

Abstract
Ketone bodies are metabolized through evolutionarily conserved pathways that support bioenergetic homeostasis, particularly in brain, heart, and skeletal muscle when carbohydrates are in short supply. The metabolism of ketone bodies interfaces with the tricarboxylic acid cycle, β-oxidation of fatty acids, de novo lipogenesis, sterol biosynthesis, glucose metabolism, the mitochondrial electron transport chain, hormonal signaling, intracellular signal transduction pathways, and the microbiome. Here we review the mechanisms through which ketone bodies are metabolized and how their signals are transmitted. We focus on the roles this metabolic pathway may play in cardiovascular disease states, the bioenergetic benefits of myocardial ketone body oxidation, and prospective interactions among ketone body metabolism, obesity, metabolic syndrome, and atherosclerosis. Ketone body metabolism is noninvasively quantifiable in humans and is responsive to nutritional interventions. Therefore, further investigation of this pathway in disease models and in humans may ultimately yield tailored diagnostic strategies and therapies for specific pathological states.
Keywords: mitochondrial function, substrate competition, HMGCS2, CoA transferase/SCOT, lipogenesis
this article is part of a collection on Nutrients and Cardiovascular Health and Disease: Glucose, Fatty Acids, and Beyond. Other articles appearing in this collection, as well as a full archive of all collections, can be found online at http://ajpheart.physiology.org/.
Introduction
Metabolism of ketone bodies is conserved among eukarya, bacteria, and archaea (8, 33, 113). Ketone bodies are synthesized in liver from acetyl-CoA derived primarily from fatty acid oxidation and are transported to extrahepatic tissues for terminal oxidation during physiological states characterized by limited carbohydrate and surplus fatty acid availability [reviewed in (137, 167); Fig. 1, A and B]. Ketone body oxidation becomes a significant contributor to overall energy metabolism within extrahepatic tissues in numerous physiological states, including the neonatal period, starvation, postexercise, and adherence to low-carbohydrate diets, when circulating ketone body concentrations increase from ∼50 μM in the normal fed state to up to 7 mM. Circulating ketone body concentrations rise to ∼1 mM after 16–20 h of fasting in healthy adult humans but can accumulate to as high as 20 mM in pathological states like diabetic ketoacidosis (33, 100, 167). Ketone body metabolism is not solely rooted in energy metabolism, as ketone bodies also serve as lipogenic and sterol biosynthetic substrates in many tissues, including the developing brain, lactating mammary gland, and liver (51, 59, 144, 168) (Fig. 1C). Furthermore, hepatic ketogenesis interfaces with fatty acid β-oxidation, the tricarboxylic acid (TCA) cycle, and gluconeogenesis (Fig. 2). Derangements of ketone body metabolism occur in numerous disease states, including types 1 and 2 diabetes and heart failure, and ketone body metabolism changes over the course of normal aging (56, 77, 115, 124, 125, 145, 158, 186, 193). In this review we examine 1) the biochemistry of ketone body metabolism and its regulation (in Ketone Body Metabolism), 2) the roles of ketone body metabolism in normal and pathological states of the myocardium (in Bioenergetics of Myocardial Ketone Body Oxidation and Ketone Bodies and Myocardial Disease), 3) the concept, and prospective cardiovascular disease relevance of extrahepatic ketone body production, which may channel ketone bodies into lipogenesis (in Reversibility of the CoA Transferase Reaction Extends Cardiovascular Disease Targets of Ketone Body Metabolism; Fig. 3), and 4) intersections among ketone body metabolism, cellular signaling pathways, and the gut microbiota (in Signaling Roles for Ketone Bodies and Ketone Body Metabolism, the Microbiota, and Cardiovascular Biology and Disease). Analysis of the known and prospective cardiovascular disease targets of ketone body metabolism is also provided (in Diagnostic and Therapeutic Targets of Ketone Body Metabolism). The effects of ketone body metabolism are summarized in Table 1.
Fig. 1.
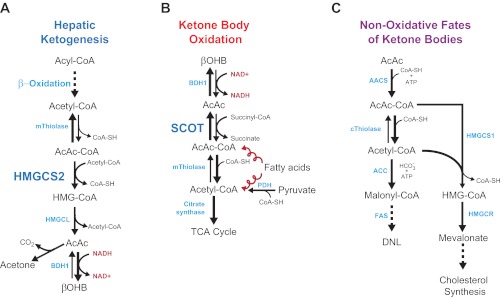
Ketone body metabolism pathways. A: ketogenesis within hepatic mitochondria is the primary source of circulating ketone bodies. B: primary metabolic fate of ketone bodies is terminal oxidation within mitochondria of extrahepatic tissues via CoA transferase [succinyl-CoA:3-oxoacid-CoA transferase (SCOT)]. Substrate competition with pyruvate-derived and fatty acyl-CoA-derived (the corkscrew arrows represent the activities of the β-oxidation spiral) acetyl-CoA are shown. C: cytoplasmic de novo lipogenesis (DNL) and cholesterol synthesis are nonoxidative metabolic fates of ketone bodies. For simplicity of C, only acetoacetate (AcAc) is depicted, although β-hydroxybutyrate (βOHB) is also a substrate for lipogenesis after it has been oxidized to AcAc via mitochondrial βOHB dehydrogenase (BDH1). AACS, acetoacetyl-CoA synthetase; ACC, acetyl-CoA carboxylase; AcAc-CoA, acetoacetyl-CoA; ATP, adenosine triphosphate; CoA-SH, free coenzyme A; FAS, fatty acid synthase; HMG-CoA, 3-hydroxymethylglutaryl-CoA; HMGCL, HMG-CoA lyase; HMGCS1, cytoplasmic HMG-CoA synthase; HMGCS2, mitochondrial HMG-CoA synthase; HMGCR, HMG-CoA reductase; NAD+(H), nicotinamide adenine dinucleotide oxidized (reduced); PDH, pyruvate dehydrogenase; TCA, tricarboxylic acid; mThiolase, mitochondrial thiolase; cThiolase, cytoplasmic thiolase. Thiolase activity is encoded by at least 6 genes: ACAA1, ACAA2 (encoding an enzyme known as T1 or CT), ACAT1 (encoding T2), ACAT2, HADHA, and HADHB.
Fig. 2.

Hepatic integration of ketogenesis. Integration of hepatic ketogenesis with hepatic TCA cycle, DNL, and pyruvate cycling/glucose metabolism. FAO, β-oxidation of fatty acids; GDH, glutamate dehydrogenase; OAA, oxaloacetate; ME, malic enzyme; PEPCK, phosphenolpyruvate carboxykinase; PC, pyruvate carboxylase; PK, pyruvate kinase; PEP, phosphenol pyruvate; CS, citrate synthase; α-KG, α-ketoglutarate.
Fig. 3.

Ketogenic flux through CoA transferase. Because CoA transferase catalyzes an equilibrium reaction, select spatiotemporal metabolic conditions may favor channeling of mitochondrial acetyl-CoA to AcAc, generating a monocarboxylate shuttle that complements the citrate-dependent tricarboxylate shuttle and permits mitochondrial efflux of β-oxidation-derived acetyl-CoA independent of the TCA cycle (TCAC). ACLY, ATP-citrate lyase; ETC, electron transport chain.
Table 1.
Implications of ketone body metabolism
|
P-to-O ratio, ATP yielded per oxygen invested; ROS, reactive oxygen species; DNL, de novo lipogenesis; GPCR, G protein-coupled receptor; GPR109A, GPCR 109A; HDAC, histone deacetylase.
Glossary
For this article we have included a short glossary of terms and concepts for the convenience of the reader.
AACS, acetoacetyl-CoA synthetase; a cytoplasmic enzyme that catalyzes the ATP-dependent covalent activation of acetoacetate (AcAc) by CoA to generate AcAc-CoA.
Anaplerosis, essential replenishment of TCA cycle intermediates that is required to preserve terminal oxidation of acetyl-CoA. The primary anaplerotic reaction is catalyzed by pyruvate carboxylase, which generates oxaloacetate from pyruvate, but oxidation of odd-chain fatty acids and amino acid catabolism also yield anaplerotic substrates.
BDH1, mitochondrial β-hydroxybutyrate (βOHB) dehydrogenase; an enzyme that catalyzes the NAD+/NADH coupled interconversion of AcAc and d-βOHB. The Keq of this reaction favors d-βOHB formation.
β-Oxidation, sequential derivation of two carbon units, in the form of acetyl-CoA, from fatty acyl chains that primarily occurs within the mitochondrial matrix. Some β-oxidation also occurs within peroxisomes.
DNL, de novo lipogenesis. The synthesis of new lipid from acetyl-CoA, dependent on the enzymes acetyl-CoA carboxylase and fatty acid synthase (FAS).
Fluxomics, analysis of metabolic fates of labeled substrates using sophisticated chemical profiling platforms including, nuclear magnetic resonance spectroscopy and mass spectrometry, coupled to statistical computing platforms.
Gnotobiotic, Greek roots: gnosis, knowledge; bios, life; a technology that allows cultivation of experimental animal models in the presence of defined microbial communities.
GPCR, G protein-coupled receptor; a plasma membrane receptor with seven transmembrane domains that mediates an intracellular signaling cascade via an interaction with a G protein.
HMGCL, HMG-CoA lyase; a mitochondrial enzyme that cleaves HMG-CoA to liberate AcAc and acetyl-CoA during ketogenesis. This enzyme mediates ketone body production from β-oxidation-derived acetyl-CoA and leucine catabolism.
HMGCS2, HMG-CoA synthase 2; a mitochondrial enzyme that condenses β-oxidation-derived AcAc-CoA and acetyl-CoA to generate HMG-CoA and liberate a CoA moiety.
Ketogenesis, production of ketone bodies. AcAc and βOHB are the predominant circulating ketone bodies.
Ketolysis, breakdown of ketone bodies either within the mitochondria for terminal oxidation or in the cytoplasm for lipogenesis.
Ketone body oxidation, contribution of acetyl-CoA derived from ketone bodies to the TCA cycle.
Micriobiota, ecosystem of ∼100 trillion prokaryotic cells that comprise a ∼1-kg organ that harbors a massive aggregate of genomes (the microbiome).
NAFLD, nonalcoholic fatty liver disease.
P-to-O ratio, the ATP yielded per oxygen invested, which depends on many factors, including the inherent energy of combustion for a substrate, the initial energetic investment required for oxidation, and the effect of oxidizing the substrate on the electrochemical potential effected by the mitochondrial electron transport chain.
Pseudoketogenesis, quantifiable dilution of isotopically labeled ketone bodies, mediated by reversibility of thiolase and CoA transferase reactions in extrahepatic tissues.
SCOT, succinyl-CoA:3-oxoacid-CoA transferase, encoded by nuclear Oxct1. A mitochondrial enzyme that catalyzes the following near equilibrium reaction: succinyl-CoA + AcAc ↔ succinate + AcAc-CoA.
Sirtuin, silent mating type information regulation 2 homolog (SIRT); In mammals, these enzymes comprise a family of seven NAD+-dependent enzymes that coordinate an array of cellular processes via deacetyalse, demalonylase, desuccinylase, and ADP-ribosylase enzymatic activities.
TCA cycle, tricarboxylic acid cycle. Also termed Krebs cycle, citric acid cycle.
Terminal oxidation, contribution of acetyl-CoA to the TCA cycle via citrate synthase enzymatic activity that results in conversion of 1 mol of acetyl-CoA to 2 mol of CO2 and 2 mol of NADH.
Thiolase, any enzyme that catalyzes the reversible condensation of acetyl-CoA to acetoacetyl-CoA. This enzymatic activity is redundant in mammals.
Ketone Body Metabolism
The fundamental biochemical and physiological roles of ketone body metabolism have been extensively studied and reviewed (68, 137, 167). In this section, we provide a brief overview of ketone body metabolism and then focus on recent studies revealing novel aspects of the pathway's regulatory mechanisms.
Hepatic ketogenesis.
In liver, 3-hydroxymethylglutaryl-CoA synthase 2 (encoded by the nuclear gene Hmgcs2) catalyzes a fate committing ketogenic reaction: condensation of β-oxidation-derived acetoacetyl-CoA (AcAc-CoA) and acetyl-CoA to generate HMG-CoA, which is cleaved by HMGCL to generate AcAc. AcAc is reduced to d-βOHB, in an NAD+/NADH-coupled near equilibrium reaction catalyzed by phosphatidylcholine-dependent mitochondrial d-βOHB dehydrogenase (BDH1), in which the Keq favors d-βOHB formation (26, 119) (Fig. 1A). Because the BDH1 catalyzed reduction/oxidation of AcAc and d-βOHB is common to the final reaction of ketogenesis and the first reaction of ketone body oxidation (Fig. 1, A and B) (119, 178), BDH1 modulates mitochondrial redox potential in liver and extrahepatic tissues, wherein the AcAc-to-βOHB ratio is directly proportional to the mitochondrial NAD+-to-NADH ratio (230). A cytoplasmic d-βOHB-dehydrogenase (BDH2) with only 20% sequence identity to BDH1 has been identified (72). More work is needed to elucidate the mechanisms by which the BDH enzymes are regulated and function in vivo. AcAc is also nonenzymatically decarboxylated to acetone. Ketone bodies are released by the liver via solute carrier 16A (SLC16A) family members 1, 6, and 7 and circulate to extrahepatic tissues where they primarily undergo terminal oxidation (75, 76, 91).
ketogenic substrates.
Ketogenesis predominantly occurs in liver mitochondria at rates proportional to β-oxidation when dietary carbohydrates are limiting and is highly integrated with the TCA cycle and gluconeogenesis (Fig. 2). Biochemical studies by pioneering investigators including Krebs, McGarry, and Foster demonstrated that hepatic metabolic fluxes of acetyl-CoA govern rates of ketogenesis [reviewed in (68, 137)]. Fatty acid β-oxidation-derived AcAc-CoA and acetyl-CoA are the primary ketogenic substrates. Glucose metabolism accounts for <1% of circulating ketone bodies in states of low-carbohydrate intake because pyruvate predominantly enters the hepatic TCA cycle via carboxylation to oxaloacetate or malate rather than decarboxylation (to acetyl-CoA) (99, 134, 142). In addition, amino acid catabolism accounts for a small percentage of circulating ketone bodies, with leucine catabolism generating up to 4% of circulating ketone bodies in the post-absorptive state (207).
Regulation of ketogenic mediators.
Key regulatory steps in ketogenesis include lipolysis of fatty acids from triacylglycerols, transport to and across the hepatocyte plasma membrane, transport into mitochondria via allosterically regulated carnitine palmitoyltransferase 1, the β-oxidation spiral, and the hormonal regulators of these processes, predominantly glucagon and insulin. These classical mechanisms have been reviewed (55, 137, 138, 194). Hepatic ketogenesis is a spillover pathway for β-oxidation-derived acetyl-CoA generated in excess of the liver's energetic needs (Fig. 2). Acetyl-CoA subsumes several roles integral to hepatic intermediary metabolism beyond ATP generation via terminal oxidation. Acetyl-CoA allosterically activates 1) pyruvate carboxylase, thereby activating a metabolic control mechanism that augments anaplerotic entry of metabolites into the TCA cycle (154, 185) and 2) pyruvate dehydrogenase kinase, which phosphorylates and inhibits pyruvate dehydrogenase (39), thereby further enhancing flow of pyruvate into the TCA cycle via anaplerosis. Furthermore, cytoplasmic acetyl-CoA, whose pool is augmented by transport mechanisms that convert mitochondrial acetyl-CoA to transportable metabolites (see CoA transferase-dependent ketogenesis in extrahepatic tissues), inhibits fatty acid oxidation: acetyl-CoA carboxylase catalyzes the conversion of acetyl-CoA to malonyl-CoA, the lipogenic substrate and an allosteric inhibitor of mitochondrial carnitine palmitoyltransferase 1, decreasing delivery of acyl chains to the mitochondrial matrix for terminal oxidation [reviewed in (102, 137)]. Thus the mitochondrial acetyl-CoA pool regulates and is regulated by the spillover pathway of ketogenesis, which orchestrates key aspects of hepatic intermediary metabolism. The studies described below focus on recently discovered mechanisms of ketogenic regulation.
Following procession through β-oxidation, ketogenic fate is determined by HMGCS2, whose regulatory mechanisms are schematized in Fig. 4A. Because of its ability to support high rates of hepatic ketogenesis, divergence of this mitochondrial HMGCS ∼500 Mya from the gene encoding cytoplasmic HMGCS1 may have supported the emergence of increasing brain weight-to-body weight ratios during vertebrate evolution (30, 43, 161). The Hmgcs2 gene and encoded protein are regulatory targets during the transition to extrauterine life; in starvation, diabetes, and aging; and during adherence to low-carbohydrate/high-fat (ketogenic) diets, and in aging (17, 33, 68, 84, 179, 186). The Hmgcs2 gene is dynamically regulated at the transcriptional level. Methylation of 5′ regulatory sequences within the Hmgcs2 gene silences its transcription in fetal liver and in nonketogenic adult tissues (12). At birth, hepatic Hmgcs2 becomes hypomethylated and thereby becomes responsive to circulating hormones (9, 12, 55). Insulin suppresses Hmgcs2 transcription, prospectively via phosphorylation-induced sequestration of FOXA2 from the nucleus, whereas glucagon induces it via activation of the cAMP regulatory element binding protein (9, 84, 162, 209, 231). In addition, free fatty acids induce Hmgcs2 in a peroxisome proliferator-activated receptor (PPAR)-α-dependent manner (169, 228). In fact, Hmgcs2 is induced in vivo in the post-absorptive state and by adherence to ketogenic diet through the activities of the fibroblast growth factor (FGF)-21/PPARα axis (16, 93, 169). HMGCS2 may also reciprocally induce Fgf21 gene expression (220). In addition, following cysteine palmitoylation, HMGCS2 translocates to the nucleus, physically interacts with PPARα and potentiates its own gene transcription (111, 140). Inhibition of mammalian target of rapamycin complex 1 signaling has been identified as a primary mechanism responsible for de-repression of PPARα-mediated transcriptional changes responsible for the induction of ketogenesis in the post-absorptive state (73, 186). Hepatic Bdh1 also exhibits a developmental expression pattern, increasing in brain and liver from birth to weaning, and is also induced by ketogenic diet in an FGF21-dependent manner (16, 242).
Fig. 4.

Regulatory mechanisms for HMGCS2 and CoA transferase (SCOT). HMGCS2 (A) and SCOT (B) both undergo transcriptional (red) and post-translational modes of regulation (blue). Some regulatory factors exhibit both classes of effects (violet). mTORC1, mammalian target of rapamycin complex 1; PPARα, peroxisome proliferator-activated receptor-α; PTM; post-translational modification; SIRT3, sirtuin (silent mating type information regulation 2 homolog) 3.
In addition to cysteine palmitoylation, HMGCS2 enzymatic activity is responsive to other forms of post-translational modification. Mitochondrial activity of HMGCS2 is inhibited through allosteric regulation by succinyl-CoA (84, 128, 164) and through succinylation of lysine residues (9, 84, 162, 209). While the regulatory mechanisms remain to be determined, desuccinylation is stimulated by glucagon (162), and recent discoveries that identify sirtuin (SIRT5) as a mitochondrial NAD+-dependent lysine desuccinylase prompt the hypothesis that SIRT5 function dynamically regulates mitochondrial HMGCS2 activity (47, 243). HMGCS2 activity is also induced in the post-absorptive state by SIRT3, a mitochondrial NAD+-dependent deacetylase (188). Because SIRT3 regulates multiple enzymatic mediators of β-oxidation, sirtuins may be multitiered regulators of ketogenesis. Genetically obese mice were recently shown to exhibit increased hepatic HMGCS2 serine phosphorylation, a post-translational modification that enhances HMGCS2 activity in vitro and was correlated with elevated serum d-βOHB levels in vivo (70).
Despite all of the described transcriptional and post-translational mechanisms that regulate HMGCS2, it is unknown whether regulation of Hmgcs2 gene expression correlates with changes in protein abundance and whether these changes in gene expression directly regulate ketogenesis. Importantly, it remains undetermined whether HMGCS2 post-translational modification itself is a primary determinant of ketogenic flux. Nonetheless, HMGCS2 activity is required for the normal ketogenic response to states of diminished carbohydrate intake, as its absence in humans yields hypoketotic hypoglycemia and fatty liver in states of diminished carbohydrate intake (7, 28, 208).
Ketone body utilization.
Ketone body catabolism generates acetyl-CoA that can be terminally oxidized within the TCA cycle or used for sterol biosynthesis and DNL. Ketone bodies are an alternative and glucose-sparing fuel source, avidly oxidized in heart and muscle. Moreover, neurons do not effectively generate high-energy phosphates from fatty acids and consequently oxidize ketone bodies during starvation and in the neonatal period [presented and reviewed in (33, 50, 167, 225, 235)]. Below we review the biochemical activities of the enzymes of ketone body utilization, focusing on recent studies that reveal the precise physiological roles of ketone body utilization in vivo.
ketone body oxidation.
Ketone bodies are extracted from the circulation by peripheral tissues via SLC16A1 and -7 (75, 76). Within mitochondria of peripheral organs, BDH1 catalyzes the oxidation of d-βOHB to AcAc. AcAc is activated to AcAc-CoA by SCOT (CoA transferase), the only mammalian CoA transferase that catalyzes a near equilibrium reaction which exchanges coenzyme A between succinate and AcAc (Fig. 1B). The free energy released by hydrolysis of AcAc-CoA is greater than that of succinyl-CoA (198). Therefore, the equilibrium of this reaction thermodynamically favors the formation of AcAc [(229), also see Reversibility of the CoA Transferase Reaction Extends Cardiovascular Disease Targets of Ketone Body Metabolism]. Thus ketone body oxidative flux occurs by mass action: an abundant supply of AcAc and rapid utilization of acetyl-CoA through citrate synthase favors AcAc-CoA formation and contribution of ketone bodies to the TCA cycle. A reversible AcAc-CoA thiolase reaction yields two molecules of acetyl-CoA that enter the TCA cycle (Fig. 1B) (61, 66, 167, 229). CoA transferase is necessary, but not sufficient, for ketone body oxidation in mice and humans [see below and (20, 40, 60, 62, 105)].
Ketone body oxidation becomes a primary contributor to bioenergetic homeostasis during ketotic states, during which ketone bodies are oxidized in proportion to their delivery in heart, brain, and muscle until saturation of either uptake or oxidation occurs (17, 33, 155, 166, 199, 225). Relative to ketogenesis, little is known about the regulation of mediators of ketone body oxidation. Perhaps paradoxically, expression of the gene encoding CoA transferase (Oxct1), CoA transferase protein abundance, and CoA transferase enzymatic activity are all diminished in rodent heart and muscle during states of sustained ketosis (53, 54, 71, 152, 213, 228). In pathological contexts such as diabetic ketoacidosis, diminution of CoA transferase abundance and activity limit ketone body disposal, whereas insulin deficiency (or severe impairment of its signaling) stimulates peripheral fatty acid mobilization and unabated hepatic ketogenesis. This mismatch of ketogenesis and peripheral disposal predisposes to the extreme hyperketonemia that can emerge in diabetic ketoacidosis.
In ketotic states, expression of Oxct1 may be negatively regulated through mechanisms involving PPARs that are abrogated by insulin [(228); Fig. 4B]. Myocardial Oxct1 expression may also be downregulated under select nonketotic metabolic states, as transgenic mice overexpressing the non-insulin-dependent glucose transporter (GLUT1/SLC2A1) in cardiomyocytes exhibit decreased Oxct1 expression and diminished ketone body oxidation (234). Future experiments are needed to determine whether regulation of Oxct1 expression occurs through transcriptional or post-transcriptional mechanisms (or both), which may help support insight into whether these events are adaptive or maladaptive. Post-translational modification of CoA transferase occurs through nonenzymatic tyrosine nitration, which has been observed in hearts of endotoxin-treated rats, streptozotocin-treated rats, and db/db mice (135, 213, 223). Tyrosine nitration correlates with impairments of enzymatic activity (Fig. 4B). Conversely, nitration of CoA transferase on tryptophan residues in aged rat hearts and kidneys appears to augment enzymatic activity (31, 163). Molecular mechanisms of residue-specific nitration or denitration designed to modulate CoA transferase activity may exist and require elucidation, as do additional prospective post-translational modifications of CoA transferase.
Although touted as energy-efficient substrates (180, 219), an energetic requirement for ketone body oxidation at the cellular level has never been demonstrated, even in states of diminished carbohydrate supply. Nonetheless, human inborn errors of ketone body oxidation do result in clinically significant disease, as ∼30 CoA transferase-deficient patients have been identified. These patients present early in life with severe ketoacidosis that results in lethargy, vomiting, and coma, requiring aggressive intravenous hydration, bicarbonate, and glucose and insulin therapy (20, 62, 105, 143, 149, 181, 192, 210). Some CoA transferase-deficient patients also present with hypoglycemia, and CoA transferase deficiency may contribute to a subset of idiopathic ketotic hypoglycemia cases (20, 27, 52, 83, 92, 108). If maintained on carbohydrate-rich diets that are mildly reduced in protein content, CoA transferase-deficient patients seem to thrive, albeit with persistent hyperketonemia. To mechanistically dissect the homeostatic roles of ketolysis, germline CoA transferase-knockout (SCOT-KO) mice were developed (40). These mice cannot oxidize ketone bodies and invariably die within 48 h of extrauterine life because of hyperketonemic hypoglycemia that is marked by hypolactatemia and an increase in the serum AcAc-to-d-βOHB ratio. The mechanisms and consequences of this unusually high ratio remain to be determined. Despite the critical nature of CoA transferase and ketone body disposal at an organismal level, recently published studies demonstrated that mice with selective loss of CoA transferase in either cardiomyocytes, neurons, or skeletal myocytes—the three greatest consumers of ketone bodies (64, 167)—survive the neonatal period and starvation as adults, with only subtle metabolic abnormalities in these two states (41). Future studies using these and related models will be important to determine the metabolic and bioenergetic adaptations to CoA transferase deficiency and the inability to derive energy from ketone bodies.
Nonoxidative metabolism of ketone bodies.
Ketone bodies also contribute to lipogenesis and sterol biosynthesis in developing brain, lactating mammary gland, and liver following enzymatic and endergonic activation of AcAc to AcAc-CoA by cytoplasmic AACS [(51, 59, 144, 168) and Fig. 1C]. While AcAc-CoA serves as a direct substrate for cytoplasmic HMGCS1, which catalyzes the fate-committing step of sterol biosynthesis, channeling of AcAc-CoA into DNL requires a thiolytic cleavage reaction (i.e., cytoplasmic ketolysis) to yield acetyl-CoA, which becomes the lipogenic substrate malonyl-CoA upon carboxylation (19, 49, 51, 67, 226). Recent studies support the physiological importance of ketone bodies as anabolic substrates. Genetic knockdown of AACS in mouse liver lowered total blood cholesterol in vivo, and in vitro AACS knockdown impaired differentiation of primary mouse embryonic neurons and inhibited adipocyte differentiation of 3T3-L1 cells [(79–81), also see Reversibility of the CoA Transferase Reaction Extends Cardiovascular Disease Targets of Ketone Body Metabolism]. While lipogenic fates of ketone bodies may support essential biological functions, these pathways do not serve as a primary pathway for disposal of ketone bodies when hepatic ketogenesis is stimulated, because CoA transferase deficiency in mice and humans, which abrogates terminal ketone body oxidation, causes severe hyperketonemia (20, 40, 60, 62, 105). Nonetheless, future studies in models of AACS disruption are merited to firmly establish the physiological roles for nonoxidative fates of ketone bodies.
Bioenergetics of Myocardial Ketone Body Oxidation
In the normal adult heart, mitochondrial oxidative phosphorylation provides more than 95% of the ATP generated for its mechanical, electrical, and homeostatic activities. Fatty acid oxidation provides up to 70% of the ATP produced by the heart, with metabolism of glucose, lactate, amino acids, and ketone bodies supplying the balance [reviewed in (127)]. Cardiomyocytes demonstrate considerable metabolic flexibility during dynamic alterations of nutrient state and hemodynamic stress, conferring an important adaptive property to the myocardium (22, 203, 204, 239). Myocardium is the highest ketone body consumer per unit mass (17, 33, 61). Cardiomyocytes oxidize ketone bodies in proportion to their delivery, at the expense of terminal fatty acid oxidation and glucose oxidation (22, 42, 65, 82, 98, 156, 206, 234). Competition between oxidation of ketone bodies and fatty acids is independent of changes in myocardial malonyl-CoA concentrations (196).
Ketone body oxidation is more energetically efficient than terminal fatty acid oxidation. While terminal fatty acid oxidation yields the highest theoretical payoff of ATP per C2 unit of all the myocardial substrates, the initial ATP investment required to activate long-chain fatty acids for oxidation (i.e., to generate a fatty acyl-CoA thioester) and fatty acid-induced expression of uncoupling proteins may actually confer greater energetic density to ketone bodies (219). Furthermore, compared with terminal fatty acid oxidation, proportionately more of the reducing equivalents generated by ketone body oxidation are delivered via NADH to complex I within the electron transport chain. Oxidation of βOHB also increases the redox span between complexes I and III by keeping mitochondrial ubiquinone oxidized. This may increase the potential energy harvested from oxidation of ketone bodies (relative to fatty acids), thereby yielding more energy available for P-to-O ratio, improving the energetic efficiency of oxidizing ketone bodies over fatty acids (104, 180, 219). Because unrestrained mitochondrial fatty acid oxidation may augment the generation of reactive oxygen species (ROS) to levels that exceed scavenging mechanisms, competitive contribution of ketone bodies to the TCA cycle in cardiomyocytes may be adaptive under conditions in which the ability to switch between fatty acids and glucose becomes impaired (104, 180, 219). Moreover, ketone bodies diminish oxidative stress by scavenging free radicals and by maintaining ubiquinone in the oxidized state (Fig. 5) (74, 130, 219).
Fig. 5.
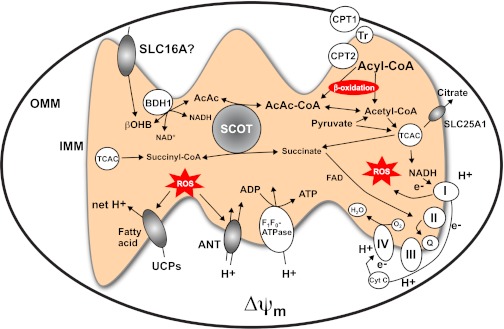
Diverse roles of CoA transferase in mitochondrial function. βOHB and AcAc cross the plasma membrane via solute ligand carrier protein (SLC) 16A (SLC16A) family members and may employ these or other transporters to enter the mitochondrial matrix. Within the mitochondria, d-βOHB is oxidized to AcAc by the inner-membrane bound and phosphatidylcholine-dependent BDH1. CoA transferase (SCOT) catalyzes a near equilibrium reaction through which CoA is exchanged between succinate and AcAc. Oxidation of ketone bodies occurs by mass action and may diminish reactive oxygen species (ROS) formation, compared with oxidation of fatty acids. See text for details. ADP, adenosine diphosphate; ANT, adenine nucleotide transporter; CPT, carnitine palmitoyltransferase; Cyt C, cytochrome c; e−, electron; H+, hydrogen ion; I, II, II, IV, complexes I-IV of the electron transport chain, respectively; IMM, inner mitochondrial membrane; OMM, outer mitochondrial membrane; Q, ubiquinone; Δψm, electrochemical potential across the inner mitochondrial membrane; Tr, translocase; UCPs, uncoupling proteins; FAD, flavin adenine dinucleotide.
CoA transferase-dependent activation of AcAc to AcAc-CoA sequesters free CoA-SH (90, 176). Rodent hearts perfused ex vivo with AcAc alone exhibit depleted free CoA-SH pools and require addition of either CoA precursors or anaplerotic substrates to maintain anaplerotic and TCA cycle flux and to prevent functional decline in cardiac performance (175–177). Although ketone bodies do not serve as the sole myocardial fuel under any in vivo circumstance, these studies underscore the important role of cardiac anaplerosis in maintaining myocardial bioenergetic homeostasis and cardiac function, a topic that was recently reviewed by Des Rosiers and colleagues (44).
Until recently, it was not known how the myocardium adapts to chronic ketosis, an important question relevant to the role that ketone body metabolism may serve in myopathic hearts. Mouse models of physiological ketotic nutritional states (24 h of fasting or 4 wk of a ketogenic diet) demonstrate that the myocardium engages a transcriptional program including, as described above, transcriptional suppression of Oxct1 and diminution of CoA transferase protein (228). Consistent with diminution of CoA transferase, nuclear magnetic resonance profiling demonstrated that maintenance on a ketogenic diet decreased myocardial 13C-enrichment of glutamate from 13C-labeled ketone bodies that were delivered in vivo or ex vivo by 25%, indicating diminished terminal oxidation of ketone bodies in the TCA cycle (101, 228). Furthermore, attenuation of ketone body oxidation correlated with failure of ketone bodies to inhibit contribution of fatty acids to the TCA cycle. Together, these results indicate that ketotic nutrient environments induce mechanisms that modulate myocardial utilization of ketone bodies. Because hemodynamic parameters were preserved in hearts of mice fed a ketogenic diet, these results suggest that modulation of ketone body metabolism may be adaptive in the setting of sustained ketosis.
Ketone Bodies and Myocardial Disease
Numerous experimental approaches have established that cardiomyopathy is associated with changes in cardiac substrate and energy metabolism and that altered energy metabolism can cause cardiomyopathy [reviewed in (1, 10, 29, 44, 89, 94, 97, 109, 118, 127)]. Both metabolic and bioenergetic lines of evidence indicate that ketone body metabolism could play a significant role in the myopathic heart. Hepatic ketogenesis is stimulated and ketone bodies circulate at increased concentrations in the setting of heart failure in a relationship directly proportional to filling pressure (115, 124, 125, 145, 158). As previously noted, the myocardium oxidizes ketone bodies at the expense of fatty acid oxidation (22, 82, 196, 218), and thus reductions of myocardial fatty acid oxidation that occur during the development of advanced cardiomyopathy may not be coupled to reductions of ketone body oxidation (1, 10, 127). A study in humans with advanced heart failure indicated that myocardial extraction of delivered ketone bodies is maintained in the failing heart but not skeletal muscle (96). Similarly, the contribution of ketone bodies to cardiac energy metabolism may be elevated in patients with dilated and hypertrophic cardiomyopathies (174). Therefore, while the data on ketone body metabolism in heart failure are currently very limited, diminished myocardial ketone body oxidation could promote pathological outcomes. Of the identified CoA transferase-deficient patients, two were reported to present with dilated cardiomyopathy (63, 126, 210). Future studies in humans that specifically measure CoA transferase function and ketone body oxidation in cardiomyopathic states, complemented by mechanistic studies using tissue-selective genetic rodent models, will be required to definitively determine how myocardial ketone body metabolism changes in pathophysiological states and the contexts in which myocardial ketone body utilization may be adaptive or maladaptive.
Studies in rodents demonstrate a possible role for ketone body metabolism in myocardial adaptation to ischemia-reperfusion injury. In two studies using ex vivo perfusion approaches in rat hearts, maintenance on low-carbohydrate diets before ischemia-reperfusion protocols gave conflicting results with regard to infarct size and hemodynamic performance (6, 221). Prospective cardioprotective effects of a low-carbohydrate diet may be attributable to an increase in the number of myocardial mitochondria or transcriptional upregulation of key mediators of oxidative phosphorylation (6, 112). Cardioprotective effects have been observed using in vivo ischemia-reperfusion approaches in rats subjected to starvation-induced ketosis, initiated through prolonged fasting, and also via intravenous injection of dl-βOHB immediately before ischemic injury, which conferred a significant decrease in both infarct size and myocardial cell death (191, 244). These studies should stimulate further investigation using genetic and pharmacological approaches to determine the validity and mechanisms of these preliminary studies and whether the observations actually relate to myocardial metabolism of ketone bodies.
Relationships among ketone body metabolism, membrane excitability, and arrhythmogenesis have also been reported. Metabolomic and proteomic analysis of atrial tissue harvested during cardiac surgeries demonstrated an increase in both myocardial βOHB content and CoA transferase abundance in patients with atrial fibrillation compared with control patients (136). Although myocardial ketone body utilization in the setting of atrial fibrillation has not been investigated, mechanisms that directly link ketone bodies to membrane excitability and arrhythmogenesis have been preliminarily assessed. βOHB blocks the transient outward K+ current in murine ventricular myocytes, causing action potential prolongation (46). However, only l-βOHB inhibits the transient outward K+ current. l-βOHB is measurable in myocardial extracts, but does not circulate and likely results from hydrolysis of the β-oxidation intermediate l-βOHB-CoA (95, 122, 165, 184). Hepatic ketogenesis only produces d-βOHB, which is the only form that is a BDH substrate, and thus a substrate for oxidation. SLC16A transporters in rat myocytes demonstrate no stereoselectivity for βOHB (222). Prospective pathophysiological significance of the l-βOHB stereoisomer requires investigation.
Reversibility of the CoA Transferase Reaction Extends Cardiovascular Disease Targets of Ketone Body Metabolism
Each enzyme involved in ketone body oxidation catalyzes a reversible reaction. Thus each tissue that oxidizes ketone bodies has the enzymatic potential to synthesize them.
Pseudoketogenesis.
Brunengraber and colleagues (57) quantified exchange between isotopically labeled fatty acids and unlabeled ketone bodies in rat hearts perfused ex vivo. Des Rosiers et al. (45) subsequently quantified dilution of labeled ketone bodies in hepatectomized dogs in vivo. This isotopic dilution by extrahepatic tissues was termed pseudoketogenesis, which occurs because CoA transferase and AcAc-CoA thiolase catalyze reversible reactions. While a physiological role for pseudoketogenesis has yet to be established, the process leads to overestimation of ketone body turnover in whole body tracer dilution studies, because its rate is up to one-third of the rate of ketone body uptake (57).
CoA transferase-dependent ketogenesis in extrahepatic tissues.
CoA transferase is expressed in all mammalian cells that harbor mitochondria, except hepatocytes, but not all cell types oxidize ketone bodies, suggesting that CoA transferase may mediate nonoxidative metabolic functions in certain tissues or physiological states (11, 64, 153, 229). Because AcAc exits mitochondria via monocarboxylate transporters, ketogenic flux through thiolase and CoA transferase may permit carbon efflux from mitochondria independent of citrate synthase, thus generating a monocarboxylate transport system that complements both citrate-dependent tricarboxylate transport (Fig. 3) and acetylcarnitine transport (172). Such a role for CoA transferase reflects its dual evolutionary history as an enzyme of ketone body production and utilization (157, 240). Ketogenic flux through mitochondrial thiolase and CoA transferase may be poised to regulate discrete cellular functions directly relevant to metabolic syndrome, diabetes, and atherosclerosis, because ketone bodies are substrates for cytoplasmic DNL and sterol biosynthesis [Fig. 3 and (19, 49, 51, 67)]. A series of studies have implicated ketogenic flux through CoA transferase as a putative mechanism through which ketone bodies act as insulin secretagogues in pancreatic β-cells (78, 131–133). The authors of these reports demonstrated that ketone bodies and ketone body precursors potentiate insulin release, possibly by contributing to formation of cytoplasmic short-chain acyl-CoAs (131–133) and that diminished CoA transferase expression correlated with impaired glucose stimulated insulin secretion in a rat insulinoma cell line (78). Future experiments will be required to determine the biological roles of ketogenic flux in cell types not classically associated with lipogenesis, including β-cells of the pancreas and cardiomyocytes.
An additional nonoxidative role for CoA transferase may emerge in macrophages, which exhibit robust CoA transferase enzymatic activity but do not terminally oxidize ketone bodies (146, 147). Recent discoveries have highlighted the importance of macrophage metabolism in cardiovascular disease [reviewed in (151)]. As macrophage-specific deficiency of the enzyme required for DNL, FAS, ameliorates diet-induced atherosclerosis in mice (182), it is intriguing to consider that anabolic procession of metabolites through mitochondrial thiolase and CoA transferase acts upstream of FAS to contribute to macrophage DNL flux. CoA transferase is also abundantly expressed in adipose tissue, in which the enzyme that catalyzes the reaction downstream from CoA transferase, AACS (Fig. 3), is dynamically regulated by PPARγ, a key mediator of adipogenesis and adipocyte function (2, 81, 233). Adipocyte-specific FAS deficiency increases baseline energy expenditure and improves diet-induced obesity in mice (123). Because ketone bodies have been proposed as quantitatively significant DNL substrates in multiple cell types (51, 238), these recent findings suggest that use of genetic models is warranted to draw mechanistic and cardiovascular disease-relevant links among CoA transferase, lipogenesis from ketone bodies, and metabolic and vascular diseases.
Signaling Roles for Ketone Bodies
Because serum ketone body concentrations vary over a large dynamic range, they may act as physiologically relevant signals for cell-surface and intracellular receptors. In fact, d-βOHB is an endogenous ligand for a niacin receptor, GPCR 109A (GPR109A), with an EC50 of 770 μM (3). Like niacin, d-βOHB can inhibit adipose tissue lipolysis, which has been proposed to create a negative feedback loop in which ketosis curtails ketogenesis by limiting delivery of nonesterified fatty acids to the liver (205, 212). GPR109A signaling also promotes reverse cholesterol transport in macrophages (129). It is unknown whether d-βOHB plays a role in this cascade.
βOHB signaling may also influence cardiovascular biology through additional GPCRs. A recent study in mice revealed that βOHB decreases sympathetic outflow and reduces heart rate and total energy expenditure by antagonizing (through an unknown mechanism) GPR41, a Gi/o-coupled receptor for short-chain fatty acids (SCFAs, e.g., acetate, propionate, and butyrate) that is abundantly expressed in sympathetic ganglia (106). Thus evidence implicating ketone bodies as signaling molecules further supports roles for ketone bodies that transcend energy metabolism, indicating that additional experimentation is required to elucidate the receptors and mechanisms through which ketone bodies serve as extracellular signals.
Recently published findings also demonstrated that d-βOHB inhibits class I histone deacetylases (HDACs), resulting in increased histone acetylation and thus increased expression of genes encoding mediators of resistance to oxidative stress (189). The molecular effects of d-βOHB were observed at high-IC50 concentrations (2.4–5.3 mM, depending on the HDAC isoform) and were recapitulated by caloric restriction, fasting, or AcAc. To determine if these effects were dependent on d-βOHB metabolism, small interfering RNAs against BDH1/2 were transfected into cells, which were then treated with d-βOHB. The authors observed that with up to 3 mM d-βOHB, the effects on histone acetylation were preserved, leading to the conclusion that most of the influence of d-βOHB on HDAC function was independent of d-βOHB metabolism. However, residual BDH persisted in the small interfering RNAs-transfected cells, and the control experiment confirming diminished d-βOHB oxidation in BDH knockdown cells was not performed. Thus it remains plausible that oxidation of d-βOHB to AcAc by BDH1 could alter mitochondrial redox potential and that CoA transferase-dependent contribution of AcAc to the TCA cycle for terminal oxidation could alter cellular energy metabolism (see ketone body oxidation and Fig. 5), each of which may contribute to the observed effects on histone acetylation. Nonetheless, these observations reveal exciting roles for ketone body metabolism in a multitude of disease states, which will benefit from the development and application of both pharmacological and genetic tools.
Ketone Body Metabolism, the Microbiota, and Cardiovascular Biology and Disease
A therapeutically tractable interface between ketone body metabolism and cardiovascular disease is offered through the dynamic community of microorganisms living on our cutaneous and mucosal surfaces, the microbiota. This ecosystem of ∼100 trillion cells, 10 times the total number of our own cells, comprises a ∼1 kg organ that harbors a massive aggregate of genomes (the microbiome) encoding millions of genes, 100 times that of the human genome, that influence the human metabolome and coordinate immune function [recently reviewed in (13, 25, 69, 85, 87, 92a, 92b, 148, 211)]. The data supporting causal links between the microbiota and host cardiovascular disease in both rodent models and humans are compelling. Gut microbial ecology undergoes marked transformation in obese and type-2 diabetic humans and rodents, and the dynamic energy-harvesting capacity of the gut microbial community plays a significant contributing role to energy homeostasis in the host (14, 15, 34–36, 120, 121, 141, 160, 214–216, 232, 241). Acting in synergy, the host's genome and microbiome coordinate supraorganismal metabolic processes, creating a suite of cometabolites that influence a panoply of pathophysiological processes including NAFLD, hypertension, insulin resistance, and atherosclerosis (38, 48, 86, 224). Furthermore, the ability of indigenous microbial communities to coordinate the functions of the host's immune system is likely a key influence over the development of cardiovascular disease [reviewed in (87)], including the development of type-1 diabetes (227). Together, comparative genomics, metabolomics, and gnotobiotic studies (Greek roots: gnosis, knowledge; bios, life; a technology that allows cultivation of experimental animal models in the presence of defined microbial communities) may usher an era that ultimately allows caregivers to administer individualized forms of therapy that configure the microbiota using probiotics and prebiotics to ameliorate cardiovascular disease.
Studies of gnotobiotic animals have revealed intriguing relationships among the microbiota, ketone bodies, and cardiovascular biology. Normalized to tibial length or body weight, hearts of germ-free mice, which are born and raised in sterile gnotobiotic isolators, are ∼15% smaller than those of normally colonized mice, but myocardial mass increases within 2 wk of microbial colonization (42). In vivo and ex vivo functional parameters of hearts of germ-free mice are normal, but myocardial glucose oxidation rates are higher. A systematic assessment of substrate delivery and metabolism in germ-free mice revealed that while circulating ketone body concentrations were not significantly different under fed conditions, ketosis was blunted during nutrient deprivation in germ-free mice because of diminished hepatic ketogenesis (42). While reduced adiposity of germ-free mice (14) is a likely contributor to blunted ketogenesis during fasting, hepatic ketogenic machinery, including expression of Hmgcs2, was curtailed in fasting germ-free mice. The reduction in cardiac size and alterations of systemic and myocardial metabolism were abrogated when germ-free mice were maintained on a ketogenic diet (42). It is also intriguing to consider the aforementioned cross talk between SCFAs and ketone bodies [(106); see Signaling Roles for Ketone Bodies]. SCFAs are produced through the fermentative actions of glycan-digesting enzymes uniquely encoded by the microbiome, contributing to the host's energy harvest and to signaling through GPR41 (88, 106). Thus the microbiome may supervise an expansive and integrated network in which ketone bodies influence cardiac metabolism, size, and physiology. Future studies using genetically modified gnotobiotic animals will permit further construction of the relationship between ketone metabolism and the microbiome in diseases of the heart and vasculature.
Diagnostic and Therapeutic Targets of Ketone Body Metabolism
The diversity of metabolic and signaling processes in which ketone body metabolism participates provides a range of diagnostic and therapeutic applications, only a subset of which is under active investigation.
Abnormalities of ketone body oxidation.
CoA transferase-deficient humans typically present early in life with severe ketoacidosis that can result in vomiting, coma, and death if intravenous glucose and insulin therapy is not rapidly instituted (20, 150, 210). Neonatal mice with complete loss of terminal ketone body oxidation (SCOT-KO mice) invariably die within the first 48 h of life in a manner that mimics human sudden infant death syndrome (SIDS) (40). Furthermore, a recent observational study that performed metabolic autopsies on 255 SIDS patients found that three exhibited underlying disorders of ketone body metabolism (159). Therefore, a small subset of cases that ultimately receive a diagnosis of SIDS may actually be attributable to latent abnormalities of CoA transferase function. However, statewide neonatal screening protocols in the United States do not currently detect newborns with isolated disorders of ketone body oxidation, who exhibit hyperketonemia, but generally normal organic acid and acylcarnitine profiles (143). Improved screening methods, measuring both AcAc and d-βOHB, may detect patients with latent deficiency of ketone body oxidation, further reduce SIDS incidence, and predict metabolic complications which may manifest later in life.
Dietary therapies: ketogenic diets, ketone esters, and odd-chain fatty acids.
Ketogenic diets are actively used for weight loss and anticonvulsant therapy and are intensively studied as potential adjunctive therapy for brain cancers and neurodegenerative diseases including Parkinson's and Alzheimer's diseases (110, 139, 187, 236, 237). Additionally, limited studies raise the possibility that humans with NAFLD could benefit from low-carbohydrate diet therapy (32, 58). An experimental ketogenic diet has been used to mitigate a mitochondrial cardiomyopathy in mice (112). The full scope of cardiovascular diseases responsive to nutritional and/or pharmacological manipulation of ketone body metabolism and the associated metabolic mechanisms remain underexplored.
Ketogenic diets are often unpalatable, which leads to poor patient compliance. Additionally, such diets raise blood cholesterol and free fatty acids, increase the risk of nephrolithiasis, and cause constipation (23, 116, 201). Therefore, Veech and Clarke developed and tested ingestible ketone ester compounds, which can rapidly generate ketoses exceeding 5 mM in rats and humans (37, 103, 195), while sparing the adverse consequences of high-fat diets. A small preliminary study indicates that oral ketone esters are safe in humans (37). Rodents fed these compounds acutely decrease food intake. Although these animals do not exhibit changes in body weight over an extended time period, they do become more insulin sensitive, possibly because of increased expression of uncoupling proteins in brain and brown adipose tissue (103, 195). Further studies will evaluate the efficacy of these compounds in the mitigation and prevention of metabolic, myocardial, and neurological diseases in rodent and human subjects.
In the last decade, studies analyzing the beneficial roles of the anaplerotic five-carbon (C5) ketone bodies and their precursor odd-chain fatty acids for the treatment of long-chain fatty acid oxidation (LCFAO) disorders have emerged (170, 171). Conventional management of LCFAO disorders consists of dietary therapy with the medium-chain fatty acid octanoate. Substitution of octanoate for the odd-chain fatty acid heptanoate (given as the triglyceride triheptanoin) further improves clinical outcomes, specifically by reducing the incidence of rhabdomyolysis and cardiomyopathy (170, 171). While only trace levels of C5 ketone bodies are normally found in human body fluids (114, 202), ingestion of odd-chain fatty acids promotes hepatic C5-ketogenesis: β-oxidation of odd-chain fatty acids yields propionyl-CoA, an anaplerotic substrate, which, in the liver, can also be packaged into C5 ketone bodies (107). Oxidation of C5-ketone bodies in peripheral tissues occurs through CoA transferase (197), regenerating propionyl-CoA. However, as oxidation of heptanoate within myocytes locally generates anaplerotic propionyl-CoA, direct evidence linking C5-ketone body metabolism to improved clinical outcome in LCFAO disorders currently remains lacking. Thus it may be useful to cross experimental models of tissue-specific CoA transferase deficiency to existing models of LCFAO defects (41, 183) to establish the metabolic roles of C5-ketone body metabolism in myocyte oxidation. Future studies of the role of these three approaches—low-carbohydrate ketogenic diets, ketone ester-containing diets, or anaplerotic odd-chain fatty acid-containing diets in cardiomyopathy—remain to be systematically performed but are warranted (44, 117).
Modifying ketone body enzymatic pathways: targeting HMGCS2 and CoA transferase.
As both nutrients and hormonal factors affect ketogenic regulation, it is not surprising that ketone body metabolism is dynamically regulated in mouse models of diet-induced obesity and in humans with metabolic syndrome. Hepatic ketogenesis becomes suppressed at later stages of the evolution of hyperinsulinemic obesity (18, 21, 179, 190, 193, 200). Because transgene-mediated Hmgcs2 overexpression within hepatocytes enhances ketogenesis and simultaneously diminishes circulating free fatty acid concentrations in vivo (217), regulation of HMGCS2 activity (See regulation of ketogenic mediators) becomes a prospective therapeutic target. Diversion of hepatic fatty acyl chains into ketone bodies could diminish carbon that would otherwise require terminal oxidation, storage, or packaging into lipoproteins for secretion, prospectively mitigating hepatic steatosis and insulin resistance.
Whole body ketone body turnover is noninvasively quantifiable in humans, as is tissue-level oxidative flux of ketone bodies via positron emission tomography using 11C-labeled ketone body tracers, which have been used for studies in brain, but not yet in other organs (24, 173). Additionally, ketone body metabolism and the mitochondrial enzyme CoA transferase are amenable to a number of nutritional and, perhaps ultimately, pharmacological therapies. Importantly, as described above, myocardial CoA transferase activity plays an important role in the regulation of substrate selection, mitochondrial ROS generation, and cardiac work and bioenergetic efficiency (Fig. 5). Therefore, analysis of animal models with sophisticated genetic manipulations of ketone body metabolism, coupled with pharmacological targeting strategies, may provide rationale for a suite of translational and clinical studies that validate ketone body metabolism as a myocardial therapeutic target. Additionally, CoA transferase and AACS activities may ultimately serve as therapeutic targets in contexts in which the ketone body-lipogenic pathways play significant roles, particularly within adipocytes, hepatocytes, and macrophages.
Conclusions
Ketone body metabolism maintains bioenergetic homeostasis when dietary carbohydrates are limiting. Defects in either the synthetic or oxidative arms of ketone body metabolism result in disease pathogenesis in humans, and ketone body oxidation is required for maintenance of glycemia and survival in neonatal mice. Ketone bodies regulate mitochondrial metabolism, energetics, and ROS production via their oxidation and may therefore have significant signaling roles within cardiomyocytes. Ketogenic flux through CoA transferase channels ketone bodies to lipogenesis, which could regulate signaling processes in many cell types, including macrophages, adipocytes, and pancreatic β-cells, which express CoA transferase abundantly but may have relatively diminished need to harvest energy from ketone bodies. Therefore, the metabolism of ketone bodies may influence numerous human disease states relevant to cardiovascular disease, including obesity, diabetes, atherosclerosis, and heart failure. Because both CoA transferase and HMGCS2 experience multitiered regulation, these enzymes may be tractable to pharmacological manipulation. Before these ends are achieved, the scope of viable therapeutic targets of ketone body metabolism must first be developed. Integrative experiments in both animal models and humans that exploit the convergence of sophisticated genetic approaches, quantitative substrate fate mapping (fluxomics), metabolomic profiling, physiological studies, and quantification of mitochondrial function are needed to elucidate the mechanisms through which ketone body metabolism influences cardiovascular pathophysiological processes.
GRANTS
This work was supported in part by National Institutes of Health Grants HL-007873 (a training grant that supports D. G. Cotter), HL-007275 (a training grant that supports R. C. Schugar), and DK-091538 (to P. A. Crawford), plus grants from the Diabetic Cardiovascular Disease Center at Washington University and the March of Dimes (both to P. A. Crawford).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
D.G.C. and P.A.C. prepared figures; D.G.C., R.C.S., and P.A.C. drafted manuscript; D.G.C., R.C.S., and P.A.C. edited and revised manuscript; D.G.C., R.C.S., and P.A.C. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Baris Ercal, Xiaojing Huang, and, in particular, the reviewers of the manuscript for helpful comments and Laura Kyro for assistance with graphics.
REFERENCES
- 1. Abel ED, Doenst T. Mitochondrial adaptations to physiological versus pathological cardiac hypertrophy. Cardiovasc Res 90: 230–242, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aguilo F, Camarero N, Relat J, Marrero PF, Haro D. Transcriptional regulation of the human acetoacetyl-CoA synthetase gene by PPARgamma. Biochem J 427: 255–264, 2010 [DOI] [PubMed] [Google Scholar]
- 3. Ahmed K, Tunaru S, Offermanns S. GPR109A, GPR109B and GPR81, a family of hydroxy-carboxylic acid receptors. Trends Pharmacol Sci 30: 557–562, 2009 [DOI] [PubMed] [Google Scholar]
- 6. Al-Zaid NS, Dashti HM, Mathew TC, Juggi JS. Low carbohydrate ketogenic diet enhances cardiac tolerance to global ischaemia. Acta Cardiol 62: 381–389, 2007 [DOI] [PubMed] [Google Scholar]
- 7. Aledo R, Zschocke J, Pie J, Mir C, Fiesel S, Mayatepek E, Hoffmann GF, Casals N, Hegardt FG. Genetic basis of mitochondrial HMG-CoA synthase deficiency. Hum Genet 109: 19–23, 2001 [DOI] [PubMed] [Google Scholar]
- 8. Aneja P, Dziak R, Cai GQ, Charles TC. Identification of an acetoacetyl coenzyme A synthetase-dependent pathway for utilization of l-(+)-3-hydroxybutyrate in Sinorhizobium meliloti. J Bacteriol 184: 1571–1577, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Arias GM, Asins G, Hegardt FG, Serra D. The effect of fasting and insulin treatment on carnitine palmitoyl transferase I and mitochondrial 3-hydroxy-3-methylglutaryl coenzyme A synthase mRNA levels in liver from suckling rats. Biochem Soc Trans 3: 493S, 1995 [DOI] [PubMed] [Google Scholar]
- 10. Ashrafian H, Frenneaux MP, Opie LH. Metabolic mechanisms in heart failure. Circulation 116: 434–448, 2007 [DOI] [PubMed] [Google Scholar]
- 11. Avogaro A, Doria A, Gnudi L, Carraro A, Duner E, Brocco E, Tiengo A, Crepaldi G, Bier DM, Nosadini R. Forearm ketone body metabolism in normal and in insulin-dependent diabetic patients. Am J Physiol Endocrinol Metab 263: E261–E267, 1992 [DOI] [PubMed] [Google Scholar]
- 12. Ayté J, Gil-Gómez G, Hegardt FG. Methylation of the regulatory region of the mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase gene leads to its transcriptional inactivation. Biochem J 295: 807–812, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Backhed F, Crawford PA. Coordinated regulation of the metabolome and lipidome at the host-microbial interface. Biochim Biophys Acta 1801: 240–245, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, Gordon JI. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA 101: 15718–15723, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Backhed F, Manchester JK, Semenkovich CF, Gordon JI. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci USA 104: 979–984, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Badman MK, Pissios P, Kennedy AR, Koukos G, Flier JS, Maratos-Flier E. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab 5: 426–437, 2007 [DOI] [PubMed] [Google Scholar]
- 17. Balasse EO, Fery F. Ketone body production and disposal: effects of fasting, diabetes, and exercise. Diabetes Metab Rev 5: 247–270, 1989 [DOI] [PubMed] [Google Scholar]
- 18. Bergman BC, Cornier MA, Horton TJ, Bessesen DH. Effects of fasting on insulin action and glucose kinetics in lean and obese men and women. Am J Physiol Endocrinol Metab 293: E1103–E1111, 2007 [DOI] [PubMed] [Google Scholar]
- 19. Bergstrom JD, Wong GA, Edwards PA, Edmond J. The regulation of acetoacetyl-CoA synthetase activity by modulators of cholesterol synthesis in vivo and the utilization of acetoacetate for cholesterogenesis. J Biol Chem 259: 14548–14553, 1984 [PubMed] [Google Scholar]
- 20. Berry GT, Fukao T, Mitchell GA, Mazur A, Ciafre M, Gibson J, Kondo N, Palmieri MJ. Neonatal hypoglycaemia in severe succinyl-CoA: 3-oxoacid CoA-transferase deficiency. J Inherit Metab Dis 24: 587–595, 2001 [DOI] [PubMed] [Google Scholar]
- 21. Bickerton AS, Roberts R, Fielding BA, Tornqvist H, Blaak EE, Wagenmakers AJ, Gilbert M, Humphreys SM, Karpe F, Frayn KN. Adipose tissue fatty acid metabolism in insulin-resistant men. Diabetologia 51: 1466–1474, 2008 [DOI] [PubMed] [Google Scholar]
- 22. Bing RJ. The metabolism of the heart. Harvey Lect 50: 27–70, 1955 [PubMed] [Google Scholar]
- 23. Bisschop PH, de Metz J, Ackermans MT, Endert E, Pijl H, Kuipers F, Meijer AJ, Sauerwein HP, Romijn JA. Dietary fat content alters insulin-mediated glucose metabolism in healthy men. Am J Clin Nutr 73: 554–559, 2001 [DOI] [PubMed] [Google Scholar]
- 24. Blomqvist G, Alvarsson M, Grill V, Von Heijne G, Ingvar M, Thorell JO, Stone-Elander S, Widen L, Ekberg K. Effect of acute hyperketonemia on the cerebral uptake of ketone bodies in nondiabetic subjects and IDDM patients. Am J Physiol Endocrinol Metab 283: E20–E28, 2002 [DOI] [PubMed] [Google Scholar]
- 25. Blumberg R, Powrie F. Microbiota, disease, and back to health: a metastable journey. Sci Transl Med 4: 137rv137, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bock H, Fleischer S. Preparation of a homogeneous soluble d-beta-hydroxybutyrate apodehydrogenase from mitochondria. J Biol Chem 250: 5774–5761, 1975 [PubMed] [Google Scholar]
- 27. Bodamer OA, Hussein K, Morris AA, Langhans CD, Rating D, Mayatepek E, Leonard JV. Glucose and leucine kinetics in idiopathic ketotic hypoglycaemia. Arch Dis Child 91: 483–486, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bouchard L, Robert MF, Vinarov D, Stanley CA, Thompson GN, Morris A, Leonard JV, Quant P, Hsu BY, Boneh A, Boukaftane Y, Ashmarina L, Wang S, Miziorko H, Mitchell GA. Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase deficiency: clinical course and description of causal mutations in two patients. Pediatr Res 49: 326–331, 2001 [DOI] [PubMed] [Google Scholar]
- 29. Boudina S, Abel ED. Diabetic cardiomyopathy, causes and effects. Rev Endocr Metab Disord 11: 31–39, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Boukaftane Y, Duncan A, Wang S, Labuda D, Robert MF, Sarrazin J, Schappert K, Mitchell GA. Human mitochondrial HMG CoA synthase: liver cDNA and partial genomic cloning, chromosome mapping to 1p12-p13, and possible role in vertebrate evolution. Genomics 23: 552–559, 1994 [DOI] [PubMed] [Google Scholar]
- 31. Bregere C, Rebrin I, Gallaher TK, Sohal RS. Effects of age and calorie restriction on tryptophan nitration, protein content, and activity of succinyl-CoA:3-ketoacid CoA transferase in rat kidney mitochondria. Free Radic Biol Med 48: 609–618, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Browning JD, Baker JA, Rogers T, Davis J, Satapati S, Burgess SC. Short-term weight loss and hepatic triglyceride reduction: evidence of a metabolic advantage with dietary carbohydrate restriction. Am J Clin Nutr 93: 1048–1052, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cahill GF., Jr Fuel metabolism in starvation. Annu Rev Nutr 26: 1–22, 2006 [DOI] [PubMed] [Google Scholar]
- 34. Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, Waget A, Delmee E, Cousin B, Sulpice T, Chamontin B, Ferrieres J, Tanti JF, Gibson GR, Casteilla L, Delzenne NM, Alessi MC, Burcelin R. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 56: 1761–1772, 2007 [DOI] [PubMed] [Google Scholar]
- 35. Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 57: 1470–1481, 2008 [DOI] [PubMed] [Google Scholar]
- 36. Cani PD, Possemiers S, Van de Wiele T, Guiot Y, Everard A, Rottier O, Geurts L, Naslain D, Neyrinck AM, Lambert DM, Muccioli GG, Delzenne NM. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut 57: 1091–1103, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Clarke K, Tchabanenko K, Pawlosky R, Carter E, Todd King M, Musa-Veloso K, Ho M, Roberts A, Robertson J, Vanitallie TB, Veech RL. Kinetics, safety and tolerability of (R)-3-hydroxybutyl (R)-3-hydroxybutyrate in healthy adult subjects. Regul Toxicol Pharmacol 63: 401–408, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Claus SP, Tsang TM, Wang Y, Cloarec O, Skordi E, Martin FP, Rezzi S, Ross A, Kochhar S, Holmes E, Nicholson JK. Systemic multicompartmental effects of the gut microbiome on mouse metabolic phenotypes. Mol Syst Biol 4: 219, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cooper RH, Randle PJ, Denton RM. Stimulation of phosphorylation and inactivation of pyruvate dehydrogenase by physiological inhibitors of the pyruvate dehydrogenase reaction. Nature 257: 808–809, 1975 [DOI] [PubMed] [Google Scholar]
- 40. Cotter DG, d'Avignon DA, Wentz AE, Weber ML, Crawford PA. Obligate role for ketone body oxidation in neonatal metabolic homeostasis. J Biol Chem 286: 6902–6910, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cotter DG, Schugar RC, Wentz AE, d'Avignon DA, Crawford PA. Successful adaptation to ketosis by mice with tissue-specific deficiency of ketone body oxidation. Am J Physiol Endocrinol Metab 304: E363–E374, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Crawford PA, Crowley JR, Sambandam N, Muegge BD, Costello EK, Hamady M, Knight R, Gordon JI. Regulation of myocardial ketone body metabolism by the gut microbiota during nutrient deprivation. Proc Natl Acad Sci USA 106: 11276–11281, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cunnane SC, Crawford MA. Survival of the fattest: fat babies were the key to evolution of the large human brain. Comp Biochem Physiol A Mol Integr Physiol 136: 17–26, 2003 [DOI] [PubMed] [Google Scholar]
- 44. Des Rosiers C, Labarthe F, Lloyd SG, Chatham JC. Cardiac anaplerosis in health and disease: food for thought. Cardiovasc Res 90: 210–219, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Des Rosiers C, Montgomery JA, Garneau M, David F, Mamer OA, Daloze P, Toffolo G, Cobelli C, Landau BR, Brunengraber H. Pseudoketogenesis in hepatectomized dogs. Am J Physiol Endocrinol Metab 258: E519–E528, 1990 [DOI] [PubMed] [Google Scholar]
- 46. Doepner B, Thierfelder S, Hirche H, Benndorf K. 3-Hydroxybutyrate blocks the transient K+ outward current in myocardial mouse cells in a stereoselective fashion. J Physiol 500: 85–94, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 47. Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, Kim J, Woo J, Kim JH, Choi BH, He B, Chen W, Zhang S, Cerione RA, Auwerx J, Hao Q, Lin H. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 334: 806–809, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dumas ME, Barton RH, Toye A, Cloarec O, Blancher C, Rothwell A, Fearnside J, Tatoud R, Blanc V, Lindon JC, Mitchell SC, Holmes E, McCarthy MI, Scott J, Gauguier D, Nicholson JK. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc Natl Acad Sci USA 103: 12511–12516, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Edmond J. Ketone bodies as precursors of sterols and fatty acids in the developing rat. J Biol Chem 249: 72–80, 1974 [PubMed] [Google Scholar]
- 50. Edmond J, Robbins RA, Bergstrom JD, Cole RA, de Vellis J. Capacity for substrate utilization in oxidative metabolism by neurons, astrocytes, and oligodendrocytes from developing brain in primary culture. J Neurosci Res 18: 551–561, 1987 [DOI] [PubMed] [Google Scholar]
- 51. Endemann G, Goetz PG, Edmond J, Brunengraber H. Lipogenesis from ketone bodies in the isolated perfused rat liver. Evidence for the cytosolic activation of acetoacetate. J Biol Chem 257: 3434–3440, 1982 [PubMed] [Google Scholar]
- 52. Fellman V, Kotarsky H. Mitochondrial hepatopathies in the newborn period. Semin Fetal Neonatal Med 16: 222–228, 2011 [DOI] [PubMed] [Google Scholar]
- 53. Fenselau A, Wallis K. 3-Oxo acid coenzyme A-transferase in normal and diabetic rat muscle. Biochem J 158: 509–512, 1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fenselau A, Wallis K. Substrate specificity and mechanism of action of acetoacetate coenzyme A transferase from rat heart. Biochemistry 13: 3884–3888, 1974 [DOI] [PubMed] [Google Scholar]
- 55. Ferre P, Satabin P, Decaux J, Escriva F, Girard J. Development and regulation of ketogenesis in hepatocytes isolated from newborn rats. Biochem J 214: 937–942, 1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fery F, Balasse EO. Ketone body production and disposal in diabetic ketosis. A comparison with fasting ketosis. Diabetes 34: 326–332, 1985 [DOI] [PubMed] [Google Scholar]
- 57. Fink G, Desrochers S, Des Rosiers C, Garneau M, David F, Daloze T, Landau BR, Brunengraber H. Pseudoketogenesis in the perfused rat heart. J Biol Chem 263: 18036–18042, 1988 [PubMed] [Google Scholar]
- 58. Foster GD, Wyatt HR, Hill JO, Makris AP, Rosenbaum DL, Brill C, Stein RI, Mohammed BS, Miller B, Rader DJ, Zemel B, Wadden TA, Tenhave T, Newcomb CW, Klein S. Weight and metabolic outcomes after 2 years on a low-carbohydrate versus low-fat diet: a randomized trial. Ann Intern Med 153: 147–157, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Freed LE, Endemann G, Tomera JF, Gavino VC, Brunengraber H. Lipogenesis from ketone bodies in perfused livers from streptozocin-induced diabetic rats. Diabetes 37: 50–55, 1988 [DOI] [PubMed] [Google Scholar]
- 60. Fukao T. 3-ketoacid CoA transferase (SCOT) deficiency. Orphanet encyclopedia, 2001. (www.orpha.net) [Google Scholar]
- 61. Fukao T, Lopaschuk GD, Mitchell GA. Pathways and control of ketone body metabolism: on the fringe of lipid biochemistry. Prostaglandins Leukot Essent Fatty Acids 70: 243–251, 2004 [DOI] [PubMed] [Google Scholar]
- 62. Fukao T, Mitchell GA, Song XQ, Nakamura H, Kassovska-Bratinova S, Orii KE, Wraith JE, Besley G, Wanders RJ, Niezen-Koning KE, Berry GT, Palmieri M, Kondo N. Succinyl-CoA:3-ketoacid CoA transferase (SCOT): cloning of the human SCOT gene, tertiary structural modeling of the human SCOT monomer, and characterization of three pathogenic mutations. Genomics 68: 144–151, 2000 [DOI] [PubMed] [Google Scholar]
- 63. Fukao T, Sakurai S, Rolland MO, Zabot MT, Schulze A, Yamada K, Kondo N. A 6-bp deletion at the splice donor site of the first intron resulted in aberrant splicing using a cryptic splice site within exon 1 in a patient with succinyl-CoA: 3-Ketoacid CoA transferase (SCOT) deficiency. Mol Genet Metab 89: 280–282, 2006 [DOI] [PubMed] [Google Scholar]
- 64. Fukao T, Song XQ, Mitchell GA, Yamaguchi S, Sukegawa K, Orii T, Kondo N. Enzymes of ketone body utilization in human tissues: protein and messenger RNA levels of succinyl-coenzyme A (CoA):3-ketoacid CoA transferase and mitochondrial and cytosolic acetoacetyl-CoA thiolases. Pediatr Res 42: 498–502, 1997 [DOI] [PubMed] [Google Scholar]
- 65. Garland PB, Newsholme EA, Randle PJ. Effect of fatty acids, ketone bodies, diabetes and starvation on pyruvate metabolism in rat heart and diaphragm muscle. Nature 195: 381–383, 1962 [DOI] [PubMed] [Google Scholar]
- 66. Garland PB, Randle PJ, Newsholme EA. Citrate as an intermediary in the inhibition of phosphofructokinase in rat heart muscle by fatty acids, ketone bodies, pyruvate, diabetes, and starvation. Nature 200: 169–170, 1963 [DOI] [PubMed] [Google Scholar]
- 67. Geelen MJ, Lopes-Cardozo M, Edmond J. Acetoacetate: a major substrate for the synthesis of cholesterol and fatty acids by isolated rat hepatocytes. FEBS Lett 163: 269–273, 1983 [DOI] [PubMed] [Google Scholar]
- 68. Girard J, Ferre P, Pegorier JP, Duee PH. Adaptations of glucose and fatty acid metabolism during perinatal period and suckling-weaning transition. Physiol Rev 72: 507–562, 1992 [DOI] [PubMed] [Google Scholar]
- 69. Gordon JI. Honor thy gut symbionts redux. Science 336: 1251–1253, 2012 [DOI] [PubMed] [Google Scholar]
- 70. Grimsrud PA, Carson JJ, Hebert AS, Hubler SL, Niemi NM, Bailey DJ, Jochem A, Stapleton DS, Keller MP, Westphall MS, Yandell BS, Attie AD, Coon JJ, Pagliarini DJ. A quantitative map of the liver mitochondrial phosphoproteome reveals posttranslational control of ketogenesis. Cell Metab 16: 672–683, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Grinblat L, Pacheco Bolanos LF, Stoppani AO. Decreased rate of ketone-body oxidation and decreased activity of d-3-hydroxybutyrate dehydrogenase and succinyl-CoA:3-oxo-acid CoA-transferase in heart mitochondria of diabetic rats. Biochem J 240: 49–56, 1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Guo K, Lukacik P, Papagrigoriou E, Meier M, Lee WH, Adamski J, Oppermann U. Characterization of human DHRS6, an orphan short chain dehydrogenase/reductase enzyme: a novel, cytosolic type 2 R-beta-hydroxybutyrate dehydrogenase. J Biol Chem 281: 10291–10297, 2006 [DOI] [PubMed] [Google Scholar]
- 73. Haas JT, Miao J, Chanda D, Wang Y, Zhao E, Haas ME, Hirschey M, Vaitheesvaran B, Farese RV, Jr, Kurland IJ, Graham M, Crooke R, Foufelle F, Biddinger SB. Hepatic insulin signaling is required for obesity-dependent expression of SREBP-1c mRNA but not for feeding-dependent expression. Cell Metab 15: 873–884, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Haces ML, Hernandez-Fonseca K, Medina-Campos ON, Montiel T, Pedraza-Chaverri J, Massieu L. Antioxidant capacity contributes to protection of ketone bodies against oxidative damage induced during hypoglycemic conditions. Exp Neurol 211: 85–96, 2008 [DOI] [PubMed] [Google Scholar]
- 75. Halestrap AP. The monocarboxylate transporter family—Structure and functional characterization. IUBMB Life 64: 1–9, 2012 [DOI] [PubMed] [Google Scholar]
- 76. Halestrap AP, Wilson MC. The monocarboxylate transporter family—role and regulation. IUBMB Life 64: 109–119, 2012 [DOI] [PubMed] [Google Scholar]
- 77. Hall SE, Wastney ME, Bolton TM, Braaten JT, Berman M. Ketone body kinetics in humans: the effects of insulin-dependent diabetes, obesity, and starvation. J Lipid Res 25: 1184–1194, 1984 [PubMed] [Google Scholar]
- 78. Hasan NM, Longacre MJ, Seed-Ahmed M, Kendrick MA, Gu H, Ostenson CG, Fukao T, Macdonald MJ. Lower succinyl-CoA:3-ketoacid-CoA transferase (SCOT) and ATP citrate lyase in pancreatic islets of a rat model of type 2 diabetes: knockdown of SCOT inhibits insulin release in rat insulinoma cells. Arch Biochem Biophys 499: 62–68, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hasegawa S, Ikeda Y, Yamasaki M, Fukui T. The role of acetoacetyl-CoA synthetase, a ketone body-utilizing enzyme, in 3T3–L1 adipocyte differentiation. Biol Pharm Bull 35: 1980–1985, 2012 [DOI] [PubMed] [Google Scholar]
- 80. Hasegawa S, Kume H, Iinuma S, Yamasaki M, Takahashi N, Fukui T. Acetoacetyl-CoA synthetase is essential for normal neuronal development. Biochem Biophys Res Commun 427: 398–403, 2012 [DOI] [PubMed] [Google Scholar]
- 81. Hasegawa S, Noda K, Maeda A, Matsuoka M, Yamasaki M, Fukui T. Acetoacetyl-CoA synthetase, a ketone body-utilizing enzyme, is controlled by SREBP-2 and affects serum cholesterol levels. Mol Genet Metab 107: 553–560, 2012 [DOI] [PubMed] [Google Scholar]
- 82. Hasselbaink DM, Glatz JF, Luiken JJ, Roemen TH, Van der Vusse GJ. Ketone bodies disturb fatty acid handling in isolated cardiomyocytes derived from control and diabetic rats. Biochem J 371: 753–760, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hawdon J. Hypoglycaemia and the neonatal brain. Eur J Pediatr 158: S9–S12, 1999 [DOI] [PubMed] [Google Scholar]
- 84. Hegardt FG. Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase: a control enzyme in ketogenesis. Biochem J 338: 569–582, 1999 [PMC free article] [PubMed] [Google Scholar]
- 85. Holmes E, Kinross J, Gibson GR, Burcelin R, Jia W, Pettersson S, Nicholson JK. Therapeutic modulation of microbiota-host metabolic interactions. Sci Transl Med 4: 137rv136, 2012 [DOI] [PubMed] [Google Scholar]
- 86. Holmes E, Loo RL, Stamler J, Bictash M, Yap IK, Chan Q, Ebbels T, De Iorio M, Brown IJ, Veselkov KA, Daviglus ML, Kesteloot H, Ueshima H, Zhao L, Nicholson JK, Elliott P. Human metabolic phenotype diversity and its association with diet and blood pressure. Nature 453: 396–400, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science 336: 1268–1273, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hoverstad T, Midtvedt T. Short-chain fatty acids in germfree mice and rats. J Nutr 116: 1772–1776, 1986 [DOI] [PubMed] [Google Scholar]
- 89. Hue L, Taegtmeyer H. The Randle cycle revisited: a new head for an old hat. Am J Physiol Endocrinol Metab 297: E578–E591, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Huelsmann WC, Siliprandi D, Ciman M, Siliprandi N. Effect of carnitine on the oxidation of alpha-oxoglutarate to succinate in the presence of acetoacetate or pyruvate. Biochim Biophys Acta 93: 166–168, 1964 [DOI] [PubMed] [Google Scholar]
- 91. Hugo SE, Cruz-Garcia L, Karanth S, Anderson RM, Stainier DY, Schlegel A. A monocarboxylate transporter required for hepatocyte secretion of ketone bodies during fasting. Genes Dev 26: 282–293, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Huidekoper HH, Duran M, Turkenburg M, Ackermans MT, Sauerwein HP, Wijburg FA. Fasting adaptation in idiopathic ketotic hypoglycemia: a mismatch between glucose production and demand. Eur J Pediatr 167: 859–865, 2008 [DOI] [PubMed] [Google Scholar]
- 92a. Human Microbiome Project Consortium A framework for human microbiome research. Nature 486: 215–221, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92b. Human Microbiome Project Consortium Structure function, and diversity of the healthy human microbiome. Nature 486: 207–214, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Inagaki T, Dutchak P, Zhao G, Ding X, Gautron L, Parameswara V, Li Y, Goetz R, Mohammadi M, Esser V, Elmquist JK, Gerard RD, Burgess SC, Hammer RE, Mangelsdorf DJ, Kliewer SA. Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab 5: 415–425, 2007 [DOI] [PubMed] [Google Scholar]
- 94. Ingwall JS, Weiss RG. Is the failing heart energy starved? On using chemical energy to support cardiac function. Circ Res 95: 135–145, 2004 [DOI] [PubMed] [Google Scholar]
- 95. Ito M, Fukui T, Kamokari M, Saito T, Tomita K. Purification and characterization of acetoacetyl-CoA synthetase from rat liver. Biochim Biophys Acta 794: 183–193, 1984 [PubMed] [Google Scholar]
- 96. Janardhan A, Chen J, Crawford PA. Altered systemic ketone body metabolism in advanced heart failure. Tex Heart Inst J 38: 533–538, 2011 [PMC free article] [PubMed] [Google Scholar]
- 97. Jaswal JS, Keung W, Wang W, Ussher JR, Lopaschuk GD. Targeting fatty acid and carbohydrate oxidation—a novel therapeutic intervention in the ischemic and failing heart. Biochim Biophys Acta 1813: 1333–1350, 2011 [DOI] [PubMed] [Google Scholar]
- 98. Jeffrey FM, Diczku V, Sherry AD, Malloy CR. Substrate selection in the isolated working rat heart: effects of reperfusion, afterload, and concentration. Basic Res Cardiol 90: 388–396, 1995 [DOI] [PubMed] [Google Scholar]
- 99. Jeoung NH, Rahimi Y, Wu P, Lee WN, Harris RA. Fasting induces ketoacidosis and hypothermia in PDHK2/PDHK4-double-knockout mice. Biochem J 443: 829–839, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Johnson RH, Walton JL, Krebs HA, Williamson DH. Post-exercise ketosis. Lancet 2: 1383–1385, 1969 [DOI] [PubMed] [Google Scholar]
- 101. Jones JG, Hansen J, Sherry AD, Malloy CR, Victor RG. Determination of acetyl-CoA enrichment in rat heart and skeletal muscle by 1H nuclear magnetic resonance analysis of glutamate in tissue extracts. Analytical biochemistry 249: 201–206, 1997 [DOI] [PubMed] [Google Scholar]
- 102. Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab 1: 15–25, 2005 [DOI] [PubMed] [Google Scholar]
- 103. Kashiwaya Y, Pawlosky R, Markis W, King MT, Bergman C, Srivastava S, Murray A, Clarke K, Veech RL. A ketone ester diet increases brain malonyl-CoA and Uncoupling proteins 4 and 5 while decreasing food intake in the normal Wistar Rat. J Biol Chem 285: 25950–25956, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Kashiwaya Y, Sato K, Tsuchiya N, Thomas S, Fell DA, Veech RL, Passonneau JV. Control of glucose utilization in working perfused rat heart. J Biol Chem 269: 25502–25514, 1994 [PubMed] [Google Scholar]
- 105. Kassovska-Bratinova S, Fukao T, Song XQ, Duncan AM, Chen HS, Robert MF, Perez-Cerda C, Ugarte M, Chartrand C, Vobecky S, Kondo N, Mitchell GA. Succinyl CoA: 3-oxoacid CoA transferase (SCOT): human cDNA cloning, human chromosomal mapping to 5p13, and mutation detection in a SCOT-deficient patient. Am J Hum Genet 59: 519–528, 1996 [PMC free article] [PubMed] [Google Scholar]
- 106. Kimura I, Inoue D, Maeda T, Hara T, Ichimura A, Miyauchi S, Kobayashi M, Hirasawa A, Tsujimoto G. Short-chain fatty acids and ketones directly regulate sympathetic nervous system via G protein-coupled receptor 41 (GPR41). Proc Natl Acad Sci USA 108: 8030–8035, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Kinman RP, Kasumov T, Jobbins KA, Thomas KR, Adams JE, Brunengraber LN, Kutz G, Brewer WU, Roe CR, Brunengraber H. Parenteral and enteral metabolism of anaplerotic triheptanoin in normal rats. Am J Physiol Endocrinol Metab 291: E860–E866, 2006 [DOI] [PubMed] [Google Scholar]
- 108. Kochar IS, Hussain K. From hyperinsulinaemic hypoglycaemia to ketotic hypoglycaemia: the range of glucose abnormalities in patients born with intrauterine growth retardation. Eur J Pediatr 166: 1003–1007, 2007 [DOI] [PubMed] [Google Scholar]
- 109. Kolwicz SC, Jr, Tian R. Glucose metabolism and cardiac hypertrophy. Cardiovasc Res 90: 194–201, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Kossoff EH, Hartman AL. Ketogenic diets: new advances for metabolism based therapies. Curr Opin Neurol 25: 173–178, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Kostiuk MA, Keller BO, Berthiaume LG. Palmitoylation of ketogenic enzyme HMGCS2 enhances its interaction with PPARα and transcription at the Hmgcs2 PPRE. FASEB J 24: 1914–1924, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Krebs P, Fan W, Chen YH, Tobita K, Downes MR, Wood MR, Sun L, Li X, Xia Y, Ding N, Spaeth JM, Moresco EM, Boyer TG, Lo CW, Yen J, Evans RM, Beutler B. Lethal mitochondrial cardiomyopathy in a hypomorphic Med30 mouse mutant is ameliorated by ketogenic diet. Proc Natl Acad Sci USA 108: 19678–19682, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Krishnakumar AM, Sliwa D, Endrizzi JA, Boyd ES, Ensign SA, Peters JW. Getting a handle on the role of coenzyme M in alkene metabolism. Microbiol Mol Biol Rev 72: 445–456, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Kuhara T, Inoue Y, Shinka T, Matsumoto I, Matsuo M. Identification of 3-hydroxy-3-ethylglutaric acid in urine of patients with propionic acidaemia. Biomed Mass Spectrom 10: 629–632, 1983 [DOI] [PubMed] [Google Scholar]
- 115. Kupari M, Lommi J, Ventila M, Karjalainen U. Breath acetone in congestive heart failure. Am J Cardiol 76: 1076–1078, 1995 [DOI] [PubMed] [Google Scholar]
- 116. Kwiterovich PO, Jr, Vining EP, Pyzik P, Skolasky R, Jr, Freeman JM. Effect of a high-fat ketogenic diet on plasma levels of lipids, lipoproteins, and apolipoproteins in children. JAMA 290: 912–920, 2003 [DOI] [PubMed] [Google Scholar]
- 117. Labarthe F, Gélinas R, Des Rosiers C. Medium-chain fatty acids as metabolic therapy in cardiac disease. Cardiovasc Drugs Ther 22: 97–106, 2008 [DOI] [PubMed] [Google Scholar]
- 118. Laing SP, Swerdlow AJ, Slater SD, Burden AC, Morris A, Waugh NR, Gatling W, Bingley PJ, Patterson CC. Mortality from heart disease in a cohort of 23,000 patients with insulin-treated diabetes. Diabetologia 46: 760–765, 2003 [DOI] [PubMed] [Google Scholar]
- 119. Lehninger ALSH, Wise JB. d-β-Hydroxybutyric dehydrogenase of mitochondria. J Biol Chem 235: 2450–2455, 1960 [PubMed] [Google Scholar]
- 120. Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci USA 102: 11070–11075, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Li JV, Ashrafian H, Bueter M, Kinross J, Sands C, le Roux CW, Bloom SR, Darzi A, Athanasiou T, Marchesi JR, Nicholson JK, Holmes E. Metabolic surgery profoundly influences gut microbial-host metabolic cross-talk. Gut 60: 1214–1223, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Lincoln BC, Des Rosiers C, Brunengraber H. Metabolism of S-3-hydroxybutyrate in the perfused rat liver. Arch Biochem Biophys 259: 149–156, 1987 [DOI] [PubMed] [Google Scholar]
- 123. Lodhi IJ, Yin L, Jensen-Urstad AP, Funai K, Coleman T, Baird JH, El Ramahi MK, Razani B, Song H, Fu-Hsu F, Turk J, Semenkovich CF. Inhibiting adipose tissue lipogenesis reprograms thermogenesis and PPARgamma activation to decrease diet-induced obesity. Cell Metab 16: 189–201, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Lommi J, Koskinen P, Naveri H, Harkonen M, Kupari M. Heart failure ketosis. J Intern Med 242: 231–238, 1997 [DOI] [PubMed] [Google Scholar]
- 125. Lommi J, Kupari M, Koskinen P, Naveri H, Leinonen H, Pulkki K, Harkonen M. Blood ketone bodies in congestive heart failure. J Am Coll Cardiol 28: 665–672, 1996 [DOI] [PubMed] [Google Scholar]
- 126. Longo N, Fukao T, Singh R, Pasquali M, Barrios RG, Kondo N, Gibson KM. Succinyl-CoA:3-ketoacid transferase (SCOT) deficiency in a new patient homozygous for an R217X mutation. J Inherit Metab Dis 27: 691–692, 2004 [DOI] [PubMed] [Google Scholar]
- 127. Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev 90: 207–258, 2010 [DOI] [PubMed] [Google Scholar]
- 128. Lowe DM, Tubbs PK. 3-Hydroxy-3-methylglutaryl-coenzyme A synthase from ox liver. Purification, molecular and catalytic properties. Biochem J 227: 591–599, 1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Lukasova M, Malaval C, Gille A, Kero J, Offermanns S. Nicotinic acid inhibits progression of atherosclerosis in mice through its receptor GPR109A expressed by immune cells. J Clin Invest 121: 1163–1173, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Maalouf M, Sullivan PG, Davis L, Kim DY, Rho JM. Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience 145: 256–264, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. MacDonald MJ. Synergistic potent insulin release by combinations of weak secretagogues in pancreatic islets and INS-1 cells. J Biol Chem 282: 6043–6052, 2007 [DOI] [PubMed] [Google Scholar]
- 132. MacDonald MJ, Hasan N, Longacre M. Studies with leucine, β-hydroxybutyrate and ATP citrate lyase-deficient beta cells support the acetoacetate pathway of insulin secretion. Biochim Biophys Acta 1780: 966–972, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. MacDonald MJ, Longacre MJ, Stoker SW, Brown LJ, Hasan NM, Kendrick MA. Acetoacetate and beta-hydroxybutyrate in combination with other metabolites release insulin from INS-1 cells and provide clues about pathways in insulin secretion. Am J Physiol Cell Physiol 294: C442–C450, 2008 [DOI] [PubMed] [Google Scholar]
- 134. Magnusson I, Schumann WC, Bartsch GE, Chandramouli V, Kumaran K, Wahren J, Landau BR. Noninvasive tracing of krebs cycle metabolism in liver. J Biol Chem 266: 6975–6984, 1991 [PubMed] [Google Scholar]
- 135. Marcondes S, Turko IV, Murad F. Nitration of succinyl-CoA:3-oxoacid CoA-transferase in rats after endotoxin administration. Proc Natl Acad Sci USA 98: 7146–7151, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Mayr M, Yusuf S, Weir G, Chung YL, Mayr U, Yin X, Ladroue C, Madhu B, Roberts N, De Souza A, Fredericks S, Stubbs M, Griffiths JR, Jahangiri M, Xu Q, Camm AJ. Combined metabolomic and proteomic analysis of human atrial fibrillation. J Am Coll Cardiol 51: 585–594, 2008 [DOI] [PubMed] [Google Scholar]
- 137. McGarry JD, Foster DW. Regulation of hepatic fatty acid oxidation and ketone body production. Annu Rev Biochem 49: 395–420, 1980 [DOI] [PubMed] [Google Scholar]
- 138. McGarry JD, Stark MJ, Foster DW. Hepatic malonyl-CoA levels of fed, fasted and diabetic rats as measured using a simple radioisotopic assay. J Biol Chem 253: 8291–8293, 1978 [PubMed] [Google Scholar]
- 139. McNally MA, Hartman AL. Ketone bodies in epilepsy. J Neurochem 121: 28–35, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Meertens LM, Miyata KS, Cechetto JD, Rachubinski RA, Capone JP. A mitochondrial ketogenic enzyme regulates its gene expression by association with the nuclear hormone receptor PPARalpha. EMBO J 17: 6972–6978, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Membrez M, Blancher F, Jaquet M, Bibiloni R, Cani PD, Burcelin RG, Corthesy I, Mace K, Chou CJ. Gut microbiota modulation with norfloxacin and ampicillin enhances glucose tolerance in mice. FASEB J 22: 2416–2426, 2008 [DOI] [PubMed] [Google Scholar]
- 142. Merritt ME, Harrison C, Sherry AD, Malloy CR, Burgess SC. Flux through hepatic pyruvate carboxylase and phosphoenolpyruvate carboxykinase detected by hyperpolarized 13C magnetic resonance. Proc Natl Acad Sci USA 108: 19084–19089, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Mitchell GA, Fukao T. Inborn errors of ketone body metabolism. In: The Online Metabolic and Molecular Bases of Inherited Diseases (OMMBID), edited by Beaudet A, Vogelstein B, Kinzler KW, Antonarakis S, Ballabio A. Columbus, OH: McGraw-Hill, 2000, part 10, chapt 102, p. 12 [Google Scholar]
- 144. Morris AA. Cerebral ketone body metabolism. J Inherit Metab Dis 28: 109–121, 2005 [DOI] [PubMed] [Google Scholar]
- 145. Neely JR, Rovetto MJ, Oram JF. Myocardial utilization of carbohydrate and lipids. Prog Cardiovasc Dis 15: 289–329, 1972 [DOI] [PubMed] [Google Scholar]
- 146. Newsholme P, Curi R, Gordon S, Newsholme EA. Metabolism of glucose, glutamine, long-chain fatty acids and ketone bodies by murine macrophages. Biochem J 239: 121–125, 1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Newsholme P, Gordon S, Newsholme EA. Rates of utilization and fates of glucose, glutamine, pyruvate, fatty acids and ketone bodies by mouse macrophages. Biochem J 242: 631–636, 1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, Pettersson S. Host-gut microbiota metabolic interactions. Science 336: 1262–1267, 2012 [DOI] [PubMed] [Google Scholar]
- 149. Niezen-Koning KE, Wanders RJ, Ruiter JP, Ijlst L, Visser G, Reitsma-Bierens WC, Heymans HS, Reijngoud DJ, Smit GP. Succinyl-CoA:acetoacetate transferase deficiency: identification of a new patient with a neonatal onset and review of the literature. Eur J Pediatr 156: 870–873, 1997 [DOI] [PubMed] [Google Scholar]
- 150. Niezen-Koning KE, Wanders RJ, Ruiter JP, Ijlst L, Visser G, Reitsma-Bierens WC, Heymans HS, Reijngoud DJ, Smit GP. Succinyl-CoA:acetoacetate transferase deficiency: identification of a new patient with a neonatal onset and review of the literature. Eur J Pediatr 156: 870–873, 1997 [DOI] [PubMed] [Google Scholar]
- 151. Odegaard JI, Chawla A. Alternative macrophage activation and metabolism. Annu Rev Pathol Mech Dis 6: 275–297, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Okuda Y, Kawai K, Ohmori H, Yamashita K. Ketone body utilization and its metabolic effect in resting muscles of normal and streptozotocin-diabetic rats. Endocr J 38: 245–251, 1991 [DOI] [PubMed] [Google Scholar]
- 153. Orii KE, Fukao T, Song XQ, Mitchell GA, Kondo N. Liver-specific silencing of the human gene encoding succinyl-CoA: 3-ketoacid CoA transferase. Tohoku J Exp Med 215: 227–236, 2008 [DOI] [PubMed] [Google Scholar]
- 154. Owen OE, Kalhan SC, Hanson RW. The key role of anaplerosis and cataplerosis for citric acid cycle function. J Biol Chem 277: 30409–30412, 2002 [DOI] [PubMed] [Google Scholar]
- 155. Owen OE, Morgan AP, Kemp HG, Sullivan JM, Herrera MG, Cahill GF., Jr Brain metabolism during fasting. J Clin Invest 46: 1589–1595, 1967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Pelletier A, Tardif A, Gingras MH, Chiasson JL, Coderre L. Chronic exposure to ketone bodies impairs glucose uptake in adult cardiomyocytes in response to insulin but not vanadate: the role of PI3-K. Mol Cell Biochem 296: 97–108, 2007 [DOI] [PubMed] [Google Scholar]
- 157. Phillips JW, Hird FJ. Ketogenesis in vertebrate livers. Comp Biochem Physiol B 57: 133–138, 1977 [DOI] [PubMed] [Google Scholar]
- 158. Pittman JG, Cohen P. The pathogenesis of cardiac cachexia. N Engl J Med 271: 403–409, 1964 [DOI] [PubMed] [Google Scholar]
- 159. Pryce JW, Weber MA, Heales S, Malone M, Sebire NJ. Tandem mass spectrometry findings at autopsy for detection of metabolic disease in infant deaths: postmortem changes and confounding factors. J Clin Pathol 64: 1005–1009, 2011 [DOI] [PubMed] [Google Scholar]
- 160. Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, Shen D, Peng Y, Zhang D, Jie Z, Wu W, Qin Y, Xue W, Li J, Han L, Lu D, Wu P, Dai Y, Sun X, Li Z, Tang A, Zhong S, Li X, Chen W, Xu R, Wang M, Feng Q, Gong M, Yu J, Zhang Y, Zhang M, Hansen T, Sanchez G, Raes J, Falony G, Okuda S, Almeida M, LeChatelier E, Renault P, Pons N, Batto JM, Zhang Z, Chen H, Yang R, Zheng W, Li S, Yang H, Wang J, Ehrlich SD, Nielsen R, Pedersen O, Kristiansen K, Wang J. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490: 55–60, 2012 [DOI] [PubMed] [Google Scholar]
- 161. Quant PA, Robin D, Robin P, Girard J, Brand MD. A top-down control analysis in isolated rat liver mitochondria: can the 3-hydroxy-3-methylglutaryl-CoA pathway be rate-controlling for ketogenesis? Biochim Biophys Acta 1156: 135–143, 1993 [DOI] [PubMed] [Google Scholar]
- 162. Quant PA, Tubbs PK, Brand MD. Glucagon activates mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase in vivo by decreasing the extent of succinylation of the enzyme. Eur J Biochem 187: 169–174, 1990 [DOI] [PubMed] [Google Scholar]
- 163. Rebrin I, Bregere C, Kamzalov S, Gallaher TK, Sohal RS. Nitration of tryptophan 372 in succinyl-CoA:3-ketoacid CoA transferase during aging in rat heart mitochondria. Biochemistry 46: 10130–10144, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Reed WD, Clinkenbeard D, Lane MD. Molecular and catalytic properties of mitochondrial (ketogenic) 3-hydroxy-3-methylglutaryl coenzyme A synthase of liver. J Biol Chem 250: 3117–3123, 1975 [PubMed] [Google Scholar]
- 165. Reed WD, Ozand PT. Enzymes of l-(+)-3-hydroxybutyrate metabolism in the rat. Arch Biochem Biophys 205: 94–103, 1980 [DOI] [PubMed] [Google Scholar]
- 166. Reichard GA, Jr, Owen OE, Haff AC, Paul P, Bortz WM. Ketone-body production and oxidation in fasting obese humans. J Clin Invest 53: 508–515, 1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Robinson AM, Williamson DH. Physiological roles of ketone bodies as substrates and signals in mammalian tissues. Physiol Rev 60: 143–187, 1980 [DOI] [PubMed] [Google Scholar]
- 168. Robinson AM, Williamson DH. Utilization of d-3-hydroxy[3–14C]butyrate for lipogenesis in vivo in lactating rat mammary gland. Biochem J 176: 635–638, 1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Rodriguez JC, Gil-Gomez G, Hegardt FG, Haro D. Peroxisome proliferator-activated receptor mediates induction of the mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase gene by fatty acids. J Biol Chem 269: 18767–18772, 1994 [PubMed] [Google Scholar]
- 170. Roe CR, Sweetman L, Roe DS, David F, Brunengraber H. Treatment of cardiomyopathy and rhabdomyolysis in long-chain fat oxidation disorders using an anaplerotic odd-chain triglyceride. J Clin Invest 110: 259–269, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Roe CR, Yang BZ, Brunengraber H, Roe DS, Wallace M, Garritson BK. Carnitine palmitoyltransferase II deficiency: successful anaplerotic diet therapy. Neurology 71: 260–264, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Rosca MG, Lemieux H, Hoppel CL. Mitochondria in the elderly: Is acetylcarnitine a rejuvenator? Adv Drug Deliv Rev 61: 1332–1342, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Roy M, Nugent S, Tremblay-Mercier J, Tremblay S, Courchesne-Loyer A, Beaudoin JF, Tremblay L, Descoteaux M, Lecomte R, Cunnane SC. The ketogenic diet increases brain glucose and ketone uptake in aged rats: a dual tracer PET and volumetric MRI study. Brain Res 1488: 14–23, 2012 [DOI] [PubMed] [Google Scholar]
- 174. Rudolph W, Schinz A. Studies on myocardial blood flow, oxygen consumption, and myocardial metabolism in patients with cardiomyopathy. Recent Adv Stud Card Struct Metab 2: 739–749, 1973 [PubMed] [Google Scholar]
- 175. Russell RR, 3rd, Taegtmeyer H. Changes in citric acid cycle flux and anaplerosis antedate the functional decline in isolated rat hearts utilizing acetoacetate. J Clin Invest 87: 384–390, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Russell RR, 3rd, Taegtmeyer H. Coenzyme A sequestration in rat hearts oxidizing ketone bodies. J Clin Invest 89: 968–973, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Russell RR, 3rd, Taegtmeyer H. Pyruvate carboxylation prevents the decline in contractile function of rat hearts oxidizing acetoacetate. Am J Physiol Heart Circ Physiol 261: H1756–H1762, 1991 [DOI] [PubMed] [Google Scholar]
- 178. Sandermann H, Jr, McIntyre JO, Fleischer S. Site-site interaction in the phospholipid activation of d-beta-hydroxybutyrate dehydrogenase. J Biol Chem 261: 6201–6208, 1986 [PubMed] [Google Scholar]
- 179. Satapati S, Sunny NE, Kucejova B, Fu X, He T, Mendez-Lucas A, Shelton JM, Perales JC, Browning JD, Burgess SC. Elevated TCA cycle function in the pathology of diet induced hepatic insulin resistance and fatty liver. J Lipid Res 53: 1080–1092, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Sato K, Kashiwaya Y, Keon CA, Tsuchiya N, King MT, Radda GK, Chance B, Clarke K, Veech RL. Insulin, ketone bodies, and mitochondrial energy transduction. FASEB J 9: 651–658, 1995 [DOI] [PubMed] [Google Scholar]
- 181. Saudubray JM, Specola N, Middleton B, Lombes A, Bonnefont JP, Jakobs C, Vassault A, Charpentier C, Day R. Hyperketotic states due to inherited defects of ketolysis. Enzyme 38: 80–90, 1987 [DOI] [PubMed] [Google Scholar]
- 182. Schneider JG, Yang Z, Chakravarthy MV, Lodhi IJ, Wei X, Turk J, Semenkovich CF. Macrophage fatty-acid synthase deficiency decreases diet-induced atherosclerosis. J Biol Chem 285: 23398–23409, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Schuler AM, Wood PA. Mouse models for disorders of mitochondrial fatty acid beta-oxidation. ILAR J 43: 57–65, 2002 [DOI] [PubMed] [Google Scholar]
- 184. Scofield RF, Brady PS, Schumann WC, Kumaran K, Ohgaku S, Margolis JM, Landau BR. On the lack of formation of l-(+)-3-hydroxybutyrate by liver. Arch Biochem Biophys 214: 268–272, 1982 [DOI] [PubMed] [Google Scholar]
- 185. Scrutton MC, Utter MF. Pyruvate carboxylase. IX. Some properties of the activation by certain acyl derivatives of coenzyme A. J Biol Chem 242: 1723–1735, 1967 [PubMed] [Google Scholar]
- 186. Sengupta S, Peterson T, Laplante M, Oh S, Sabatini D. mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature 468: 1100–1104, 2010 [DOI] [PubMed] [Google Scholar]
- 187. Seyfried TN, Kiebish MA, Marsh J, Shelton LM, Huysentruyt LC, Mukherjee P. Metabolic management of brain cancer. Biochim Biophys Acta 1807: 577–594, 2011 [DOI] [PubMed] [Google Scholar]
- 188. Shimazu T, Hirschey MD, Hua L, Dittenhafer-Reed KE, Schwer B, Lombard DB, Li Y, Bunkenborg J, Alt FW, Denu JM, Jacobson MP, Verdin E. SIRT3 deacetylates mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 and regulates ketone body production. Cell Metab 12: 654–661, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, Grueter CA, Lim H, Saunders LR, Stevens RD, Newgard CB, Farese RV, Jr, de Cabo R, Ulrich S, Akassoglou K, Verdin E. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 339: 211–214, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Skrha J, Kunesova M, Hilgertova J, Weiserova H, Krizova J, Kotrlikova E. Short-term very low calorie diet reduces oxidative stress in obese type 2 diabetic patients. Physiol Res 54: 33–39, 2005 [DOI] [PubMed] [Google Scholar]
- 191. Snorek M, Hodyc D, Sedivy V, Durisova J, Skoumalova A, Wilhelm J, Neckar J, Kolar F, Herget J. Short-term fasting reduces the extent of myocardial infarction and incidence of reperfusion arrhythmias in rats. Physiol Res 61: 567–574, 2013 [DOI] [PubMed] [Google Scholar]
- 192. Snyderman SE, Sansaricq C, Middleton B. Succinyl-CoA:3-ketoacid CoA-transferase deficiency. Pediatrics 101: 709–711, 1998 [DOI] [PubMed] [Google Scholar]
- 193. Soeters MR, Sauerwein HP, Faas L, Smeenge M, Duran M, Wanders RJ, Ruiter AF, Ackermans MT, Fliers E, Houten SM, Serlie MJ. Effects of insulin on ketogenesis following fasting in lean and obese men. Obesity (Silver Spring) 17: 1326–1331, 2009 [DOI] [PubMed] [Google Scholar]
- 194. Srere PA, Foster DW. On the proposed relation of citrate enzymes to fatty acid synthesis and ketosis in starvation. Biochem Biophys Res Commun 26: 556–561, 1967 [DOI] [PubMed] [Google Scholar]
- 195. Srivastava S, Kashiwaya Y, King MT, Baxa U, Tam J, Niu G, Chen X, Clarke K, Veech RL. Mitochondrial biogenesis and increased uncoupling protein 1 in brown adipose tissue of mice fed a ketone ester diet. FASEB J 26: 2351–2362, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Stanley WC, Meadows SR, Kivilo KM, Roth BA, Lopaschuk GD. β-Hydroxybutyrate inhibits myocardial fatty acid oxidation in vivo independent of changes in malonyl-CoA content. Am J Physiol Heart Circ Physiol 285: H1626–H1631, 2003 [DOI] [PubMed] [Google Scholar]
- 197. Stern JR, Coon MJ, Del Campillo A. Enzymes of fatty acid metabolism. III. Breakdown and synthesis of beta-keto fatty acids. J Biol Chem 221: 1–14, 1956 [PubMed] [Google Scholar]
- 198. Stern JR, Coon MJ, Del Campillo A, Schneider MC. Enzymes of fatty acid metabolism. IV. Preparation and properties of coenzyme A transferase. J Biol Chem 221: 15–31, 1956 [PubMed] [Google Scholar]
- 199. Sultan AM. d-3-hydroxybutyrate metabolism in the perfused rat heart. Mol Cell Biochem 79: 113–118, 1988 [DOI] [PubMed] [Google Scholar]
- 200. Sunny NE, Satapati S, Fu X, He TT, Mehdibeigi R, Spring-Robinson C, Duarte J, Potthoff MJ, Browning JD, Burgess SC. Progressive adaptation of hepatic ketogenesis in mice fed a high-fat diet. Am J Physiol Endocrinol Metab 298: E1226–E1235, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Suzuki J, Shen WJ, Nelson BD, Selwood SP, Murphy GM, Jr, Kanehara H, Takahashi S, Oida K, Miyamori I, Kraemer FB. Cardiac gene expression profile and lipid accumulation in response to starvation. Am J Physiol Endocrinol Metab 283: E94–E102, 2002 [DOI] [PubMed] [Google Scholar]
- 202. Sweetman L, Weyler W, Nyhan WL, de Cespedes C, Loria AR, Estrada Y. Abnormal metabolites of isoleucine in a patient with propionyl-CoA carboxylase deficiency. Biomed Mass Spectrom 5: 198–207, 1978 [DOI] [PubMed] [Google Scholar]
- 203. Taegtmeyer H, Hems R, Krebs HA. Utilization of energy-providing substrates in the isolated working rat heart. Biochem J 186: 701–711, 1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Taegtmeyer H, McNulty P, Young ME. Adaptation and maladaptation of the heart in diabetes: Part I: general concepts. Circulation 105: 1727–1733, 2002 [DOI] [PubMed] [Google Scholar]
- 205. Taggart AK, Kero J, Gan X, Cai TQ, Cheng K, Ippolito M, Ren N, Kaplan R, Wu K, Wu TJ, Jin L, Liaw C, Chen R, Richman J, Connolly D, Offermanns S, Wright SD, Waters MG. (D)-beta-Hydroxybutyrate inhibits adipocyte lipolysis via the nicotinic acid receptor PUMA-G. J Biol Chem 280: 26649–26652, 2005 [DOI] [PubMed] [Google Scholar]
- 206. Tardif A, Julien N, Pelletier A, Thibault G, Srivastava AK, Chiasson JL, Coderre L. Chronic exposure to beta-hydroxybutyrate impairs insulin action in primary cultures of adult cardiomyocytes. Am J Physiol Endocrinol Metab 281: E1205–E1212, 2001 [DOI] [PubMed] [Google Scholar]
- 207. Thomas LK, Ittmann M, Cooper C. The role of leucine in ketogenesis in starved rats. Biochem J 204: 399–403, 1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Thompson GN, Hsu BY, Pitt JJ, Treacy E, Stanley CA. Fasting hypoketotic coma in a child with deficiency of mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase. N Engl J Med 337: 1203–1207, 1997 [DOI] [PubMed] [Google Scholar]
- 209. Thumelin SF, Girard J, Pegorier JP. Developmental changes in mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase gene expression in rat liver, intestine and kidney. Biochem J 292: 493–496, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Tildon JT, Cornblath M. Succinyl-CoA: 3-ketoacid CoA-transferase deficiency. A cause for ketoacidosis in infancy. J Clin Invest 51: 493–498, 1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Tremaroli V, Backhed F. Functional interactions between the gut microbiota and host metabolism. Nature 489: 242–249, 2012 [DOI] [PubMed] [Google Scholar]
- 212. Tunaru S, Kero J, Schaub A, Wufka C, Blaukat A, Pfeffer K, Offermanns S. PUMA-G and HM74 are receptors for nicotinic acid and mediate its anti-lipolytic effect. Nat Med 9: 352–355, 2003 [DOI] [PubMed] [Google Scholar]
- 213. Turko IV, Marcondes S, Murad F. Diabetes-associated nitration of tyrosine and inactivation of succinyl-CoA:3-oxoacid CoA-transferase. Am J Physiol Heart Circ Physiol 281: H2289–H2294, 2001 [DOI] [PubMed] [Google Scholar]
- 214. Turnbaugh PJ, Backhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 3: 213–223, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444: 1027–1031, 2006 [DOI] [PubMed] [Google Scholar]
- 216. Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med 1: 6ra14, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Valera A, Pelegrin M, Asins G, Fillat C, Sabater J, Pujol A, Hegardt FG, Bosch F. Overexpression of mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase in transgenic mice causes hepatic hyperketogenesis. J Biol Chem 269: 6267–6270, 1994 [PubMed] [Google Scholar]
- 218. Vanoverschelde JL, Wijns W, Kolanowski J, Bol A, Decoster PM, Michel C, Cogneau M, Heyndrickx GR, Essamri B, Melin JA. Competition between palmitate and ketone bodies as fuels for the heart: study with positron emission tomography. Am J Physiol Heart Circ Physiol 264: H701–H707, 1993 [DOI] [PubMed] [Google Scholar]
- 219. Veech RL. The therapeutic implications of ketone bodies: the effects of ketone bodies in pathological conditions: ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins Leukot Essent Fatty Acids 70: 309–319, 2004 [DOI] [PubMed] [Google Scholar]
- 220. Vila-Brau A, Sousa-Coelho ALD, Mayordomo C, Haro D, Marrero PF. Human HMGCS2 regulates mitochondrial fatty acid oxidation and FGF21 expression in HepG2 cell line. J Biol Chem 286: 20423–20430, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Wang P, Tate JM, Lloyd SG. Low carbohydrate diet decreases myocardial insulin signaling and increases susceptibility to myocardial ischemia. Life Sci 83: 836–844, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Wang X, Levi AJ, Halestrap AP. Substrate and inhibitor specificities of the monocarboxylate transporters of single rat heart cells. Am J Physiol Heart Circ Physiol 270: H476–H484, 1996 [DOI] [PubMed] [Google Scholar]
- 223. Wang Y, Peng F, Tong W, Sun H, Xu N, Liu S. The nitrated proteome in heart mitochondria of the db/db mouse model: Characterization of nitrated tyrosine residues in SCOT. J Proteome Res 9: 4254–4263, 2010 [DOI] [PubMed] [Google Scholar]
- 224. Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, Wu Y, Schauer P, Smith JD, Allayee H, Tang WH, DiDonato JA, Lusis AJ, Hazen SL. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 472: 57–63, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Ward Platt M, Deshpande S. Metabolic adaptation at birth. Semin Fetal Neonatal Med 10: 341–350, 2005 [DOI] [PubMed] [Google Scholar]
- 226. Webber RJ, Edmond J. Utilization of L(+)-3-hydroxybutyrate, D(-)-3-hydroxybutyrate, acetoacetate, and glucose for respiration and lipid synthesis in the 18-day-old rat. J Biol Chem 252: 5222–5226, 1977 [PubMed] [Google Scholar]
- 227. Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, Hu C, Wong FS, Szot GL, Bluestone JA, Gordon JI, Chervonsky AV. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature 455: 1109–1113, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Wentz AE, d'Avignon DA, Weber ML, Cotter DG, Doherty JM, Kerns R, Nagarajan R, Sambandam N, Crawford PA. Adaptation of myocardial substrate metabolism to a ketogenic nutrient environment. J Biol Chem 285: 24447–24456, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Williamson DH, Bates MW, Page MA, Krebs HA. Activities of enzymes involved in acetoacetate utilization in adult mammalian tissues. Biochem J 121: 41–47, 1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Williamson DH, Lund P, Krebs HA. The redox state of free nicotinamide-adenine dinucleotide in the cytoplasm and mitochondria of rat liver. Biochem J 103: 514–527, 1967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Wolfrum C, Asilmaz E, Luca E, Friedman JM, Stoffel M. Foxa2 regulates lipid metabolism and ketogenesis in the liver during fasting and in diabetes. Nature 432: 1027–1032, 2004 [DOI] [PubMed] [Google Scholar]
- 232. Wu X, Ma C, Han L, Nawaz M, Gao F, Zhang X, Yu P, Zhao C, Li L, Zhou A, Wang J, Moore JE, Millar BC, Xu J. Molecular characterisation of the faecal microbiota in patients with type II diabetes. Curr Microbiol 61: 69–78, 2010 [DOI] [PubMed] [Google Scholar]
- 233. Yamasaki M, Hasegawa S, Suzuki H, Hidai K, Saitoh Y, Fukui T. Acetoacetyl-CoA synthetase gene is abundant in rat adipose, and related with fatty acid synthesis in mature adipocytes. Biochem Biophys Res Commun 335: 215–219, 2005 [DOI] [PubMed] [Google Scholar]
- 234. Yan J, Young ME, Cui L, Lopaschuk GD, Liao R, Tian R. Increased glucose uptake and oxidation in mouse hearts prevent high fatty acid oxidation but cause cardiac dysfunction in diet-induced obesity. Circulation 119: 2818–2828, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Yang SY, He XY, Schulz H. Fatty acid oxidation in rat brain is limited by the low activity of 3-ketoacyl-coenzyme a thiolase. J Biol Chem 262: 13027–11303, 1987 [PubMed] [Google Scholar]
- 236. Yang X, Cheng B. Neuroprotective and anti-inflammatory activities of ketogenic diet on MPTP-induced neurotoxicity. J Mol Neurosci 42: 145–153, 2010 [DOI] [PubMed] [Google Scholar]
- 237. Yao J, Chen S, Mao Z, Cadenas E, Brinton R. 2-Deoxy-d-glucose treatment induces ketogenesis, sustains mitochondrial function, and reduces pathology in female mouse model of Alzheimer's disease. PLoS One 6: e21788, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Yoo H, Stephanopoulos G, Kelleher JK. Quantifying carbon sources for de novo lipogenesis in wild-type and IRS-1 knockout brown adipocytes. J Lipid Res 45: 1324–1332, 2004 [DOI] [PubMed] [Google Scholar]
- 239. Young ME, McNulty P, Taegtmeyer H. Adaptation and maladaptation of the heart in diabetes: Part II: potential mechanisms. Circulation 105: 1861–1870, 2002 [DOI] [PubMed] [Google Scholar]
- 240. Zammit VA, Beis A, Newsholme EA. The role of 3-oxo acid-CoA transferase in the regulation of ketogenesis in the liver. FEBS Lett 103: 212–215, 1979 [DOI] [PubMed] [Google Scholar]
- 241. Zhang H, DiBaise JK, Zuccolo A, Kudrna D, Braidotti M, Yu Y, Parameswaran P, Crowell MD, Wing R, Rittmann BE, Krajmalnik-Brown R. Human gut microbiota in obesity and after gastric bypass. Proc Natl Acad Sci USA 106: 2365–2370, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Zhang WW, Churchill S, Churchill P. Developmental regulation of d-β-hydroxybutyrate dehydrogenase in rat liver and brain. FEBS Lett 256: 71–74, 1989 [DOI] [PubMed] [Google Scholar]
- 243. Zhang Z, Tan M, Xie Z, Dai L, Chen Y, Zhao Y. Identification of lysine succinylation as a new post-translational modification. Nat Chem Biol 2011: 58–63, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Zou Z, Sasaguri S, Rajesh KG, Suzuki R. dl-3-Hydroxybutyrate administration prevents myocardial damage after coronary occlusion in rat hearts. Am J Physiol Heart Circ Physiol 283: H1968–H1974, 2002 [DOI] [PubMed] [Google Scholar]