

Abstract
A DTPA-Gd containing polymer was grown in the interior of a heat shock protein cage resulting in T1 particle relaxivities of 4,200mM-1 sec-1 for the 12nm particle. Relaxivity parameters were determined and this analysis suggests that the rotational correlation time has been optimized while the water exchange life time is slower than optimal. This synthetic approach holds much promise for the development of next generation contrast agents and this report will aid in their design.
Designing a magnetic resonance (MR) contrast agent (CA) on the scaffold of a protein cage holds the promise for increasing image contrast of target tissues. While the high resolution of MR affords excellent anatomical detail, the ability to detect low copy number tissues is limited by the low sensitivity of the technique. CA are used to increase the sensitivity of MR to tissues of interest by accelerating the NMR relaxation rate of water proteins in the vicinity of the target tissue.[1-4] Chelated gadolinium (Gd3+) ions are commonly used as T1 weighted CA to increase the relaxation rate of water protons and this effect is called paramagnetic relaxation enhancement (PRE). The utility of CA is currently limited by the inability to detect nanomolar concentrations of CA in vivo. Here, the capability to image a target is limited by the sensitivity of detecting the location of the CA rather than by the resolution of MR. Designing contrast agents that are more sensitive to MR scanners will allow for the detection of smaller and lower copy number targets.
Protein cages, such as viral capsids, heat shock proteins (HSP), and ferritin-like proteins are good candidates as scaffolds to which chelated Gd groups can be anchored resulting in designer contrast agents with near ideal relaxivity properties.[5-10] The large surface area and interior volume of protein cages can accommodate large numbers of Gd ions that directly increases the particle relaxivity of these agents. Particle relaxivity is defined as the relaxivity at a particle concentration of 1 mM. By anchoring the Gd chelate to the protein cage the mobility of the Gd ion is decreased, which is advantageous for efficient relaxation, and therefore the relaxation rate for each Gd or the ionic relaxivity (relaxivity per mM Gd ion) is increased.[8, 10] Finally, targeting groups can be presented on the exterior of the protein cage to direct localization of the protein cages to receptors of interest resulting in novel MR probes to increase conspicuity of tissues of interest.[11-25] We have developed a protein cage - branched polymer hybrid particle containing a high density of Gd ions inside the cage, that displays extremely efficient relaxivity properties. This class of particles holds great promise for further development as targeted MR probes.
There are a few parameters that must be optimized with regard to development of Gd based contrast agents.[1-4] The first consideration is that Gd is toxic and must therefore be used in the form of a tightly associated metal ion.[26, 27] Since the Gd based relaxivity is a spin-lattice effect, direct water interaction with the Gd ion is necessary through an inner sphere mechanism. The number of metal-bound water molecules that can occupy a coordination sphere of Gd is referred to as “q” and in general, as q increases the stability of the chelator-metal interaction decreases. Therefore a balance must be struck between increasing the number of water coordination sites and the overall stability of the metal-chelator interaction.
Another parameter that must be optimized is the residence time of the Gd-bound water, referred to as τM. It is important for the water to remain bound to Gd long enough for dipole-dipole interaction and relaxation to occur. However, when τM is too high, and the water molecule remains bound to Gd long after the spins have relaxed, the result is an effective decrease in the “duty cycle” of the Gd ion resulting in a decrease of the overall relaxivity. The τM value can be modulated by varying the chemical nature of the metal chelator. The final parameter that is important for efficient PRE is the rotational correlation time of the Gd ion (τR). Larger Gd complexes result in greater τR values which yields more efficient PRE. Figure 1 shows the parameters that effect PRE.
Figure 1.
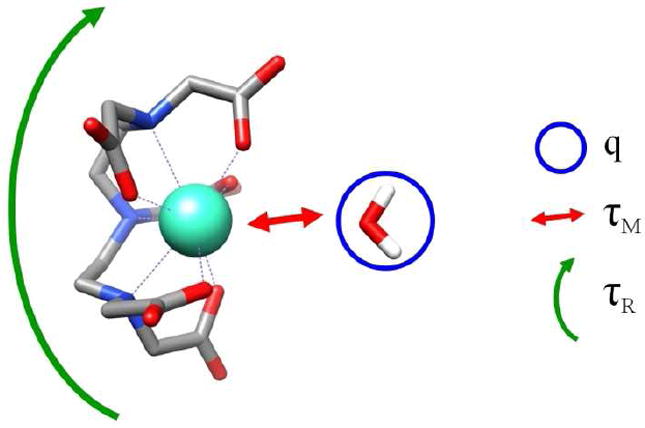
An schematic of DTPA-Gd and the important physical parameters that effect the relaxivity; including the number of water molecules that directly interact with the Gd ion (q), the exchange lifetime of the metal bound water molecule (τM), and the rotational correlation time of the Gd chelate (τR).
To date several groups have reported the utilization of viral capsids or other protein cages structures as MRI contrast agents. In the first report of this kind, Allen and coworkers utilized the endogenous metal binding site in the plant virus Cowpea chlorotic mottle virus (CCMV) and bound Gd3+ ions to endogenous Ca-binding sites in the capsid.[5] Since the metal ion in this capsid is situated at the interface of three subunits it is likely that the τR for Gd is similar to τR of the entire cage therefore this Gd-protein cage construct should exhibit near ideal τR values. This construct has a relatively weak affinity for the Gd ion (Kd = 31 μM), which along with analysis of the crystal structure of the metal binding pocket both suggest that q is likely to be greater than one. Recently we performed fitting of the CCMV-Gd complex r1 NMRD profile to the Solomon-Bloembergen-Morgan (SBM) analytical model for relaxivity returned values of q = 4, τM = 3 · 10-9 seconds, τR > 10-7 seconds, all of which are highly desirable (cf. Supporting Information). Not surprisingly, the optimal properties of this construct resulted in extremely high ionic relaxivities (r1 = 202 mM-1 sec-1, 62 MHz). Unfortunately the low Gd affinity of this construct made the clinical applicability impractical.
Several groups have developed a range of next generation constructs which can now be divided into three categories, 1) endogenous metal binding sites[5], 2) genetic insertion of a metal binding peptide[8], 3) chemical attachment of small molecule chelates[6-10]. Of these three categories the first two are less significant from a clinical standpoint due to their lower Gd binding affinities. The third category is the most common approach with three groups reporting similar results. Anderson et al attached DTPA-Gd to lysine residues on the surface of the MS2 capsid (MS2 DTPA ITC)[6]. Despite the relatively low ionic relaxivity of this construct the high density of Gd labeling resulted in the highest particle relaxivity for constructs with sufficient Gd binding stability and is approximately three orders of magnitude higher than the small molecule contrast agent (Gd-DTPA). Prasuhn et al produced a similar construct by using click chemistry to attach DOTA-Gd to CPMV and Q-beta protein cages and this scheme also produced a construct with a relatively low ionic relaxivity (r1 = 16 mM-1 sec-1, 62 MHz) however high levels of Gd ions were attached.[9] A CCMV-DOTA-Gd construct and a MS2 Bis (HOPO) TAM constructs both have higher ionic relaxivities (r1 = 46 mM-1 sec-1, r1 = 31 mM-1 sec-1 respectively, both at 62 MHz) but lower density of Gd labeling resulted in lower particle relaxivities.[7]
To optimize the relaxivity properties of Gd based contrast agents it is helpful to have quantitative information of the parameters important to PRE. Since τR, τM and q have different dependencies on the external magnetic field, it is possible to extract information of these values from a plot of relaxivities obtained for one contrast agent taken over a range of field strengths. These plot are referred to as nuclear magnetic resonance dispersion (NMRD) plots and they can be fit to the Solomon-Bloembergen-Morgan (SBM) analytical model for PRE and values for τR, τM and q can be obtained.[28]
Francis et al labeled the MS2 viral capsids on the interior surface and on the exterior surface with a novel Gd chelator.[7] A fit of their NMRD data with a modified SBM model suggest that the τM for this construct is near optimal and q is greater than one.[10, 29] However local mobility in the chelator attachment scheme partially negated the ideal rotational properties of this large construct. Furthermore, these authors reported a relaxivity difference between the interior and exterior labeled versions of MS2 construct and attributed this to a difference in the rigidity of the these two anchoring schemes. The τR values were lower for the interior labeled construct that has higher relaxivity values suggesting local mobility can dampen the ideal rotational effects of protein cages. These reports of protein cage – Gd based CA have inspired us to develop a novel protein cage branched polymer hybrid system with T1 ionic relaxivities of 25mM-1 sec-1 and that contain up to 159 Gd per particle. These CA have high particle relaxivities (4,200mM-1 sec-1) and have extremely high relaxivity densities (r1 / particle volume).
A strategy for the synthesis of these protein cage – branched polymer hybrids has previously been reported and consisted of synthesizing a branched polymer structure, via azide - alkyne “click” cycloaddition reactions.[30] The branched polymer projects into the interior cavity of the HSP cage from Methanococcus jannaschii, Figure 2, and spans the cavity of the protein cage.[31] Recombinant Hsp self-assembles in an E. coli expression system from 24 identical protein subunits forming a spherical protein cage of 12 nm exterior diameter and 6.5 nm interior diameter.[32] The Hsp cage is relatively porous and robust making it a good model system for internal modification.[30, 33, 34] The presence of eight 3 nm pores at the 3-fold and six 1.7 nm pores at the 4-fold axes allows free access of small molecules to the interior cavity. The cage is stable up to ~60 °C and in the pH range of 5-8. The G41C mutant of Hsp introduces a unique cysteine residue at position 41 resulting in an internally directed initiation point for polymer growth.[30, 33, 34]
Figure 2.
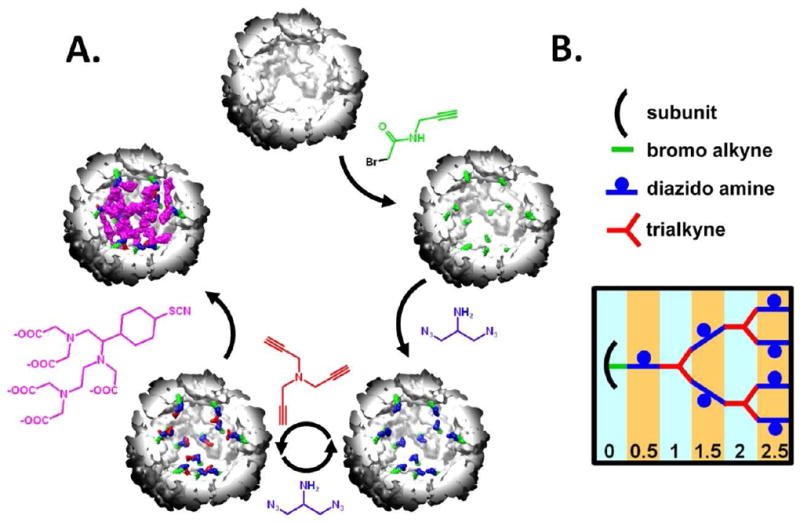
(A) The synthetic scheme to generate a branched polymer inside the Hsp cage and this synthesis is shown to scale with a cut-away view of Hsp. Hsp, G0.0, G0.5, G1.0 and G1.5 with DTPA are shown starting at the top and moving clockwise. First, an alkyne containing(green) molecule is anchored to the interior of the protein cage. Next a diazido amine compound(blue) is clicked to the alkyne molecule on the interior of the cage. This is followed by a series of click reactions with the trialkyne(red) and diazido amine molecules and these reactions create a series of increasing generations of the Hsp – branched polymer hybrid. Next, the introduced amines on the diazido amine compound were reacted with an amine reactive form of the DTPA-Gd chelate. (B) This represents an idealized growth of the polymer at each generation and assumes 100% reaction yields and no crosslinking between various polymer branches
A branched polymer was grown in a stepwise fashion within the internal cavity of the Hsp cage (Figure 2A). The polymer was initiated by reaction of N-propargyl bromoacetamide with the genetically inserted cysteine, located on the interior of the protein cage. This alkyne derivative is referred to as generation zero (G0.0). The exposed alkyne was subsequently reacted with 2-azido-1-azidomethyl ethylamine (diazido-amine) via a Cu(I) catalyzed ‘click’ reaction to yield G0.5. Exposed azide functional groups on G0.5 were subsequently ‘clicked’ with tripropargyl amine (trialkyne) to generate a branched structure (G1.0). Iterative stepwise reactions with di-azido-amine and tri-alkyne were then undertaken to produce generations G1.5, G2.0, G2.5, G3.0 and G3.5.
These protein cage - branched polymer hybrid materials (Hsp-BP) are thermally stable, to >120°C, due to the internal cross-linking of the branched polymer. Every generation of this branched polymer adds additional derivatizable amine functional groups, up to 200 on the fully crosslinked cage. After the polymer synthesis the Hsp-BP cages maintain their native shape and size distribution.[30] This increased stability dramatically expands the synthetic range and the potential in vivo utility of these biological templates. Surface exposed amines on lysine residues were reacted, in one preparation, with N-hydroxysuccinimide acetate. This reaction “passivated” the endogenous amines and in doing so we hoped to decouple the relaxivity properties of Gd chelates that were attached to either the endogenous amines or to the amines introduced during the polymer growth.
The diazido-amine, which is incorporated into the branched polymer, contains a primary amine which has been shown to be chemically addressable after polymer formation on the interior of a protein cage.[30] After the polymer was grown in the cage an amine reactive form of the DTPA-Gd contrast agent (2-(4-isothiocyanatobenzyl)-DTPA) was reacted with G0, G0.5, G1.5, G2.5 and G3.5. Increased concentrations of the Gd3+ were detected with increasing generations up to a maximum of 159 Gd3+ per cage at G2.5 (Figure 4a). This data is in general agreement with a similar branched polymer preparation that was labeled with a fluorescein isothiocyanate and produced 208 labels per cage at G2.5.[30] In both cases the maximum labeling was observed at G2.5 with no increase in labeling at G3.5. Steric crowding, shown in the modeled structures (Figure 3.), is the mostly likely explanation for the loss of amine reactivity in G3.5.
Figure 4.
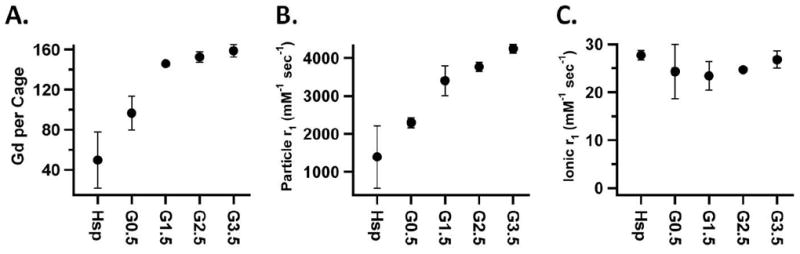
Gd ions per Hsp cage for the cage hybrid construct (A), particle r1 relaxivity (B) and ionic r1 relaxivity (C) for the hybrid system at 31MHz (0.73 T) for generations G0.0 to G3.5. Calculations of Gd ions per Hsp cage was based on ICP-MS data (Gd3+) and BCA protein quantitation (Hsp). The three plots were produced by averaging the data from Batch 1 and 2 of the non passivated constructs.
Figure 3.
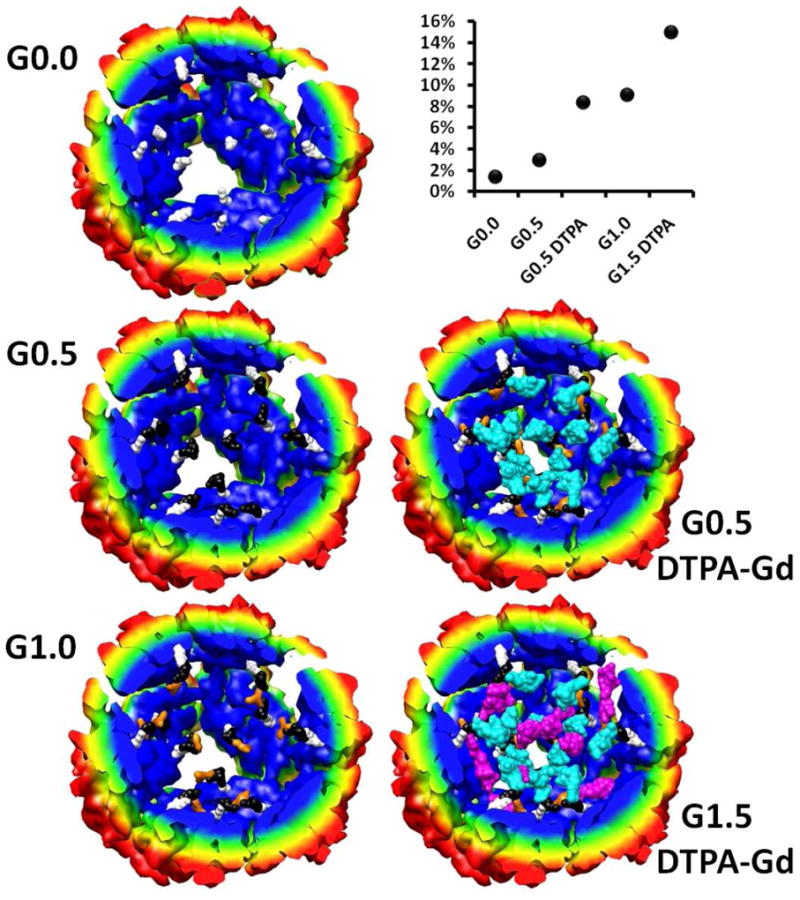
Structural models of branched polymer inside Hsp for generations G0.0, G0.5, G1.0 shown on the left. Shown on the right are models of G0.5 and G1.5 with DTPA-Gd coupled through the primary amine of the branched polymer. Here the bromo alkyne molecule is shown in white, the diazido amine (black), trialkyne (orange), DTPA-Gd attached G0.5 (cyan) and DTPA-Gd attached G1.5 (purple). The degree of polymerization and DTPA-Gd labeling was based on mass spectrometry data. The percent of the interior volume of Hsp that is occupied by the polymer and Gd-DTPA is plotted as a function of polymer growth points on the upper right corner of the figure. (cf. Supporting Information for details on the construction of this model).
To aid in the understanding of the polymer packing in the interior cavity of the protein cage a physical model of the branched polymer structure was constructed within structurally defined Hsp cage (pdb file: 1shs) for generations G0.0, G0.5 (± DTPA-Gd), G1.0 and G1.5 with DTPA-Gd, Figure 3. The degree of polymer growth was determined from mass spectrometry and this data guided the creation of the branched polymer network model (cf. Supporting Information). While only 15% of the interior volume of the cage is occupied at G1.5 with DTPA-Gd, visual inspection shows that the interior is significantly crowded.
Relaxivity measurements were collected on DTPA-Gd coupled to Hsp-BP for generations G0.0, G0.5, G1.5, G2.5 and G3.5. The particle relaxivity increased as additional Gd was loaded into each successive generation to a maximum value of 4,200 sec-1 mM-1 (31MHz), Figure 4b. This corresponds to the second highest value for a protein cage construct with clinically relevant binding. Dramatic changes in the ionic relaxivity were not observed across the generations and the generational average value was 25mM-1 sec-1 (31MHz), that was above average for protein cage constructs and is much higher than DTPA-Gd which has a r1 = 4mM-1 sec-1 (42MHz), Figure 4c.
We were interested in determining the parameters that influence relaxivity (τM, τR and q) and therefore r1 NMRD profiles of the branched polymer constructs were collected at field strengths ranging from 4 to 62 MHz. Twenty individual NMRD profiles were collected containing two preparations of generations G0.0, G0.5, G1.5, G2.5 and G3.5 of the passivated and non-passivated hybrid constructs.
The individual NMRD profiles were similar for all preparations (cf. Supporting Information) indicating that no drastic changes in the relaxivity occurred as the chelator was attached to increasing generations of the polymer. This also suggests that there is not a drastic difference between the relaxivity properties of the Gd chelates that are anchored to endogenous lysine residues or to the polymer backbone. Therefore, the twenty NMRD profiles were averaged (Figure 5). This averaged NMRD profile is indicative of Gd attached to a relatively large species with a peak at 31 MHz, consistent with the intact Hsp-branched polymer conjugate. By fitting this profile to the SBM model for Gd based PRE values for τM, τR and q were obtained (τM = 5.6 · 10-7 seconds, τR = 7.6 · 10-9 seconds and q = 1.1) (cf. Supporting Information).
Figure 5.
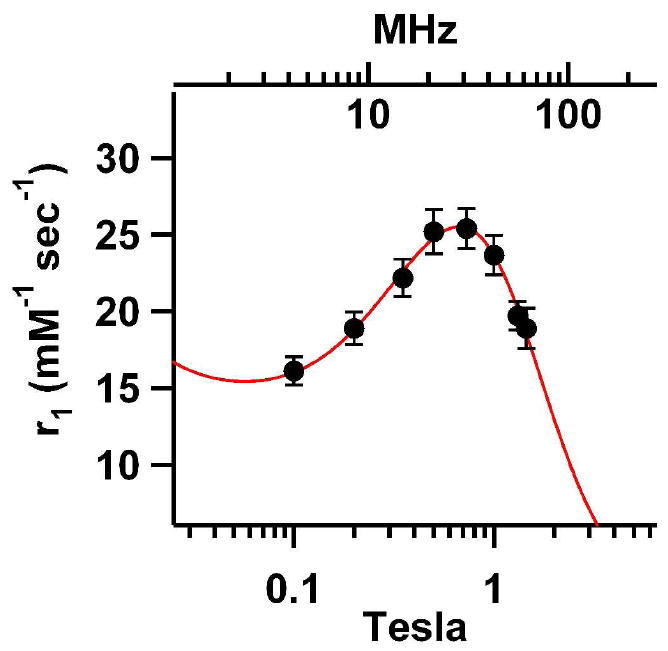
NMRD profile for the cage polymer hybrid construct. Twenty measurements were averaged for each data point consisting of two prepartations of both passivated and unpassivated samples for the following generations G0.0, G0.5, G1.5, G2.5, G3.5. The error bars represent 1 standard deviation. A SBM fit was performed on these data resulting in τM = 5.6 · 10-7 seconds, τR = 7.6 · 10-9 seconds and q = 1.1.
The experimentally determined τR value (7.6 · 10-9 seconds) was lower than what was expected for the entire Hsp cage, which can be calculated from the Einstein Stokes equation (τR = 2.2 · 10-7 seconds) (cf. Supporting Information). This result is consistent with the idea that the linker connecting the metal to the cage is flexible. However, by tethering the chelated metal to the cage the mobility is significantly decreased when compared to the τR obtained for DTPA-Gd alone (τR ≈ 7 · 10-11 seconds).[35] The SBM model predicts that increasing the τR values above 10-8 seconds does not have a dramatic effect on PRE (cf. Supporting Information). Therefore the τR for the hybrid polymer-protein system is within an optimal range for these materials. This effect is shown Figure 6A-B where no significant difference in PRE is observed when τR is longer than the value determined by the SBM fit to the experimental data. However when τR is set to a smaller value a reduction in PRE is predicted by the SMB model.
Figure 6.
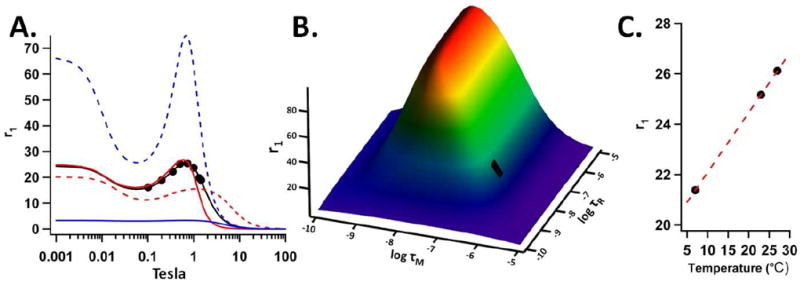
(A) The experimental relaxivity data (black points) and a SBM fit (solid black line) to these data. NMRD profiles with the q and τM equal to the fit values and a lower τR value (τR = 7.6 · 10-10, red dotted line) and higher value (τR = 7.6 · 10-8, red solid line). Also profiles with the q and τR equal to the fit values and a lower τM value (τM = 7.6 · 10-10, blue dotted line) and higher value (τR = 7.6 · 10-8, blue solid line). (B) A plot of the relaxivity as calculated by the SBM model as a function of τR and τM at 0.73Tesla (31MHz) and q = 1.1. The black rectangle indicates the region corresponding to τR and τM values returned by the SBM fit to the experimental data. (C) A plot of r1 vs. temperature for the Hsp hybrid construct at 0.5Tesla (21MHz). Each experimental data point in (A) and (C) was produced by averaging both batches of the passivated and non-passivated preparations for G0.0, G0.5, G1.5, G2.5 and G3.5 and r1 is the ionic relaxivity with units of mM-1 sec-1.
The fit of the NMRD data for the Hsp-BP-Gd-DTPA construct indicates a water exchange lifetime of 5.6 · 10-7 seconds. By comparison to DTPA-Gd (τM = 2.4 · 10-7) the water exchange for the chelate is significantly slower when docked on the polymer-cage hybrid system.[36] The SBM model predicts that the water exchange lifetime of the Hsp-BP-Gd-DTPA is not optimal (too long) and significant PRE increases would result from a faster water exchange rate. When the τM value is set to a larger value the PRE is reduced and as the water exchange rate is increased a dramatic increase in PRE is predicted by the SBM model.
Water exchange lifetimes for many Gd chelators are longer than the optimal value for maximum PRE as predicted by analytical models for PRE and this is the case for DTPA-Gd. The water exchange rate can be increased by raising the temperature of the sample and this should lead to a positive effect on PRE. However heating the sample also results in a shorter rotational correlation time, which should negatively impact PRE. Since heating the sample has these two opposite effects on PRE, it was possible to ascertain the limiting factor for efficient PRE, τR or τM, in the Hsp-BP-Gd-DTPA construct. As shown in Figure 6C, a direct correlation between relaxivity and temperature was observed for the Hsp-BP-Gd-DTPA construct, suggesting that the positive effect of a shorter exchange lifetime out-competed the negative effect of a shorter rotational correlation time. The analysis of the temperature dependence is in agreement with the SBM fitting results since both analyses suggest the τR is at an optimal value while the τM is higher than the ideal value for maximum PRE.
Although drastic changes in the NMRD profiles were not observed across generations there were slight differences. The most obvious difference was that the peak centered at 31MHz was shifted slightly to the left as the generations increased (cf. Supporting Information). Therefore, we analyzed the SBM fits to individual generations to glean additional information from this system. Both τM and q remained relatively constant across all generations, Figure 7A,B. This suggests that there is not a drastic decrease in the permeability of water through the protein cage polymer hybrid construct with increasing polymer generation.
Figure 7.
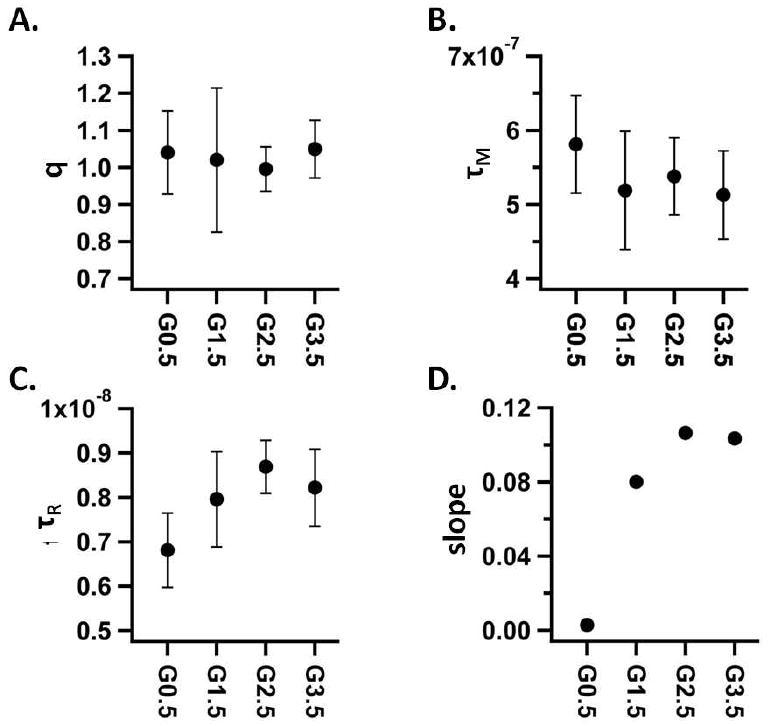
Results of SBM fits of individual generations, showing the generational change for q (A), τM (B), τR (C) and the slope of a linear fit to a plot of r1 vs. temperature (D) for generations G0.5, G1.5, G2.5 G3.5. Four experimental points were averaged for each data point shown in plots A-C, comprised of two experimental points from the non-passivated preparations and two from passivated preparations. The error bars are ± one standard deviation for plots A-C.
An increase in τR was observed as the polymeric network was grown (Figure 7C) through increasing generations. Previous studies suggest that local mobility in the linker has a large role in determining the relaxivity of protein cage – Gd chelate systems.[8, 10] The generational increase in τR shown in Figure 7C could result from decreased local mobility due to increasing rigidity of the polymer imparted by cross-linking at higher generations. It is known that the hybrid protein cage branched polymer constructs become more stable to heat treatment at higher generations. It is possible that local rigidity of the polymer coincides with the global increase in stability of the higher generations.[30]
Furthermore, the relaxivity showed an altered dependence on temperature across the generations. A positive dependence on temperature holds true for all generations however, the slope of this positive dependence (r1 vs. temperature) increased from G0.0 to a plateau at G2.5 (Figure 7D) (cf. Supporting Information). Two physical models could explain this observation. First, since τR is higher for higher generations, heating the lower generation samples may cause τR to decrease past a critical point where the relaxivity is significantly decreased. In this scenario the higher generation samples may have a τR value that is further up the plateau in the r1 vs. τR plot such that a reduction in τR due to heating would not significantly decrease the relaxivity. A second possible explanation for this change of slope (r1 vs. temperature) for the generations was that at higher generations there was a decrease in the water flux through the protein cage and this results in decreased relaxivity. Increasing the temperature allows more water to pass through the protein cage that, in this scenario, has more of an affect for the higher generation samples. The higher generations would therefore show a greater dependence on an increase in temperature when compared to the lower generations. This second explanation is not corroborated by the SBM fitting results since q and τM do not change which would be expected if the flux of water were changing across generations. However, the accuracy and resolution of the values obtained from the SBM fit may be too low to conclusively rule out a generational change in q or τM and this second explanation should not be disregarded.
The most promising aspect of this work is the development of a method to take advantage of the interior volume of the protein cage for development of a T1 CA. To date only the surface of protein cages including viral capsids have been modified to carry Gd chelators by single step chemical labeling. Here we expand the chemical modification to fill the interior volume of the protein cage with a DTPA-Gd labeled polymer network. This results in a particle that has a per particle relaxivity of 4,200mM-1 sec-1, which while lower than the MS2-DTPA-Gd (7,200mM-1 sec-1), exhibits relaxivity properties that are clinically significant. The MS2 construct is significantly larger than the Hsp, occupying twelve times the volume per particle. When the relaxivity behavior of these materials are compared on a volume basis, the Hsp construct exhibits a relaxivity that is seven times that of the MS2 construct. Until now the relaxivity properties of wtCCMV-Gd seemed unattainable in systems with high affinity chelators. However, we have achieved this by filling the interior cavity of the Hsp with DTPA-Gd and the per volume relaxivity or the “relaxivity density” values are equal to that of the wtCCMV-Gd construct (cf. Supporting Information).
This work represents a paradigm shift in the employment of protein cages as functional medical materials. We show that there are significant benefits, namely increased stability and efficient particle relaxivity properties that result from a synthetic approach that takes full advantage of the interior volume of protein cages. This strategy can be extended to larger protein cages and it will be interesting to see if the ionic relaxivity remains constant in larger systems. Currently we are developing similar protein cage-polymer hybrid constructs with Gd chelators that have more desirable q and τM values to see how these particles perform. Also, using this strategy of labeling protein cage with a branched polymer containing moieties with other desired properties such as drug delivery, radiation dose and hyperthermia, should be pursued in an attempt to develop high performance clinical agents. Finally, successful application of these functionalized protein cages as imaging or drug delivery agents is likely to require coupling of in vivo targeting functionality and therefore we are hopeful that targeting functionality of protein cages is developed further.
In conclusion we report the successful generation of a Gd chelate-containing branched polymer on the interior of a protein cage. Parameters important to relaxivity were determined by fitting NMRD profiles to the SBM model. These results suggest that while the exchange life time is lower than optimal, the rotational correlation time of the Gd ions has been optimized.
Supplementary Material
Acknowledgments
This research was supported in part by grants from the National Institutes of Health (R21EB005364), the National Science Foundation (CBET-0709358), and the Office of Naval Research (N00014-03-1-0692).
References
- 1.Lauffer RB. Paramagnetic Metal-Complexes as Water Proton Relaxation Agents for Nmr Imaging - Theory and Design. Chemical Reviews. 1987;87(5):901–927. [Google Scholar]
- 2.Caravan P, et al. Gadolinium(III) chelates as MRI contrast agents: Structure, dynamics, and applications. Chemical Reviews. 1999;99(9):2293–2352. doi: 10.1021/cr980440x. [DOI] [PubMed] [Google Scholar]
- 3.Aime S, et al. Insights into the use of paramagnetic Gd(III) complexes in MR-molecular imaging investigations. J Magn Reson Imaging. 2002;16(4):394–406. doi: 10.1002/jmri.10180. [DOI] [PubMed] [Google Scholar]
- 4.Caravan P. Strategies for increasing the sensitivity of gadolinium based MRI contrast agents. Chem Soc Rev. 2006;35(6):512–23. doi: 10.1039/b510982p. [DOI] [PubMed] [Google Scholar]
- 5.Allen M, et al. Paramagnetic viral nanoparticles as potential high-relaxivity magnetic resonance contrast agents. Magnetic Resonance in Medicine. 2005;54(4):807–812. doi: 10.1002/mrm.20614. [DOI] [PubMed] [Google Scholar]
- 6.Anderson EA, et al. Viral nanoparticles donning a paramagnetic coat: conjugation of MRI contrast agents to the MS2 capsid. Nano Lett. 2006;6(6):1160–4. doi: 10.1021/nl060378g. [DOI] [PubMed] [Google Scholar]
- 7.Hooker JM, et al. Magnetic resonance contrast agents from viral capsid shells: a comparison of exterior and interior cargo strategies. Nano Lett. 2007;7(8):2207–10. doi: 10.1021/nl070512c. [DOI] [PubMed] [Google Scholar]
- 8.Liepold L, et al. Viral capsids as MRI contrast agents. Magn Reson Med. 2007;58(5):871–9. doi: 10.1002/mrm.21307. [DOI] [PubMed] [Google Scholar]
- 9.Prasuhn DE, Jr, et al. Viral MRI contrast agents: coordination of Gd by native virions and attachment of Gd complexes by azide-alkyne cycloaddition. Chem Commun (Camb) 2007;(12):1269–71. doi: 10.1039/b615084e. [DOI] [PubMed] [Google Scholar]
- 10.Datta A, et al. High relaxivity gadolinium hydroxypyridonate-viral capsid conjugates: nanosized MRI contrast agents. J Am Chem Soc. 2008;130(8):2546–52. doi: 10.1021/ja0765363. [DOI] [PubMed] [Google Scholar]
- 11.Raja KS, Wang Q, Finn MG. Icosahedral virus particles as polyvalent carbohydrate display platforms. Chembiochem. 2003;4(12):1348–51. doi: 10.1002/cbic.200300759. [DOI] [PubMed] [Google Scholar]
- 12.Raja KS, et al. Hybrid virus-polymer materials. 1. Synthesis and properties of PEG-decorated cowpea mosaic virus. Biomacromolecules. 2003;4(3):472–6. doi: 10.1021/bm025740+. [DOI] [PubMed] [Google Scholar]
- 13.Rae CS, et al. Systemic trafficking of plant virus nanoparticles in mice via the oral route. Virology. 2005;343(2):224–35. doi: 10.1016/j.virol.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 14.Flenniken ML, et al. Melanoma and lymphocyte cell-specific targeting incorporated into a heat shock protein cage architecture. Chem Biol. 2006;13(2):161–70. doi: 10.1016/j.chembiol.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 15.Uchida M, et al. Targeting of cancer cells with ferrimagnetic ferritin cage nanoparticles. J Am Chem Soc. 2006;128(51):16626–33. doi: 10.1021/ja0655690. [DOI] [PubMed] [Google Scholar]
- 16.Destito G, et al. Folic acid-mediated targeting of cowpea mosaic virus particles to tumor cells. Chem Biol. 2007;14(10):1152–62. doi: 10.1016/j.chembiol.2007.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaiser CR, et al. Biodistribution studies of protein cage nanoparticles demonstrate broad tissue distribution and rapid clearance in vivo. Int J Nanomedicine. 2007;2(4):715–33. [PMC free article] [PubMed] [Google Scholar]
- 18.Koudelka KJ, et al. Interaction between a 54-kilodalton mammalian cell surface protein and cowpea mosaic virus. J Virol. 2007;81(4):1632–40. doi: 10.1128/JVI.00960-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kovacs EW, et al. Dual-surface-modified bacteriophage MS2 as an ideal scaffold for a viral capsid-based drug delivery system. Bioconjug Chem. 2007;18(4):1140–7. doi: 10.1021/bc070006e. [DOI] [PubMed] [Google Scholar]
- 20.Singh P, et al. Bio-distribution, toxicity and pathology of cowpea mosaic virus nanoparticles in vivo. J Control Release. 2007;120(1-2):41–50. doi: 10.1016/j.jconrel.2007.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaltgrad E, et al. On-virus construction of polyvalent glycan ligands for cell-surface receptors. J Am Chem Soc. 2008;130(14):4578–9. doi: 10.1021/ja077801n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Prasuhn DE, Jr, et al. Plasma clearance of bacteriophage Qbeta particles as a function of surface charge. J Am Chem Soc. 2008;130(4):1328–34. doi: 10.1021/ja075937f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koudelka KJ, et al. Endothelial targeting of cowpea mosaic virus (CPMV) via surface vimentin. PLoS Pathog. 2009;5(5):e1000417. doi: 10.1371/journal.ppat.1000417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shriver LP, Koudelka KJ, Manchester M. Viral nanoparticles associate with regions of inflammation and blood brain barrier disruption during CNS infection. J Neuroimmunol. 2009;211(1-2):66–72. doi: 10.1016/j.jneuroim.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steinmetz NF, Manchester M. PEGylated viral nanoparticles for biomedicine: the impact of PEG chain length on VNP cell interactions in vitro and ex vivo. Biomacromolecules. 2009;10(4):784–92. doi: 10.1021/bm8012742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rocklage SM, Worah D, Kim SH. Metal-Ion Release from Paramagnetic Chelates - What Is Tolerable. Magnetic Resonance in Medicine. 1991;22(2):216–221. doi: 10.1002/mrm.1910220211. [DOI] [PubMed] [Google Scholar]
- 27.Penfield JG, Reilly RF., Jr What nephrologists need to know about gadolinium. Nat Clin Pract Nephrol. 2007;3(12):654–68. doi: 10.1038/ncpneph0660. [DOI] [PubMed] [Google Scholar]
- 28.Helm L. Relaxivity in paramagnetic systems: Theory and mechanisms. Progress in Nuclear Magnetic Resonance Spectroscopy. 2006;49(1):45–64. [Google Scholar]
- 29.Lipari, Szabo Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J Am Chem Soc. 1982;104(17):4546–4559. [Google Scholar]
- 30.Abedin MJ, et al. Synthesis of a cross-linked branched polymer network in the interior of a protein cage. J Am Chem Soc. 2009;131(12):4346–54. doi: 10.1021/ja8079862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim KK, Kim R, Kim SH. Crystal structure of a small heat-shock protein. Nature. 1998;394(6693):595–9. doi: 10.1038/29106. [DOI] [PubMed] [Google Scholar]
- 32.Kim KK, et al. Purification, crystallization, and preliminary X-ray crystallographic data analysis of small heat shock protein homolog from Methanococcus jannaschii, a hyperthermophile. J Struct Biol. 1998;121(1):76–80. doi: 10.1006/jsbi.1998.3969. [DOI] [PubMed] [Google Scholar]
- 33.Flenniken ML, et al. Selective attachment and release of a chemotherapeutic agent from the interior of a protein cage architecture. Chem Commun (Camb) 2005;(4):447–9. doi: 10.1039/b413435d. [DOI] [PubMed] [Google Scholar]
- 34.Flenniken ML, et al. A library of protein cage architectures as nanomaterials. Curr Top Microbiol Immunol. 2009;327:71–93. doi: 10.1007/978-3-540-69379-6_4. [DOI] [PubMed] [Google Scholar]
- 35.Eldik Rv. In: Advances in Inorganic Chemistry: Relaxometry of water-metal ion interactions. Eldik Rv., editor. Vol. 57. Academic Press; 2005. p. 536. [Google Scholar]
- 36.Helm L, Nicolle GM, Merbach AE. Water and Proton Exchange Processes on Metal Ions. ChemInform. 2006;37(14) [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.