

Abstract
Mitochondrial dysfunction has a significant role in the development of diabetic cardiomyopathy. Mitochondrial oxidant stress has been accepted as the singular cause of mitochondrial DNA (mtDNA) damage as an underlying cause of mitochondrial dysfunction. However, separate from a direct effect on mtDNA integrity, diabetic-induced increases in oxidant stress alter mitochondrial topoisomerase function to propagate mtDNA mutations as a contributor to mitochondrial dysfunction. Both glucose-challenged neonatal cardiomyocytes and the diabetic Goto-Kakizaki (GK) rat were studied. In both the GK left ventricle (LV) and in cardiomyocytes, chronically elevated glucose presentation induced a significant increase in mtDNA damage that was accompanied by decreased mitochondrial function. TTGE analysis revealed a number of base pair substitutions in the 3' end of COX3 from GK LV mtDNA that significantly altered the protein sequence. Mitochondrial topoisomerase DNA cleavage activity in isolated mitochondria was significantly increased in the GK LV compared with Wistar controls. Both hydroxycamptothecin, a topoisomerase type 1 inhibitor, and doxorubicin, a topoisomerase type 2 inhibitor, significantly exacerbated the DNA cleavage activity of isolated mitochondrial extracts indicating the presence of multiple functional topoisomerases in the mitochondria. Mitochondrial topoisomerase function was significantly altered in the presence of H2O2 suggesting that separate from a direct effect on mtDNA, oxidant stress mediated type II diabetes-induced alterations of mitochondrial topoisomerase function. These findings are significant in that the activation/inhibition state of the mitochondrial topoisomerases will have important consequences for mtDNA integrity and the well being of the diabetic myocardium.
Keywords: diabetic cardiomyopathy, type 2 diabetes, mitochondrial DNA damage, mitochondrial dysfunction
cardiovascular disease is responsible for a higher incidence of mortality in diabetics than the general population. Diabetic cardiomyopathy (DCM) is characterized by the development of a myopathy that manifests initially as diastolic dysfunction, but evolves into increased cavitary dilation and mural thinning, which is reflective of decompensated eccentric hypertrophy. DCM is considered to be independent of atherosclerosis or hypertension but is exacerbated by either (94). Mitochondrial dysfunction has long been known to have a significant role in the development and complications of DCM (32, 46, 66, 82, 92). Mitochondrial dysfunction is also associated with other pathologies including cancer, skeletal muscle disorders, and neurodegenerative diseases such as Wolfram syndrome or Leber's hereditary optic neuropathy (LHON) (18, 36, 54, 104).
Separate from inborn errors, mitochondrial DNA (mtDNA) mutations are thought to accumulate as a function of the aging process and believed to be responsible for the decline in mitochondrial function. The mtDNA mutation rate is higher in diabetic patients than euglycemic individuals (86). Mitochondrial DNA is thought to be at a greater risk because of its close proximity to the electron transport chain (ETC) and the higher levels of reactive oxygen species (ROS) that it generates. Also mtDNA does not contain introns and any mutation is likely to alter coding sequence for one of the 13 proteins encoded by the mtDNA. In postmitotic cells, the mitochondrial genome continues to replicate about once a month (13). Diabetic-induced mtDNA mutations that are not corrected by mitochondrial DNA repair pathways are fixed in the mitochondrial genome upon replication and this may account for their greater accumulation in diabetes.
Clinically, chronic hyperglycemia and poor glycemic control are negative prognosticators for diabetic individuals. Hyperglycemia is common to many animal models of type II diabetes, including the Goto-Kakizaki (GK) rat. GK rats are nonobese, and in contrast to other models of type 2 diabetes (T2D), the GK rats do not have hyperlipidemia or hypercholesterolemia. As a result they present as a milder form of T2D that is predominantly a model of hyperglycemia.
Less clear are the underlying mechanisms leading to failure. Mitochondrial dependent reactive oxygen species (ROS) generation has been accepted as the singular cause of mtDNA damage in diabetes and other pathophysiological states. However, the mitochondria have developed defense mechanisms to handle ROS and short-term hyperglycemia has been shown to activate antioxidant defenses (39). Second, mitochondria do possess DNA repair pathways. Third, glucose is not the preferred substrate of the myocardium and diabetes is known to further decrease glycolysis in the myocardium which should lower intracellular glucose concentrations (1, 45, 64). In contrast, significant increases in pentose shunt pathway activity have been reported that may significantly increase intracellular ROS (44, 70, 105). As a result it is unclear to what extent hyperglycemia may be a factor for promoting mitochondrial dysfunction as an underlying cause of diabetic cardiomyopathy. Using the H9c2 cell line, we have previously reported that even mild increases in glucose presentation compromised mitochondrial function as a result of a decline in mtDNA integrity. Separate from a direct impact of oxidative stress on mtDNA, ROS-induced alteration of mitochondrial topoisomerase activity exacerbated and propagated increases in mtDNA damage (80).
These findings are significant in that the functional state of the mitochondrial topoisomerases has important consequences for mtDNA integrity and the well being of the cell. So it is against this backdrop we have tested the hypothesis in a model of type 2 diabetes that chronic hyperglycemia-induced dysfunction of mitochondrial topoisomerase propagates mtDNA mutations as a contributor to mitochondrial dysfunction.
METHODS
Male euglycemic Wistar and diabetic GK rats (3–6 mo old) were used throughout this study, in addition to primary neonatal cardiomyocyte cultures. The GK animals were originally received courtesy of R. Farese (University of South Florida for Health Sciences) (115). Experimental protocols had institutional approval, and animals were maintained in accordance with the American Physiological Society's Guiding Principles in the Care and Use of Animals and the Guide for the Care and Use of Laboratory Animals (National Research Council, Revised 1996).
Cell culture.
Neonatal cardiomyocytes from newborn Wistar animals were prepared using collagenase IV, as we have previously described (22, 23). Following preparation cells were plated overnight in LG-DMEM + 10% FBS+ 0.1 mmol/l BrdU + 1.0 mmol/l d-valine overnight before switching to the experimental media (LG-DMEM + 1% FBS + 1x NEAA + 2 mmol/l glutamine) and glucose set to 5.5, 16.5, or 33.0 mmol/l for up to 13 days. Media was changed on alternate days and where indicated drug additions made at that time. Media osmolarity was balanced using mannitol.
Cellular and mitochondrial function.
ATP production was measured by the CellTiter-Glo luminescent assay (Promega, Madison WI). GSH levels were measured using the GSH-Glo kit (Promega, Madison, WI). Cytochrome oxidase (complex IV) was measured by the oxidation of reduced cytochrome c as we have previously described (80). To separate the subsarcolemmal fraction (SSF) and intermyofibrillar fraction (IMF), the homogenized tissue was centrifuged (800 g, 10 min at 4°C times 1). The supernatant was passed through a stainless steel strainer and the SSF mitochondria were collected by centrifugation (10,000 g, 10 min at 4°C). The pellet from the 800 g spin was resuspended in a Nagarse buffer and kept on ice for 10 min (100 mmol/l KCl, 50 mmol/l MOPS pH 7.4, 2 mmol/l EGTA, 1 mmol/l ATP, 0.2% BSA, and 50 μg/ml Nagarse). The IMF mitochondria were then isolated by centrifugation (800 g, 10 min at 4°C times 1, 10,000 g 10 min at 4°C times 1). Following isolation both mitochondrial fractions were resuspended in buffer (100 mmol/l KCl, 10 mmol/l MOPS pH 7.4, 0.2% BSA).
Glucose-6-phosphate dehydrogenase (G6Pdh) activity was measured as described by Sepillion et al. (105). ROS production was examined by measurement of hydrogen peroxide (H2O2) or superoxide ion production. H2O2 production was determined by the ABTS-HRP spectrophotometric method described by Higuchi et al. (49). Superoxide generation was determined using 5 μmol/l lucigenin chemiluminescence protocol as previously described (40). To estimate the potential contributions of different sources of superoxide, NAD(P)H and NADH oxidase activities were measured in the presence of 200 μmol/l NAD(P)H or 200 μmol/l NADH, respectively. Mitochondrial superoxide was determined using 2.5 μmol/l MitoSox (Invitrogen, Carslbad, CA). The development of fluorescence was measured using a Tecan M200 plate reader (ex/em: 510/580). Values presented are means ± SE and normalized to control (5.5 mmol/l glucose) of arbitrary optical density or fluorescence units. Measurement of cytosolic ROS was made using dihydroethidium (DHE). In brief, cells were loaded with 20 μM DHE for 30 min at 37°C, leaving one well blank as a negative control. Cells were washed with PBS before being trypsinized and transferred to a centrifuge tube. Cells were washed twice in PBS by pelleting them at 800 g for 5 min. Cells were analyzed using a Guava EasyCyte Mini (Millipore, Billerica, MA). DHE was detected in the PM2 (red) channel and gates were set using the unstained cells and excluded cells less than 10 μm.
Mitochondrial isolation.
Mitochondria were isolated by differential centrifugation as described previously (21, 80). In brief, tissues from animals were minced using fine scissors before being put into the dounce homogenizer. Cultured cells were collected in ice-cold PBS and centrifuged (300 g, 5 min at 4°C). The minced tissues or cell pellets were suspended in mitochondrial isolation buffer (250 mmol/l sucrose, 10 mmol/l Tris-Cl pH 7.5, 1 mmol/l EDTA, 1 mmol/l EGTA, 1.5 mmol/l MgCl2, 10 mmol/l KCl, 1 mmol/l DTT, 1 mmol/l PMSF, 1× protease inhibitors; Sigma P-8340) and homogenized using a dounce homogenizer (ten strokes “A” pestle and 10 strokes “B” pestle) on ice. The extracts were then subjected to successive rounds of centrifugation [1) 300 g, 5 min at 4°C times 3; 2) 1,000 g, 5 min at 4°C times 2; 3) 2,000 g 5 min at 4°C times 1; 4) 13,000 g, 10 min at 4°C times 3]. Mitochondrial pellets were resuspended in buffer (50 mmol/l Tris-Cl pH 7.5, 0.5 mmol/l EDTA, 0.5 mmol/l EGTA, 1 mmol/l DTT, 10% glycerol, 1× protease inhibitors) and lysed on ice for 30 min before protein concentration was determined by the Bradford method (Bio-Rad, Hercules CA).
mtDNA damage.
Separate protocols were used to evaluate mtDNA integrity: 1) long-range PCR (LRPCR) to assess DNA strand breaks (21), 2) mitochondrial copy number, 3) reverse random polynucleotide polymorphism analysis to identify specific sequence changes, and 4) TTGE analysis to scan for sequence changes. Total DNA was extracted from cultured cardiomyocytes or 1–10 mg left ventricle using Sigma Extract-n-Amp (Sigma, St. Louis, MO) or DirectPCR (Viagen, Los Angeles, CA). The first approach used a LRPCR protocol to assess mtDNA damage by real-time QPCR using a Stratagene 3000Mx as we have previously described (21, 80). In brief, any lesion (strand breaks, base modifications, and apurinic sites) will stop a thermostable DNA polymerase capable of generating a long DNA product (21). The Ct derived from this amplification was compared with the Ct of a short PCR (SRPCR) product (150–250 bp) that is unlikely to contain any lesions. SRPCR was done using a Brilliant QPCR Master Mix, while LRPCR was done using PfuUltra II Fusion HS DNA polymerase (Stratagene, La Jolla, CA). The primers for the LRPCR reaction were 5'-GCCAGGACCAAACCTTT GTGTTTA-3' forward and 5'-GGACTAGCC CATTCACTAC-3' reverse; other primers used were as previously described (21). Quantification of mtDNA damage and mitochondrial copy number were derived by the 2ΔCt method, from the comparison of LRPCR:SRPCR and SRPCR:β-actin, respectively. To determine if specific sites were altered, a reverse random polynucleotide polymorphism analysis (rRFLP) was used. The rationale for this approach is that any diabetic-induced alteration of an enzyme recognition sequence will blunt its restriction and increase yield upon QPCR amplification. LV DNA was prepared as described above and cut with Bsr1 or MboI. An aliquot of the restriction digest was taken and primers amplifying across the cytochrome B (Bsr1) or D-Loop (Mbo1) regions of mtDNA were used in a QPCR protocol. The goal of this approach was to determine if glucose-induced damage to mtDNA resulted in nucleotide substitutions in the sequence recognized by the restriction enzyme. Quantification of the restriction site integrity was derived by the 2ΔCt method, from the comparison of cut:uncut paired samples.
Temporal temperature gradient analysis (TTGE).
A PCR reaction product was amplified that included a small portion of the mitochondrial genome. The PCR products are loaded onto a denaturing polyacrylamide (5% acrylamide/9 mol/l urea) gel, and during electrophoresis the temperature of the gel is increased at a fixed rate (70 V; 20 h/ 48–66°C at 1°C/h). Primers were used to amplify a 543-bp fragment within the 3' end of subunit III of cytochrome oxidase. Following electrophoresis, the bands were cutout, and the DNA extracted and used as a template for another round of PCR amplification to derive sufficient material to permit sequencing. Sequencing of DNA was outsourced to Genewiz (Plainfield, NJ).
Mitochondrial topoisomerase DNA assay.
Two separate assays were performed to determine DNA cleavage and DNA relaxation activity. DNA cleavage was determined by degradation of linear DNA as described by Kao et al. (56) and modified from our previous description (80) to use a Cy5-fluorescent probe. In brief, a linear mtDNA was amplified by a PCR reaction using an internally labeled Cy5-labeled primer, forward 5'-AAATTTCCCGACACAAAATCTT TCC(Cy5)TCCTAACTA AACCCTCTTTACTTGC-3' and reverse 5'-ctcttggtaagtaaatttctttctcc-3', using mtDNA as the template to generate a 1274-bp probe. Isolated mitochondria (1–10 μg protein) were incubated in buffer (50 mmol/l Tris-Cl pH 7.5, 100 mmol/l KCl, 0.5 mM EDTA, 0.5 mmol/l DTT, 30 μg/ml BSA) to a final volume of 20 μl. The reaction was cooled on ice and at time zero the labeled DNA was added. The reaction was incubated for 30 min at 37°C to which 4 μl loading buffer was added. Ten microliters was electrophoresed on a 4% polyacrylamide gel and visualized using a Storm 840 PhosphoImager (Molecular Dynamics, Sunnyvale, CA). Band density was quantified using AlphaEaseFC software (AlphaInnotech, San Leando, CA). For topoisomerase inhibitor experiments, 10 μg mitochondrial extracts were incubated for 60 min at 37°C with those reagents (60 μmol/l M H2O2, 100 μmol/l hydroxycamptothecin, 0.5 mmol/l doxorubicin) or vehicle before the addition of DNA and then incubated for a further 30 min at 37°C. The DNA relaxation protocol was modified from that described by Low et al. (72). In brief, mitochondrial extracts (1–10 μg) were incubated in buffer (50 mM Tris-Cl pH 8.0, 120 mM KCl, 10 mM MgCl2, 0.5 mM ATP, 0.5 mM DTT) and to this 500 ng supercoiled pBR322 plasmid was added to a total volume of 20 μl. The mixture was incubated at 37°C for 10 min and the reaction stopped by the addition of 4 μl of a 5% sarcosine/6× SDS buffer and the reaction placed on ice until 5 μl was loaded onto a 1% agarose/TAE gel. Bands were imaged at 260 nm using a ChemiImager 5550 (AlphaInnotech, San Leando, CA).
Western blot analysis.
Western blot analysis on cell homogenates was performed as previously described (98): the primary antibody COX 4 subunit (1:1,000 dilution: Proteintech, Chicago, IL), with the secondary antibody being an anti-rabbit HRP (1:4,000 dilution; Amersham, Buckinghamshire, UK), and the primary antibody COX 3 subunit (1:500 dilution: Santa Cruz Biotech, Santa Cruz, CA), with the secondary antibody being an anti-goat HRP (1:5000 dilution: Santa Cruz Biotech), used in combination with a Pierce ECL kit. For band quantification, care was taken to ensure that band density remained within the linear range of the film and did not saturate the film, by performing exposures of different times. Band density was quantified using AlphaEaseFC software (AlphaInnotech, San Leando, CA).
Statistical analysis.
ANOVA analyses were performed using NCSS Software (NCSS, Kaysville, UT). Post hoc analysis was done using a Fisher's LSD analysis. Values presented are means ± SE, and statistical significance was set at P < 0.05, unless otherwise indicated.
RESULTS
Thirteen days of elevated glucose presentation compromised cardiomyocyte function as reflected by significant decreases in ATP production and cytochrome oxidase activity (Fig. 1). In conjunction with this, we observed a significant increase in mtDNA damage (Fig. 1). Using primers to amplify across the length of the mtDNA we did not observe distinct deletion products in the LRPCR products from glucose challenged cardiomyocytes. These findings indicate that glucose induced mitochondrial dysfunction in cardiomyocytes and that the balance between mtDNA repair and mtDNA damage shifted to the latter.
Fig. 1.

Thirteen days of elevated glucose increased mitochondrial DNA (mtDNA) damage. Cardiomyocytes were exposed to 5.5, 16.5, or 33 mM glucose in DMEM/1% FBS for 13 days. Measurements were made as described in methods. mtDNA damage was assessed using the long-range PCR (LRPCR) protocol. Values are normalized to 5.5 mM glucose and are means ± SE. *P < 0.05 compared with the 5.5 mM control.
Chronically elevated glucose compromised the oxidant buffering capacity of the cardiomyocytes as reflected by a significant decrease in the amount of reduced GSH determined and this was followed by significant increases in MitoSox fluorescence (Fig. 1). In the diabetic heart, glucose usage has been shown to shift away from glycolysis and toward an increased flux through the pentose phosphate pathway (1, 8, 70, 84, 105, 118). In agreement with those reports, we observed that 13 days of elevated glucose presentation resulted in a significant increase in cardiomyocyte glucose-6-phosphate dehydrogenase (G6PdH) activity (Fig. 2A) and to a lesser extent in H9c2 cells (5.5 mM glucose, 100 ± 5.0%; 33 mM glucose, 115.3 ± 4.8%; P < 0.05). The inclusion of dehydroepiandrosterone (DHEA), an inhibitor of the G6PdH, blocked glucose-induced increases in cytosolic ROS (Fig. 2B). DHEA appeared to have a protective effect and blocked glucose-induced mtDNA damage (Fig. 2C) and restored cardiomyocyte ATP production (Fig. 2D). Similar to DHEA, 6-aminonicotinamide, a G6PdH competitive inhibitor, restored ATP production in hyperglycemic cardiomyocytes (data not shown). Left ventricle (LV) G6PdH activity was significantly increased in the diabetic GK LV compared with the Wistar LV (Fig. 3A). G6PdH is the first and rate limiting step in the pentose shunt pathway, and increased NADPH generation is a product of this reaction. Previously, we have reported that NADPH oxidase-dependent superoxide production was significantly increased in the aorta of GK rats (42). Similar to that finding, we observed a significant increase in myocardial NADPH-driven chemiluminescence generation (Fig. 3B) as well as H2O2 production (Fig. 3C). In contrast, NADH-driven chemiluminescence generation (Fig. 3B) was significantly depressed. These findings suggest that extramitochondrial superoxide generation provided a significant source of oxidant stress in the diabetic heart.
Fig. 2.

Elevated glucose increased glucose-6-phosphate activity to increase cytosolic reactive oxygen species (ROS) and induce mtDNA damage. Cardiomyocytes were maintained in DMEM/1% FBS and exposed to 5.5, 16.5, or 33 mM glucose for 13 days. A: elevated glucose increased glucose-6-phosphate dehydrogenase (G6PdH) activity in cardiomyocytes. B: dehydroepiandrosterone (DHEA) counteracted glucose-induced increases in cytosolic ROS. Cells were treated with 20 μM DHE for 30 min before being washed, and cells greater than 10 μm were counted in a Guava Easycyte Cytometer. C: DHEA prevented mtDNA damage from increased glucose exposure. mtDNA damage was assessed using the LRPCR protocol. D: DHEA restored ATP production in glucose-challenged cardiomyocytes. Values are normalized to 5.5 mM glucose or 5.5. mM vehicle and are means ± SE. *P < 0.05 compared with the 5.5 mM glucose control; #P < 0.05 compared with 33 mM glucose/vehicle.
Fig. 3.
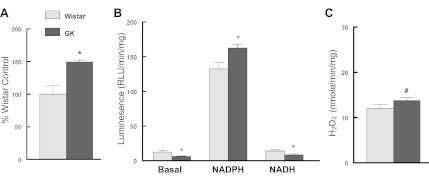
A: diabetes increased glucose-6-phosphate dehydrogenase activity in the diabetic left ventricle (LV). B: diabetes increased NADPH-driven chemiluminescence, but not NADH-driven chemiluminescence, in the diabetic LV. C: diabetes increased LV H2O2 production. LV extracts were prepared and analyzed for activity as described in methods. G6PdH was analyzed following the protocol of Serpillon et al. (105), while superoxide was determined by a lucigenin chemiluminescence protocol (40). GK, Goto-Kakizaki. Values are mean ± SE' *P < 0.05, #P < 0.10 compared with the respective control.
Mitochondrial dysfunction has been observed in both animal models of diabetes and in diabetic patients (86). In agreement with those observations, cytochrome oxidase activity was significantly decreased in LV extracts as well as in both the SSF and IMF mitochondrial fractions (Fig. 4A). In conjunction with this, we observed significant decreases in cytochrome oxidase subunit IV (nuclear encoded) and subunit III (mitochondrial encoded) from the GK LV (Fig. 4B).
Fig. 4.
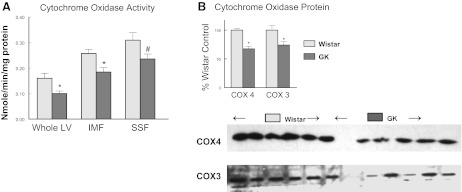
Diabetes decreased cytochrome oxidase activity and subunit 3 and 4 expression in the diabetic LV. A: cytochrome oxidase activity was determined by oxidation of reduced cytochrome c as described in methods (98). SSF, subsarcolemmal fraction; IMF, intermyofibrillar fraction. B: quantification of subunit 3 and 4 in LV. Values are means ± SE: *P < 0.05, #P < 0.10 compared with Wistar control.
Nomiyama et al. (86) reported that the mtDNA mutation rate was significantly higher in diabetic patients than in healthy individuals. Similarly, in both H9c2 cells (80) and neonatal cardiomyocytes (Fig. 1), mtDNA damage was significantly increased as a function of elevated glucose presentation. In vivo a significant increase in mtDNA damage was also observed in the diabetic GK LV (Fig. 5A), without a change in mitochondrial copy number (Fig. 5B). Although mitochondrial deletions have been reported in models of aging, we did not see any consistent evidence of deletions in the GK LV (Fig. 5C). Using a reverse RFLP approach with the restriction endonucleases Bsr1 (the cytochrome B region) or MboI (D-Loop region), we did not find any evidence of diabetic-induced sequence specific alterations (data not shown).
Fig. 5.
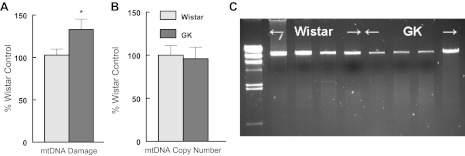
A: diabetes damages mtDNA in GK left ventricle compared with Wistar LV. mtDNA damage was assessed using the LRPCR protocol as described in methods. B: diabetes did not influence LV mitochondrial copy number. mtDNA copy number by PCR and is the ratio of mtDNA:actin DNA. Values are normalized to Wistar control and are means ± SE *P < 0.05. C: representative LRPCR mtDNA products from Wistar and GK LV.
DNA damage may take several forms including nucleotide substitution, insertions, deletions, or breakpoints as our LRPCR approach has focused on. Several methodologies have been applied in an attempt to permit comprehensive scanning of the mitochondrial genome. These include RFLP, SSCP, allele-specific oligonucleotide dot blot hybridization, or denaturing gradient gel electrophoresis (DGGE) analyses. More recently, temporal temperature gradient electrophoresis (TTGE) has been developed. A small section of the mitochondrial genome is amplified and during electrophoresis the temperature of the gel is increased at a fixed rate. This brings about strand dissociation and allows for resolution of DNA fragments that differ by as little as a single nucleotide substitution. This approach permits screening for either homoplasmic or heteroplasmic mutations (11, 85). Using a TTGE analysis, no changes in mtDNA sequence were observed in the six Wistar LV samples analyzed. In contrast, four of six GK LV exhibited mtDNA mutations within the COX 3 subunit region. Three of four GK LV samples had mutations that included base pair substitutions or insertions which altered the 3' end of the COX3 coding sequence resulting in early termination. In a fourth animal (GK355), a conservative single amino acid substitution was observed exchanging a glutamine to threonine, both of which are polar uncharged amino acids. Table 1 illustrates the translated consequences of the mtDNA changes for the cytochrome oxidase subunit III protein sequence.
Table 1.
TTGE analysis of cytochrome oxidase subunit III
| n | Fasting Glucose, mol/l | Total Errors in COX III | Average Errors per Animal | No. of Animals With Errors | |
|---|---|---|---|---|---|
| Wistar | 6 | 4.17 ± 0.11 | 0 | 0 | 0 |
| GK | 6 | 7.11 ± 0.44 | 13 | 2.2 | 4/6 |
| Wild type | |||||
| MTHQTHAYHMVNPSPWPLTGALSALLLTSGLVMWFHYNSTILLSLGLLTNILTMYQWWRDIIREGTYQGHHTPIVQKGLRY | |||||
| GMILFIVSEVFFFAGFFWAFYHSSLVPTHDLGGCWPPTGITPLNPLEVPLLNTSVLLASGVSITWAHHSLMEGNRNHMNQALLITILLG | |||||
| YFTILQASEYFETSFSISDGIYGSTFFMATGFHGLHVIIGSTFLIVCLLRQLKFHFTSKHHFGFEAAAWYWHFVDVVWLFLYVSIYWWG | |||||
| SYS | |||||
| Goto-Kakizaki: | |||||
| GK312 | |||||
| MTHQTHAYHMVNPSPWPLTGALSALLLTSGLVMWFHYNSTILLSLGLLTNILTMYQWWRDIIREGTYQGHHTPIVQKGLRY | |||||
| GMILFIVSEVFFFAGFFWAFYHSSLVPTHDLGGCWPPTGITPLNPLEVPLLNTSVLLASGVSITWAHHSLMEGNRNHMNQGPTNHHSL | |||||
| GK322 | |||||
| MTHQTHAYHMVNPSPWPLTGALSALLLTSGLVMWFHYNSTILLSLGLLTNILTMYQWWRDIIREGTYQGHHTPIVQKGLRY | |||||
| GMILFIVSEVFFFAGFFWAFYHSSLVPTHDLGGCWPPTGITPLNPLEVPLLNTSVLLASGVSITWAHHSLMEGNRSHMSQALLITILLG | |||||
| LYFTILQASKMFRNIIFYL | |||||
| GK318 | |||||
| MTHQTHAYHMVNPSPWPLTGALSALLLTSGLVMWFHYNSTILLSLGLLTNILTMYQWWRDIIREGTYQGHHTPIVQKGLRY | |||||
| GMILFIVSEVFFFAGFFWAFYHSSLVPTHDLGGCWPPTGITPLNPLEVPLLNTSVLLASGVSITWAHHSLMEGNRNHMNQALLITILLG | |||||
| YFTILQASEYFETSFSISDGIYGSTFFMATGFHGLHVIIGSTFLIVCLLRTTKIPLHIKTSFRIWSRSMMLTLR | |||||
| GK355 | |||||
| MTHQTHAYHMVNPSPWPLTGALSALLLTSGLVMWFHYNSTILLSLGLLTNILTMYQWWRDIIREGTYQGHHTPIVQKGLRY | |||||
| GMILFIVSEVFFFAGFFWAFYHSSLVPTHDLGGCWPPTGITPLNPLEVPLLNTSVLLASGVSITWAHHSLMEGNRNHMNQALLITILLG | |||||
| YFTILQASEYFETSFSISDGIYGSTFFMATGFHGLHVIIGSTFLIVCLLRTLKFHFTSKHHFGFEAAAWYWHFVDVVWLFLYVSIYWWG | |||||
| SYS | |||||
GK, Goto-Kakizaki diabetic rat; TTGE, temporal temperature gradient analysis. Italicized and underlined sequences are the altered amino sequence resulting from the altered mtDNA sequence.
Topoisomerases have complex reaction mechanisms in which the enzyme covalently binds to the DNA and induces strand breakage before DNA torsion stress is relieved. This is followed by strand religation of the DNA before the enzyme disengages. Relief of torsional stress is demonstrated in vitro by the DNA relaxation assay using supercoiled plasmid DNA. As shown in Fig. 6A, the mitochondrial extracts generated the same isopane pattern of relaxation as purified nuclear topoisomerase 1 (lane 4). When isolated mitochondria were passed through a G200 Sephadex column, DNA cleavage assay of the size-separated fractions found a significant increase in DNA cleavage activity in the 150- to 160-kDa fractions corresponding to mitochondrial topoisomerase IIβ (data not shown). Many topoisomerase inhibitors increase DNA strand breaks without a concomitant increase in ligation as detected by the DNA cleavage activity. When isolated mitochondria were incubated in the presence of the topoisomerase inhibitors hydroxycamptothecin (type 1) or doxorubicin (type 2), both drugs enhanced myocardial mitochondrial topoisomerase DNA cleavage activity (Fig. 6, B and C). Diabetes induced significant increases in mitochondrial topoisomerase DNA cleavage activity in mitochondrial extracts derived from the diabetic GK LV compared with control LV extracts (Fig. 6D). Although not specific for any of the three unique mitochondrial topoisomerases, the increases in DNA cleavage suggest that mitochondrial topoisomerase function was collectively altered by diabetes.
Fig. 6.

A: DNA relaxation of supercoiled plasmid DNA. Lane 1, no extract; lane 2, 1.0 μg mitochondrial extract; lane 3, 0.5 μg mitochondrial extract; lane 4, 2.5 U purified topoisomerase 1; lane 5, linearized pBR322. B: 10 μM hydroxycamptothecin exacerbated DNA cleavage of LV mitochondrial extracts. Values are means ± SE and are normalized to no mitochondria (No Mito) control. *P < 0.05 compared with No Mito control; #P < 0.05 compared with mitochondria alone (+Mito). C: 0.5 mM doxorubicin exacerbated DNA cleavage of LV mitochondrial extracts. Values are means ± SE and are normalized to No Mito. D: diabetes increased myocardial mitochondrial topoisomerase DNA cleavage activity. Myocardial mitochondrial extracts were incubated with linear mtDNA for 30 min before being electrophoresed, and cleavage products were quantified by band intensity. Values are means ± SE and are normalized to Wistar DNA cleavage. *P < 0.05 compared with Wistar LV mitochondrial extract.
Cai et al. (7) reported an increase in cytosolic oxidant stress soon after exposure to increased glucose. Li et al. (68) reported that oxidant stress altered nuclear topoisomerase 1 function to depress the re-ligation step. This suggests that with diabetes the exaggerated hyperglycemic response to a meal may be further prolonged by an oxidant-induced alteration of mitochondrial topoisomerase function. Alone 60 μM H2O2 did not alter DNA cleavage indicating that this concentration was below the threshold for a direct effect on DNA. Also it was unlikely that a Fenton reaction participated since the reactions were performed in the presence of the iron chelator, EDTA. When mitochondrial extracts were incubated in the presence of 60 μM H2O2, mitochondrial topoisomerase DNA cleavage was significantly increased beyond that of cleavage in the absence of H2O2 (Fig. 7A). Topoisomerase IIB has known phosphorylation sites and that phosphorylation of nuclear topoisomerase IIB significantly altered its activity (73, 76, 129). These sequences are conserved in the mitochondrial topoisomerase IIB. Using alkaline phosphatase to modify the phosphorylation state of mitochondrial topoisomerases, we observed that DNA cleavage activity was significantly decreased compared with vehicle-treated mitochondria (Fig. 7B). This suggests that phosphorylation state may also regulate mitochondrial topoisomerase function.
Fig. 7.

Regulation of myocardial mitochondrial topoisomerase activity. A: H2O2 increases mitochondrial topoisomerase-dependent DNA cleavage. Mitochondria were preincubated in the absence or presence of 60 μM H2O2 prior to the addition of DNA. *P < 0.05 compared with respective no mitochondria/H2O control; #P < .0.05 compared with +mitochondria/H2O. B: dephosphorylation alters mitochondrial topoisomerase DNA cleavage activity. No Mito: no extract; +Mito: untreated mitochondria; CIP trt, mitochondria treated with 1 U calf intestinal phosphatase for 30 min at 37°C before DNA was added. Values are means ± SE and are normalized to no Mito or no Mito/H2O; *P < 0.05 compared with No Mito; #P < 0.05 compared with untreated mitochondria.
DISCUSSION
Mitochondrial dysfunction has a significant role in the development and complications of DCM (32, 66, 82, 92). The major findings of our studies are that in the diabetic heart, separate from a direct impact of oxidative stress on mtDNA, ROS-induced alteration of mitochondrial topoisomerase activity propagates an increase in mtDNA damage. These findings are significant in that the activation/inhibition state of the mitochondrial topoisomerases will have important consequences for mtDNA integrity and maintaining myocardial function.
The GK rat is a nonobese model of type II diabetes that is essentially a model of hyperglycemia without significant hyperlipidemia or hypercholesterolemia. The GK rats develop elevated fasting glucose, exhibit decreased glucose intolerance, and increased HbA1c levels at an early age (37, 40, 42, 124, 127). GK rats present with many diabetes-related complications observed in human diabetic patients, including reduced nerve conduction velocity and progressive renal involvement (53, 83, 93, 115, 124–126). D'Souza reported in the GK ventricle an increase in wall thickness that was associated with increased myocyte diameter and collagen matrix proliferation as well as increased expression of the fetal gene pattern, characteristic of an adaptive response to overload (19). Although not hypertensive, the GK rats exhibit a small increase in MAP and systolic blood pressure compared with their Wistar controls (12). By echocardiography, we did not observe significant differences in the GK fractional shortening or ejection fraction under basal conditions (data not shown). However, El-Omar et al. (26) did observe that in response to a hypoxic challenge, diastolic function was significantly altered in the GK hearts, a finding consistent with DCM.
Mitochondrial-dependent ROS generation has been accepted as the singular cause of mtDNA damage in diabetes and other pathophysiological states. Several lines of evidence point toward a glucose-induced increase in mitochondrial ROS including ESR studies, image analysis using MitoSox, as well as studies using yellow fluorescent protein (cpYFP) to measure superoxide flashes (4, 7, 47, 48, 74, 78, 119). A common limitation is that they only examined changes as an early event. We and others that have examined a longer time frame have observed changes in mitochondrial ROS to be a transient event in response to chronically increased glucose or palmitate presentation Further, Herlein et al. have argued the position that in mild diabetes or prediabetes, mitochondrial superoxide may not be elevated in contrast to its decided presence in more severe diabetic states (6, 48, 78). These events point toward other pathways participating in the prediabetic state or mildly diabetic state to propagate mitochondrial dysfunction.
The role of radical oxygen species (ROS) in cellular metabolism is evolving. Historically perceived as a by-product of aerobic metabolism, the cell's ability to generate specific different oxidant species indicates a significant and useful purpose (9, 15). More recently, this has taken the concept of redox signaling and the appreciation that the cell is not a dilute solution but a crowded highly organized space (9, 33, 114). Compartmentalization of ROS is both by physical separation as well as activation of distinct enzyme complexes. Aside from the mitochondria, the NADPH oxidase system and NOS enzymes generate a significant portion of cellular ROS, while other molecules serve to act as buffers. Although clearly shown to have a destructive effect, a concentration-dependent biphasic effect of ROS is apparent within the myocardium. Low concentrations of ROS appear to serve as signaling molecules, while higher levels propagate a destructive outcome (10, 60, 65). The threshold for this biphasic effect appears in part to be dependent on the oxidant buffering capacity of the cell, as decreases in glutathione levels appear to lower the threshold for stress-induced damage within the cell (28, 78, 88, 89). GSH, an oxidant scavenger, is depleted by diabetes across different tissues and models of diabetes (5, 27, 34, 61, 106, 107, 123). Consistent with those reports we observed significant decreases in GSH as an early change that preceded increases in mitochondrial ROS or mtDNA damage. Although increased G6PdH activity might be thought to increase GSH levels, that is not the case here and suggests that depletion of oxidant buffers may be an early step in the downward spiral of mitochondrial function.
Diabetes increases flux through the pentose shunt pathway in the heart, and this may be a significant source of oxidant stress within the cytosol (43, 57, 105). Glucose-6-phosphate dehydrogenase (G6PdH) is rate limiting in this pathway and in the present study its inhibition reduced glucose-induced mtDNA damage and restored cellular ATP production. In a STZ model of diabetes, Ozdemir et al. (90) also observed significant increases in G6PdH and 6-PGD activity and these changes were reversed by treatment with the ANG II antagonist, candesartan-cilexetil. Many papers have shown that angiotensin blockade counteracts the diabetic state (3, 29, 40, 87, 90, 95, 100). Not just an endocrine but also an autocrine, elevated glucose increased ANG II expression and release from cardiomyocytes (31). Ricci et al. (99) reported an increase in mtDNA damage in response to 1 h of 1 nM ANG II in cultured cardiomyocytes. However, the increase in mtDNA damage and drop in mitochondrial ΔΨm lagged behind the rapid rise in cytosolic superoxide levels but preceded detectable increases in mitochondrial superoxide. This suggests that mtDNA damage and mitochondrial dysfunction occurred by activation of an alternative pathway or production of an alternative oxidant species. Aside from glucose-6-phosphate, the major reaction product of G6PdH is NADPH, the substrate for NADPH oxidase. Diabetes increases myocardial Nox4 expression, and its localization to the outer membrane of the mitochondria is significant (4, 38, 75). Unlike Nox1, Nox4 does not require p47 or p67 for activation, and biochemical studies suggest that it preferentially generates H2O2 over superoxide (16, 79, 108). Block et al. (4) demonstrated in mesangial cells that a siNOX4 probe abrogated glucose-induced increased superoxide generation. Both in the diabetic heart and in cultured cardiomyocytes N-acetyl-l-cysteine (NAC), a cytosolic localized antioxidant, has been reported to decrease oxidative stress markers (30, 59). These reports are all consistent with our findings that NADPH oxidase activity and cytosolic H2O2 activity were increased in the GK hearts, while NADH-driven oxidase activity was not. And it argues against mitochondrial-driven NADH oxidase as being the singular or even predominant source of superoxide in the GK heart. Collectively these findings suggest that a substantial increase in cytosolic-derived ROS that has access to the myocardial mitochondrial compartment may be the predominant source of oxidant stress in this hyperglycemic model of diabetes.
Diabetes induces many changes that are cell and tissue specific. While cardiomyocyte glycolysis is decreased with diabetes, the opposite is observed in endothelial cells (96). The results in neonatal cardiomyocytes and in diabetic hearts were similar to our earlier findings in the H9c2 cell line; chronic increases in glucose presentation led to a degradation of mitochondrial function. In that study, individual ETC complexes containing mtDNA-encoded sequences were compromised by hyperglycemia, while the Complex II (a mitochondrial complex that is wholly nuclear encoded) was not (80). Separate from hyperglycemia, chronically elevated palmitate presentation also significantly decreased ATP levels (97). Our findings are also similar to streptozotocin-induced diabetes, in which significant decreases in the levels of the mitochondrially encoded COX I or IV were observed (55). In the present study mitochondrial dysfunction was linked to the degradation of the ETC proteins.
The concept of increased mtDNA replication as a means to overcome mtDNA mutations has been advanced by a number of studies (17, 25, 52, 101, 113). However it remains unclear if this is a stable or transient event, and whether it expands or dilutes the mtDNA mutation pool. Recently Tewari et al. (110a) demonstrated that elevated glucose presentation or diabetes decreased expression of the nuclear encoded mitochondrial transcriptional machinery and that its restoration by relief of oxidant stress or by overexpression of POLG1 ameliorated mitochondrial dysfunction. Their data suggest that decreased mtDNA transcription was a function of lower expression rather than mtDNA damage within the D-Loop region. Red ragged fiber syndrome is a function of mtDNA mutations and increased copy numbers have been observed in aged muscle (36). This is thought to be an attempt by the cell to compensate for the loss of ATP generation capability. Santos et al. (102) observed that the increase in copy number was a transient event, being elevated at 2 mo but then depressed at 6 and 12 mo. In the present study we did not observe significant changes in mtDNA copy number in the GK heart. Our use of the LRPCR protocol identified DNA strand breaks or other obstructions that blocked progression of the DNA polymerases. That the Pfu DNA polymerase used in the LRPCR analysis was unable to amplify across DNA breaks has some important consequences. Both mammalian DNA and RNA polymerases are unable to read across DNA breaks (109). As well, these polymerases are also unable to progress when blocked by a topoisomerase-DNA complex (109). Thus both replication and transcription may suffer as a function of mtDNA damage and altered topoisomerase activity. Thus the differences in reported copy numbers among these studies may lie in the different models used; the STZ is a severe form of type 1 diabetes vs. GK, which is a mild form of type 2 diabetes.
The reverse RFLP approach used in the present study determined that specific sequences were unaltered by diabetes. Since this approach only scans 6 bp segments, it is likely that this approach did not have sufficient sensitivity to identify significant damage. The TTGG protocol scanned across distinct regions of mtDNA and identified point mutations that altered the primary mtDNA sequence. Importantly, we did not observe a consistent pattern of mutations or deletions. The data suggest that diabetic induced damage did not target specific sequences of the mtDNA and that damage occurred in a random rather than a patterned manner. Others have reported the presence of hepatic mtDNA deletions from alcoholic patients (77). As well, the lack of deletions is different for those studies that have examined the effects of aging on mtDNA states (36, 117). Collectively, these findings suggest that the underlying mechanisms for diabetic induction of mtDNA damage might differ from other pathophysiological states.
The changes in mtDNA sequence varied but each significantly altered the coding sequence resulting in early termination or mutation of the C-terminal region. Although our data only examined the C-terminal region of COX III, the results are likely to be reflective across much of the mtDNA. The impact of altered mtDNA primary sequence has been more dramatically demonstrated by the mtDNA-Poldef transgenic mice in which the mtDNA polymerase lacked “proofreading” capability. In those mice, cardiac cells accumulated mtDNA mutations at a rate of more than 20-fold compared with controls, had a significant increase in apoptosis, and presented with significant heart failure (130, 131). Despite this, there was not a significant change in mitochondrial function; the P/O ratio and respiratory control index were similar in mtDNA-Poldef and controls. Interestingly, markers for ROS were not increased, suggesting that oxidative stress was not an obligate mediator of mtDNA mutations. Although not diabetic, this model suggests that any increase in mtDNA mutations may serve as an underlying cause of mitochondrial dysfunction. Others have also reported that ROS production and oxidative damage were not increased in different tissues of mice with depletion (117) or point mutations of mtDNA (50, 58, 111). An alternative conclusion to those studies was that the accelerated apoptosis rid the heart of dysfunctional cells and those remaining cells did not yet have demonstrable ROS. Interestingly, the mtDNA-Poldef mice lacked significant mtDNA deletions, to which the authors argue that point mutations and not deletions are involved in the accelerated defective phenotype (20). They observed decreases in the presence of fully assembled Complex I and IV but not Complex II and our results concur. Separate from those studies, two papers examined the impact of mtDNA mutations on OXPHOS assembly (35, 91). A mutation in cytochrome b (A15533G) was observed in a patient presenting with lactic acidosis and mild mental decay. Mimicking the mutation in transmutational cybrids resulted in significant alterations in the rate of Complex assembly (35). Using a similar approach and examining mutations common to LHON (Leber's hereditary optic neuropathy), Pello et. al. observed significant differences in the rates of ETC complex assembly (91). Another considerable problem generated by point mutations is the interdependency of the mitochondrial complexes not only for their assembly but also stability within the mitochondrial membrane (2, 20, 62, 112). Collectively these studies point to the critical role that mtDNA fidelity has on the mitochondrial function.
In postmitotic cells, the mitochondrial genome is thought to replicate about once per month and resolving the topology of mtDNA replication would be almost insurmountable in the absence of the mitochondrial topoisomerases (13). Three unique topoisomerases have been demonstrated in the mitochondria; topoisomerase IIβ (a type II), mtTOP1 (a type IB), and TOP3α (a type IA) (72, 80, 120, 132). mtTOP1 is a paralog of the nuclear isoforms and likely arose by a gene duplication event. mtTOP1 has a lower DNA binding affinity, and it has been suggested that this may serve to relieve supercoiling of newly formed RNA transcripts rather than participate in mtDNA replication (14, 103). We have previously reported that immunoprecipitation of mtTOP1 topoisomerases from isolated mitochondrial preparations decreased DNA cleavage activity indicating the presence of functional mtTOP1 in the mitochondria (80). Wu et al. (122) reported that in Drosophila knockout of the mtTOP3α gene resulted in a decline of mtDNA suggesting that it participates in the maintenance of the mitochondrial genome. The functional role of mitochondrial topoisomerase IIβ also remains unclear; however, it has been suggested to participate in decatenating newly synthesized mtDNA circles (72). Because the topoisomerase IIB produces double strand breaks, it may also be responsible for the deletions observed in some tissues but not in others. In the present study, we have demonstrated that diabetes significantly increased the DNA cleavage activity of the mitochondrial topoisomerases. The data indicate the participation of all three proteins in mtDNA cleavage but at present it is not possible to determine if one mitochondrial topoisomerase dominates the diabetic-induced increases in myocardial mtDNA damage.
Several posttranslational modifications exist that may modulate enzyme function, and very little is known about regulation of mitochondrial topoisomerase. Most of what is suspected has been inferred from studies of nuclear topoisomerases. In addition to changes in glycolysis and the pentose shunt pathway function, hyperglycemia is also thought to alter the activity of the hexosamine pathway. GlcNacylation is increased by hyperglycemia/diabetes and a mitochondrially directed splice variant of O-GlcNacylation has been reported (51, 71), suggesting that this pathway may also be important in the mitochondrion. In STZ diabetes, increased TOP1 GlcNacylation was associated with a decrease in nuclear TOP1 DNA relaxation activity (67). Both protein kinase C and casein kinase I δ/ϵ activity have been shown to modulate DNA cleavage activity of nuclear topoisomerase IIα (41, 128). Incubation of alkaline phosphate (to promote dephosphorylation) decreased nuclear topoisomerase I relaxation of supercoiled (SC) DNA (110). Both Top1 and mtTop1 have a common phosphorylated site Tyr288 within the core region of the enzyme, and modification at this site has been shown to alter camptothecin sensitivity (129). Others have shown that Tyr723 phosphorylation alters its preference for supercoiled or relaxed DNA (73, 76). The present study makes similar observations in that we observed that phosphatase treatment decreased DNA cleavage activity of the mitochondrial topoisomerases, suggesting a significant role for posttranslational regulation of mitochondrial topoisomerase function.
Summary
Studies examining mitochondrial dysfunction and mtDNA damage have focused on elevated mitochondrial oxidant stress as a singular cause for diabetic cardiomyopathy as well as other pathologies such as alcoholism, cancer, neurodegeneration, and radiation-induced mitochondrial dysfunction. However, antioxidant therapy studies have yielded results that range from disappointing to a potentially detrimental effect of antioxidants (24, 69, 81, 116). Other approaches that raise or lower mitochondrial antioxidant capacity have also yielded conflicting results (63, 121). These findings point to a more complex interaction of ROS within the mitochondria and suggest that alternative pathways may mediate the effect of ROS on mitochondria and mtDNA integrity. Mitochondria do have DNA repair capability, and our findings suggest that with diabetes or chronic hyperglycemia there may be limits to functional recovery. In part this may be due to exhaustion of the endogenous oxidant buffering systems. Alternatively, it may be that a transient signal is prolonged by activation of a pathway to exacerbate the initial insult. These findings are important since heretofore it has been thought that mtDNA damage was only as a result of a direct attack of ROS on mtDNA. Our finding of only transient increases in mitochondrial ROS levels in the H9c2 cell line suggested activation of alternative processes (80). Separate from a direct impact of oxidative stress on mtDNA, ROS-induced alteration of myocardial mitochondrial topoisomerase activity accelerated and propagated increases in mtDNA damage. These observations also now include the diabetic-induced increases in mitochondrial topoisomerase activity derived from the diabetic GK left ventricle. These findings are significant in that the activation/inhibition state of the mitochondrial topoisomerases will have important consequences for mtDNA integrity and the well being of the myocardium. Topoisomerases are the focal point for many antibiotic and antineoplastic reagents, and their regulation is central to the clinical management of different diseases. Understanding the regulation of the mitochondrial topoisomerases is critical for protection of myocardial mtDNA, not only for the management of diabetes but also for the many other clinical treatments that target the topoisomerases.
Innovation
Mitochondrial oxidant stress has been widely accepted as the singular cause of diabetic-induced mtDNA damage leading to mitochondrial dysfunction. The present manuscript takes exception to this idea on two levels. First, separate from a direct impact of oxidative stress on mtDNA, ROS-induced alteration of myocardial mitochondrial topoisomerase function accelerated and propagated increases in mtDNA damage. Second, in this model of type II diabetes, the cytosol generates significant oxidant stress separate from and in addition to the mitochondrial sources.
GRANTS
This study was supported in part by National Institutes of Health Grants HD-065551 (J. G. Edwards), HL-43023 (J. G. Edwards), and HL-08535206 (S. A. Gupte) and by the New York Medical College Research Endowment Fund (J. G. Edwards).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: S.H., N.L., B.P., D.L., J.M., and J.G.E. performed experiments; S.H., N.L., B.P., D.L., J.M., and J.G.E. analyzed data; S.H., N.L., and J.G.E. interpreted results of experiments; S.H., S.A.G., and J.G.E. edited and revised manuscript; S.H., N.L., B.P., D.L., J.M., S.A.G., and J.G.E. approved final version of manuscript; J.G.E. conception and design of research; J.G.E. prepared figures; J.G.E. drafted manuscript.
REFERENCES
- 1. Aasum E, Hafstad AD, Severson DL, Larsen TS. Age-dependent changes in metabolism, contractile function, and ischemic sensitivity in hearts from db/db mice. Diabetes 52: 434–441, 2003 [DOI] [PubMed] [Google Scholar]
- 2. Acin-Perez R, Bayona-Bafaluy MP, Fernandez-Silva P, Moreno-Loshuertos R, Perez-Martos A, Bruno C, Moraes CT, Enriquez JA. Respiratory complex III is required to maintain complex I in mammalian mitochondria. Mol Cell 13: 805–815, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alfarano C, Sartiani L, Nediani C, Mannucci E, Mugelli A, Cerbai E, Raimondi L. Functional coupling of angiotensin II type 1 receptor with insulin resistance of energy substrate uptakes in immortalized cardiomyocytes (HL-1 cells). Br J Pharmacol 153: 907–914, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Block K, Gorin Y, Abboud HE. Subcellular localization of Nox4 and regulation in diabetes. Proc Natl Acad Sci USA 106: 14385–14390, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bouderba S, Sanz MN, Sanchez-Martin C, El-Mir MY, Villanueva GR, Detaille D, Koceir EA. Hepatic mitochondrial alterations and increased oxidative stress in nutritional diabetes-prone Psammomys obesus model. Exp Diabetes Res 2012: 430176 doi:10.1155/2012/430176. [Epub 2012 May 17] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, Aziz S, Johnson JI, Bugger H, Zaha VG, Abel ED. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes 56: 2457–2466, 2007 [DOI] [PubMed] [Google Scholar]
- 7. Cai L, Li W, Wang G, Guo L, Jiang Y, Kang YJ. Hyperglycemia-induced apoptosis in mouse myocardium: mitochondrial cytochrome C-mediated caspase-3 activation pathway. Diabetes 51: 1938–1948, 2002 [DOI] [PubMed] [Google Scholar]
- 8. Carley AN, Severson DL. Fatty acid metabolism is enhanced in type 2 diabetic hearts. Biochim Biophys Acta 1734: 112–126, 2005 [DOI] [PubMed] [Google Scholar]
- 9. Chen K, Craige SE, Keaney JF., Jr Downstream targets and intracellular compartmentalization in Nox signaling. Antioxid Redox Signal 11: 2467–2480, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen QM, Tu VC, Wu Y, Bahl JJ. Hydrogen peroxide dose dependent induction of cell death or hypertrophy in cardiomyocytes. Arch Biochem Biophys 373: 242–248, 2000 [DOI] [PubMed] [Google Scholar]
- 11. Chen TJ, Boles RG, Wong LJ. Detection of mitochondrial DNA mutations by temporal temperature gradient gel electrophoresis. Clin Chem 45: 1162–1167, 1999 [PubMed] [Google Scholar]
- 12. Cheng ZJ, Vaskonen T, Tikkanen I, Nurminen K, Ruskoaho H, Vapaatalo H, Muller D, Park JK, Luft FC, Mervaala EM. Endothelial dysfunction and salt-sensitive hypertension in spontaneously diabetic Goto-Kakizaki rats. Hypertension 37: 433–439, 2001 [DOI] [PubMed] [Google Scholar]
- 13. Cortopassi G, Wang E. Modelling the effects of age-related mtDNA mutation accumulation; complex I deficiency, superoxide and cell death. Biochim Biophys Acta 1271: 171–176, 1995 [DOI] [PubMed] [Google Scholar]
- 14. Dalla Rosa I, Goffart S, Wurm M, Wiek C, Essmann F, Sobek S, Schroeder P, Zhang H, Krutmann J, Hanenberg H, Schulze-Osthoff K, Mielke C, Pommier Y, Boege F, Christensen MO. Adaptation of topoisomerase I paralogs to nuclear and mitochondrial DNA. Nucleic Acids Res 37: 6414–6428, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Darwin C. The Origins of Species. London: John Murray, 1859 [Google Scholar]
- 16. Dikalova AE, Bikineyeva AT, Budzyn K, Nazarewicz RR, McCann L, Lewis W, Harrison DG, Dikalov SI. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ Res 107: 106–116, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dillon LM, Williams SL, Hida A, Peacock JD, Prolla TA, Lincoln J, Moraes CT. Increased mitochondrial biogenesis in muscle improves aging phenotypes in the mtDNA mutator mouse. Hum Mol Genet 21: 2288–2297, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Domenech E, Gomez-Zaera M, Nunes V. Wolfram/DIDMOAD syndrome, a heterogenic and molecularly complex neurodegenerative disease. Pediatr Endocrinol Rev 3: 249–257, 2006 [PubMed] [Google Scholar]
- 19. D'Souza A, Howarth FC, Yanni J, Dobryznski H, Boyett MR, Adeghate E, Bidasee KR, Singh J. Left ventricle structural remodelling in the prediabetic Goto-Kakizaki rat. Exp Physiol 96: 875–888 [DOI] [PubMed] [Google Scholar]
- 20. Edgar D, Shabalina I, Camara Y, Wredenberg A, Calvaruso MA, Nijtmans L, Nedergaard J, Cannon B, Larsson NG, Trifunovic A. Random point mutations with major effects on protein-coding genes are the driving force behind premature aging in mtDNA mutator mice. Cell Metab 10: 131–138, 2009 [DOI] [PubMed] [Google Scholar]
- 21. Edwards JG. Quantification of mitochondrial DNA (mtDNA) damage and error rates by real-time QPCR. Mitochondrion 9: 31–35, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Edwards JG, Bahl J, Flink I, Cheng S, Morkin E. Thyroid hormone influences expression of the beta myosin heavy chain gene. Biochem Biophys Res Commun 199: 1482–1488, 1994 [DOI] [PubMed] [Google Scholar]
- 23. Edwards JG, Bahl JJ, Flink I, Milavetz J, Morkin E. A repressor region in the human beta myosin heavy chain gene that has a partial position dependency. Biochem Biophys Res Commun 189: 504–510, 1992 [DOI] [PubMed] [Google Scholar]
- 24. Eidelman RS, Hollar D, Hebert PR, Lamas GA, Hennekens CH. Randomized trials of vitamin E in the treatment and prevention of cardiovascular disease. Arch Intern Med 164: 1552–1556, 2004 [DOI] [PubMed] [Google Scholar]
- 25. Ekstrand MI, Falkenberg M, Rantanen A, Park CB, Gaspari M, Hultenby K, Rustin P, Gustafsson CM, Larsson NG. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum Mol Genet 13: 935–944, 2004 [DOI] [PubMed] [Google Scholar]
- 26. El-Omar MM, Yang ZK, Phillips AO, Shah AM. Cardiac dysfunction in the Goto-Kakizaki rat. A model of type II diabetes mellitus. Basic Res Cardiol 99: 133–141, 2004 [DOI] [PubMed] [Google Scholar]
- 27. Erejuwa OO, Sulaiman SA, Wahab MS, Sirajudeen KN, Salleh MS, Gurtu S. Differential responses to blood pressure and oxidative stress in streptozotocin-induced diabetic Wistar-Kyoto rats and spontaneously hypertensive rats: effects of antioxidant (honey) treatment. Int J Mol Sci 12: 1888–1907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Esposti MD, Hatzinisiriou I, McLennan H, Ralph S. Bcl-2 and mitochondrial oxygen radicals. New approaches with reactive oxygen species-sensitive probes. J Biol Chem 274: 29831–29837, 1999 [DOI] [PubMed] [Google Scholar]
- 29. Ferriss JB, O'Hare JA, Kelleher CC, Sullivan PA, Cole MM, Ross HF, O'Sullivan DJ. Diabetic control and the renin-angiotensin system, catecholamines, and blood pressure. Hypertension 7: II58–II63, 1985 [DOI] [PubMed] [Google Scholar]
- 30. Fiordaliso F, Bianchi R, Staszewsky L, Cuccovillo I, Doni M, Laragione T, Salio M, Savino C, Melucci S, Santangelo F, Scanziani E, Masson S, Ghezzi P, Latini R. Antioxidant treatment attenuates hyperglycemia-induced cardiomyocyte death in rats. J Mol Cell Cardiol 37: 959–968, 2004 [DOI] [PubMed] [Google Scholar]
- 31. Fiordaliso F, Leri A, Cesselli D, Limana F, Safai B, Nadal-Ginard B, Anversa P, Kajstura J. Hyperglycemia activates p53 and p53-regulated genes leading to myocyte cell death. Diabetes 50: 2363–2375, 2001 [DOI] [PubMed] [Google Scholar]
- 32. Frustaci A, Kajstura J, Chimenti C, Jakoniuk I, Leri A, Maseri A, Nadal-Ginard B, Anversa P. Myocardial cell death in human diabetes. Circ Res 87: 1123–1132, 2000 [DOI] [PubMed] [Google Scholar]
- 33. Fulton AB. How crowded is the cytoplasm? Cell 30: 345–347, 1982 [DOI] [PubMed] [Google Scholar]
- 34. Ghosh S, Pulinilkunnil T, Yuen G, Kewalramani G, An D, Qi D, Abrahani A, Rodrigues B. Cardiomyocyte apoptosis induced by short-term diabetes requires mitochondrial GSH depletion. Am J Physiol Heart Circ Physiol 289: H768–H776, 2005 [DOI] [PubMed] [Google Scholar]
- 35. Gil Borlado MC, Moreno Lastres D, Gonzalez Hoyuela M, Moran M, Blazquez A, Pello R, Marin Buera L, Gabaldon T, Garcia Penas JJ, Martin MA, Arenas J, Ugalde C. Impact of the mitochondrial genetic background in complex III deficiency. PLoS One 5: e12801, 2010. doi:10.1371/journal.pone.0012801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gokey NG, Cao Z, Pak JW, Lee D, McKiernan SH, McKenzie D, Weindruch R, Aiken JM. Molecular analyses of mtDNA deletion mutations in microdissected skeletal muscle fibers from aged rhesus monkeys. Aging Cell 3: 319–326, 2004 [DOI] [PubMed] [Google Scholar]
- 37. Goto Y, Kakizaki M, Masaki N. Production of a spontaneous diabetic rats by repetition of selective breeding. Tohoku J Exp Med 119: 85–90, 1976 [DOI] [PubMed] [Google Scholar]
- 38. Graham KA, Kulawiec M, Owens KM, Li X, Desouki MM, Chandra D, Singh KK. NADPH oxidase 4 is an oncoprotein localized to mitochondria. Cancer Biol Ther 10: 223–231, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Green K, Brand MD, Murphy MP. Prevention of mitochondrial oxidative damage as a therapeutic strategy in diabetes. Diabetes 53, Suppl 1: S110–S118, 2004 [DOI] [PubMed] [Google Scholar]
- 40. Grijalva J, Hicks S, Zhao X, Medikayala S, Kaminski PM, Wolin MS, Edwards JG. Exercise training enhanced myocardial endothelial nitric oxide synthase (eNOS) function in diabetic Goto-Kakizaki (GK) rats. Cardiovasc Diabetol 7: 34, 2008. doi:10.1186/1475-2840-7-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Grozav AG, Chikamori K, Kozuki T, Grabowski DR, Bukowski RM, Willard B, Kinter M, Andersen AH, Ganapathi R, Ganapathi MK. Casein kinase I delta/epsilon phosphorylates topoisomerase IIalpha at serine-1106 and modulates DNA cleavage activity. Nucleic Acids Res 37: 382–392, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gupte R, Labinskyy N, Csiszar A, Ungvari Z, Edwards JG. Role of NAD(P)H oxidase in superoxide generation and endothelial dysfunction in diabetic Goto-Kakizaki (GK) rats. PLos One 5: 311800, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gupte RS, Floyd BC, Kozicky M, George S, Ungvari ZI, Neito V, Wolin MS, Gupte SA. Synergistic activation of glucose-6-phosphate dehydrogenase and NAD(P)H oxidase by Src kinase elevates superoxide in type 2 diabetic, Zucker fa/fa, rat liver. Free Radic Biol Med 47: 219–228, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gupte SA, Levine RJ, Gupte RS, Young ME, Lionetti V, Labinskyy V, Floyd BC, Ojaimi C, Bellomo M, Wolin MS, Recchia FA. Glucose-6-phosphate dehydrogenase-derived NADPH fuels superoxide production in the failing heart. J Mol Cell Cardiol 41: 340–349, 2006 [DOI] [PubMed] [Google Scholar]
- 45. Hafstad AD, Solevag GH, Severson DL, Larsen TS, Aasum E. Perfused hearts from Type 2 diabetic (db/db) mice show metabolic responsiveness to insulin. Am J Physiol Heart Circ Physiol 290: H1763–H1769, 2006 [DOI] [PubMed] [Google Scholar]
- 46. Hall JC, Sordahl LA, Stefko PL. The effect of insulin on oxidative phosphorylation in normal and diabetic mitochondria. J Biol Chem 235: 1536–1539, 1960 [PubMed] [Google Scholar]
- 47. Herlein JA, Fink BD, Henry DM, Yorek MA, Teesch LM, Sivitz WI. Mitochondrial superoxide and Coenzyme Q in insulin deficient rats: Increased electron leak. Am J Physiol Regul Integr Comp Physiol 301: R1616–R1624, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Herlein JA, Fink BD, Sivitz WI. Superoxide production by mitochondria of insulin-sensitive tissues: mechanistic differences and effect of early diabetes. Metabolism 59: 247–257, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Higuchi Y, Shoin S, Matsukawa S. Enhancement of the antitumor effect of glucose oxidase by combined administration of hydrogen peroxide decomposition inhibitors together with an oxygenated fluorocarbon. Jpn J Cancer Res 82: 942–949, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hiona A, Sanz A, Kujoth GC, Pamplona R, Seo AY, Hofer T, Someya S, Miyakawa T, Nakayama C, Samhan-Arias AK, Servais S, Barger JL, Portero-Otin M, Tanokura M, Prolla TA, Leeuwenburgh C. Mitochondrial DNA mutations induce mitochondrial dysfunction, apoptosis and sarcopenia in skeletal muscle of mitochondrial DNA mutator mice. PLoS One 5: e11468, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hu Y, Suarez J, Fricovsky E, Wang H, Scott BT, Trauger SA, Han W, Oyeleye MO, Dillmann WH. Increased enzymatic O-GlcNAcylation of mitochondrial proteins impairs mitochondrial function in cardiac myocytes exposed to high glucose. J Biol Chem 284: 547–555, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ikeuchi M, Matsusaka H, Kang D, Matsushima S, Ide T, Kubota T, Fujiwara T, Hamasaki N, Takeshita A, Sunagawa K, Tsutsui H. Overexpression of mitochondrial transcription factor a ameliorates mitochondrial deficiencies and cardiac failure after myocardial infarction. Circulation 112: 683–690, 2005 [DOI] [PubMed] [Google Scholar]
- 53. Janssen U, Riley SG, Vassiliadou A, Floege J, Phillips AO. Hypertension superimposed on type II diabetes in Goto Kakizaki rats induces progressive nephropathy. Kidney Int 63: 2162–2170, 2003 [DOI] [PubMed] [Google Scholar]
- 54. Kagan J, Srivastava S. Mitochondria as a target for early detection and diagnosis of cancer. Crit Rev Clin Lab Sci 42: 453–472, 2005 [DOI] [PubMed] [Google Scholar]
- 55. Kakimoto M, Inoguchi T, Sonta T, Yu HY, Imamura M, Etoh T, Hashimoto T, Nawata H. Accumulation of 8-hydroxy-2'-deoxyguanosine and mitochondrial DNA deletion in kidney of diabetic rats. Diabetes 51: 1588–1595, 2002 [DOI] [PubMed] [Google Scholar]
- 56. Kao YH, Hsieh HP, Chitlimalla SK, Pan WY, Kuo CC, Tsai YC, Lin WH, Chuang SE, Chang JY. A novel peroxisome proliferator-activated receptor alpha/gamma agonist, BPR1H0101, inhibits topoisomerase II catalytic activity in human cancer cells. Anticancer Drugs 19: 151–158, 2008 [DOI] [PubMed] [Google Scholar]
- 57. Kitahara A, Toyota T, Kakizaki M, Goto Y. Activities of hepatic enzymes in spontaneous diabetes rats produced by selective breeding of normal Wistar rats. Tohoku J Exp Med 126: 7–11, 1978 [DOI] [PubMed] [Google Scholar]
- 58. Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van Remmen H, Sedivy JM, Yamasoba T, Tanokura M, Weindruch R, Leeuwenburgh C, Prolla TA. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309: 481–484, 2005 [DOI] [PubMed] [Google Scholar]
- 59. Kumar S, Sitasawad SL. N-acetylcysteine prevents glucose/glucose oxidase-induced oxidative stress, mitochondrial damage and apoptosis in H9c2 cells. Life Sci 84: 328–336, 2009 [DOI] [PubMed] [Google Scholar]
- 60. Kwon SH, Pimentel DR, Remondino A, Sawyer DB, Colucci WS. H2O2 regulates cardiac myocyte phenotype via concentration-dependent activation of distinct kinase pathways. J Mol Cell Cardiol 35: 615–621, 2003 [DOI] [PubMed] [Google Scholar]
- 61. Laher I, Beam J, Botta A, Barendregt R, Sulistyoningrum D, Devlin A, Rheault M, Ghosh S. Short-term exercise worsens cardiac oxidative stress and fibrosis in 8-month old db/db mice by depleting cardiac glutathione. Free Radic Res, 2012 [DOI] [PubMed] [Google Scholar]
- 62. Lamantea E, Carrara F, Mariotti C, Morandi L, Tiranti V, Zeviani M. A novel nonsense mutation (Q352X) in the mitochondrial cytochrome b gene associated with a combined deficiency of complexes I and III. Neuromuscul Disord 12: 49–52, 2002 [DOI] [PubMed] [Google Scholar]
- 63. Larosche I, Choumar A, Fromenty B, Letteron P, Abbey-Toby A, Van Remmen H, Epstein CJ, Richardson A, Feldmann G, Pessayre D, Mansouri A. Prolonged ethanol administration depletes mitochondrial DNA in MnSOD-overexpressing transgenic mice, but not in their wild type littermates. Toxicol Appl Pharmacol 234: 326–338, 2009 [DOI] [PubMed] [Google Scholar]
- 64. Lautamaki R, Airaksinen KE, Seppanen M, Toikka J, Luotolahti M, Ball E, Borra R, Harkonen R, Iozzo P, Stewart M, Knuuti J, Nuutila P. Rosiglitazone improves myocardial glucose uptake in patients with type 2 diabetes and coronary artery disease: a 16-week randomized, double-blind, placebo-controlled study. Diabetes 54: 2787–2794, 2005 [DOI] [PubMed] [Google Scholar]
- 65. Lee HC, Yin PH, Lu CY, Chi CW, Wei YH. Increase of mitochondria and mitochondrial DNA in response to oxidative stress in human cells. Biochem J 348: 425–432, 2000 [PMC free article] [PubMed] [Google Scholar]
- 66. Lee HK, Song JH, Shin CS, Park DJ, Park KS, Lee KU, Koh CS. Decreased mitochondrial DNA content in peripheral blood precedes the development of non-insulin-dependent diabetes mellitus. Diabetes Res Clin Pract 42: 161–167, 1998 [DOI] [PubMed] [Google Scholar]
- 67. Levi I, Segev Y, Priel E. Type 1 diabetes affects topoisomerase I activity and GlcNAcylation in rat organs: kidney liver and pancreas. Glycobiology 22: 704–713, 2012 [DOI] [PubMed] [Google Scholar]
- 68. Li TK, Chen AY, Yu C, Mao Y, Wang H, Liu LF. Activation of topoisomerase II-mediated excision of chromosomal DNA loops during oxidative stress. Genes Dev 13: 1553–1560, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lohrke B, Xu J, Weitzel JM, Kruger B, Goldammer T, Viergutz T. N-acetylcysteine impairs survival of luteal cells through mitochondrial dysfunction. Cytometry A 77: 310–320, 2011 [DOI] [PubMed] [Google Scholar]
- 70. Lopaschuk GD, Folmes CD, Stanley WC. Cardiac energy metabolism in obesity. Circ Res 101: 335–347, 2007 [DOI] [PubMed] [Google Scholar]
- 71. Love DC, Kochan J, Cathey RL, Shin SH, Hanover JA. Mitochondrial and nucleocytoplasmic targeting of O-linked GlcNAc transferase. J Cell Sci 116: 647–654, 2003 [DOI] [PubMed] [Google Scholar]
- 72. Low RL, Orton S, Friedman DB. A truncated form of DNA topoisomerase IIbeta associates with the mtDNA genome in mammalian mitochondria. Eur J Biochem 270: 4173–4186, 2003 [DOI] [PubMed] [Google Scholar]
- 73. Lynn RM, Bjornsti MA, Caron PR, Wang JC. Peptide sequencing and site-directed mutagenesis identify tyrosine-727 as the active site tyrosine of Saccharomyces cerevisiae DNA topoisomerase I. Proc Natl Acad Sci USA 86: 3559–3563, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ma Q, Fang H, Shang W, Liu L, Xu Z, Ye T, Wang X, Zheng M, Chen Q, Cheng H. Superoxide flashes: early mitochondrial signals for oxidative stress-induced apoptosis. J Biol Chem 286: 27573–27581, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Maalouf RM, Eid AA, Gorin YC, Block K, Escobar GP, Bailey S, Abboud HE. Nox4-derived reactive oxygen species mediate cardiomyocyte injury in early type 1 diabetes. Am J Physiol Cell Physiol 302: C597–C604, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Madden KR, Stewart L, Champoux JJ. Preferential binding of human topoisomerase I to superhelical DNA. EMBO J 14: 5399–5409, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Mansouri A, Fromenty B, Berson A, Robin MA, Grimbert S, Beaugrand M, Erlinger S, Pessayre D. Multiple hepatic mitochondrial DNA deletions suggest premature oxidative aging in alcoholic patients. J Hepatol 27: 96–102, 1997 [DOI] [PubMed] [Google Scholar]
- 78. Mariappan N, Elks CM, Sriramula S, Guggilam A, Liu Z, Borkhsenious O, Francis J. NF-kappaB-induced oxidative stress contributes to mitochondrial and cardiac dysfunction in type II diabetes. Cardiovasc Res 85: 473–483, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Martyn KD, Frederick LM, von Loehneysen K, Dinauer MC, Knaus UG. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell Signal 18: 69–82, 2006 [DOI] [PubMed] [Google Scholar]
- 80. Medikayala S, Piteo B, Zhao X, Edwards JG. Chronically elevated glucose compromises myocardial mitochondrial DNA integrity by alteration of mitochondrial topoisomerase function. Am J Physiol Cell Physiol 300: C338–C348, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Miller ER, 3rd, Pastor-Barriuso R, Dalal D, Riemersma RA, Appel LJ, Guallar E. Meta-analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Ann Intern Med 142: 37–46, 2005 [DOI] [PubMed] [Google Scholar]
- 82. Morino K, Petersen KF, Dufour S, Befroy D, Frattini J, Shatzkes N, Neschen S, White MF, Bilz S, Sono S, Pypaert M, Shulman GI. Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J Clin Invest 115: 3587–3593, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Murakawa Y, Zhang W, Pierson CR, Brismar T, Ostenson CG, Efendic S, Sima AA. Impaired glucose tolerance and insulinopenia in the GK-rat causes peripheral neuropathy. Diabetes Metab Res Rev 18: 473–483, 2002 [DOI] [PubMed] [Google Scholar]
- 84. Nakao M, Matsubara T, Sakamoto N. Effects of diabetes on cardiac glycogen metabolism in rats. Heart Vessels 8: 171–175, 1993 [DOI] [PubMed] [Google Scholar]
- 85. Nollau P, Wagener C. Methods for detection of point mutations: performance and quality assessment. IFCC Scientific Division, Committee on Molecular Biology Techniques. Clin Chem 43: 1114–1128, 1997 [PubMed] [Google Scholar]
- 86. Nomiyama T, Tanaka Y, Piao L, Hattori N, Uchino H, Watada H, Kawamori R, Ohta S. Accumulation of somatic mutation in mitochondrial DNA and atherosclerosis in diabetic patients. Ann NY Acad Sci 1011: 193–204, 2004 [DOI] [PubMed] [Google Scholar]
- 87. Olivares-Reyes JA, Arellano-Plancarte A, Castillo-Hernandez JR. Angiotensin II and the development of insulin resistance: implications for diabetes. Mol Cell Endocrinol 302: 128–139, 2009 [DOI] [PubMed] [Google Scholar]
- 88. Oyama K, Takahashi K, Sakurai K. Cardiomyocyte H9c2 cells exhibit differential sensitivity to intracellular reactive oxygen species generation with regard to their hypertrophic vs. death responses to exogenously added hydrogen peroxide. J Clin Biochem Nutr 45: 361–369, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Oyama K, Takahashi K, Sakurai K. Hydrogen peroxide induces cell cycle arrest in cardiomyoblast H9c2 cells, which is related to hypertrophy. Biol Pharm Bull 34: 501–506, 2011 [DOI] [PubMed] [Google Scholar]
- 90. Ozdemir S, Tandogan B, Ulusu NN, Turan B. Angiotensin II receptor blockage prevents diabetes-induced oxidative damage in rat heart. Folia Biol (Praha) 55: 11–16, 2009 [DOI] [PubMed] [Google Scholar]
- 91. Pello R, Martin MA, Carelli V, Nijtmans LG, Achilli A, Pala M, Torroni A, Gomez-Duran A, Ruiz-Pesini E, Martinuzzi A, Smeitink JA, Arenas J, Ugalde C. Mitochondrial DNA background modulates the assembly kinetics of OXPHOS complexes in a cellular model of mitochondrial disease. Hum Mol Genet 17: 4001–4011, 2008 [DOI] [PubMed] [Google Scholar]
- 92. Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, DiPietro L, Cline GW, Shulman GI. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science 300: 1140–1142, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Phillips AO, Baboolal K, Riley S, Grone H, Janssen U, Steadman R, Williams J, Floege J. Association of prolonged hyperglycemia with glomerular hypertrophy and renal basement membrane thickening in the Goto Kakizaki model of non-insulin-dependent diabetes mellitus. Am J Kidney Dis 37: 400–410, 2001 [DOI] [PubMed] [Google Scholar]
- 94. Poornima IG, Parikh P, Shannon RP. Diabetic cardiomyopathy: the search for a unifying hypothesis. Circ Res 98: 596–605, 2006 [DOI] [PubMed] [Google Scholar]
- 95. Privratsky JR, Wold LE, Sowers JR, Quinn MT, Ren J. AT1 blockade prevents glucose-induced cardiac dysfunction in ventricular myocytes: role of the AT1 receptor and NADPH oxidase. Hypertension 42: 206–212, 2003 [DOI] [PubMed] [Google Scholar]
- 96. Quijano C, Castro L, Peluffo G, Valez V, Radi R. Enhanced mitochondrial superoxide in hyperglycemic endothelial cells: direct measurements and formation of hydrogen peroxide and peroxynitrite. Am J Physiol Heart Circ Physiol 293: H3404–H3414, 2007 [DOI] [PubMed] [Google Scholar]
- 97. Rachek LI, Musiyenko SI, LeDoux SP, Wilson GL. Palmitate induced mitochondrial deoxyribonucleic acid damage and apoptosis in l6 rat skeletal muscle cells. Endocrinology 148: 293–299, 2007 [DOI] [PubMed] [Google Scholar]
- 98. Rafalski K, Abdourahman A, Edwards JG. Early adaptations to training: upregulation of alpha-myosin heavy chain gene expression. Med Sci Sports Exerc 39: 75–82, 2007 [DOI] [PubMed] [Google Scholar]
- 99. Ricci C, Pastukh V, Leonard J, Turrens J, Wilson G, Schaffer D, Schaffer SW. Mitochondrial DNA damage triggers mitochondrial-superoxide generation and apoptosis. Am J Physiol Cell Physiol 294: C413–C422, 2008 [DOI] [PubMed] [Google Scholar]
- 100. Roe ND, Thomas DP, Ren J. Inhibition of NADPH oxidase alleviates experimental diabetes-induced myocardial contractile dysfunction. Diabetes Obes Metab 13: 465–473, 2011 [DOI] [PubMed] [Google Scholar]
- 101. Santos JM, Tewari S, Kowluru RA. A compensatory mechanism protects retinal mitochondria from initial insult in diabetic retinopathy. Free Radic Biol Med 53: 1729–1737, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Santos JM, Tewari S, Kowluru RA. A compensatory mechanism protects retinal mitochondria from initial insult in diabetic retinopathy. Free Radic Biol Med 53: 1729–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Schultz MC, Brill SJ, Ju Q, Sternglanz R, Reeder RH. Topoisomerases and yeast rRNA transcription: negative supercoiling stimulates initiation and topoisomerase activity is required for elongation. Genes Dev 6: 1332–1341, 1992 [DOI] [PubMed] [Google Scholar]
- 104. Sciacco M, Prelle A, Fagiolari G, Bordoni A, Crimi M, Di Fonzo A, Ciscato P, Lamperti C, D'Adda E, Jann S, Bresolin N, Comi GP, Moggio M. A case of CPT deficiency, homoplasmic mtDNA mutation and ragged red fibers at muscle biopsy. J Neurol Sci 239: 21–24, 2005 [DOI] [PubMed] [Google Scholar]
- 105. Serpillon S, Floyd BC, Gupte RS, George S, Kozicky M, Neito V, Recchia F, Stanley W, Wolin MS, Gupte SA. Superoxide production by NAD(P)H oxidase and mitochondria is increased in genetically obese and hyperglycemic rat heart and aorta before the development of cardiac dysfunction. The role of glucose-6-phosphate dehydrogenase-derived NADPH. Am J Physiol Heart Circ Physiol 297: H153–H162, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Shanmugam K, Mallikarjuna K, Kesireddy N, Reddy K. Neuroprotective effect of ginger on anit-oxidant enzymes in STZ-induced rats. Food Chem Toxicol 49: 893–897, 2011 [DOI] [PubMed] [Google Scholar]
- 107. Shen D, Dalton TP, Nebert DW, Shertzer HG. Glutathione redox state regulates mitochondrial reactive oxygen production. J Biol Chem 280: 25305–25312, 2005 [DOI] [PubMed] [Google Scholar]
- 108. Sirker A, Zhang M, Shah AM. NADPH oxidases in cardiovascular disease: insights from in vivo models and clinical studies. Basic Res Cardiol 106: 735–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Sordet O, Larochelle S, Nicolas E, Stevens EV, Zhang C, Shokat KM, Fisher RP, Pommier Y. Hyperphosphorylation of RNA polymerase II in response to topoisomerase I cleavage complexes and its association with transcription- and BRCA1-dependent degradation of topoisomerase I. J Mol Biol 381: 540–549, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. St-Amant C, Lussier S, Lehoux J, Laberge RM, Boissonneault G. Altered phosphorylation of topoisomerase I following overexpression in an ovarian cancer cell line. Biochem Cell Biol 84: 55–66, 2006 [DOI] [PubMed] [Google Scholar]
- 110a. Tewari S, Santos JM, Kowluru RA. Damaged mitochondrial DNA replication system and the development of diabetic retinopathy. Antioxid Redox Signal 17: 493–504, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Trifunovic A, Hansson A, Wredenberg A, Rovio AT, Dufour E, Khvorostov I, Spelbrink JN, Wibom R, Jacobs HT, Larsson NG. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc Natl Acad Sci USA 102: 17993–17998, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Ugalde C, Triepels RH, Coenen MJ, van den Heuvel LP, Smeets R, Uusimaa J, Briones P, Campistol J, Majamaa K, Smeitink JA, Nijtmans LG. Impaired complex I assembly in a Leigh syndrome patient with a novel missense mutation in the ND6 gene. Ann Neurol 54: 665–669, 2003 [DOI] [PubMed] [Google Scholar]
- 113. Ungvari Z, Labinskyy N, Gupte S, Chander PN, Edwards JG, Csiszar A. Dysregulation of mitochondrial biogenesis in vascular endothelial and smooth muscle cells of aged rats. Am J Physiol Heart Circ Physiol 294: H2121–H2128, 2008 [DOI] [PubMed] [Google Scholar]
- 114. Ushio-Fukai M. Compartmentalization of redox signaling through NADPH oxidase-derived ROS. Antioxid Redox Signal 11: 1289–1299, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Vesely DL, Gower WR, Jr, Dietz JR, Overton RM, Clark LC, Antwi EK, Farese RV. Elevated atrial natriuretic peptides and early renal failure in type 2 diabetic Goto-Kakizaki rats. Metabolism 48: 771–778, 1999 [DOI] [PubMed] [Google Scholar]
- 116. Vivekananthan DP, Penn MS, Sapp SK, Hsu A, Topol EJ. Use of antioxidant vitamins for the prevention of cardiovascular disease: meta-analysis of randomised trials. Lancet 361: 2017–2023, 2003 [DOI] [PubMed] [Google Scholar]
- 117. Wanagat J, Cao Z, Pathare P, Aiken JM. Mitochondrial DNA deletion mutations colocalize with segmental electron transport system abnormalities, muscle fiber atrophy, fiber splitting, and oxidative damage in sarcopenia. FASEB J 15: 322–332, 2001 [DOI] [PubMed] [Google Scholar]
- 118. Wang P, Lloyd SG, Zeng H, Bonen A, Chatham JC. Impact of altered substrate utilization on cardiac function in isolated hearts from Zucker diabetic fatty rats. Am J Physiol Heart Circ Physiol 288: H2102–H2110, 2005 [DOI] [PubMed] [Google Scholar]
- 119. Wang W, Fang H, Groom L, Cheng A, Zhang W, Liu J, Wang X, Li K, Han P, Zheng M, Yin J, Mattson MP, Kao JP, Lakatta EG, Sheu SS, Ouyang K, Chen J, Dirksen RT, Cheng H. Superoxide flashes in single mitochondria. Cell 134: 279–290, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Wang Y, Lyu YL, Wang JC. Dual localization of human DNA topoisomerase IIIalpha to mitochondria and nucleus. Proc Natl Acad Sci USA 99: 12114–12119, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Wheeler MD, Nakagami M, Bradford BU, Uesugi T, Mason RP, Connor HD, Dikalova A, Kadiiska M, Thurman RG. Overexpression of manganese superoxide dismutase prevents alcohol-induced liver injury in the rat. J Biol Chem 276: 36664–36672, 2001 [DOI] [PubMed] [Google Scholar]
- 122. Wu J, Feng L, Hsieh TS. Drosophila topo IIIalpha is required for the maintenance of mitochondrial genome and male germ-line stem cells. Proc Natl Acad Sci USA 107: 6228–6233, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Xiong Y, Uys JD, Tew KD, Townsend DM. S-glutathionylation: from molecular mechanisms to health outcomes. Antioxid Redox Signal 15: 233–270, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Yagihashi S, Goto Y, Kakizaki M, Kaseda N. Thickening of glomerular basement membrane in spontaneously diabetic rats. Diabetologia 15: 309–312, 1978 [DOI] [PubMed] [Google Scholar]
- 125. Yagihashi S, Kaseda N, Kakizaki M, Goto Y. Evolution of glomerular lesions in rats with spontaneous diabetes. Tohoku J Exp Med 127: 359–367, 1979 [DOI] [PubMed] [Google Scholar]
- 126. Yagihashi S, Tonosaki A, Yamada K, Kakizaki M, Goto Y. Peripheral neuropathy in selectively-inbred spontaneously diabetic rats: electrophysiological, morphometrical and freeze-replica studies. Tohoku J Exp Med 138: 39–48, 1982 [DOI] [PubMed] [Google Scholar]
- 127. Yasuda K, Nishikawa W, Iwanaka N, Nakamura E, Seino Y, Tsuda K, Ishihara A. Abnormality in fibre type distribution of soleus and plantaris muscles in non-obese diabetic Goto-Kakizaki rats. Clin Exp Pharmacol Physiol 29: 1001–1008, 2002 [DOI] [PubMed] [Google Scholar]
- 128. Yoshida K, Yamaguchi T, Shinagawa H, Taira N, Nakayama KI, Miki Y. Protein kinase C delta activates topoisomerase IIalpha to induce apoptotic cell death in response to DNA damage. Mol Cell Biol 26: 3414–3431, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Yu D, Khan E, Khaleque MA, Lee J, Laco G, Kohlhagen G, Kharbanda S, Cheng YC, Pommier Y, Bharti A. Phosphorylation of DNA topoisomerase I by the c-Abl tyrosine kinase confers camptothecin sensitivity. J Biol Chem 279: 51851–51861, 2004 [DOI] [PubMed] [Google Scholar]
- 130. Zhang D, Mott JL, Chang SW, Denniger G, Feng Z, Zassenhaus HP. Construction of transgenic mice with tissue-specific acceleration of mitochondrial DNA mutagenesis. Genomics 69: 151–161, 2000 [DOI] [PubMed] [Google Scholar]
- 131. Zhang D, Mott JL, Chang SW, Stevens M, Mikolajczak P, Zassenhaus HP. Mitochondrial DNA mutations activate programmed cell survival in the mouse heart. Am J Physiol Heart Circ Physiol 288: H2476–H2483, 2005 [DOI] [PubMed] [Google Scholar]
- 132. Zhang H, Meng LH, Pommier Y. Mitochondrial topoisomerases and alternative splicing of the human TOP1mt gene. Biochimie 89: 474–481, 2007 [DOI] [PubMed] [Google Scholar]