchronized HeLa cells treated by either C4, at the concentration of 3 µM, for 4 (A), 8 (B) and 10 (C) hours in comparison to non-treated cells. DNA was stained with propidium iodine and the samples were analyzed with the BD Accuri C6 flow cytometer (BD Biosciences, US). The percentage of cells in the different phases is indicated in supplementary material Table S1.
We wonder whether chromosome compaction defects could be a consequence of the absence of histone H3 phosphorylation, at mitosis entry. To address this question, we synchronized cells in prometaphase and we treated them by C4 at two separated stages: before and (or) after prometaphase (Fig. 6). Cells treated by C4 upon prometaphase release had a normal phospho-histone H3 level but exhibited a high percentage of anaphase chromosome compaction defects (around 40%). Conversely, cells that entered into mitosis in the presence of C4, and were released from prometaphase in its absence, recovered and showed only 11.5% of lagging chromosomes in anaphase (Fig. 6A). Consequently the absence of histone H3 phosphorylation in prophase and the chromosome compaction defects in anaphase were two independent actions of the molecule C4. As aurora kinase B is the main kinase phosphorylating histone H3 on Ser 10 and 28 and is also involved in chromosome compaction (Mora-Bermúdez et al., 2007), we hypothesised that C4 targeted a low ATP dependent activity of aurora kinase B. If so, the observed defects could be reproduced with other aurora kinase inhibitors at low concentrations.
In cells treated by C4 (3 µM), only 4% of the cells in prophase were positive for histone H3 phosphorylation, 67% had few positive dots, and 29% were negative whereas all the cells in metaphase were scored positive (Fig. 6B). When cells were treated by a low concentration of benzo[e]pyridoindole C1 (0.5 µM), a known aurora kinase inhibitor (Hoang et al., 2009), we observed a decrease of histone H3 phosphorylation in prophase (60% were negative and 40% scored intermediate) whereas histone H3 was fully phosphorylated in metaphase (Fig. 6B). Taking into account that C1 and C4 belong to the same family of molecules and in order to avoid the possible involvement of a common off-target, we reproduced the experiment with varying concentrations of VX-680, a reference ATP-competitive aurora kinase inhibitor (Harrington et al., 2004). At 50 nM and 100 nM, VX-680 prevented histone H3 phosphorylation in prophase but not in metaphase (Fig. 6B). We searched for chromosome compaction defects, at anaphase onset, upon VX-680 (0.1 µM) treatment (Fig. 6A) and found that 58% of the anaphasic cells exhibited lagging chromosomes. Again these defects were independent of the inhibition of H3 phosphorylation at mitotic entry and appeared when prometaphasic cells were released in the presence of VX-680 (around 48% of abnormal anaphases, Fig. 6A).
As benzo[e]pyridoindoles C1 and C4 as well as VX-680 are pan-aurora kinase inhibitors, we checked whether AZD-1152, a selective inhibitor for aurora kinase B, could fully reproduce the observed effects. At the concentration of 25 nM, AZD-1152 induced a delay in mitosis exit (Fig. 7A) but cells could then start a new cycle. When cells entered into mitosis, histone H3 (S10) was not phosphorylated (Fig. 7B), the phosphorylation signal was then detected near centromeres (Fig. 7C) and it was found maximal in metaphase (Fig. 7B). We have noted the presence of lagging chromosomes and the partial mis-localization of aurora kinase in late anaphase (Fig. 7D). When AZD-1152 was added upon metaphasic release, around 30% of the anaphase exhibited lagging chromosomes. Therefore the mitotic defects induced by C4 were mostly accounted for by aurora kinase B. However, the possible participation of aurora kinase A in the anaphase compaction defects upon C4 treatment cannot be dismissed.
Fig. 7. Effect of low concentrations of AZD1152.
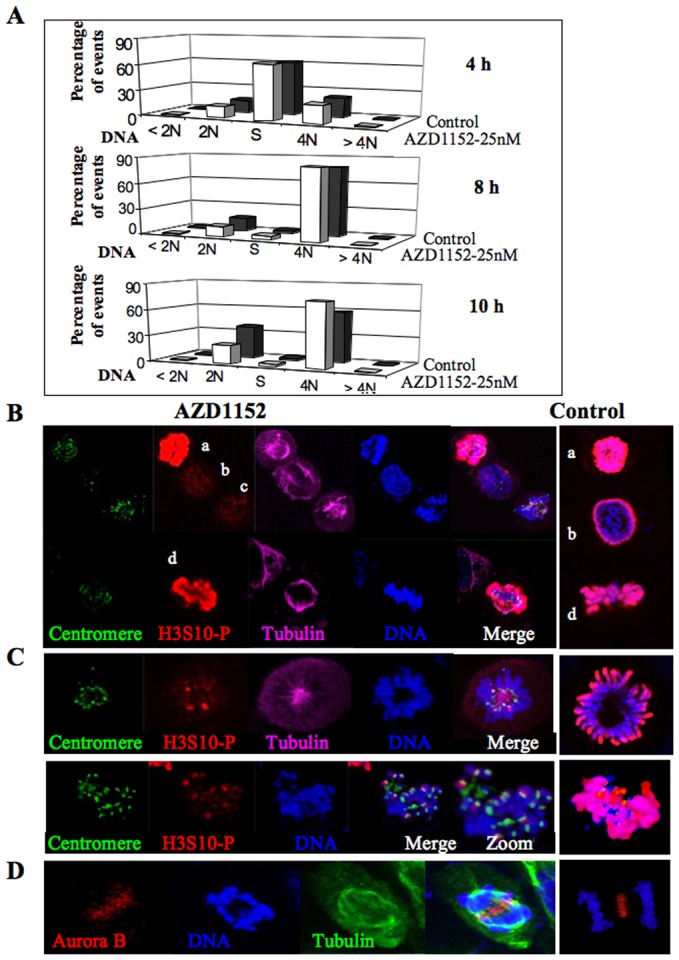
(A) Cell cycle progression upon AZD1152 treatment FACS analyses show the repartition of synchronized HeLa cells treated by either AZD1152, at the concentration of 25 nM, for 4, 8 and 10 hours in comparison to non-treated cells. DNA was stained with propidium iodide and the samples were analyzed with the BD Accuri C6 flow cytometer (BD Biosciences, US). (B) Immunofluorescence of HeLa cells treated by AZD1152, for 15 hours, at the concentration of 25 nM. Centromeres are revealed in green, histone H3(S10) phosphorylation (H3S10-P) is in red, α-tubulin in far red and DNA in blue. Control cells are shown on the right, H3(S10) phosphorylation (H3S10-P) is in red and DNA in blue. Cells a and d are in metaphase, b in prophase and c in prometaphase. (C) Same experiments as in B. Centromeres are detected by a specific antibody and represented in green. Two different prometaphasic cells are imaged. For an easy visualisation of the colocalization, an enlarged merge is shown in one case. Control cells are shown on the right, H3(S10) phosphorylation (H3S10-P) is in red and DNA in blue. (D) Cells were synchronized by nocodazole and imaged 3 hours after release. Most of the cells in anaphase (around 30%) present compaction defects. Aurora kinase B is revealed in red, DNA in blue, α-tubulin in green. A control cell is shown on the right, aurora kinase B is in red, DNA in blue.
Our data suggest that aurora kinase B is submitted to activation waves. Previously we showed, by FRAP experiments, that the chromosomal passenger complex could adopt different conformations (Delacour-Larose et al., 2007; Delacour-Larose et al., 2004). Whereas aurora kinase B is immobile all along mitosis, survivin is only mobile in (pro)metaphase. As shown in Fig. 8, INCENP was found mobile in G2/prophase when the partners were immobile. Then, in prometaphase and metaphase, survivin was mobile conversely to others, and finally the complex was stuck on microtubules. Therefore the binary data analysis, which defined mobile and immobile proteins, reveals that the basal activity of aurora kinase B correlates to conformational changes within the chromosome passenger complex. The low-ATP activity is first observed when the CPC moves from pericentromeric chromatin to centromeres and similarly, shortly after its transfer to microtubules (Fig. 8).
Fig. 8. Compared mobilities of passenger proteins in G2/M-prophase, metaphase and anaphase.

FRAP experiments were performed on HeLa cells stably expressing GFP-INCENP or survivin-GFP or aurora kinase B-GFP. The bleached zone is indicated by the dotted line. The recovery of fluorescence was registered at the times indicated and expressed as relative intensities. The kinetics of recovery are shown on the right side of the cell images. Time is in seconds. Survivin and aurora recovering are already published (Delacour-Larose et al., 2004; Delacour-Larose et al., 2007) and recalled in the figure for comparison. The localization of passenger proteins during mitosis progression is indicated as well as the timing of efficiency of C4 (from G2/M to prometaphase and at anaphase onset). The proposed variations of aurora kinase B affinity for ATP are visualized with arrows.
Discussion
Successful cell division requires the temporal and spatial integration of chromosomal and cytoskeletal events. One key integrator is the chromosomal passenger complex (CPC) composed of INCENP, aurora kinase B, borealin, and survivin (reviewed by van der Waal et al., 2012). Spatially regulated activity of the CPC is essential for the correction of kinetochore microtubule attachment errors, bipolar spindle stability, and completion of cytokinesis (van der Waal et al., 2012). Aurora kinase B is the unique enzymatic member within the complex; it phosphorylates numerous substrates, during mitosis, such as histones H3 and CenP-A, as well as its partners (INCENP and survivin). Aurora B is known to reach different levels of activations through interactions with CPC members and by phosphorylations (Honda et al., 2003; Jelluma et al., 2008; Petsalaki et al., 2011; Rosasco-Nitcher et al., 2008; Yasui et al., 2004). How these different activation steps are related to the mitotic activities is poorly documented. Recently, by incorporating mutant INCENP, Xu et al. revealed that the full activation is at least required for transferring the CPC to the midzone at anaphase onset (Xu et al., 2009).
When studying the benzo[e]pyridoindole aurora kinase inhibitors, we noticed that some compounds like C4 only inhibited histone H3 phosphorylation in prophase. Then phospho-histone H3 reached its normal level in prometaphase. We reproduced this transitory inhibition by decreasing the concentration of potent aurora kinase inhibitors. These inhibitors being ATP competitive inhibitors, it means that, in prometaphase, a shift occurring in the catalytic domain of aurora B modifies its affinity for ATP. We noticed that the compound C4 (8-H) differed from the counterpart C1 (8-Et) by the absence of an alkyl group on the summit 8. The largest molecule was found to better fit with the ATP binding site. The activation loop of aurora kinase B contains a DFG (Asp-Phe-Gly) motif like most AGC kinases. But it seems that in aurora kinases, this ATP-binding motif could adopt more complex conformations; a DFG-up conformation was described for aurora kinase A in addition to the active DFG-in and the inactive DFG-out states (Dodson et al., 2010; Martin et al., 2012). The existence of this unusual conformation in aurora B and its possible involvement in the observed shift of affinity for ATP remain to be investigated.
Conversely to what was observed when BubR1 or Melk are inhibited, inhibition of aurora kinase did not accelerate the timing of mitotic entry (Park et al., 2009). We observed that cells still assembled chromosomes in the absence of phospho-histone H3. The role of this epigenetic mark is still in debate. In vitro assays demonstrated that the phosphorylation is not required for chromosome condensation in Xenopus egg extracts (de la Barre et al., 2001). In vivo experiments, in Drosophila, revealed a very weak correlation between the level of histone H3 phosphorylation and the degree of chromosome compaction (Adams et al., 2001). Our group has postulated that the mark was a ready-to-go label that implied no relationship between chromosome condensation and histone H3 phosphorylation (Hans and Dimitrov, 2001; Prigent and Dimitrov, 2003). Once the cell has arrived in metaphase its chromosomes should be phosphorylated, and this is independent of their state of condensation (Hans and Dimitrov, 2001). In this study we observed the spreading of the phosphorylation on well-organized chromosomes. The absence of mitotic consequences of this delayed phosphorylation favours the label hypothesis for the role of H3 phosphorylation.
Benzo[e]pyridoindole C4 as well as low concentrations of C1 or VX-680 or AZD-1152 revealed the basal activity of aurora kinase B. The two independent consequences of the inhibition of basal activity are the absence of phosphorylation in prophase and chromosome compaction defects in anaphase. Aurora kinase B full activation was not required for H3 phosphorylation, this fits with the observations of Xu and collaborators (Xu et al., 2009). By preventing INCENP expression, they mildly decreased histone H3 phosphorylation. Xu et al. reported a gradient of kinase activity when mitosis progressed whereas Tan and Kapoor described the existence of gradients of phosphorylation of CPC substrates (Tan and Kapoor, 2011; Xu et al., 2009; Xu et al., 2010). The use of competitive inhibitors suggests at least two activation waves of aurora kinase B, the basal activity seems sufficient in prophase and post anaphase onset. Similarly, low level of cyclin B1–cdk1 activity was found to be required during prophase compared to later mitotic steps (Gavet and Pines, 2010). As a consequence aurora kinase B exhibited a full activity in prometaphase and metaphase as required for transferring the CPC from centromere to microtubule (Meldi and Brickner, 2011; Xu et al., 2009). The observed chromosome compaction defects, in anaphase, in the presence of low-ATP competitors, revealed a shift to basal activity when the kinase lies on microtubules. These different steps correspond to different CPC conformations as revealed by FRAP experiments.
The induction of lagging chromosomes at anaphase onset by low concentration of kinase inhibitors is somewhat worrying. The consequences might be the loss of midbody proteins, a non-identical repartition of proteins, the induction of aneuploidy or DNA mislocalization in the daughter cells. These drawbacks are important since the inaccurate localization of DNA in the nucleus impairs gene expressions (Meldi and Brickner, 2011). This situation may be encountered, at distance from the treatment, in patients treated by aurora kinase inhibitors if long lived compounds are used.
In conclusion, the perturbation of protein kinases with small inhibitors is a powerful approach to dissect kinase function in complex biological systems. In the presence of low-ATP competitors of aurora kinase, cells entered into mitosis without the main mitotic epigenetic mark, the phosphorylation of histone H3, confirming its non-essential role in chromosome assembly. Aurora kinase B activity was found at basal level in prophase, fully activated on centromere and reduced again on microtubules. These waves of activation/deactivation correspond to different conformations of the CPC as described by FRAP.
Materials and Methods
Materials
VX-680 was purchased from Kava Technology, Inc. MG132, thymidine, nocodazole, phalloidin-TRITC, propidium iodide and AZD-1152 were from Sigma–Aldrich. RNase A was supplied by Euromedex. Benzo[e]pyridoindole synthesis and characterization were already described (Hoang et al., 2009; Nguyen et al., 1992).
Cell lines
HeLa cells and Hek-293 were cultured in growth medium (DMEM 1 g/l of glucose with 10% heat-inactivated foetal bovine serum (Gibco–Invitrogen), 2 mM L-glutamine, 100 UI/ml penicillin–streptomycin) at 37°C in a humidified atmosphere with 5% CO2.
HeLa cells stably expressing survivin-GFP or aurora kinase B-GFP or INCENP-GFP and Hek-293 expressing histone H2A-GFP were already described (Delacour-Larose et al., 2007; Delacour-Larose et al., 2004; Hoang et al., 2009).
HeLa cells were synchronized at S phase by serum deprivation for 2 days followed by a thymidine block (3 mM) for 20 hours. After PBS washing, cells were then either allowed to grow in normal conditions or arrested at mitosis entry by MG132 treatment (3 hours, 10 µM). HeLa cells were synchronized in prometaphase by nocodazole (0.33 µM).
Immunofluorescence
Hela cells grown on glass coverslips were treated by C4 (3 µM), AZD1152 (25 nM) or DMSO (control) in different trials and then fixed by formaldehyde 4% for 10 minutes at 37°C. Immunofluorescence was performed as described previously (Delacour-Larose et al., 2004). Coverslips were incubated with the antibodies directed against the following antigens: phospho-histone H3-Serine10 (Upstate, 1:2000); phospho-histone H3-Serine 28 (Upstate, 1:2000); aurora kinase B (Epitomics, 1:2000); phospho-MPM2 (Milipore, 1:1000); human anti-centromere HCT-0100 (Immunovision, 1:2000); aurora kinase A (Abcam, 1:1000); α-tubulin (Sigma, 1:1500). Actin was stained by phalloidin-rhodamin (1 µg/ml). DNA was visualized with Hoechst 33342 (Sigma, 0.5 µg/ml). Images were collected with a ZEISS 710 Laser Scanning Confocal microscope with a 63× immersion oil objective. Slices of 0.5 µm are shown.
Western Blot
Synchronized cells were treated by C4 (3 µM) or DMSO (control) for 3, 4, 6 and 8 hours, then harvested and lyzed in 9M urea, finally supplemented with Laemmli sample buffer as described previously. Western blots were performed using the following antibodies: rabbit anti-phospho-histone H3 (Ser10) (Upstate, 1:2000); rabbit anti-phospho-histone H3 (Ser28) (Upstate, 1:1000); rabbit anti-aurora B (Epitomics, 1:5000); rabbit anti-phospho-aurora A (Thr288) (Cell Signaling, 1:1000); rabbit anti-phospho-CENP-A (Ser 7) (Cell Signaling, 1:1000). Bands were visualized by horseradish peroxidase labelled antibodies and ECL technique (Amersham Bioscience). Images were observed by ChemiDoc MP system (Biorad).
Cell cycle analysis
HeLa cells were synchronized at S phase. After release, they were treated by C4 (3 µM), AZD1152 (25 nM) or DMSO (control) for 4, 8, 10, 12, 25 and 30 hours. For determination of cell cycle profiles, cells were fixed by ice-cold 70% ethanol, for 1 hour, and then, incubated with propidium iodide solution (50 µg/ml) in the presence of 0.2 mg/ml RNase A for 15 minutes, at 37°C. DNA content was measured using the BD Accuri C6 flow cytometer (BD Biosciences, US) and CFlow Plus software.
Time lapse
Ex vivo experiments were conducted on cells grown on Lab-Tek chambered coverglass (Nalge Nunc International) and maintained under standard culture conditions (37°C, 5% CO2). Images were acquired on a Zeiss dynascope confocal microscope using a PlanApochromat 40× water immersion objective. Images were analyzed with the Zen software provided by Zeiss.
FRAP
Cells were grown on Lab-Tek chambered coverglass (Nalge Nunc International). For imaging, cells were maintained at 37°C on a temperature and CO2 controlled stage. Photobleaching was performed, as described (Delacour-Larose et al., 2004; Delacour-Larose et al., 2007), on a ZEISS LSM510 system using a PlanApochromat 40× water immersion objective. GFP was excited with a 488-nm Argon2 laser (power varying from 0.1 to 1%). For FRAP (Fluorescence Recovery After Photobleaching) experiments, outlined regions were bleached by 10 iterations of a full power laser and recovery was monitored every 20 seconds for ∼4–5 minutes. Fluorescence intensities were quantified with homemade software and bleaching due to the acquisition was corrected. It was less than 10% in all experiments. Arbitrarily, the intensity of the region prior to bleaching was set at 1 while that of the background was set at 0. Relative intensities are represented as a function of time. Data were recovered in two independent experiments and 8 to 10 cells were followed in each mitotic stage. In mitotic cells, movement of fluorescent objects could be wrongly interpreted as a recovery of fluorescence. Therefore, as already described (Delacour-Larose et al., 2004), we performed 3D-reconstitution all along the experiment.
Supplementary Material
Acknowledgments
L.-T.-T.L. and H.-L.V. were funded by a Vietnam/French program. This work was supported by INSERM, UJF, CNRS, Institut Curie. The authors greatly thank Dr Stéfan Dimitrov for his encouragement during this work. Microscopy experiments were conducted on the IBISA platform of the CRI INSERM/UJF U823.
Footnotes
Competing interests: The authors have no competing interests to declare.
References
- Adams R. R., Maiato H., Earnshaw W. C., Carmena M. (2001). Essential roles of Drosophila inner centromere protein (INCENP) and Aurora B in histone H3 phosphorylation, metaphase chromosome alignment, kinetochore disjunction, and chromosome segregation. J. Cell Biol. 153, 865–880 10.1083/jcb.153.4.865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek S. H. (2011). When signaling kinases meet histones and histone modifiers in the nucleus. Mol. Cell 42, 274–284 10.1016/j.molcel.2011.03.022 [DOI] [PubMed] [Google Scholar]
- Carmena M., Ruchaud S., Earnshaw W. C. (2009). Making the Auroras glow: regulation of Aurora A and B kinase function by interacting proteins. Curr. Opin. Cell Biol. 21, 796–805 10.1016/j.ceb.2009.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheetham G. M. T., Charlton P. A., Golec J. M. C., Pollard J. R. (2007). Structural basis for potent inhibition of the Aurora kinases and a T315I multi-drug resistant mutant form of Abl kinase by VX-680. Cancer Lett. 251, 323–329 10.1016/j.canlet.2006.12.004 [DOI] [PubMed] [Google Scholar]
- de la Barre A.–E., Angelov D., Molla A., Dimitrov S. (2001). The N-terminus of histone H2B, but not that of histone H3 or its phosphorylation, is essential for chromosome condensation. EMBO J. 20, 6383–6393 10.1093/emboj/20.22.6383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delacour–Larose M., Molla A., Skoufias D. A., Margolis R. L., Dimitrov S. (2004). Distinct dynamics of Aurora B and Survivin during mitosis. Cell Cycle 3, 1418–1426 10.4161/cc.3.11.1203 [DOI] [PubMed] [Google Scholar]
- Delacour–Larose M., Thi M. N., Dimitrov S., Molla A. (2007). Role of survivin phosphorylation by Aurora B in mitosis. Cell Cycle 6, 1878–1885 10.4161/cc.6.15.4482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson C. A., Kosmopoulou M., Richards M. W., Atrash B., Bavetsias V., Blagg J., Bayliss R. (2010). Crystal structure of an Aurora-A mutant that mimics Aurora-B bound to MLN8054: insights into selectivity and drug design. Biochem. J. 427, 19–28 10.1042/BJ20091530 [DOI] [PubMed] [Google Scholar]
- Garuti L., Roberti M., Bottegoni G. (2009). Small molecule Aurora kinases inhibitors. Curr. Med. Chem. 16, 1949–1963 10.2174/092986709788682227 [DOI] [PubMed] [Google Scholar]
- Gavet O., Pines J. (2010). Progressive activation of CyclinB1-Cdk1 coordinates entry to mitosis. Dev. Cell 18, 533–543 10.1016/j.devcel.2010.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girdler F., Gascoigne K. E., Eyers P. A., Hartmuth S., Crafter C., Foote K. M., Keen N. J., Taylor S. S. (2006). Validating Aurora B as an anti-cancer drug target. J. Cell Sci. 119, 3664–3675 10.1242/jcs.03145 [DOI] [PubMed] [Google Scholar]
- Girdler F., Sessa F., Patercoli S., Villa F., Musacchio A., Taylor S. (2008). Molecular basis of drug resistance in Aurora kinases. Chem. Biol. 15, 552–562 10.1016/j.chembiol.2008.04.013 [DOI] [PubMed] [Google Scholar]
- Hannak E., Kirkham M., Hyman A. A., Oegema K. (2001). Aurora-A kinase is required for centrosome maturation in Caenorhabditis elegans. J. Cell Biol. 155, 1109–1116 10.1083/jcb.200108051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hans F., Dimitrov S. (2001). Histone H3 phosphorylation and cell division. Oncogene 20, 3021–3027 10.1038/sj.onc.1204326 [DOI] [PubMed] [Google Scholar]
- Harrington E. A., Bebbington D., Moore J., Rasmussen R. K., Ajose–Adeogun A. O., Nakayama T., Graham J. A., Demur C., Hercend T., Diu–Hercend A.et al. (2004). VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat. Med. 10, 262–267 10.1038/nm1003 [DOI] [PubMed] [Google Scholar]
- Hégarat N., Smith E., Nayak G., Takeda S., Eyers P. A., Hochegger H. (2011). Aurora A and Aurora B jointly coordinate chromosome segregation and anaphase microtubule dynamics. J. Cell Biol. 195, 1103–1113 10.1083/jcb.201105058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang T. M.–N., Favier B., Valette A., Barette C., Nguyen C. H., Lafanechère L., Grierson D. S., Dimitrov S., Molla A. (2009). Benzo[e]pyridoindoles, novel inhibitors of the Aurora kinases. Cell Cycle 8, 765–772 10.4161/cc.8.5.7879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda R., Körner R., Nigg E. A. (2003). Exploring the functional interactions between Aurora B, INCENP, and survivin in mitosis. Mol. Biol. Cell 14, 3325–3341 10.1091/mbc.E02-11-0769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelluma N., Brenkman A. B., van den Broek N. J. F., Cruijsen C. W. A., van Osch M. H. J., Lens S. M. A., Medema R. H., Kops G. J. P. L. (2008). Mps1 phosphorylates Borealin to control Aurora B activity and chromosome alignment. Cell 132, 233–246 10.1016/j.cell.2007.11.046 [DOI] [PubMed] [Google Scholar]
- Kollareddy M., Zheleva D., Dzubak P., Brahmkshatriya P. S., Lepsik M., Hajduch M. (2012). Aurora kinase inhibitors: progress towards the clinic. Invest. New Drugs 30, 2411–2432 10.1007/s10637-012-9798-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lens S. M. A., Medema R. H. (2003). The survivin/Aurora B complex: its role in coordinating tension and attachment. Cell Cycle 2, 507–510 10.4161/cc.2.6.559 [DOI] [PubMed] [Google Scholar]
- Martin M. P., Zhu J.–Y., Lawrence H. R., Pireddu R., Luo Y., Alam R., Ozcan S., Sebti S. M., Lawrence N. J., Schönbrunn E. (2012). A novel mechanism by which small molecule inhibitors induce the DFG flip in Aurora A. ACS Chem. Biol. 7, 698–706 10.1021/cb200508b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meldi L., Brickner J. H. (2011). Compartmentalization of the nucleus. Trends Cell Biol. 21, 701–708 10.1016/j.tcb.2011.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza M., Norden C., Durrer K., Rauter H., Uhlmann F., Barral Y. (2009). A mechanism for chromosome segregation sensing by the NoCut checkpoint. Nat. Cell Biol. 11, 477–483 10.1038/ncb1855 [DOI] [PubMed] [Google Scholar]
- Mora–Bermúdez F., Gerlich D., Ellenberg J. (2007). Maximal chromosome compaction occurs by axial shortening in anaphase and depends on Aurora kinase. Nat. Cell Biol. 9, 822–831 10.1038/ncb1606 [DOI] [PubMed] [Google Scholar]
- Nguyen C. H., Bisagni E., Lavelle F., Bissery M. C., Huel C. (1992). Synthesis and antitumor properties of new 4-methyl-substituted- pyrido[4,3-b]indoles (gamma-carbolines). Anticancer Drug Des. 7, 219–233. [PubMed] [Google Scholar]
- Park S.–Y., Kim S., Cho H., Kwon S.–H., Chae S., Kang D., Seong Y.–S., Cho H. (2009). Depletion of BubR1 promotes premature centrosomal localization of cyclin B1 and accelerates mitotic entry. Cell Cycle 8, 1754–1764 10.4161/cc.8.11.8671 [DOI] [PubMed] [Google Scholar]
- Petsalaki E., Akoumianaki T., Black E. J., Gillespie D. A. F., Zachos G. (2011). Phosphorylation at serine 331 is required for Aurora B activation. J. Cell Biol. 195, 449–466 10.1083/jcb.201104023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prigent C., Dimitrov S. (2003). Phosphorylation of serine 10 in histone H3, what for? J. Cell Sci. 116, 3677–3685 10.1242/jcs.00735 [DOI] [PubMed] [Google Scholar]
- Rosasco–Nitcher S. E., Lan W., Khorasanizadeh S., Stukenberg P. T. (2008). Centromeric Aurora-B activation requires TD-60, microtubules, and substrate priming phosphorylation. Science 319, 469–472 10.1126/science.1148980 [DOI] [PubMed] [Google Scholar]
- Sardon T., Peset I., Petrova B., Vernos I. (2008). Dissecting the role of Aurora A during spindle assembly. EMBO J. 27, 2567–2579 10.1038/emboj.2008.173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saurin A. T., van der Waal M. S., Medema R. H., Lens S. M. A., Kops G. J. P. L. (2011). Aurora B potentiates Mps1 activation to ensure rapid checkpoint establishment at the onset of mitosis. Nat. Commun. 2, 316 10.1038/ncomms1319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki A., Coppinger J. A., Jang C.–Y., Yates J. R., Fang G. (2008). Bora and the kinase Aurora A cooperatively activate the kinase Plk1 and control mitotic entry. Science 320, 1655–1658 10.1126/science.1157425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen S., Zhou H., White R. A. (1997). A putative serine/threonine kinase encoding gene BTAK on chromosome 20q13 is amplified and overexpressed in human breast cancer cell lines. Oncogene 14, 2195–2200 10.1038/sj.onc.1201065 [DOI] [PubMed] [Google Scholar]
- Sessa F., Mapelli M., Ciferri C., Tarricone C., Areces L. B., Schneider T. R., Stukenberg P. T., Musacchio A. (2005). Mechanism of Aurora B activation by INCENP and inhibition by hesperadin. Mol. Cell 18, 379–391 10.1016/j.molcel.2005.03.031 [DOI] [PubMed] [Google Scholar]
- Soncini C., Carpinelli P., Gianellini L., Fancelli D., Vianello P., Rusconi L., Storici P., Zugnoni P., Pesenti E., Croci V.et al. (2006). PHA-680632, a novel Aurora kinase inhibitor with potent antitumoral activity. Clin. Cancer Res. 12, 4080–4089 10.1158/1078-0432.CCR-05-1964 [DOI] [PubMed] [Google Scholar]
- Tan L., Kapoor T. M. (2011). Examining the dynamics of chromosomal passenger complex (CPC)-dependent phosphorylation during cell division. Proc. Natl. Acad. Sci. USA 108, 16675–16680 10.1073/pnas.1106748108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vader G., Lens S. M. A. (2008). The Aurora kinase family in cell division and cancer. Biochim. Biophys. Acta 1786, 60–72 10.1016/j.bbcan.2008.07.003 [DOI] [PubMed] [Google Scholar]
- van der Waal M. S., Hengeveld R. C., van der Horst A., Lens S. M. A. (2012). Cell division control by the Chromosomal Passenger Complex. Exp. Cell Res. 318, 1407–1420 10.1016/j.yexcr.2012.03.015 [DOI] [PubMed] [Google Scholar]
- Vu H. L., Hoang T. M. N., Favier B., Molla A. (2010). Aurora kinases and passenger proteins as targets for cancer therapy: an update. Current Enzyme Inhibition 6, 19–25 10.2174/157340810790712041 [DOI] [Google Scholar]
- Xu Z., Ogawa H., Vagnarelli P., Bergmann J. H., Hudson D. F., Ruchaud S., Fukagawa T., Earnshaw W. C., Samejima K. (2009). INCENP-Aurora B interactions modulate kinase activity and chromosome passenger complex localization. J. Cell Biol. 187, 637–653 10.1083/jcb.200906053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z., Vagnarelli P., Ogawa H., Samejima K., Earnshaw W. C. (2010). Gradient of increasing Aurora B kinase activity is required for cells to execute mitosis. J. Biol. Chem. 285, 40163–40170 10.1074/jbc.M110.181545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasui Y., Urano T., Kawajiri A., Nagata K.–i., Tatsuka M., Saya H., Furukawa K., Takahashi T., Izawa I., Inagaki M. (2004). Autophosphorylation of a newly identified site of Aurora-B is indispensable for cytokinesis. J. Biol. Chem. 279, 12997–13003 10.1074/jbc.M311128200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.