

Abstract
The second member of the transient receptor potential-melastatin channel family (TRPM2) is expressed in the heart and vasculature. TRPM2 channels were expressed in the sarcolemma and transverse tubules of adult left ventricular (LV) myocytes. Cardiac TRPM2 channels were functional since activation with H2O2 resulted in Ca2+ influx that was dependent on extracellular Ca2+, was significantly higher in wild-type (WT) myocytes compared with TRPM2 knockout (KO) myocytes, and inhibited by clotrimazole in WT myocytes. At rest, there were no differences in LV mass, heart rate, fractional shortening, and +dP/dt between WT and KO hearts. At 2–3 days after ischemia-reperfusion (I/R), despite similar areas at risk and infarct sizes, KO hearts had lower fractional shortening and +dP/dt compared with WT hearts. Compared with WT I/R myocytes, expression of the Na+/Ca2+ exchanger (NCX1) and NCX1 current were increased, expression of the α1-subunit of Na+-K+-ATPase and Na+ pump current were decreased, and action potential duration was prolonged in KO I/R myocytes. Post-I/R, intracellular Ca2+ concentration transients and contraction amplitudes were equally depressed in WT and KO myocytes. After 2 h of hypoxia followed by 30 min of reoxygenation, levels of ROS were significantly higher in KO compared with WT LV myocytes. Compared with WT I/R hearts, oxygen radical scavenging enzymes (SODs) and their upstream regulators (forkhead box transcription factors and hypoxia-inducible factor) were lower, whereas NADPH oxidase was higher, in KO I/R hearts. We conclude that TRPM2 channels protected hearts from I/R injury by decreasing generation and enhancing scavenging of ROS, thereby reducing I/R-induced oxidative stress.
Keywords: ischemic cardiomyopathy, voltage-independent Ca2+ channels, oxidative cardiac injury, excitation-contraction coupling
transient receptor potential (TRP) channels consist of a superfamily of monovalent and divalent cation-permeable ion channels with six transmembrane domains and are homologs of the Drosophila melanogaster TRP channel, a Ca2+-permeable channel that is essential for phototransduction (31). To date, the mammalian TRP superfamily consists of 28 members grouped into 6 subfamilies based on amino acid sequence homology: canonical, vanilloid, melastatin, polycystin, mucolipin, and ankyrin. To form a functional channel, TRP proteins assemble into either homo- or heterotetramers (11, 17, 25).
The TRP-melastatin (TRPM) subfamily contains eight mammalian members (TRPM1–TRPM8), and some of them have short splice variants (43, 44). TRPM channels have important roles in cell proliferation and survival (1, 8). Focusing on TRPM2 (previously LTRPC2) channels, the murine TRPM2 gene contains 34 exons, spans ∼61 kb, and encodes a protein of 1,507 amino acids with a predicted molecular mass of ∼170 kDa (42). The human TRPM2 gene consists of 32 exons, spans ∼90 kb, and encodes a protein of 1,503 amino acids with a predicted molecular mass of ∼170 kDa (30). Human and mouse TRPM2 ion channels have amino acid sequences that are 82% identical. Both are widely expressed, and both can be activated by oxidative stress. Both human and rodent TRPM2 channels are sensitive to the TRPM2 inhibitors clotrimazole (15) and 2-aminoethoxydiphenyl borate (38).
TRPM2 channels are expressed in many cell types, including brain, hematopoietic, heart, vascular smooth muscle, and endothelial cells (14, 29). TRPM2 channels are activated by ADP ribose (ADPR) (10) and facilitated by cyclic (c)APDR (22), H2O2 (22), and intracellular Ca2+ concentration ([Ca2+]i) (7, 39). When open, TRPM2 channels are permeable to Ca2+, Na+, and K+. The function of TRPM2 channels in the heart is unknown.
In tissues other than the heart, TRPM2 channels have been shown to play an essential role in the susceptibility to oxidative stress (12, 29). The classical hypothesis is that ROS enhance ADPR production, which activates TRPM2 (12, 20, 23, 32), and that TRPM2-mediated Ca2+ influx contributes to cytokine production, inflammation, and cell death (36). In the heart, ROS are produced physiologically during respiration by the mitochondrial electron transport chain, and increased ROS levels have been observed in myocytes stimulated with β-adrenergic agonists (2) and in pathological conditions such as ischemia-reperfusion (I/R) (40) and doxorubicin exposure (21, 33). ROS play a major role in myocyte injury through protein oxidation, lipid peroxidation, DNA oxidation, and mutagenesis. A significant increase in intracellular cADPR in cardiac myocytes occurs during hypoxia/reoxygenation (simulated I/R in vitro), which may activate TRPM2 channels and leads to elevations in [Ca2+]i (48). Abrogation of Ca2+ influx via TRPM2 channels should theoretically confer protection on myocytes subjected to oxidative stress. The present study was undertaken to critically test whether genetic elimination of TRPM2 channels would protect hearts from I/R injury.
METHODS
Generation of global TRPM2 knockout mice.
Briefly, exons 21 and 22, encoding transmembrane domains 5 and 6 and the putative Ca2+ pore of TRPM2 gene (42), were flanked by loxP recombination sites (Fig. 1A). TRPM2fx/+ mice (C57BL/6 background) were bred to Ella-Cre mice (C57BL/6 background), which express Cre recombinase ubiquitously (24). TRPM2+/0:Cre+/0 mice were bred with wild-type (WT) mice to generate heterozygous TRPM2+/0:Cre0/0 mice, which were then crossed to generate homozygous TRPM20/0:Cre0/0 mice. Littermates that were TRPM2+/+:Cre0/0 served as control “WT” mice. Using specific primers, the absence of WT TRPM2 was demonstrated by genomic PCR (Fig. 1B). By RT-PCR, absence of the WT TRPM2 transcript was confirmed in TRPM2 knockout (KO) hearts (Fig. 1C).
Fig. 1.

Generation of constitutive global transient receptor potential-melastatin 2 (TRPM2) knockout (KO) mice. A: exons 21 and 22 encoding transmembrane domain (TM)5 and TM6 and the putative Ca2+ pore were flanked by loxP recombination sites (conditional TRPM2 KO), and constitutive global TRPM2 KO mice were generated as described in methods. B: for the TRPM2 wild-type (WT) group, the following primers were used: primer 726, forward 5′-ggC TCT gCC TCA TCC CCA gAA TC-3′; and primer 727, reverse 5′-CCg gAT ACA gAT gCA ggA TgC Tg-3′. For the TRPM2 KO group, the following primers were used: primer 726, forward 5′-ggC TCT gCC TCA TCC CCA gAA TC-3′; and primer 728, reverse 5′-CTg AAg gTC CTg AgT TTg AAT CCC A-3′. Genomic PCR demonstrated the absence of WT TRPM2 in both bone marrow (M) and the heart (H) in TRPM2 KO mice. C: for RT-PCR, the following set of primers was used: primer 753, 5′-TgT TCT gTC TCC gTC TCA TgC ACA-3′; and primer 754, 5′-TCT Tgg CAG ggA TCT TCA ggA CAA 3′. Our results demonstrated the absence of WT TRPM2 transcripts in the hearts of TRPM2 KO mice.
For brevity, throughout this report, TRPM2 KO is abbreviated to KO, whether applied to mice, hearts, myocytes, or left ventricular (LV) homogenates.
Homozygous adult littermates (∼8–10 wk old) were used in this study. Mice were housed and fed on a 12:12-h light-dark cycle at either the Temple University or The Pennsylvania State University Animal Facility supervised by full-time veterinarian staff members. Standard care was provided to all mice used for experiments. All protocols and procedures applied to the mice in this study were approved and supervised by the Institutional Animal Care and Use Committees of Temple University and The Pennsylvania State University.
I/R surgery in mice.
I/R surgery was performed as previously described (9, 26). Briefly, male WT and KO mice (8–10 wk) were anesthetized with 2% isoflurane, and the heart was exposed through a left thoracotomy at the fifth intercostal space. The slipknot was tied around the left anterior descending coronary artery (LAD) 2–3 mm from its origin, and the heart was immediately returned to the chest cavity followed by evacuation of the pneumothorax and closure of muscle and skin layers. The slipknot was released after 30 min of ischemia to allow reperfusion. Sham-operated (sham) animals were subjected to the same surgical procedures except that the slipknot was not tied. Animals recovered from anesthesia within 5 min after the completion of surgery and received ibuprofen (10 mg/50 ml drinking water) for 48 h as postsurgery analgesia. Experiments on survivors were performed on days 2–3 postsurgery.
Infarct size measurement.
The myocardium was stained with 2% 2,3,5-triphenyltetrazolium (TTC) to measure infarct size as previously described (9, 26). Briefly, 72 h after I/R, the slipknot around the LAD was retied followed by an injection of 2% Evans blue dye (0.2 ml). Hearts were excised, and the LV was sliced into five equally thick sections perpendicular to the short axis of the heart and incubated in PBS containing TTC. After 15 min at room temperature, slices were digitally photographed. The Evans blue-stained area (area not at risk), TTC-negative area (infarcted myocardium), and area at risk (AAR; including both TTC-negative and -positive areas) were measured with computer-based image analyzer SigmaScan Pro 5.0 (SPSS Science, Chicago, IL). The AAR was expressed as percentage of the total LV, whereas the infarcted myocardium was expressed as a percentage of the AAR.
Echocardiographic and hemodynamic analyses of cardiac function.
Transthoracic two-dimensional echocardiography was performed in anesthetized (2% inhaled isoflurane) WT or KO mice with a 12-MHz probe as previously described (34, 45–47). For in vivo hemodynamic measurements, a 1.4-Fr micromanipulator-tipped catheter (SPR-671, Millar Instruments) was inserted into the right carotid artery and advanced into the LV of lightly anesthetized (tribromoethanol-amylene hydrate, Avertin, 2.5% wt/vol, 8 μl/g ip) mice with spontaneous respirations, which were placed on a heated (37°C) pad. Hemodynamics, including heart rate, LV end-diastolic pressure, and the maximal first time derivative of the LV pressure rise (+dP/dt) and fall (−dP/dt), were recorded in closed-chest mode, both at baseline and in response to increasing doses of isoproterenol (Iso; 0.1, 0.5, 1, 5, and 10 ng) (34, 45–47).
Isolation of adult murine ventricular myocytes.
Cardiac myocytes were isolated from the LV free wall and septum of WT and KO mice according to the protocol of Zhou et al. (53) and modified by us (34, 35, 41, 45–47). Myocytes were seeded onto laminin-coated coverslips and used within 2–8 h of isolation.
Immunolocalization of TRPM2 in adult LV myocytes.
Freshly isolated WT mouse LV myocytes were plated on laminin-coated coverslips, allowed to adhere for 3 h, and washed three times with PBS containing 2 mM EGTA (PBS-EGTA). Myocytes were fixed for 30 min in 4% paraformaldehyde in PBS-EGTA. After two rinses with PBS-EGTA, myocytes were permeabilized for 2 min with 0.05% Triton X-100. Myocytes were rinsed two times with PBS-EGTA and once with Blotto (5% nonfat dry milk, 0.1 M NaCl, and 50 mM Tris·HCl; pH 7.4). Alexa fluor 488-conjugated monoclonal antibody against the α1-subunit of Na+-K+-ATPase (Millipore, Temecula, CA, 1:250) with or without primary antibodies against TRPM2 (Bethyl Labs, Montgomery, TX, 1:50) diluted in Blotto were added to the cells, incubated at room temperature in the dark for 60 min, and rinsed three times with Blotto. Secondary antibodies (Alexa fluor 594-labeled goat anti-rabbit IgG, Invitrogen, Eugene, OR, 1:50) diluted in Blotto were added to the cells, incubated in the dark for 30 min, and followed by three PBS-EGTA rinses. Coverslips were mounted to slides with Prolong gold antifade mounting solution (Invitrogen). Confocal images (×63 oil objective, 510 Meta, Carl Zeiss) were acquired at 594-nm excitation and 617-nm emission for TRPM2 and 488-nm excitation and 510-nm emission for Na+-K+-ATPase.
Measurement of [Ca2+]i in cardiac myocytes.
Fura-2-loaded (0.67 μM fura-2 AM, 15 min, 37°C) myocytes attached to laminin-coated coverslips were incubated in medium 199 containing 1.8 mM extracellular Ca2+ concentration ([Ca2+]o) and 1 μM verapamil, and exposed to excitation light (360 and 380 nm) only during data acquisition. Epifluorescence (510 nm) was monitored at baseline and at 1, 3, 5, 10, and 15 min after the addition of H2O2 (200 μM). Some WT myocytes were incubated in 0 Ca2+ buffer [containing (in mM) 118 NaCl, 4.7 KCl, 1.2 MgSO4, 1.2 KH2PO4, 20 Na-HEPES, 4.0 EGTA, and 0.001 verapamil] or pretreated with clotrimazole (50 μM, 10 min) in medium 199 before being exposed to H2O2. In a second series of experiments, fura-2-loaded myocytes were field stimulated to contract (2 Hz, 37°C) in medium 199 (1.8 mM [Ca2+]o and no verapamil), and [Ca2+]i transients were measured. Daily calibration of fura-2 signals and [Ca2+]i transient analyses were performed as previously described (34, 35, 41, 45–47).
Myocyte shortening measurements.
Myocytes adherent to coverslips were bathed in 0.7 ml of air- and temperature-equilibrated (37°C), HEPES-buffered (20 mM, pH 7.4) medium 199 (1.8 mM [Ca2+]o) and paced to contract (2 Hz). Myocyte images were captured by a charge-coupled device video camera, and myocyte motion was analyzed offline with an edge detection algorithm as previously described (34, 35, 41, 45–47).
ROS measurements in isolated cardiac myocytes.
To simulate I/R in vitro, LV myocytes isolated from WT and KO hearts were plated on laminin-coated glass coverslips and exposed to either 21% O2-5% CO2 (normoxic) or 1% O2-5% CO2 (hypoxic) for 2 h followed by 30 min of reoxygenation. Myocytes were incubated in Krebs-Henseleit bicarbonate buffer containing 5 mM pyruvate as a substrate (4). During reoxygenation, myocytes were loaded with 5-(and 6-)chloromethyl-2′, 7′-dichlorofluorescein diacetate acetyl ester (5 μM, 30 min). Coverslips containing myocytes were mounted in an open perfusion microincubator (PDMI-2, Harvard Apparatus) at 37°C and imaged. Confocal images were obtained at 488-nm excitation and 515-nm emission. Images were analyzed, and the mean dichlorofluorescein fluorescence was quantified using Image J software (National Institutes of Health).
Electrophysiological measurements.
Na+/Ca2+ exchange current (INaCa) (34, 35, 45, 46, 50), Na+-K+-ATPase current (Ipump; pipette Na+ concentration: 80 mM and extracellular K+ concentration: 18 mM) (34, 35, 46, 47), and action potential (AP) (34, 41, 45, 46) were measured in isolated LV myocytes (30°C) with whole cell patch-clamp. Fire-polished pipettes (tip diameter: 4–6 μm) with resistances of 0.8–1.4 MΩ when filled with pipette solutions were used. The composition of pipette and bathing solutions and protocols are provided in the figures.
Immunoblot analysis.
Mouse LV homogenates were prepared as previously described (41). For the detection of α1- and α2-subunits of Na+-K+-ATPase, the α1c-subunit of the L-type Ca2+ channel (Cav1.2), sarco(endo)plasmic reticulum Ca2+-ATPase 2 (SERCA2), calsequestrin (7.5% SDS-PAGE, reducing conditions with 5% β-mercaptoethanol), Na+/Ca2+ exchanger 1 (NCX1; 7.5% SDS-PAGE, nonreducing conditions with 10 mM N-ethylmaleimide), and cardiac ryanodine receptor 2 phosphorylated at Ser2808 (pRyR2; 6% SDS-PAGE, reducing conditions), commercially available antibodies were used as previously described (34, 35, 41, 45–47). For the detection of hypoxia-inducible factor (HIF)-1α, forkhead box transcription factors (FoxO1 and FoxO3a), SODs (SOD1 and SOD2), and NADPH oxidase (NOX)4, proteins were separated by 10% or 15% (SOD1 and SOD2) SDS-PAGE followed by a transfer to Hybond-C Extra nitrocellulose (Amersham Biosciences, Piscataway, NJ). Blots were incubated with anti-HIF-1α (Abcam, Cambridge, MA, 1:250), anti-FoxO1 (Cell Signaling, Danvers, MA, 1:1,000), anti-FoxO3a (Cell Signaling, 1:300), anti-SOD1 (Calbiochem, Merck, Dormstad, Germany, 1:3,000), anti-SOD2 (Abcam, 1:400,000), or anti-NOX4 (Abcam, 1:500) antibodies. Blots were washed and incubated with the appropriate secondary antibody conjugated to horseradish peroxidase. Enhanced chemiluminescence (Amersham) was used for the detection of signals. Calsequestrin (Fitzgerald Industries, Acton, MA, 1:15,000) was used as the loading control.
Statistics.
All results are expressed as means ± SE. For analysis of [Ca2+]i as a function of group and H2O2 treatment, INaCa as a function of group and voltage, and in vivo hemodynamic parameters as a function of group and Iso treatment, two-way ANOVA was used. For analysis of echocardiographic parameters, Ipump, systolic and diastolic [Ca2+]i, [Ca2+]i transient amplitudes, myocyte contraction amplitudes, AP parameters, ROS levels, infarct sizes, AARs, and protein abundance, one-way ANOVA was used. A commercially available software package (JMP version 7, SAS Institute, Cary, NC) was used. In all analyses, P values of <0.05 were taken to be statistically significant.
RESULTS
TRPM2 channels are expressed in sarcolemma and transverse tubules and mediate Ca2+ influx in adult cardiac myocytes exposed to H2O2.
Our global KO mice grew to adulthood and were fertile. PCR (Fig. 1B) and RT-PCR (Fig. 1C) confirmed the absence of the WT TRPM2 gene and transcript, respectively, in KO hearts. Immunolocalization experiments indicated that both TRPM2 channels and the α1-subunit of Na+-K+-ATPase (sarcolemma marker) were expressed in the sarcolemma and transverse tubules in adult LV myocytes (Fig. 2). To ascertain whether TRPM2 channels were functional in the heart, myocytes isolated from WT and KO hearts were exposed to H2O2. Verapamil was present to block Ca2+ entry via L-type Ca2+ channels. There were no differences in baseline [Ca2+]i between WT and KO myocytes, and [Ca2+]i remained stable throughout 15 min of observation (Fig. 3A). After 15 min of H2O2 exposure, however, [Ca2+]i was significantly (P < 0.008) higher in WT compared with KO myocytes (Fig. 3A). The [Ca2+]i increase in H2O2-treated WT myocytes was dependent on extracellular Ca2+ (Fig. 3A), suggesting that Ca2+ influx via TRPM2 channels accounted for the [Ca2+]i increase. This interpretation is supported by the observation that clotrimazole (50 μM), which inhibits TRPM2 current in neonatal rat cardiomyocytes (49), blocked the H2O2-induced [Ca2+]i increase in WT myocytes (Fig. 3B). The small increase in [Ca2+]i in H2O2-treated KO myocytes (Fig. 3A) was likely due to nonspecific leakage pathways since 1) the increase was dependent on extracellular Ca2+ and the L-type Ca2+ channel was blocked and TRPM2 was genetically absent in KO myocytes and 2) in clotrimazole-pretreated WT myocytes exposed to H2O2 and verapamil, modest increases in [Ca2+]i above baseline were observed.
Fig. 2.
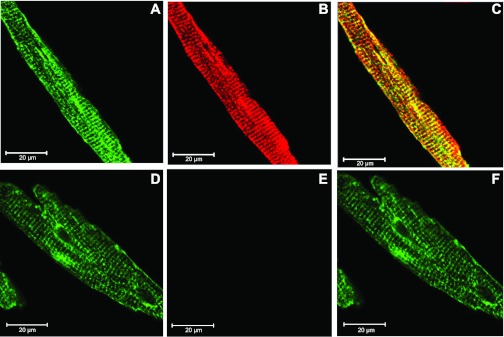
TRPM2 channels are expressed in the sarcolemma and transverse tubules of adult cardiac myocytes. Confocal images of adult WT mouse ventricular myocytes labeled with anti-α1-subunit of Na+-K+-ATPase antibody (A and D), with (B) or without (E) rabbit polyclonal anti-TRPM2 antibody, and merged images (C and F) are shown. Note the yellow (red + green) color in C, suggesting some degree of colocalization of the TRPM2 channel with the α1-subunit of Na+-K+-ATPase antibody. Scale bar = 20 μm.
Fig. 3.

Activated TRPM2 channels mediate Ca2+ influx in adult cardiac myocytes. A: fura-2-loaded myocytes were exposed to either H2O2 (200 μM) or control medium 199 (extracellular Ca2+ concentration: 1.8 mM), and intracellular Ca2+ concentration ([Ca2+]i) was measured at the times indicated. Verapamil (1 μM) was included to block Ca2+ entry via L-type Ca2+ channels. There were 13 WT (open circles) and 13 KO (solid circles) myocytes exposed to H2O2 and 6 WT (open squares) and 6 KO (open triangles) myocytes exposed to control medium 199. There were also 6 WT (solid inverted triangles) myocytes exposed to H2O2 but in the absence of extracellular Ca2+ (no added Ca2+ and 4 mM EGTA). Error bars are not shown if they fell within the boundaries of the symbol. *P < 0.008, WT vs. KO myocytes exposed to H2O2. B: fura-2-loaded WT myocytes were pretreated with or without clotrimazole (50 μM, 10 min) before H2O2 exposure, and [Ca2+]i in medium 199 (containing verapamil and clotrimazole) was measured at the times indicated. There were 17 WT myocytes without clotrimazole pretreatment (open circles) and 7 WT myocytes pretreated with clotrimazole (open squares). Error bars are not shown if they fell within the boundaries of the symbol. *P < 0.035, clotrimazole vs. no clotrimazole.
Cardiac performance in WT and KO mice.
At 8–10 wk of age, there were no differences in body weights, LV masses, and heart rates (Table 1). Echocardiography and in vivo hemodynamic measurements demonstrated normal baseline fractional shortening (FS) and +dP/dt (Fig. 4 and Table 1), respectively, in KO sham hearts compared with WT sham hearts. Both WT sham and KO sham hearts responded similarly to Iso stimulation (Fig. 4), as confirmed by the absence of group (P < 0.65) and group × Iso interaction (P < 0.90) effects.
Table 1.
Echocardiographic parameters of TRPM2 KO mice
| WT Sham |
WT I/R |
KO Sham |
KO I/R |
|||||
|---|---|---|---|---|---|---|---|---|
| Means ± SE | No. of mice/group | Means ± SE | No. of mice/group | Means ± SE | No. of mice/group | Means ± SE | No. of mice/group | |
| Body weight, g | 24.8 ± 0.8 | 4 | 25.7 ± 0.4 | 10 | 27.3 ± 1.1 | 4 | 25.9 ± 0.7 | 9 |
| LV mass, mg | 72.2 ± 13.5 | 70.1 ± 4.2 | 63.3 ± 5.5 | 76.0 ± 4.2 | ||||
| Heart rate, beats/min | 452 ± 33 | 462 ± 19 | 426 ± 20 | 474 ± 16 | ||||
| LVID at diastole, mm | 3.73 ± 0.43 | 3.79 ± 0.13 | 3.57 ± 0.17 | 3.99 ± 0.13 | ||||
| LVID at systole, mm | 2.18 ± 0.27 | 2.56 ± 0.10 | 2.11 ± 0.16 | 2.88 ± 0.08*† | ||||
| Ejection fraction, % | 73.7 ± 1.3 | 61.5 ± 1.5* | 72.9 ± 2.2 | 54.1 ± 1.9*† | ||||
| Fractional shortening, % | 41.9 ± 0.9 | 32.5 ± 1.0* | 41.2 ± 1.8 | 27.2 ± 1.3*† | ||||
| +dP/dt, mmHg/s | 7,183 ± 596 | 4 | 5,812 ± 365* | 8 | 7,023 ± 269 | 4 | 5,539 ± 356*† | 7 |
| Maximal +dP/dt, mmHg/s | 13,401 ± 793 | 10,579 ± 267* | 13,130 ± 453 | 9,428 ± 677*† | ||||
WT, wild type; sham, sham operation; KO, transient receptor potential-melastatin channel member 2 (TRPM2) knockout; I/R, ischemia-reperfusion; LVID, left ventricular (LV) internal dimension; +dP/dt, first time derivative of the LV pressure rise; maximal +dP/dt, +dP/dt after injection of 10 ng isoproterenol.
P < 0.0005, sham vs. I/R in either WT or KO myocytes;
P < 0.025, WT I/R vs. KO I/R.
Fig. 4.

Cardiac performance after ischemia-reperfusion (I/R) is worse in TRPM2 KO compared with WT hearts. Hearts were subjected to sham operation (sham) or 30 min of ischemia followed by reperfusion. Top: after 2 days of reperfusion, fractional shortening (FS) was determined by echocardiography. There were n = 4 WT sham, 10 WT I/R, 4 KO sham, and 9 KO I/R mice, respectively. *P < 0.0002, sham vs. I/R for both WT and KO hearts; #P < 0.009, WT I/R vs. KO I/R hearts. Bottom: after 3 days of reperfusion, in vivo catheterization was performed in anesthetized mice. The first time derivatives of the left ventricular (LV) pressure rise (+dP/dt) and fall (−dP/dt) were continuously measured, both at baseline and at increasing doses of isoproterenol. There were n = 4 WT sham (open circles), 4 KO sham (open squares), 8 WT-I/R (solid diamonds) and 7 KO-I/R (solid squares) mice, respectively. Error bars are not shown if they fell within the boundaries of the symbol. Two-way ANOVA indicated P < 0.0001 for WT sham vs. WT I/R and KO sham vs. KO I/R mice and P < 0.0075 for WT I/R vs. KO I/R mice.
Effects of I/R on infarct size, cardiac performance, and perioperative mortality in WT and KO mice.
Three days post-I/R, both AARs (%LV) and infarct sizes (%AAR) were similar between WT and KO hearts (Fig. 5). While both WT and KO hearts suffered significant decreases in FS (P < 0.0002) and +dP/dt (group effect, P < 0.0001) 2–3 days post-I/R compared with their sham controls, KO I/R hearts had significantly lower FS (P < 0.001) and +dP/dt (group effect, P < 0.008) compared with WT I/R hearts (Fig. 4 and Table 1), despite having similar AARs and infarct sizes. In the present series of experiments, mortality at 3 days post-I/R was 0 of 7 mice in the WT sham group, 7 of 56 mice (12.5%) in the WT I/R group, 0 of 8 mice in the KO sham group, and 6 of 57 mice (10.5%) in the KO I/R group.
Fig. 5.

Infarct sizes in WT and TRPM2 KO hearts subjected to I/R. Hearts were subjected to 30 min of ischemia followed by reperfusion for 3 days. Top: area at risk [AAR; 2,3,5-triphenyltetrazolium (TTC) positive and TTC negative], infarct area (TTC negative), and area not at risk (Evans blue dye stained) were determined for both WT and KO hearts. Bottom: summary for AAR and infarct size for 7 WT I/R and 7 KO I/R hearts.
Effects of I/R on the expression of selected proteins involved in excitation-contraction coupling in WT and KO hearts.
compared with WT I/R hearts, KO I/R hearts exhibited significantly increased expression of NCX1 (P < 0.004) and decreased expression of the α1-subunit of Na+-K+-ATPase (P < 0.003; Fig. 6 and Table 2). There were no differences in the expression of SERCA2, the α2-subunit of Na+-K+-ATPase, the α1c-subunit of Cav1.2, and pRyR2 channels between WT I/R and KO I/R hearts. Expression of Cav1.2 (P < 0.035) and pRyR2 (P < 0.0025) was higher in WT I/R compared with WT sham hearts. Compared with KO sham hearts, only NCX1 expression was significantly (P < 0.003) higher in KO I/R hearts. Finally, KO sham hearts had higher (P < 0.02) pRyR2 compared with WT sham hearts.
Fig. 6.

Effects of I/R on the expression of selected proteins involved in cardiac excitation-contraction coupling. WT or KO hearts were subjected to either sham operation or 30 min of ischemia followed by reperfusion for 3 days. LV homogenates were prepared and subjected to SDS-PAGE followed by Western blot analysis. Protein loading for ryanodine receptor phosphorylated at Ser2808 (pRyR2), sarco(endo)plasmic reticulum Ca2+-ATPase 2 (SERCA2), α1- and α2-subunits of Na+-K+-ATPase, Na+/Ca2+ exchanger 1 (NCX1), L-type Ca2+ channel (Cav1.2), and calsequestrin (CLSQ) was 50 μg/lane. Composite results are shown in Table 2.
Table 2.
Effects of TRPM2 KO and I/R on levels of selected proteins
| WT Sham | WT I/R | KO Sham | KO I/R | |
|---|---|---|---|---|
| pRyR2 | 23.5 ± 3.7 | 40.4 ± 1.9‡ | 36.5 ± 1.9§ | 32.6 ± 3.0 |
| SERCA2 | 155.0 ± 48.4 | 90.6 ± 6.7 | 137.2 ± 30.0 | 118.8 ± 15.3 |
| α1-Subunit of Na+-K+-ATPase | 53.6 ± 22.9 | 90.4 ± 8.3 | 48.5 ± 21.8 | 36.7 ± 10.5* |
| α2-Subunit of Na+-K+-ATPase | 132.7 ± 47.2 | 91.0 ± 17.4 | 100.1 ± 39.7 | 96.3 ± 19.1 |
| NCX1 | 62.2 ± 12.9 | 51.0 ± 3.8 | 39.6 ± 3.6 | 86.7 ± 8.7*† |
| Cav1.2 | 49.3 ± 5.7 | 82.8 ± 8.1‡ | 55.5 ± 7.5 | 81.9 ± 9.8 |
Values (in arbitrary units) are means ± SE; n = 3 hearts in the WT sham group; 6 hearts in the WT I/R group, 4 hearts in the KO sham group, and 6 hearts in the KO I/R group. pRyR2, cardiac ryanodine receptor phosphorylated at Ser2808; SERCA2, sarco(endo)plasmic reticulum Ca2+-ATPase 2; NCX1, cardiac Na+/Ca2+ exchanger 1; Cav1.2, α-subunit of the L-type Ca2+ channel.
P < 0.05, WT I/R vs. KO I/R;
P < 0.025, KO sham vs. KO I/R;
P < 0.03, WT sham vs. WT I/R;
P < 0.025, WT sham vs. KO sham.
Effects of I/R on INaCa, Ipump, and AP in WT and KO myocytes.
In agreement with the NCX1 expression results (Fig. 6), INaCa in myocytes isolated from KO I/R hearts was the highest among the four groups (group effect, P < 0.0001; group × voltage interaction effect, P < 0.0001; Fig. 7). Interestingly, I/R had no effect on INaCa in WT myocytes (WT sham vs. WT I/R: group effect, P < 0.14).
Fig. 7.

I/R increases Na+/Ca2+ exchange current (INaCa) in TRPM2 KO but not WT myocytes. LV myocytes were isolated from WT and KO hearts previously subjected to sham operation or 30 min of ischemia followed by 3 days of reperfusion. The standard patch-clamp whole cell configuration was used to measure INaCa. The pipette solution contained (in mM) 100 Cs+ glutamate, 7.25 NaCl, 1 MgCl2, 20 HEPES, 2.5 Na2ATP, 10 EGTA, and 6 CaCl2 (pH 7.2). Free Ca2+ in the pipette solution was 205 nM, as measured fluorimetrically with fura-2. The external solution contained (in mM) 130 NaCl, 5 CsCl, 1.2 MgSO4, 1.2 NaH2PO4, 5 CaCl2, 10 HEPES, 10 Na+ HEPES, and 10 glucose (pH 7.4). Verapamil (1 μM) was used to block L-type Ca2+ current. Our measurement conditions were biased toward measuring outward (3 Na+ out:1 Ca2+ in) INaCa. A: after the myocyte had been held at the calculated reversal potential (−73 mV) of INaCa for 5 min [to minimize fluxes through NCX1 and thus allow intracellular Na+ concentration ([Na+]i) and [Ca2+]i to equilibrate with those in pipette solution), INaCa (30°C) was measured in WT and TRPM2 KO myocytes using a descending (from +100 to −120 mV, 500 mV/s) to ascending (from −120 to +100 mV, 500 mV/s) voltage ramp, first in the absence and then in the presence of 1 mM NiCl2. B: raw currents measured in a WT myocyte. INaCa was defined as the difference current measured in the absence and presence of Ni+ during the descending voltage ramp. Note that with the exception of small contamination of the ascending ramp by cardiac Na+ current, there were little to no differences in currents measured between the descending and ascending voltage ramps. This suggests that [Ca2+]i and [Na+]i sensed by NCX1 did not appreciably change by NCX1 fluxes during the brief (880 ms) voltage ramp. INaCa was divided by membrane capacitance before comparisons. C: current-voltage relationships of INaCa (means ± SE) from WT sham (open circles; n = 7), KO sham (open triangles; n = 9), WT I/R (open squares; n = 8), and KO I/R (open diamonds; n = 12) myocytes are shown. The reversal potential of INaCa was approximately −60 mV, close to the theoretical reversal potential of −73 mV. Error bars are not shown if they fell within the boundaries of the symbol. Two-way ANOVA indicated P < 0.0001 for KO I/R vs. WT I/R or KO sham myocytes.
Although Ipump was not different between WT sham and KO sham myocytes, post-I/R Ipump due to the α1-subunit but not α2-subunit of Na+-K+-ATPase was significantly (P < 0.005) decreased in both WT and KO myocytes (Fig. 8). In addition, Ipump due to the α1-subunit of Na+-K+-ATPase was further reduced (P < 0.048) in KO I/R compared with WT I/R myocytes (Fig. 8), in agreement with decreased expression of the α1-subunit of Na+-K+-ATPase in KO I/R myocytes (Fig. 6). In response to Iso (1 μM) stimulation, the increase in Ipump due to the α1-subunit of Na+-K+-ATPase over baseline was significantly (P < 0.012) higher in WT I/R compared with KO I/R myocytes (Fig. 8). Iso stimulation resulted in little to no increase in Ipump due to the α2-subunit of Na+-K+-ATPase in both WT I/R and KO I/R myocytes.
Fig. 8.

I/R decreases Na+-K+-ATPase current (Ipump) more in TRPM2 KO myocytes. WT or KO hearts were subjected to either sham operation or 30 min ischemia of followed by 3 days of reperfusion before myocyte isolation. LV myocytes were held at 0 mV and 30°C. The pipette solution contained (in mM) 70 Na+-aspartate, 20 K+-aspartate, 8 CsOH, 7 MgSO4, 11 EGTA, 10 tetraethylammonium Cl, 1 CaCl2, 5 HEPES, 5 Na2ATP, and 0.2 GTP (pH 7.2). The external solution contained (in mM) 137.7 NaCl, 18 KCl, 2.3 NaOH, 1 MgCl2, 2 BaCl2, 1 CdCl2, 5 HEPES, and 10 glucose (pH 7.4). Top: raw current from a WT myocyte. After baseline current had been recorded, dihydroouabain (DHO; 5 μM) was added, and the difference was taken to be current due to the α2-subunit of Na+-K+-ATPase (Iα2). DHO (1 mM) was then added, and the additional decrease in current was taken to be due to the α1-subunit of Na+-K+-ATPase (Iα1). After DHO washout, Ipump recovered to baseline levels. In some experiments, isoproterenol (Iso; 1 μM) was added, and Iα1 and Iα2 were separated as before. Bottom left: current densities (means ± SE) of Iα1 and Iα2 from WT sham (open bars; n = 3), WT I/R (solid bars; n = 10), KO sham (open bars; n = 5), and KO I/R (solid bars; n = 11) myocytes. *P < 0.005, sham vs. I/R for both WT and KO myocytes; #P < 0.05, WT I/R vs. KO I/R myocytes. Bottom right: increase in Iα1 (open bars) and Iα2 (solid bars) over baseline (ΔIpump) after the addition of Iso (1 μM) to WT I/R (n = 8) and KO I/R (n = 4) myocytes. *P < 0.015, WT I/R vs. KO I/R myocytes.
There were no differences in resting membrane potential, AP amplitude, and AP duration (APD) at 90% repolarization among WT sham, WT I/R, KO sham, and KO I/R myocytes (Fig. 9). The most dramatic difference was a doubling of APD at 50% repolarization in KO I/R myocytes compared with the other groups (P < 0.0001).
Fig. 9.

I/R prolongs action potential (AP) duration (APD) more in TRPM2 KO myocytes. LV myocytes were isolated from WT and KO hearts previously subjected to either sham operation or 30 min ischemia followed by 3 days of reperfusion. Myocytes were paced at 1 Hz. The pipette solution consisted of (in mM) 125 KCl, 4 MgCl2, 0.06 CaCl2, 10 HEPES, 5 K+-EGTA, 3 Na2ATP, and 5 Na2-creatine phosphate (pH 7.2). The external solution consisted of (in mM) 132 NaCl, 5.4 KCl, 1.8 CaCl2, 1.8 MgCl2, 0.6 NaH2PO4, 7.5 HEPES, 7.5 Na+-HEPES, and 5 glucose (pH 7.4). Top: representative APs from WT sham, WT I/R, KO sham, and KO I/R myocytes recorded using the current-clamp configuration at 1.5× threshold stimulus, 4-ms duration, and 30°C. Bottom: means ± SE of resting membrane potential (Em), AP amplitude, APD at 50% (APD50), and APD at 90% repolarization (APD90) from 8 WT sham, 9 KO sham, 21 WT I/R, and 25 KO I/R myocytes. *P < 0.0001, KO I/R vs. KO sham or WT I/R myocytes.
Effects of I/R on [Ca2+]i transients and myocyte contractility in WT and KO myocytes.
Systolic [Ca2+]i was significantly (P < 0.0001) lower in both WT and KO myocytes subjected to I/R compared with their respective sham controls (Table 3). There were no differences in diastolic [Ca2+]i among WT sham, WT I/R, KO sham, and KO I/R myocytes. As a result, [Ca2+]i transient amplitudes were significantly (P < 0.0001) lower in WT I/R and KO I/R myocytes compared with their respective sham controls. There were no differences in systolic [Ca2+]i and [Ca2+]i transient amplitudes between WT I/R and KO I/R myocytes.
Table 3.
Effects of TRPM2 KO and I/R on [Ca2+]i transients and contractility
| WT Sham |
WT I/R |
KO Sham |
KO I/R |
|||||
|---|---|---|---|---|---|---|---|---|
| Means ± SE | No. of myocytes/group | Means ± SE | No. of myocytes/group | Means ± SE | No. of myocytes/group | Means ± SE | No. of myocytes/group | |
| Systolic [Ca2+]i, nM | 273 ± 16 | 31 | 190 ± 8* | 44 | 290 ± 13 | 29 | 180 ± 6* | 44 |
| Diastolic [Ca2+]i, nM | 105 ± 6 | 108 ± 3 | 107 ± 4 | 103 ± 3 | ||||
| [Ca2+]i transient amplitude, % | 28.9 ± 1.6 | 15.0 ± 0.7* | 27.1 ± 1.4 | 14.8 ± 0.5* | ||||
| Contraction amplitude, % | 6.8 ± 0.4 | 28 | 5.0 ± 0.3* | 43 | 6.6 ± 0.4 | 22 | 4.8 ± 0.3* | 44 |
The intracellular Ca2+ concentration ([Ca2+]i) transient amplitude is given as the percent increase in the fura 2 signal. The contraction amplitude is given as a percentage of the resting cell length.
P < 0.0006, I/R vs. sham in both WT and KO myocytes.
Alterations in [Ca2+]i homeostasis would be expected to affect myocyte contractility. Indeed, maximal contraction amplitude was decreased by I/R in both WT and KO myocytes (Table 3). There were no differences in maximal contraction amplitudes between WT I/R and KO I/R myocytes.
Effects of hypoxia/reoxygenation on ROS levels in WT and KO myocytes.
In the heart, both ROS (40) and cADPR (48) production are increased after I/R. Since both ROS and cADPR can activate TRPM2, we measured ROS levels in WT and KO myocytes, both under normoxic conditions and after hypoxia/reoxygenation to simulate I/R in vitro. There were no differences (P < 0.38) in ROS levels between WT and KO myocytes incubated under normoxic conditions (Fig. 10). ROS levels were significantly (P < 0.0001) higher in hypoxic/reoxygenated myocytes compared with their respective normoxic controls (Fig. 10). Importantly, after hypoxia/reoxygenation, ROS levels in KO were significantly (P < 0.0001) higher than those present in WT myocytes (Fig. 10).
Fig. 10.

ROS generation is higher in TRPM2 KO myocytes subjected to hypoxia-reoxygenation. WT and KO myocytes incubated in Krebs-Henseleit bicarbonate buffer containing pyruvate (5 mM) as the sole substrate were either exposed to hypoxia (1% O2-5% CO2) for 2 h followed by 30 min of reoxygenation [simulated I/R (sI/R)] or to control normoxic (21% O2-5% CO2) conditions. During the 30 min of reoxygenation, myocytes were loaded with 5-(and 6-)chloromethyl-2′, 7′-dichlorofluorescein (DCF) diacetate acetyl ester (5 μM). Top: DCF fluorescence from WT sI/R and KO sI/R myocytes. Bottom: DCF fluorescence of WT normoxic (black bar; n = 23), WT sI/R (blue bar; n = 35), KO normoxic (black bar; n = 30), and KO sI/R (blue bar; n = 31) myocytes. The photomultiplier was set at the same gain for all four groups of myocytes. *P < 0.0001, normoxic vs. sI/R for both WT and KO myocytes; #P < 0.0001, WT sI/R vs. KO sI/R myocytes.
Effects of I/R on oxygen free radical scavenging enzymes in KO myocytes.
Since ROS levels were higher in KO myocytes after hypoxia/reoxygenation, we measured oxygen free radical scavenging enzymes in WT I/R and KO I/R hearts. Both SOD1 and SOD2 were significantly lower in KO I/R hearts compared with WT I/R hearts (Fig. 11). Upstream regulators of SOD expression, HIF-1α, FoxO1, and FoxO3a were also significantly lower in KO I/R hearts. Finally, NOX4, which is involved in the generation of oxygen free radicals, was significantly higher in KO I/R hearts.
Fig. 11.

After I/R, TRPM2 KO hearts have a reduced capability to cope with oxidative stress. WT or KO hearts were subjected to 30 min of ischemia followed by reperfusion for 3 days. A; LV homogenates were subjected to SDS-PAGE followed by Western blot analysis to detect hypoxia-inducible factor (HIF)-1α, forkhead box transcription factors (FoxO1 and FoxO3a), SODs (SOD1 and SOD2), and NAPDH oxidase 4 (NOX4). Protein loading was normalized against CLSQ. B: composite results of A. *P < 0.05.
DISCUSSION
Although TRPM2 mRNA and protein are expressed in cardiac myocytes (49), studies on the functional significance of TRPM2 channels in the heart remain relatively sparse. Using a global constitutive TRPM2 KO mouse (C57BL/6 background) generated by deleting exons 19 and 20 encoding transmembrane domains 3 and 4, Zhang et al. (52) demonstrated increased glucose uptake, enhanced insulin signaling, and decreased calmodulin in TRPM2 KO hearts. In cultured neonatal rat cardiomyocytes, H2O2 (100 μM) activated TRPM2 through increased ADPR/NAD+ formation, leading to Ca2+ and Na+ overload in mitochondria, myocyte apoptosis through mitochondrial membrane disruption, cytochrome c release, and caspase-3-dependent chromatin condensation and fragmentation (49). The results of these two studies suggest that TRPM2 channels may play a significant role in cardiac I/R injury by inducing apoptosis and altering energy metabolism. In addition, TRPM2-mediated Ca2+ influx stimulates chemokine production by inflammatory cells (monocytes, macrophages, and granulocytes) (37) and may further contribute to tissue damage post-I/R. Therefore, the major hypothesis of the present study was that inhibiting or eliminating TRPM2 channels should confer cardiac protection against oxidative injury induced by I/R.
This is the first study that has demonstrated that TRPM2 channels are localized in the sarcolemma and transverse tubules in adult mouse LV myocytes (Fig. 2) and that the cardiac TRPM2 channels were functional since activation by H2O2 resulted in significantly higher [Ca2+]i in WT compared with KO myocytes (Fig. 3A). The [Ca2+]i increase in H2O2-treated WT myocytes was absolutely dependent on extracellular Ca2+ (Fig. 3A) and blocked by clotrimazole (Fig. 3B), indicating that Ca2+ influx via activated TRPM2 channels was the mechanism of the H2O2-induced [Ca2+]i increase. The time course of the [Ca2+]i increase in adult LV myocytes treated with H2O2 was similar to that observed in cultured neonatal rat cardiomyocytes exposed to H2O2 in that [Ca2+]i started to increase ∼10 min and reached half-maximal ∼20 min after H2O2 addition (49).
The second major finding is that genetic depletion of TRPM2 channels did not result in measurable differences in contractile function between WT sham and KO sham hearts (Fig. 4). In agreement with in vivo cardiac function measurements, single myocyte [Ca2+]i dynamics and contraction amplitudes (Table 3), AP amplitudes and APD (Fig. 9), INaCa (Fig. 7), and Ipump (Fig. 8) were also similar between WT sham and KO sham myocytes. In addition, expression of most of the proteins involved in excitation-contraction coupling was similar except for pRyR2, which was significantly higher in KO sham compared with WT sham myocytes (Fig. 6). Elevated pRyR2 has been associated with increased sarcoplasmic reticulum Ca2+ leak in heart failure (27), although this point remains controversial (19). Our data suggest that the increased energy expenditure, low cardiac calmodulin, and enhanced insulin sensitivity observed in TRPM2 KO hearts (albeit different exons of TRPM2 were deleted in the generation of TRPM2 KO mice) appear to have little to no effect on the mechanical performance of hearts and myocyte excitation-contraction coupling examined under baseline conditions.
Our 30-min cardiac ischemia followed by reperfusion in WT animals resulted in ∼13% LV infarct (Fig. 4), ∼22% decrease in FS (Fig. 5), ∼21% reduction in maximal +dP/dt (Fig. 5), ∼44% reduction in [Ca2+]i transient amplitudes, and ∼27% decrease in single myocyte contraction amplitudes (Table 3). Michael et al. (28) reported 13.7 ± 2.4% LV infarct after 30 min of occlusion followed by 24 h of reperfusion in male FVB mice. Li et al. (26) reported 21.6% reduction in maximal +dP/dt after 30 min of ischemia followed by 28 days of reperfusion in male C57BL/6 mice. Our results are similar and indicate that our I/R model was valid.
The most dramatic result of the present study was that contrary to the observation that TRPM2 mediates oxidative stress-induced apoptosis and cell death in cultured neonatal cardiomyocytes (49), TRPM2 deficiency was associated with aggravation, rather than amelioration, of in vivo cardiac contractile dysfunction post-I/R injury: a model associated with increased oxidative stress (40). Despite similar AARs and infarct sizes between WT I/R and KO I/R hearts (Fig. 5), FS and +dP/dt were clearly better preserved in WT I/R compared with KO I/R hearts (Fig. 4).
In KO-I/R hearts, decreased myocardial contractility compared with WT I/R hearts may be due to increased myocyte maladaptations postinjury. Compared with WT I/R myocytes, KO I/R myocytes had higher NCX1 expression (Fig. 6), larger INaCa (Fig. 7), decreased expression of the α1-subunit of Na+-K+-ATPase (Fig. 6), lower Ipump (Fig. 8), and significantly prolonged APD at 50% repolarization (Fig. 9), suggesting alterations in repolarizing K+ currents. Enhanced NCX1 expression and activity (13), reduced Na+-K+-ATPase expression and activity (6), and prolonged APD (51) have been observed in ischemic cardiomyopathy. A critical observation is that in WT I/R and KO I/R myocytes, [Ca2+]i dynamics and myocyte shortening mechanics were equally depressed post-I/R (Table 3). Therefore, the inferior myocardial contractility in KO I/R compared with WT I/R hearts (Fig. 4) is unlikely to be mediated by modest differences in the expression of ion transporters associated with excitation-contraction coupling (Fig. 6) and [Ca2+]i regulation.
In KO I/R myocytes, expression of NOX4 was higher, whereas that of SOD was lower, compared with WT I/R myocytes (Fig. 11). In addition, two upstream regulators of SOD expression, such as HIF-1α and FoxOs, were lower in KO-I/R myocytes (Fig. 11). These observations suggest that KO I/R myocytes may generate more ROS while simultaneously having less capacity to cope with oxidative stress. Indeed, when isolated LV myocytes were subjected to hypoxia followed by reoxygenation to simulate I/R, ROS levels were much higher in previously hypoxic KO compared with WT myocytes (Fig. 10).
A very recent publication using an independent TRPM2 KO mouse (C57BL/6 background) reported very different results (16). Specifically, after 45 min of ischemia followed by 24 h of reperfusion in vivo, neutrophil infiltration was less, infarct size was smaller, and +dP/dt was higher in KO compared with WT hearts. In addition, pretreatment with econazole (10 μM) to inhibit TRPM2 channels in WT neutrophils reduced infarct size in isolated perfused TRPM2 KO hearts subjected to 60 min of ischemia followed by 2 h of reperfusion. The authors suggested that increased neutrophil adhesion to endothelial cells mediated by TRPM2 channels may cause increased damage post-I/R. The reasons for the different results between our study and those of Hiro et al. (16) are not obvious but may include the following: 1) TRPM2 was knocked out by targeting different exons, 2) 45 versus 30 min of ischemia resulted in much larger infarcts (45% vs. 27% AAR), 3) different anesthesia (pentobarbital vs. isoflurane for I/R surgery, pentobarbital vs. avertin for in vivo hemodynamic measurements), 4) different surgical techniques used in measuring in vivo hemodynamics (opening the chest followed by LV puncture vs. catheterizing the right carotid artery in a closed-chest preparation), and 5) increased heat dissipation in open-chest mice compared with closed-chest mice. The reason for choosing 45 min of in vivo ischemia is not clear since it has been known for well over 3 decades that 40 min of ischemia results in irreversible cardiac injury and reperfusion is followed by the prompt development (within 2 min) of explosive cell swelling, contraction bands, and calcification of mitochondria (18). It may be difficult to demonstrate salutatory effects of cardiac TRPM2 channels on myocyte survival when irreversible injury has already occurred. It is also difficult to reconcile the results of Hiro et al. (16) with the report of Di et al. (5), which showed that TRPM2 channels had beneficial effects on endotoxin-induced lung inflammation by dampening NOX-mediated ROS production in WT but not KO phagocytes. Finally, cumulative evidence weighs against a pivotal role of neutrophils as a causative factor in most forms of I/R injury in the heart and brain (3).
There are caveats to the present study. The first caveat is that our study design using global TRPM2 KO mice could not unequivocally differentiate whether the beneficial effects of TRPM2 channels on myocardial contractility post-I/R was due to better capability of WT myocytes to cope with oxidative stress or to reduced inflammatory response secondary to less ROS production by infiltrating WT monocytes and macrophages (5). However, a direct causal association between infiltrating inflammatory cells and myocardial injury remains unproven (3). Future studies using cardiac-specific, conditional TRPM2 KO mice would provide an unequivocal answer. The second caveat is that our experiments did not differentiate whether increased ROS scavenging capacity or reduced ROS generation was the dominant mechanism by which TRPM2 channels protected hearts from I/R injury. The third caveat is that we only evaluated cardiac function and mortality at 3 days post-I/R and thus cannot comment whether short-term improvement in myocardial function would impact on long-term survival. Finally, the mechanisms by which enhanced capacity to cope with oxidative stress translate into improved myocardial contractility postischemia need to be further elucidated. Our data indicate that the improvement in cardiac function in KO I/R hearts is unlikely due to better [Ca2+]i dynamics and single myocyte shortening mechanics.
In conclusion, TRPM2 channels protected hearts from I/R injury. The protection was associated with a previously unknown function of TRPM2 channels in reducing ROS generation and increasing ROS scavenging capability in adult cardiac myocytes. Together with the demonstrated ability of TRPM2 channels to reduce phagocyte ROS production (5), the modest activation of TRPM2 channels may represent a novel therapeutic approach in ameliorating cardiac ischemic injury.
GRANTS
This work was supported in part by National Institutes of Health Grants RO1-DK-46778 (to B. A. Miller), RO1-HL-56205, RO1-HL-61690, RO1-HL-85503, PO1-HL-75443, and PO1-HL-91799 (to W. J. Koch), RO1-HL-86699 (to M. Madesh), PO1-HL-91799 (Project 2; to A. M. Feldman), and RO1-HL-58672 and RO1-HL-74854 (to J. Y. Cheung).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: B.A.M., J.W., I.H.-L., A.M.F., and J.Y.C. conception and design of research; B.A.M., J.W., I.H.-L., E.G., J.S., X.-Q.Z., M.M., K.M., T.G., S.-j.C., K.K., K.C., and J.Y.C. performed experiments; B.A.M., J.W., I.H.-L., E.G., J.S., X.-Q.Z., M.M., K.M., T.G., S.-j.C., K.K., K.C., and J.Y.C. analyzed data; B.A.M., J.W., I.H.-L., E.G., J.S., X.-Q.Z., W.J.K., M.M., K.M., S.-j.C., K.K., K.C., A.M.F., and J.Y.C. interpreted results of experiments; B.A.M., J.W., X.-Q.Z., M.M., S.-j.C., and J.Y.C. prepared figures; B.A.M., J.W., W.J.K., A.M.F., and J.Y.C. edited and revised manuscript; B.A.M., J.W., I.H.-L., E.G., J.S., X.-Q.Z., W.J.K., M.M., K.M., T.G., S.-j.C., K.K., K.C., A.M.F., and J.Y.C. approved final version of manuscript; J.Y.C. drafted manuscript.
REFERENCES
- 1. Aarts M, Iihara K, Wei WL, Xiong ZG, Arundine M, Cerwinski W, MacDonald JF, Tymianski M. A key role for TRPM7 channels in anoxic neuronal death. Cell 115: 863–877, 2003 [DOI] [PubMed] [Google Scholar]
- 2. Andersson DC, Fauconnier J, Yamada T, Lacampagne A, Zhang SJ, Katz A, Westerblad H. Mitochondrial production of reactive oxygen species contributes to the β-adrenergic stimulation of mouse cardiomycytes. J Physiol 589: 1791–1801, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baxter GF. The neutrophil as a mediator of myocardial ischemia-reperfusion injury: time to move on. Basic Res Cardiol 97: 268–275, 2002 [DOI] [PubMed] [Google Scholar]
- 4. Cheung JY, Thompson IG, Bonventre JV. Effects of extracellular calcium removal and anoxia on isolated rat myocytes. Am J Physiol Cell Physiol 243: C184–C190, 1982 [DOI] [PubMed] [Google Scholar]
- 5. Di A, Gao XP, Qian F, Kawamura T, Han J, Hecquet C, Ye RD, Vogel SM, Malik AB. The redox-sensitive cation channel TRPM2 modulates phagocyte ROS production and inflammation. Nat Immunol 13: 29–34, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dixon IMC, Hata T, Dhalla NS. Sarcolemmal Na+-K+-ATPase activity in congestive heart failure due to myocardial infarction. Am J Physiol Cell Physiol 262: C664–C671, 1992 [DOI] [PubMed] [Google Scholar]
- 7. Du J, Xie J, Yue L. Intracellular calcium activates TRPM2 and its alternative spliced isoforms. Proc Natl Acad Sci USA 106: 7239–7244, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Duncan LM, Deeds J, Hunter J, Shao J, Holmgren LM, Woolf EA, Tepper RI, Shyjan AW. Down-regulation of the novel gene melastatin correlates with potential for melanoma metastasis. Cancer Res 58: 1515–1520, 1998 [PubMed] [Google Scholar]
- 9. Gao E, Lei YH, Shang X, Huang ZM, Zuo L, Boucher M, Fan Q, Chuprun JK, Ma XL, Koch WJ. A novel and efficient model of coronary artery ligation and myocardial infarction in the mouse. Circ Res 107: 1445–1453, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gasser A, Glassmeier G, Fliegert R, Langhorst MF, Meinke S, Hein D, Kruger S, Weber K, Heiner I, Oppenheimer N, Schwarz JR, Guse AH. Activation of T cell calcium influx by the second messenger ADP-ribose. J Biol Chem 281: 2489–2496, 2006 [DOI] [PubMed] [Google Scholar]
- 11. Goel M, Sinkins WG, Schilling WP. Selective association of TRPC channel subunits in rat brain synaptosomes. J Biol Chem 277: 48303–48310, 2002 [DOI] [PubMed] [Google Scholar]
- 12. Hara Y, Wakamori M, Ishii M, Maeno E, Nishida M, Yoshida T, Yamada H, Shimizu S, Mori E, Kudoh J, Shimizu N, Kurose H, Okada Y, Imoto K, Mori Y. LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol Cell 9: 163–173, 2002 [DOI] [PubMed] [Google Scholar]
- 13. Hasenfuss G. Alteration of calcium-regulatory proteins in heart failure. Cardiovasc Res 37: 279–289, 1998 [DOI] [PubMed] [Google Scholar]
- 14. Hecquet CM, Ahmmed GU, Vogel SM, Malik AB. Role of TRPM2 channel in mediating H2O2-induced Ca2+ entry and endothelial hyperpermeability. Circ Res 102: 347–355, 2008 [DOI] [PubMed] [Google Scholar]
- 15. Hill K, McNulty S, Randall AD. Inhibition of TRPM2 channels by the antifungal agents clotrimazole and econazole. Naunyn Schmiedebergs Arch Pharmacol 370: 227–237, 2004 [DOI] [PubMed] [Google Scholar]
- 16. Hiroi T, Wajima T, Negoro T, Ishii M, Nakano Y, Kiuchi Y, Mori Y, Shimizu S. Neutrophil TRPM2 channels are implicated in the exacerbation of myocardial ischemia/reperfusion injury. Cardiovasc Res 97: 271–281, 2012 [DOI] [PubMed] [Google Scholar]
- 17. Hofmann T, Schaefer M, Schultz G, Gudermann T. Subunit composition of mammalian transient receptor potential channels in living cells. Proc Natl Acad Sci USA 99: 7461–7466, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jennings RB, Hawkins HK, Lowe JE, Hill ML, Klotman S, Reimer KA. Relation between high energy phosphate and lethal injury in myocardial ischemia in the dog. Am J Pathol 92: 187–214, 1978 [PMC free article] [PubMed] [Google Scholar]
- 19. Jiang MT, Lokuta AJ, Farrell EF, Wolff MR, Haworth RA, Valdivia HH. Abnormal Ca2+ release, but normal ryanodine receptors, in canine and human heart failure. Circ Res 91: 1015–1022, 2002 [DOI] [PubMed] [Google Scholar]
- 20. Kaneko S, Kawakami S, Hara Y, Wakamori M, Itoh E, Minami T, Takada Y, Kume T, Katsuki H, Mori Y, Akaike A. A critical role of TRPM2 in neuronal cell death by hydrogen peroxide. J Pharmacol Sci 101: 66–76, 2006 [DOI] [PubMed] [Google Scholar]
- 21. Kang YJ, Chen Y, Epstein PN. Suppression of doxorubicin cardiotoxicity by overexpression of catalase in the heart of transgenic mice. J Biol Chem 271: 12610–12616, 1996 [DOI] [PubMed] [Google Scholar]
- 22. Kolisek M, Beck A, Fleig A, Penner R. Cyclic ADP-ribose and hydrogen peroxide synergize with ADP-ribose in the activation of TRPM2 channels. Mol Cell 18: 61–69, 2005 [DOI] [PubMed] [Google Scholar]
- 23. Kuhn FJ, Heiner I, Luckhoff A. TRPM2: a calcium influx pathway regulated by oxidative stress and the novel second messenger ADP-ribose. Pflügers Arch 451: 212–219, 2005 [DOI] [PubMed] [Google Scholar]
- 24. Lakso M, Pichel JG, Gorman JR, Sauer B, Okamoto Y, Lee E, Alt FW, Westphal H. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc Natl Acad Sci USA 93: 5860–5865, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li M, Jiang J, Yue L. Functional characterization of homo- and heteromeric channel kinases TRPM6 and TRPM7. J Gen Physiol 127: 525–537, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li X, Mikhalkova D, Gao E, Zhang J, Myers V, Zincarelli C, Lei Y, Song J, Koch WJ, Peppel K, Cheung JY, Feldman AM, Chan TO. Myocardial injury after ischemia-reperfusion in mice deficient in Akt2 is associated with increased cardiac macrophage density. Am J Physiol Heart Circ Physiol 301: H1932–H1940, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 101: 365–376, 2000 [DOI] [PubMed] [Google Scholar]
- 28. Michael LH, Entman ML, Hartley CJ, Youker KA, Zhu J, Hall SR, Hawkins HK, Berens K, Ballantyne CM. Myocardial ischemia and reperfusion: a murine model. Am J Physiol Heart Circ Physiol 269: H2147–H2154, 1995 [DOI] [PubMed] [Google Scholar]
- 29. Miller BA, Zhang W. TRP channels as mediators of oxidative stress. Adv Exp Med Biol 704: 531–544, 2011 [DOI] [PubMed] [Google Scholar]
- 30. Nagamine K, Kudoh J, Minoshima S, Kawasaki K, Asakawa S, Ito F, Shimizu N. Molecular cloning of a novel putative Ca2+ channel protein (TRPC7) highly expressed in brain. Genomics 54: 124–131, 1998 [DOI] [PubMed] [Google Scholar]
- 31. Ramsey IS, Delling M, Clapham DE. An introduction to TRP channels. Annu Rev Physiol 68: 619–647, 2006 [DOI] [PubMed] [Google Scholar]
- 32. Sano Y, Inamura K, Miyake A, Mochizuki S, Yokoi H, Matsushime H, Furuichi K. Immunocyte Ca2+ influx system mediated by LTRPC2. Science 293: 1327–1330, 2001 [DOI] [PubMed] [Google Scholar]
- 33. Singal PK, Iliskovic N. Doxorubicin-induced cardiomyopathy. N Engl J Med 339: 900–905, 1998 [DOI] [PubMed] [Google Scholar]
- 34. Song J, Gao E, Wang J, Zhang XQ, Chan TO, Koch WJ, Shang X, Joseph JI, Peterson BZ, Feldman AM, Cheung JY. Constitutive overexpression of phospholemman S68E mutant results in arrhythmias, early mortality and heart failure: potenial involvement of Na+/Ca2+ exchanger. Am J Physiol Heart Circ Physiol 302: H770–H781, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Song J, Zhang XQ, Wang J, Cheskis E, Chan TO, Feldman AM, Tucker AL, Cheung JY. Regulation of cardiac myocyte contractility by phospholemman:Na+/Ca2+ exchange vs. Na+-K+-ATPase. Am J Physiol Heart Circ Physiol 295: H1615–H1625, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sumoza-Toledo A, Penner R. TRPM2: a multifunctional ion channel for calcium signalling. J Physiol 589: 1515–1525, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Takahashi N, Kozai D, Kobayashi R, Ebert M, Mori Y. Roles of TRPM2 in oxidative stress. Cell Calcium 50: 279–287, 2011 [DOI] [PubMed] [Google Scholar]
- 38. Togashi K, Inada H, Tominaga M. Inhibition of the transient receptor potential cation channel TRPM2 by 2-aminoethoxydiphenyl borate (2-APB). Br J Pharmacol 153: 1324–1330, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tong Q, Zhang W, Conrad K, Mostoller K, Cheung JY, Peterson BZ, Miller BA. Regulation of the transient receptor potential channel TRPM2 by the Ca2+ sensor calmodulin. J Biol Chem 281: 9076–9085, 2006 [DOI] [PubMed] [Google Scholar]
- 40. Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol 301: H2181–H2190, 2011 [DOI] [PubMed] [Google Scholar]
- 41. Tucker AL, Song J, Zhang XQ, Wang J, Ahlers BA, Carl LL, Mounsey JP, Moorman JR, Rothblum LI, Cheung JY. Altered contractility and [Ca2+]i homeostasis in phospholemman-deficient murine myocytes: role of Na+/Ca2+ exchange. Am J Physiol Heart Circ Physiol 291: H2199–H2209, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Uemura T, Kudoh J, Noda S, Kanba S, Shimizu N. Characterization of human and mouse TRPM2 genes: identification of a novel N-terminal truncated protein specifically expressed in human striatum. Biochem Biophys Res Commun 328: 1232–1243, 2005 [DOI] [PubMed] [Google Scholar]
- 43. Vazquez E, Valverde MA. A review of TRP channels splicing. Semin Cell Dev Biol 17: 607–617, 2006 [DOI] [PubMed] [Google Scholar]
- 44. Venkatachalam K, Montell C. TRP channels. Annu Rev Biochem 76: 387–417, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang J, Chan TO, Zhang XQ, Gao E, Song J, Koch WJ, Feldman AM, Cheung JY. Induced overexpression of Na+/Ca2+ exchanger transgene: altered myocyte contractility, [Ca2+]i transients, SR Ca2+ contents and action potential duration. Am J Physiol Heart Circ Physiol 297: H590–H601, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang J, Gao E, Rabinowitz J, Song J, Zhang XQ, Koch WJ, Tucker AL, Chan TO, Feldman AM, Cheung JY. Regulation of in vivo cardiac contractility by phospholemman: role of Na+/Ca2+ exchange. Am J Physiol Heart Circ Physiol 300: H859–H868, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang J, Gao E, Song J, Zhang XQ, Li J, Koch WJ, Tucker AL, Philipson KD, Chan TO, Feldman AM, Cheung JY. Phospholemman and β-adrenergic stimulation in the heart. Am J Physiol Heart Circ Physiol 298: H807–H815, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xie GH, Rah SY, Yi KS, Han MK, Chae SW, Im MJ, Kim UH. Increase of intracellular Ca2+ during ischemia/reperfusion injury of heart is mediated by cyclic ADP-ribose. Biochem Biophys Res Commun 307: 713–718, 2003 [DOI] [PubMed] [Google Scholar]
- 49. Yang KT, Chang WL, Yang PC, Chien CL, Lai MS, Su MJ, Wu ML. Activation of the transient receptor potential M2 channel and poly(ADP-ribose) polymerase is involved in oxidative stress-induced cardiomyocyte death. Cell Death Differ 13: 1815–1826, 2006 [DOI] [PubMed] [Google Scholar]
- 50. Zhang XQ, Ahlers BA, Tucker AL, Song J, Wang J, Moorman JR, Mounsey JP, Carl LL, Rothblum LI, Cheung JY. Phospholemman inhibition of the cardiac Na+/Ca2+ exchanger. Role of phosphorylation. J Biol Chem 281: 7784–7792, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhang XQ, Zhang LQ, Palmer BM, Ng YC, Musch TI, Moore RL, Cheung JY. Sprint training shortens prolonged action potential duration in postinfarction rat myocyte: mechanisms. J Appl Physiol 90: 1720–1728, 2001 [DOI] [PubMed] [Google Scholar]
- 52. Zhang Z, Zhang W, Jung DY, Ko HJ, Lee Y, Friedline RH, Lee E, Jun J, Ma Z, Kim F, Tsitsilianos N, Chapman K, Morrison A, Cooper MP, Miller BA, Kim JK. TRPM2 Ca2+ channel regulates energy balance and glucose metabolism. Am J Physiol Endocrinol Metab 302: E807–E816, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhou YY, Wang SQ, Zhu WZ, Chruscinski A, Kobilka BK, Ziman B, Wang S, Lakatta EG, Cheng H, Xiao RP. Culture and adenoviral infection of adult mouse cardiac myocytes: methods for cellular genetic physiology. Am J Physiol Heart Circ Physiol 279: H429–H436, 2000 [DOI] [PubMed] [Google Scholar]