

Abstract
Congenital cystic diseases of the lung are a rare but significant cause of morbidity in children and young adults presenting with respiratory distress and repeated chest infections. They consist of cystic adenomatoid malformation, bronchogenic cyst, pulmonary sequestration, and congenital lobar emphysema. Surgical treatment is a safe and an effective method of treatment. Chest X-ray and computed tomography are the key imaging modalities used for diagnosis.
Keywords: Bronchogenic cyst, congenital lung malformation, cystic adenomatoid malformation
INTRODUCTION

Aditi Jain
Congenital cystic diseases of the lung consist of cystic adenomatoid malformation (CAM), bronchogenic cyst, pulmonary sequestration (PS), and congenital lobar emphysema (CLE).[1] These show close relationship in terms of embryology and clinical presentation. Some cause life threatening respiratory distress at birth while others appear late in life as incidental finding on radiograph. Majority of symptomatic patients present with progressive respiratory distress or repeated chest infections with cystic changes in the chest X-ray.
Congenital cystic adenomatoid malformation
Congenital cystic adenomatoid malformations (CCAMs) present as cystic or solid lung masses restricted to part of one lung.[2] Most CAMs present with respiratory distress or compromise during infancy or recurrent pneumonias in later years, including adulthood. The pathological feature of CAMs is adenomatoid proliferation of the bronchioles that forms a cyst at the expense of alveoli. Three types of CAMs are recognized based on cyst size, number, and pathology.[3] Type 1 consists of multiple large cyst (2-10 cm), at least one cyst will be dominant with smaller cysts seen along its periphery. The cyst wall is lined with ciliated pseudostratified columnar epithelium, contains elastic tissue beneath the epithelium, smooth muscle, and fibrovascular connective tissue, including cartilage. Type 2 consists of small and more uniform size cysts (0.5-2 cm). They are lined with cuboidal to columnar epithelium and have only a thin fibromuscular wall. This type has associated anomalies – renal, cardiac, skeletal, intestinal, extra-lobar sequestration. Type 3 lesions are bulky solid lesions that usually involve entire lobe of the lung. There is slight modification of the classification of CCAM with now five types being recognized out of which only three types are identified on imaging as mentioned above. It is now called as congenital pulmonary airway malformations since all the types are not cystic.[4] Additional types are, Type 0, which has either no cyst or very small ones (<0.5 cm), and is incompatible with life and Type 4 CCAM which consists of large cysts up to 10 cm in size.[5]
The radiological features depend on the content, size, and number of cysts. Chest radiograph reveals multiple air-filled thin-walled cysts of varying sizes. At birth, these cysts are filled with fluid, later air enters into the cysts as they communicate with tracheobrochial tree and with each other. Thus may contain only air or air-fluid levels [Figure 1a].[6] Conversely only fluid filled cysts may be seen due to retained secretions, hemorrhage, or secondary to infection [Figure 3c and d].[7]
Figure 1.
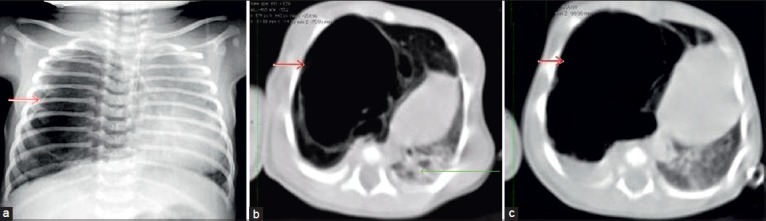
(a) Chest radiograph shows a large air filled thin walled cyst (red arrow) in the right lung with herniation of the lung to the contralateral side, (b) Computed tomography chest shows multilocular cysts (red arrow) of variable sizes in the right upper and middle lobe, largest cyst measuring 64 × 42 mm with herniation of the lung and mediastinum shift to the contralateral side. There was partial compression collapse of the right lower lobe and the left lung (green arrow). No fluid, hemorrhage is seen within the cyst (c) Computed tomography scan of chest (axial view) shows shift of mediastinum to the left due to mass effect. A large cyst (red arrow) is visible in the right lung parenchyma.
Figure 3.
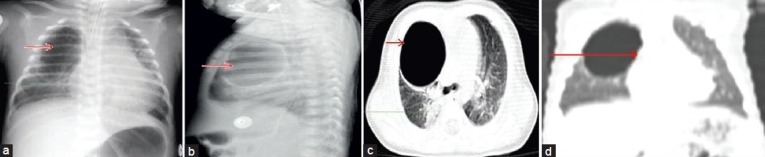
(a) Chest radiograph anteroposterior view shows large thick walled air filled unilocular cyst (red arrow) in the right lung upper and mid zones. Consolidation is seen adjacent to the cyst (green arrow), (b) Lateral view of chest radiograph shows large thick walled unilocular cyst (red arrow) in the right upper and mid zone, (c) Computed tomography chest (lung window) reveals a large unilocular thick walled air filled cavity (red arrow) in the right upper lobe with compression of the adjacent lung parenchyma. Consolidation is noted in the right lower lobe (green arrow). No fluid is seen within the cyst. No obvious communication of the cyst with bronchial tree is visible, (d) Computed tomography coronal reconstruction scan shows no obvious communication with bronchial tree (red arrow).
Computed tomography (CT) also shows similar findings as radiographs [Figure 1b, c]. In another case, the chest radiograph showed well-defined homogenous opacity [Figure 2a]. CT images also show fluid filled lesion [Figure 2b–d] The main role of CT is to differentiate these cysts from bronchogenic cysts, lobar emphysema, and from sequestration, to accurately localize the site of lesion within the lung segments for surgical planning.[6] Treatment consists of simple resection of the involved tissue or lobectomy in CCAM.
Figure 2.
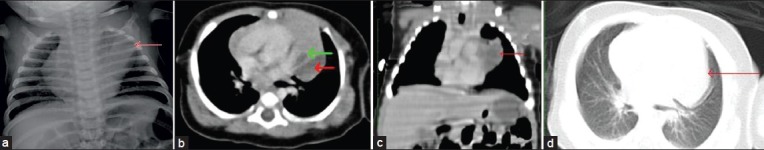
(a) Chest radiograph anteroposterior view reveals well defined homogenous opacity in the left upper zone (red arrow) which could not be differentiated from normal thymic shadow, (b) Computed tomography mediastinal window reveals a well-defined fluid attenuating cyst (red arrow) with septations in the middle mediastinum posterior to the thymus. The fat planes with the thymus (green arrow) were not well delineated, (c) Computed tomography coronal reconstruction of chest (lung window) shows fluid attenuating lesion (red arrow) adherent to the thymus, (d) In the computed tomography scan of chest (lung window) cannot differentiate whether lesion was mediastinal or lung parenchymal (red arrow) lesion.
Bronchogenic cyst
Bronchogenic cysts arise from abnormal budding of the tracheobronchial tree during airway development. This abnormal bud subsequently differentiates into a fluid-filled blind-end pouch. More often present in the mediastinum, one third can occur in the lung parenchyma, usually within the lower lobes.[2] These cysts may contain air, fluid, or both. Clinical manifestations are related to various mass effects or secondary infection of the cyst. Lined by ciliated columnar or cuboidal epithelium and are surrounded by tissues similar to those of the normal bronchus; including cartilage, smooth muscle, elastic tissue, and mucous glands.[2]
Intrapulmonary bronchogenic cyst are rare (14%), most of them are fluid attenuating. Only 36% of intrapulmonary bronchogenic cyst contains air, which occurs when the cyst communicates with the bronchial tree. Rogers and Osmer [Figure 2a][8] described their radiographic findings as a sharply defined, solitary, uncalcified round or oval density presenting as one of three categories: A cyst with a homogeneous density filled with fluid, an air-filled cyst, or a cyst containing an air and fluid [Figure 3a and b]. CT images shows the cyst as a well-defined spherical mass with or without mass effect and with attenuation of water or soft-tissue. In one case ,the radiograph shows thick walled cyst with no communication with bronchial tree [Figure 3c and d]. A fluid filled cyst is seen in the radiograph [Figure 4a]. CT images show fluid cyst [Figure 4b–d] The CT attenuation values of the intrapulmonary bronchogenic cysts are same as of mediastinal bronchogenic cysts.[2,8,9]
Figure 4.
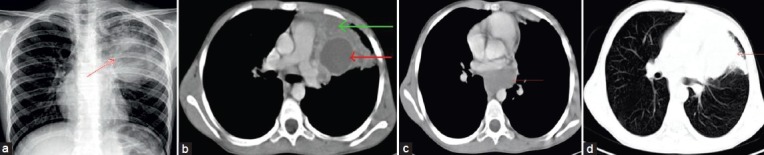
(a) Chest radiograph shows inhomogeneous opacity (red arrow) in the left upper and mid zone with well-defined inferior margin, (b) Computed tomography chest shows large well defined unilocular fluid containing cyst (red arrow) in the left upper lobe with thick enhancing walls. Surrounding lung shows consolidation (green arrow), (c) Computed tomography scan chest (mediastinal window) shows another fluid filled cyst (red arrow) in the posterior mediastinum of the subcarinal region, (d) Computed tomography chest (lung window) shows consolidation (red arrow) in the left upper lobe. Rest of the lung is clear.
The high attenuation on unenhanced CT scans is caused by hemorrhage, proteinaceous content, or calcium.[10–13] Complications include infection; compressive symptoms, such as dysphagia or arrhythmia; malignant transformation; and the rare but fatal air embolism.[10] The appropriate treatment is surgery, especially in patients with repeated infections. Occurrence of both mediastinal and parenchymal cysts together is very rare.[20]
Congenital lobar emphysema
Congenital lobar emphysema (CLE) is a disease that causes breathing difficulties in newborn or infants. Furthermore, called as infantile lobar emphysema.[9,14] The incidence of CLE is 1 in 20,000 to 1 in 30,000.[15] Hence, early recognition and treatment is essential. It is reported predominantly in male children[16] affecting left upper lobe in 43%, followed by right middle lobe in 32% of the cases.[17] Exact etiopathogenesis is still a dilemma, however, changes in bronchial cartilage have known to occur in most cases.
On chest X-ray, there is over inflation of the diseased lobe resulting in shift of mediastinum to contralateral side and compression of ipsilateral lung with attenuation of vascular markings [Figure 5a]. CT findings help to confirm these findings [Figure 5b]. CLE is commonly associated with congenital heart disease such as ventricular septal defect and patent ductus arterirous. Differential diagnosis includes CCAM, pulmonary hypoplasia, and pnuemothorax. Treatment includes lobectomy of affected lung.
Figure 5.
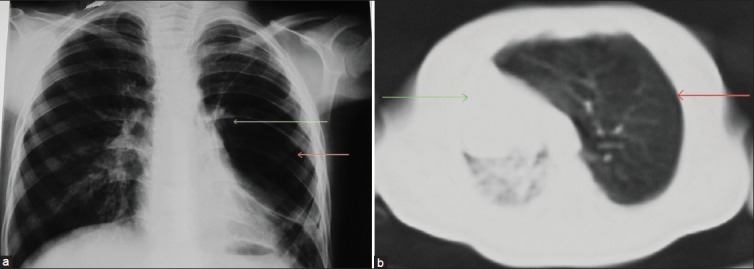
(a) Chest X-ray posteroanterior view shows a large hyperlucent area (red arrow) in the left upper, mid, and lower zone with attenuated vascular markings within the lucency (green arrow), (b) Computed tomography lung window (axial section) shows hyperinflated left upper lobe with attenuated vascular marking (red arrow). There is hernination of left lung field, mediastinal shift to the contralateral side with collapse of right lung (green arrow).
Pulmonary sequestration
Pulmonary sequestration (PS) is a disorder characterized by a non-functioning lung parenchyma, which does not communicate with tracheo-bronchial tree and receives blood supply from systemic artery. Sequestration is believed to be due to abnormal budding of primitive foregut. Anatomically it can be classified as intra-lobar and extra-lobar sequestration. Clinically, sequestration present as recurrent pneumonia in adults and adolescents, in case of intra-lobar and respiratory distress, chronic cough in infancy, in case of extra-lobar sequestration. Intra-lobar sequestration lies within visceral pleura and usually located in posterobasal segment of the lung. Arterial supply is from abdominal aorta or thoracic aorta and venous drainage through pulmonary veins into left atrium. It communicates with adjacent lung parenchyma through pore of Kohn, which allows infection to occur and resolution is incomplete or slow due to inadequate bronchial drainage.[18] Extra-lobar sequestration is surrounded by its own pleura, usually located on the left side of the lower chest. Extra-lobar sequestration is usually associated with other congenital anomalies like diaphragmatic hernia, congenital heart disease, and CCAM. Radiologically sequestration on a chest radiograph demonstrated by a soft-tissue opacity in chest base [Figure 6]. Bronchi ecstasies, cystic areas, sub-segmental atelectasis, mediastinal shift, and prominence of ipsilateral hila are other radiological findings that arise due to recurrent infection. CT scan show cystic areas containing air or fluid, focal emphysema, and atelectasis. The arterial supply and venous drainage, which are pathognomic features of sequestration are better demonstrated by contrast enhanced CT scan. Magnetic resonance scan can be used for better characterization of the lesion by differentiating the content as solid, cystic, hemorrhagic, or containing mucus components.
Figure 6.
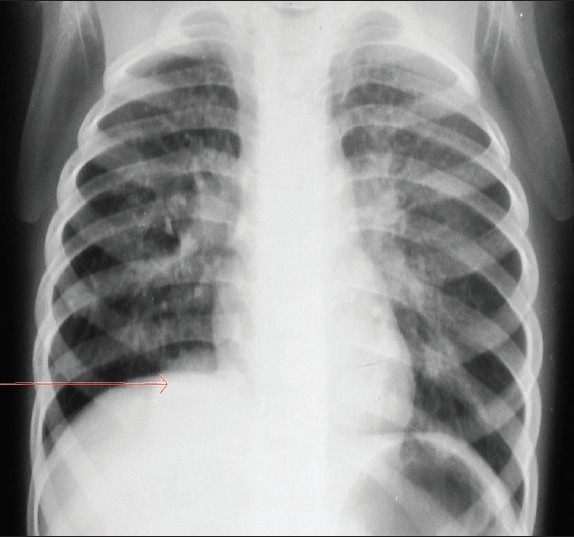
Chest X-ray anteroposterior view shows an homogenous dense opacity (red arrow) in right cardiophernic angle.
Radiological differential diagnosis that need to be considered with this condition are CCAM, diaphragmatic hernia, terotoma, and accessory spleen.
CONCLUSION
Congenital cystic diseases of the lung are a group of lesions that share similar clinical and embryological features. The exact incidence is not known.[1] Respiratory distress, cyanosis, and repeated chest infections are common symptoms. It is important to recognize and diagnose congenital cystic lung diseases. In view of long-term complications of infections and malignancy there is growing consensus that even asymptomatic infants should undergo elective surgery.[19]
Footnotes
Available FREE in open access from: http://www.clinicalimagingscience.org/text.asp?2013/3/1/5/106620
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Coran AG, Drongowski R. Congenital cystic disease of the tracheobronchial tree in infants and children. Experience with 44 consecutive cases. Arch Surg. 1994;129:521–7. doi: 10.1001/archsurg.1994.01420290067010. [DOI] [PubMed] [Google Scholar]
- 2.Nuchtern JG, Harberg FJ. Congenital lung cysts. Semin Pediatr Surg. 1994;3:233–43. [PubMed] [Google Scholar]
- 3.Stocker JT, Madawell JE, Drake RM. Congenital cystic adenomatoid malformation of lung: Classification and morphological spectrum. Hum Pathol. 1977;8:155–71. doi: 10.1016/s0046-8177(77)80078-6. [DOI] [PubMed] [Google Scholar]
- 4.Stocker JT. Congenital pulmonary airway malformation. A new name for an expanded classification of congenital cystic adenomatoid malformation of lung. Symposium : Non neoplastic lung disease. Histopathology. 2002;41:424–31. [Google Scholar]
- 5.al-Salem AH, Adu-Gyamfi Y, Grant CS. Congenital lobar emphysema. Can J Anaesth. 1990;37:377–9. doi: 10.1007/BF03005595. [DOI] [PubMed] [Google Scholar]
- 6.Spencer H. Congenital abnormalities of lung, pulmonary vessels and lymphatics. In: Spencer H, editor. Pathology of Lung. Oxford: Pergamon; 1985. pp. 79–130. [Google Scholar]
- 7.Shackelford GD, Siegel MJ. CT appearance of cystic adenomatoid malformations. J Comput Assist Tomogr. 1989;13:612–6. doi: 10.1097/00004728-198907000-00010. [DOI] [PubMed] [Google Scholar]
- 8.Rogers LF, Osmer JC. Bronchogenic cyst. A review of 46 cases. Am J Roentgenol Radium Ther Nucl Med. 1964;91:273–90. [PubMed] [Google Scholar]
- 9.Suen HC, Mathisen DJ, Grillo HC, LeBlanc J, McLoud TC, Moncure AC, et al. Surgical management and radiological characteristics of bronchogenic cysts. Ann Thorac Surg. 1993;55:476–81. doi: 10.1016/0003-4975(93)91022-f. [DOI] [PubMed] [Google Scholar]
- 10.McAdams HP, Kirejczyk WM, Rosado-de-Christenson ML, Matsumoto S. Bronchogenic cyst: Imaging features with clinical and histopathologic correlation. Radiology. 2000;217:441–6. doi: 10.1148/radiology.217.2.r00nv19441. [DOI] [PubMed] [Google Scholar]
- 11.Lyon RD, McAdams HP. Mediastinal bronchogenic cyst: Demonstration of a fluid-fluid level at MR imaging. Radiology. 1993;186:427–8. doi: 10.1148/radiology.186.2.8421745. [DOI] [PubMed] [Google Scholar]
- 12.Nakata H, Sato Y, Nakayama T, Yoshimatsu H, Kobayashi T. Bronchogenic cyst with high CT number: Analysis of contents. J Comput Assist Tomogr. 1986;10:360. doi: 10.1097/00004728-198603000-00044. [DOI] [PubMed] [Google Scholar]
- 13.Yernault JC, Kuhn G, Dumortier P, Rocmans P, Ketelbant P, De Vuyst P. “Solid” mediastinal bronchogenic cyst: Mineralogic analysis. AJR Am J Roentgenol. 1986;146:73–4. doi: 10.2214/ajr.146.1.73. [DOI] [PubMed] [Google Scholar]
- 14.Landing BH, Dixon LG. Congenital malformations and genetic disorder of the respiratory tract. Am Rev Respir Dis. 1979;120:151–85. doi: 10.1164/arrd.1979.120.1.151. [DOI] [PubMed] [Google Scholar]
- 15.Thakral CL, Maji DC, Sajwani MJ. Congenital lobar emphysema: Experience with 21 cases. Pediatr Surg Int. 2001;17:88–91. doi: 10.1007/s003830000506. [DOI] [PubMed] [Google Scholar]
- 16.Ward CF. Diseases of infants. In: Katz J, Benumof JL, Kadis LB, editors. Anesthesia and uncommon diseases. Philadelphia: WB Saunders Co; 1990. p. 199. [Google Scholar]
- 17.Kravitz RM. Congenital malformations of the lung. Pediatr Clin North Am. 1994;41:453–72. doi: 10.1016/s0031-3955(16)38765-x. [DOI] [PubMed] [Google Scholar]
- 18.Stocker JT. Cystic lung disease in infants and children. Fetal Pediatr Pathol. 2009;28:155–84. doi: 10.1080/15513810902984095. [DOI] [PubMed] [Google Scholar]
- 19.Azizkhan RG, Crombleholme TM. Congenital cystic lung disease: Contemporary antenatal and postnatal management. Pediatr Surg Int. 2008;24:643–57. doi: 10.1007/s00383-008-2139-3. [DOI] [PubMed] [Google Scholar]
- 20.Berrocal T, Madrid C, Novo S, Gutiérrez J, Arjonilla A, Gómez-León N. Congenital anomalies of the tracheobronchial tree, lung, and mediastinum: Embryology, radiology, and pathology. RadioGraphics. 2004;24:e17. doi: 10.1148/rg.e17. [DOI] [PubMed] [Google Scholar]