

Abstract
Anomalous origin of the left coronary artery from the pulmonary artery (ALCAPA) is a rare congenital anomaly. It is associated with early infant mortality and sudden death in adults. Traditionally, ALCAPA has been diagnosed by angiography or autopsy; however, the development of cardiac computed tomography (CT) and magnetic resonance imaging (MRI) has allowed noninvasive evaluation of the coronary anatomy by direct visualization of the origin of the left coronary artery (LCA) from the pulmonary artery. We report a case of 10-year-old girl who has been on follow up for dilated cardiomyopathy for 4 years. The definitive diagnosis of ALCAPA is reached by multislice computed tomography (MSCT). The MSCT scan showed an anomalous origin of LCA from the pulmonary trunk, with a tortuous and dilated right coronary artery and right-to-left collateralization. Consequently, the patient was successfully treated with surgery.
Keywords: Anomalous origin of the left coronary artery from the pulmonary artery, coronary anomaly, computed tomography angiography
INTRODUCTION

Guray Oncel
Anomalous origin of the left coronary artery from the pulmonary artery (ALCAPA) is a rare congenital anomaly occurring in 1 of 300.000 births. It is also known as Bland-White-Garland syndrome and accounts for 0.25%-0.5% of congenital cardiac disease.[1–3] It is associated with early infant mortality and adult sudden death. It usually manifests as an isolated defect, but in 5% of cases it may be associated with other cardiac anomalies such as atrial septal defect, ventricular septal defect, and aortic coarctation.[2] The origin of the left coronary artery (LCA) from the pulmonary artery is well tolerated in fetal life because pulmonary arterial pressure equals systemic pressure, which leads to antegrade flow in the anomalous LCA.[2] Shortly after birth, when pulmonary arterial pressure decreases, antegrade flow and oxygen content in the LCA also decrease leading to myocardial ischemia and infarction. When collateral circulation between the right and left coronary systems develops, left coronary artery flow reverses and enters the pulmonary trunk due to the low pulmonary arterial pressure. This is called “coronary steal” phenomenon, in which a left-to-right shunt leads to abnormal left ventricular perfusion.[3]
ALCAPA syndrome is one of the most common causes of myocardial ischemia and infarction in children and may also present as cardiomyopathy.[2,3] Without treatment, approximately 90% of patients die within the first year of life.[2,3] However, in some cases collateral blood supply from the right coronary artery may be well-established and the patients can reach to adulthood.[2–4] In patients who live to adulthood, ALCAPA syndrome may cause myocardial infarction, left ventricular dysfunction and mitral regurgitation, malignant ventricular arrhythmias generated from myocardial scar tissue or silent myocardial ischemia, which can lead to sudden cardiac death.[2–4]
Traditionally, ALCAPA has been diagnosed by angiography or autopsy; however, the development of cardiac CT and MRI has allowed noninvasive evaluation of the coronary anatomy by direct visualization of the origin of the LCA from the pulmonary artery.[2,5–7] Both modalities may be also used to assess left ventricular (LV) systolic function.[5]
The definitive treatment for ALCAPA is surgical intervention.[2,8] Early diagnosis and prompt surgical intervention have excellent results and lead to gradual myocardial recovery.[2,8]
We report a case of 10-year-old girl who has been on follow up for dilated cardiomyopathy for 4 years. The definitive diagnosis of ALCAPA is reached by multislice computed tomography (MSCT).
CASE REPORT
A 10-year-old girl was admitted to our hospital with complaints of syncope and shortness of breath during exercise. No detailed medical record could be reached but her parents declaimed that she has been on follow up for dilated cardiomyopathy in another hospital for 4 years and she has been taking digoxin since that time.
On physical examination, a parasternal 2/6 sysytolic murmur was detected on auscultation. She was thin, weak and seemed to be much younger than her age. The chest X-ray revealed cardiomegaly and signs of pulmonary congestion. The transthoracic 2-D echocardiography showed dilated left ventricle, mild mitral and tricuspid insufficiency and decreased ejection fraction. She was then referred to MSCT for evaluation of coronary anatomy.
CT coronary angiography was performed using a 64 slice CT scanner (Acquilion 64 Toshiba, Japan). Scan parameters were as follows: Slice collimation, 64 × 0.5 mm; rotation time, 0.40 mins; tube voltage, 80 kV; tube current, 300 mA; and pitch, 0.2. The scan parameters were modified to decrease the patient dose. The average heart rate was 85 bpm during the scan. The patient did not receive a b-blocker prior to the CT examination for heart rate regulation.
CT angiography was triggered automatically by the arrival of the main contrast bolus (automatic bolus tracking). A pre-scan was taken at the level of the aortic root and a region of interest (ROI) was identified on the descending aorta. As soon as the signal density level in the descending aorta reached the predefined threshold of 170 Hounsfield units (HU), the scan started. We injected 50 ml nonionic contrast medium (Iomeron 350/ml; Iomeprol, Bracco, Italy) at a flow rate of 3 ml/s in the left antecubital vein. This was followed by a 30 ml saline chaser bolus at a flow rate of 3 ml/s to wash out contrast from the right ventricle.
During the scan, the ECG was recorded simultaneously. The continuous data acquisition allows slice reconstruction at different time points in the cardiac cycle. The reconstruction intervals for both right and left coronary arteries with the fewest motion artifacts were determined (images at 50% and75% of the R-R interval, respectively) and used for further analysis. For reconstruction of axial images, we used a slice thickness of 0.5 mm and a slice width of 0.3 mm. A medium soft-tissue reconstruction kernel (FC 43) was used for reconstruction.
For post-processing, an external workstation (Aquarius Intuition, Tera Recon Inc., USA) was used. In addition to the transverse source images, multiplanar reformations (MPRs), curved MPR images, maximum intensity projections (MIPs), and volume rendered (VR) images were utilized for the evaluation. VR reconstructions depicted the vascular anatomy well and were used for 3D orientation.
In the CT angiography, the left atrium and left ventricle were shown to be markedly dilated and the left ventricular wall and interventricular septum were hypertrophic. Also CT angiography revealed an enormously dilated, tortuous and dominant right coronary artery (RCA) [Figure 1] emerging from the right aortic sinus [Figure 2] with a large number of collateral vessels feeding the left coronary system, which showed enlarged and tortuous vessels [Figure 3]. Left main coronary artery (LCA) was emerging from pulmonary artery trunk [Figure 4]. The left anterior descending artery (LAD) and left circumflex artery (LCX) were branching from LCA. Both LAD, LCX and their diagonal and obtuse branches were dilated and tortious.
Figure 1.
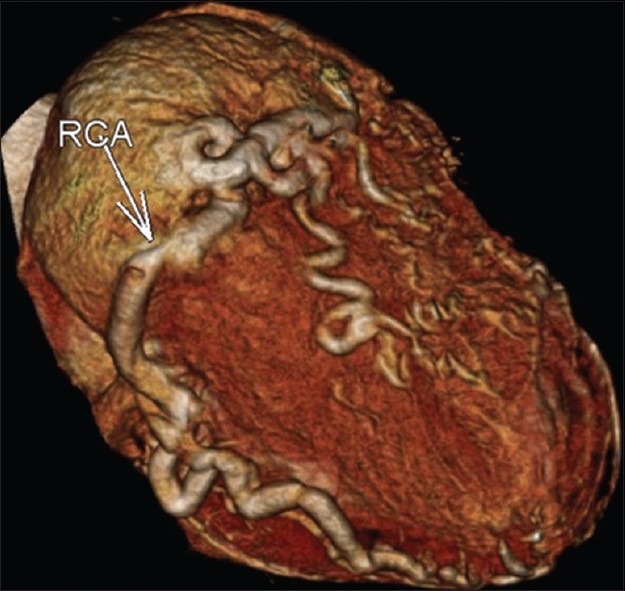
CT angiography volume rendered reformation image. The right coronary artery (RCA) is enormously dilated and tortuous with a large number of collateral vessels. Normal course of a very large right coronary artery is seen.
Figure 2.
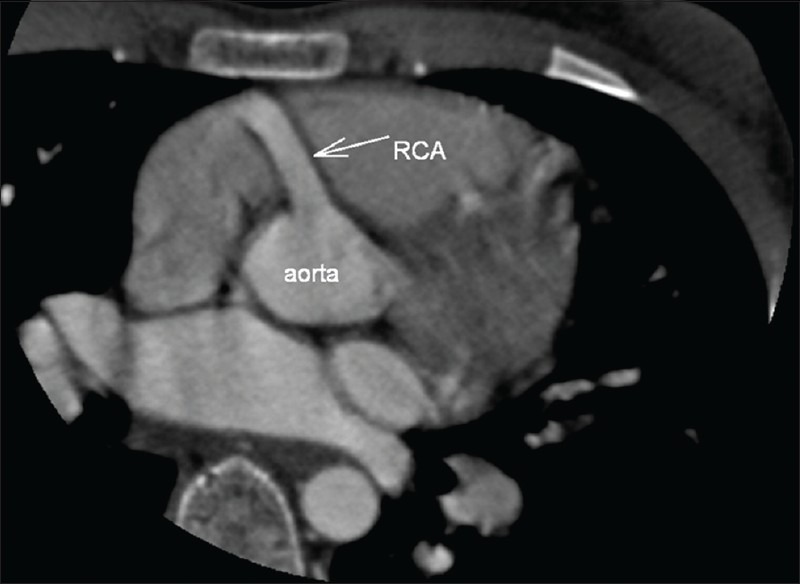
CT angiography multiplanar reformatted image. The dilated right coronary artery is emerging from the right aortic sinus.
Figure 3.
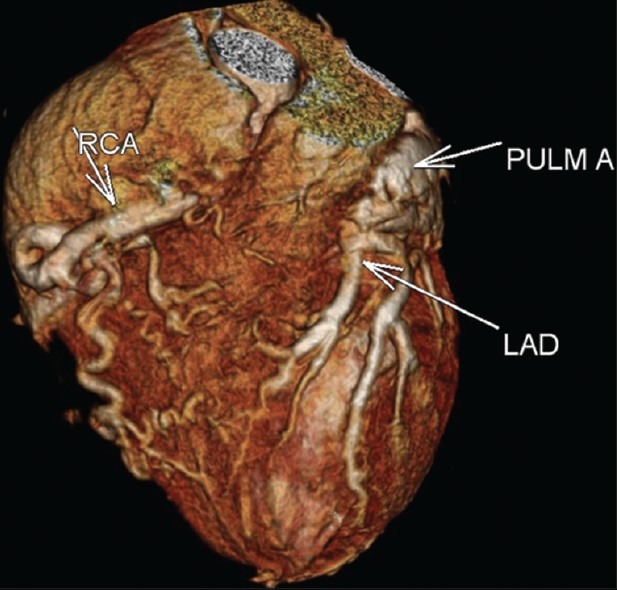
CT angiography volume rendered image. Right coronary artery is dilated and tortious with a large number of collaterals feeding the left coronary system. Left coronary artery is emerging from pulmonary artery trunk. The left anterior descending artery and their diagonal branches are also dilated and tortuous.
Figure 4.
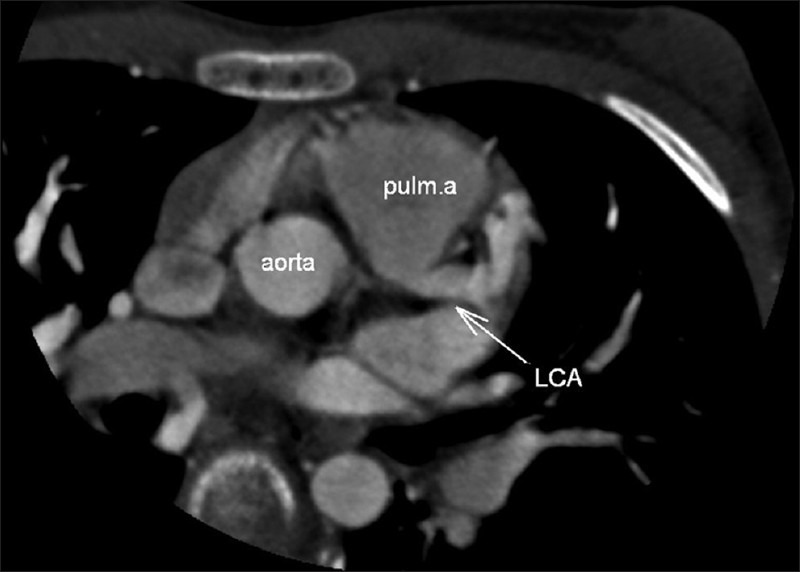
CT angiography multiplanar reformatted image. Left coronary artery is emerging from pulmonary artery trunk.
After we reported ALCAPA with CT angiography, the patient was transferred to the cardiac catheterization laboratory by the cardiovascular surgeons not only to confirm the diagnosis but also to plan the surgical procedure. In fact, CT angiography images were enough for the diagnosis of ALCAPA and a further invasive diagnostic procedure with radiation exposure was unnecessary.
Coronary angiography confirmed the diagnosis and showed an anomalous left coronary artery arising from the pulmonary artery (ALCAPA) with retrograde filling through collaterals from an enlarged right coronary artery. It revealed no other additional information.
Consequently, the patient underwent an uncomplicated aortic reimplantation of the left main coronary artery. No perioperative or postoperative complication occurred. The patient is now on follow up, with no major clinical symptoms in the fourth postoperative month.
DISCUSSION
Origin of the left coronary artery from the pulmonary artery, or Bland-White-Garland syndrome, is a rare anomaly which is frequently lethal in children and adults.[1,2]
Symptoms usually occur in infants 1-2 months after birth because of left-to-right shunting from the higher pressure left coronary arterial system to the lower pressure pulmonary arterial system. Without treatment, approximately 90% of infants die within the first year of life.[2,3] Death is usually due to circulatory insufficiency, myocardial infarction, or life-threatening cardiac dysrhythmias.[2,3] However, the patients may survive into childhood and even adulthood with clinical presentations varying from symptomatic chronic mitral insufficiency or global ischemic cardiomyopathy. Factors that may lead to survival beyond infancy include the development of abundant intercoronary collateral arteries.[2,3–5]
In a review of 25 infant and adult cases, Kaunitz confirmed that symptom onset often coincided with closure of the ductus arteriosus 2 months after birth.[9] He also noted in the adult cases, RCA dilation and significant development of collateralization from the RCA to the LCA. This became the first distinction proposed between infants who die in the first year and those who survive to adulthood.
Until recently, conventional coronary angiography was the diagnostic method of choice for detecting coronary anomalies. However, angiography is invasive and associated with a low but well-known risk of complications. Also, the precise course and the exact anatomy of the anomalous vessel may be difficult to delineate due to its complex geometry.[2,3]
MSCT angiography and MR imaging are valuable noninvasive modalities that can be used to to identify and define anomalous coronary arteries and their course with a very high accuracy.[2,5]
MSCT is a noninvasive imaging technique which is fast and offers excellent spatial resolution, which is required to assess small vessels such as the coronary arteries. The short investigation time, relative noninvasiveness of the procedure, simple preparation and minimal aftercare make MSCT coronary angiography advantageous over conventional coronary angiography. The main disadvantages of MSCT angiography are its relatively high radiation dose and its inability to assess flow.[2,5,6]
MR imaging does not use ionizing radiation. But the main disadvantages of MR imaging in comparison with CT are its relatively long examination times and its low spatial resolution.[2,5,7]
Cardiac CT and MRI are useful not only in diagnosis, but may also offer prognostic information, allow for risk stratification, and be utilized for long-term follow up imaging.
The definitive treatment for ALCAPA is surgical intervention. In cases of ALCAPA several surgical treatment options have been described.[8] In infants, most of the patients with corrected ALCAPA show normalization of both ventricular function and mitral valve insufficiency. Estimated long-term survival at 20 years was recently shown to be 94.8%.[10] No long-term studies of large populations of adults with corrected ALCAPA are available, but the prognosis is generally good. The late outcome after revascularization mainly depends on the extent of irreversible impaired left ventricular function and the presence of myocardial scar tissue.[2,8]
In our patient, CT angiography provided direct visualization of the LCA arising from the main pulmonary artery which is the diagnostic hallmark of ALCAPA syndrome. Also it revealed dilated and tortious right coronary artery and dilated intercoronary collateral arteries along the epicardial surface of the heart. They represent the collateral pathways between the RCA and the LCA to supply the left ventricle flow. Left ventricular hypertrophy and dilatation resulting from chronic myocardial ischemia were also clearly depicted. Reaching the correct diagnosis of ALCAPA provided the patient a surgical treatment option with a potential promise of excellent prognosis.
CONCLUSION
ALCAPA is a rare and life-threatening condition. Early diagnosis is very important because surgical treatment with optimal timing generally results in an excellent prognosis. MSCT provides correct diagnosis with remarkable anatomical detail non-invasively with short investigation time and minimal aftercare. Also it does not necessitate general anesthesia in children. Therefore it can be considered as the modality of choice in the diagnostic work up of ALCAPA as well as the other congenital coronary abnormalities and no further invasive diagnostic procedure with radiation exposure is necessary. Also in children with dilated cardiomyopathy, CT angiography should be taken into consideration early in the course of diagnostic investigations.
Footnotes
Available FREE in open access from: http://www.clinicalimagingscience.org/text.asp?2013/3/1/4/106618
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Hauser M. Congenital anomalies of the coronary arteries. Heart. 2005;91:1240–5. doi: 10.1136/hrt.2004.057299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pena E, Nguyen ET, Merchant N, Dennie G. ALCAPA syndrome: Not just a pediatric disease. Radiographics. 2009;29:553–65. doi: 10.1148/rg.292085059. [DOI] [PubMed] [Google Scholar]
- 3.Kristensen T, Kofoed KF, Helqvist S, Helvind M, Søndergaard L. Anomalous origin of the left coronary artery from the pulmonary artery (ALCAPA) presenting with ventricular fibrillation in an adult: A case report. J Cardiothorac Surg. 2008;3:33. doi: 10.1186/1749-8090-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takimura CK, Nakamoto A, Hotta VT, Campos MF, Malamo M, Otsubo R. Anomalous origin of the left coronary artery from the pulmonary artery: Report of an adult case. Arq Bras Cardiol. 2002;78:309–14. doi: 10.1590/s0066-782x2002000300006. [DOI] [PubMed] [Google Scholar]
- 5.Khanna A, Torigian DA, Ferrari VA, Bross RJ, Rosen MA. Anomalous origin of the left coronary artery from the pulmonary artery in adulthood on CT and MRI. AJR Am J Roentgenol. 2005;185:326–9. doi: 10.2214/ajr.185.2.01850326. [DOI] [PubMed] [Google Scholar]
- 6.Nacif MS, Luz JH, Moreira DM, Rochitte CE, Oliveira AC., Júnior Anomalous origin of coronary artery (ALCAPA) in 64-channel TC scanner. Arq Bras Cardiol. 2010;94:143–6. doi: 10.1590/s0066-782x2010000600022. [DOI] [PubMed] [Google Scholar]
- 7.Takenaga M, Matsuda J, Miyamoto N, Ikushima I, Koiwaya Y. Magnetic resonance imaging of Bland-White-Garland syndrome-a case of anomalous origin of the left coronary artery form the pulmonary trunk in a 22-year-old woman. Jpn Circ J. 1998;62:219–21. doi: 10.1253/jcj.62.219. [DOI] [PubMed] [Google Scholar]
- 8.Dodge-Khatami A, Mavroudis C, Backer CL. Anomalous origin of the left coronary artery from the pulmonary artery: Collective review of surgical therapy. Ann Thorac Surg. 2002;74:946–55. doi: 10.1016/s0003-4975(02)03633-0. [DOI] [PubMed] [Google Scholar]
- 9.Kaunitz PE. Origin of left coronary artery from pulmonary artery: Review of the literature and report of two cases. Am Heart J. 1947;33:182–206. doi: 10.1016/0002-8703(47)90005-7. [DOI] [PubMed] [Google Scholar]
- 10.Lange R, Vogt M, Hörer J, Cleuziou J, Menzel A, Holper K, et al. Long-term results of repair of anomalous origin of the left coronary artery from the pulmonary artery. Ann Thorac Surg. 2007;83:1463–71. doi: 10.1016/j.athoracsur.2006.11.005. [DOI] [PubMed] [Google Scholar]