

Abstract
Epigenetic regulators represent a promising new class of therapeutic targets for cancer. Enhancer of zeste homolog 2 (EZH2), a subunit of Polycomb repressive complex 2 (PRC2), silences gene expression via its histone methyltransferase activity. Here we report that the oncogenic function of EZH2 in castration-resistant prostate cancer (CRPC) is independent of its role as a transcriptional repressor. Instead, it involves the ability of EZH2 to act as a co-activator for critical transcription factors including the androgen receptor (AR). This functional switch is dependent on phosphorylation of EZH2, and requires an intact methyltransferase domain. Hence, targeting the non-PRC2 function of EZH2 may have significant therapeutic efficacy for treating metastatic, hormone-refractory prostate cancer.
Factors involved in maintaining the epigenetic state of the cell are frequently altered in cancer and are promising therapeutic targets. The expression of Enhancer of zeste homolog 2 (EZH2) is correlated with prostate cancer progression, especially to its lethal castration-resistant state (CRPC) (1). EZH2 is the catalytic subunit of Polycomb repressive complex 2 (PRC2) that silences transcription through trimethylation of histone H3 on lysine 27 (H3K27me3) (2). Most studies have focused on PRC2-mediated repression as the oncogenic mechanism of EZH2. In addition, tumor suppressors such as DAB2IP have been reported as EZH2/PRC2 targets (3). However, substantial studies have indicated that both Drosophila Enhancer of zeste [E(z)] and EZH2 have potential functions other than that of a transcriptional repressor (4-6), although the mechanisms are unclear.
We utilized the LNCaP cell line as a model of androgen-dependent prostate cancer and LNCaP-abl (abl), its androgen-independent derivative (7), to study EZH2 function in the progression of prostate cancer to CRPC. As is the case for clinical tumors (1), EZH2 levels in abl cells are significantly higher than in LNCaP (Fig. 1A). EZH2 silencing has a more profound effect on the androgen-independent growth of abl cells than on the androgen-dependent growth of LNCaP (Fig. 1B and fig. S1). The requirement of EZH2 for androgen-independent growth was confirmed in an in vivo mouse xenograft CRPC model employing CWR22Rv1 cells (Fig. 1C).
Fig. 1. Overexpression of EZH2-stimulated genes in clinical CRPC samples.
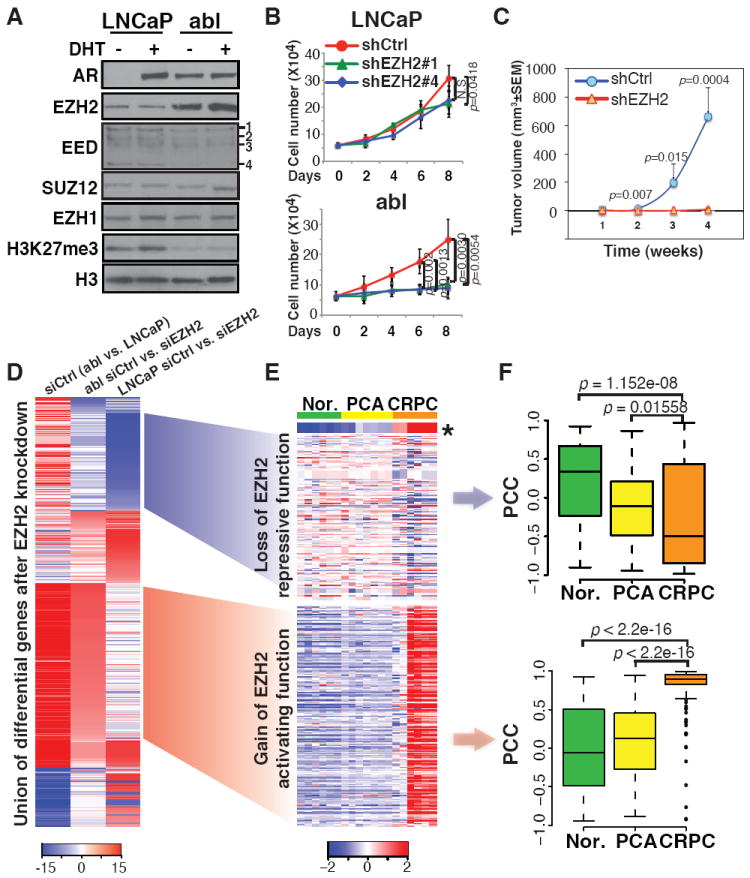
(A) Immunoblot of nuclear extracts from LNCaP and abl cells treated without (-) or with (+) 5α-dihydrotestosterone (DHT) to control for effects of cell growth on protein expression. EED isoforms are numbered (22). (B) Growth of cells transduced with lentiviral shRNAs targeting scrambled control (shCtrl) or EZH2 (shEZH2#1 and #4). (C) Tumor growth curve of castrated male scid mice injected with CWR22Rv1 cells with or without EZH2 silencing. (D) Clustering of the union of differentially expressed genes in LNCaP and abl after transfection with siRNAs against control (siCtrl) or EZH2 (siEZH2). (E and F) Heat map of expression levels (E) and box plots of Pearson correlation coefficients (PCC) (F) of EZH2-repressed (top) and -stimulated (bottom) genes with EZH2 level in the Varambally cohort. Nor., normal tissues; PCA, primary tumors. *, EZH2 level.
We then explored EZH2-dependent genes in LNCaP and abl cells. Although similar numbers of genes are up- or down-regulated following EZH2 silencing in LNCaP, many more genes were significantly down-regulated upon EZH2 depletion in abl, and these EZH2-stimulated genes are highly expressed in abl (Fig. 1D). EZH2 silencing using two independent siRNAs confirmed the de-repression of the EZH2-repressed gene DAB2IP in LNCaP, and down-regulation of several EZH2-stimulated genes in abl (fig. S2A). We found similar results in two other hormone-refractory cell lines, C4-2B and CWR22Rv1 (fig. S2B). We then examined the profiles of EZH2-dependent genes in two clinical prostate cancer cohorts (8, 9). Although the set of EZH2-repressed genes in LNCaP exhibit lower expression in CRPC and marginal negative correlation with EZH2 level, the set of EZH2-stimulated genes identified in abl have significantly higher expression level and positive correlation with EZH2 in these metastatic, hormone-refractory prostate tumors (Fig. 1, E and F, and fig. S3). These results suggest a potentially important functional switch of EZH2 from transcriptional repression to gene activation in CRPC.
To determine whether the gene activation function of EZH2 is the effect of direct binding, we conducted ChIP-seq of EZH2 and H3K27me3. Although EZH2 and H3K27me3 co-localized at the majority of sites in both LNCaP and abl, we identified a subset of EZH2 sites that lack nearby H3K27me3 in abl (Fig. 2A). These EZH2 sites lacking H3K27me3 were validated using four different EZH2 antibodies (fig. S4A) and by EZH2 silencing (fig. S4B). We defined EZH2 “ensemble” peaks as those with both EZH2 and H3K27me3 enrichment, and “solo” peaks as those with only EZH2 binding. A majority of both ensemble and solo binding sites were located at the promoter regions or gene bodies (fig. S5A). Although ensemble peaks in LNCaP and abl overlap significantly, very few solo peaks overlap between these two cell lines (fig. S5B). This difference is even more dramatic when examining the genes nearby EZH2 binding sites (fig. S5C). This finding suggests that EZH2 gains a unique set of chromatin binding sites that lack H3K27me3 in abl. In addition, the solo peaks are enriched for the active histone marks H3K4 dimethylation, trimethylation (H3K4me2 and H3K4me3) and RNA polymerase II (Pol II) (Fig. 2B), suggesting the potential function of these peaks in gene activation. Indeed, although ensemble binding is enriched near the transcription start sites (TSS) of EZH2-repressed genes, solo binding is enriched near EZH2-stimulated genes (Fig. 2C and fig. S6). Strikingly, EZH2 depletion decreases the levels of the active marks at these solo sites (fig. S7), indicating a mechanism of EZH2 in gene activation via modulation of active chromatin states. Genes directly activated by EZH2 in abl are significantly over expressed in gene signatures derived from independent metastatic, hormone-refractory prostate tumors (fig. S8A), and survival analysis supported the prognostic power only of EZH2-activated genes with solo peak binding in abl (Fig. 2D (8) and fig. S8, B-D (10)). Taken together, these data support the importance of EZH2 gene activation function in CRPC.
Fig. 2. EZH2 binding without H3K27me3 is associated with gene activation in CRPC.
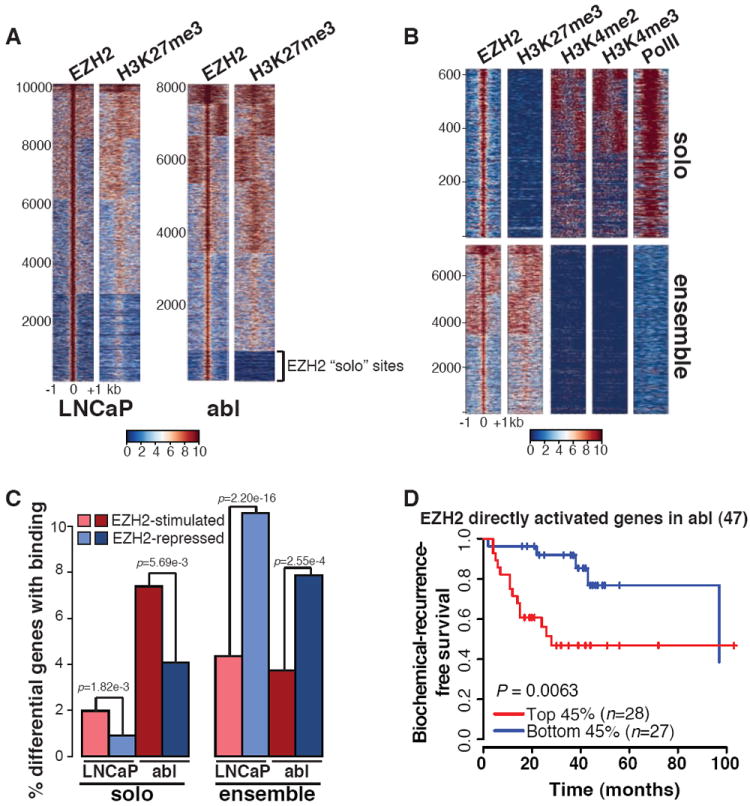
(A) Heat maps of EZH2 and H3K27me3 ChIP-seq signal ±1 kb around the EZH2 peak summit in LNCaP and abl. The color scale indicates average signal using a 10 bp window. The numbered index of EZH2 peaks is shown to the left. (B) Heat maps of EZH2, H3K27me3, H3K4me2, H3K4me3, and PolII ChIP-seq signal ±1 kb around EZH2 solo or ensemble peak summit in abl. (C) Percentages of differentially expressed genes upon EZH2 depletion in LNCaP or abl cells containing EZH2 solo or ensemble peaks within 20 kb around transcription start sites (TSS). (D) Kaplan Meier plots of EZH2 directly activated genes in the Yu cohort. The number of genes is indicated at the top of each plot. n, numbers of patients.
We then tested the involvement of another PRC2 subunit SUZ12 in the EZH2 solo peaks in CRPC. SUZ12 ChIP-seq signals displayed significant correlation with both H3K27me3 (0.57) and EZH2 ensemble peaks (0.66), but little correlation with EZH2 solo peaks (0.27) (fig. S9A). Silencing of SUZ12 drastically reduced EZH2 binding at ensemble peaks, but had no effect at the solo peaks (fig. S9B). Silencing of either SUZ12 or EED increased the expression of the EZH2-repressed gene DAB2IP, but had no effects on EZH2-activated genes (fig. S9C). These results indicate that EZH2 solo peaks are independent of the PRC2 complex. Results from gel filtration chromatography demonstrated that EZH2 is present in complexes other than PRC2 in abl, since a significant fraction of EZH2 elutes as a broad peak distinct from SUZ12 or EED, although in LNCaP the majority of EZH2 co-elutes with other PRC2 subunits (Fig. 3A). We next asked whether the methyltransferase activity of EZH2 is required despite the lack of the other PRC2 components. We replaced the endogenous EZH2 in abl cells with either the wild-type (Esr-WT) or two enzymatically inactive mutants [SET domain deletion (Esr-ΔSET) (11) and H694A/F672I double-point mutation (Esr-DM) (12, 13)] (fig. S10A). Ectopic re-expression of the wild-type EZH2, but not the catalytically inactive mutants, can rescue the effects of EZH2 silencing on both gene activation (Fig. 3B) and androgen-independent growth of CRPC cells (Fig. 3C and fig. S10B). We also found that EZH2 over expression was sufficient to promote the androgen-independent growth of LNCaP cells and that the enzymatic activity was required (Fig. 3D). Under these conditions, the expression of EZH2-activated target genes was elevated to comparable levels as in abl by wild-type EZH2, but not by the activity-dead mutants (fig. S10C). These results suggest that EZH2 utilizes a PRC2-independent methyltransferase activity for both gene activation and androgen-independent growth.
Fig. 3. Requirement for the methyltransferase activity and interaction with AR for the EZH2 transactivation function.
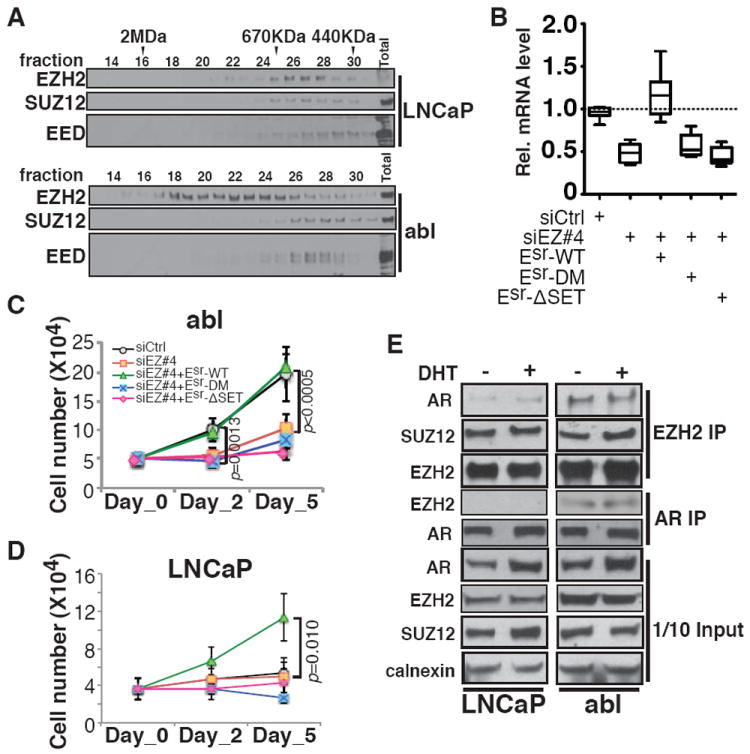
(A) Immunoblot of nuclear extracts from LNCaP and abl cells after gel filtration fractionation. Molecular mass standards are indicated. (B) Box plots of minimum to maximum RT-PCR values for EZH2-activated genes in abl after replacement with wild-type or mutant EZH2 as indicated. (C and D) Growth of abl (C) and LNCaP (D) in hormone-depleted medium, following replacement with wild-type or mutant EZH2 as indicated. (E) Co-immunoprecipitation of EZH2 and AR in LNCaP and abl without (-) or with (+) DHT.
To determine how EZH2 might be targeted to solo peaks, we conducted motif analysis and found significant enrichment of the androgen receptor (AR) binding motif at EZH2 solo peaks in abl (fig. S11A). AR chromatin binding was enriched at the center of EZH2 solo peaks, but not at ensemble peaks (fig. S11B). Co-immunoprecipitation detected a robust physical interaction between EZH2 and AR in abl (Fig. 3E). The interaction between AR and EZH2 was lost when the endogenous EZH2 was replaced with Domain I or SET domain deletion mutants (fig. S12), suggesting the requirement of these two domains for the interaction. EZH2 solo peaks in abl significantly overlaps with AR global binding, and EZH2- and AR-activated genes also overlap significantly in abl (fig. S13). However, not all solo peaks contain AR motif or overlap with AR binding, suggesting that other factors in addition to AR may contribute to EZH2 recruitment to solo sites. Notably EZH2 depletion does not change AR mRNA or protein levels, but does decrease AR-associated lysine methylation and this requires an intact EZH2 enzymatic activity (fig. S14). This suggests that EZH2 exerts its activation function not through modulating AR level, but rather through alterations in methylation of AR or AR-associated proteins. Silencing of EZH2 decreased AR recruitment to solo sites bound by both AR and EZH2 (fig. S15A), and had no significant effects on AR binding to other sites (fig. S15B). Similarly, knockdown of AR decreased EZH2 binding to the co-localized solo peaks (fig. S15C). Depletion of both EZH2 and AR led to a more marked reduction in the expression of co-regulated genes than silencing of either alone (fig. S15D). These results indicate that EZH2 and AR cooperate to activate target genes through their cooperative recruitment.
To identify what factors determine the functional switch of EZH2 from a repressor to an activator, we examined the phosphorylation status of EZH2 that has been reported to alter its enzymatic activity towards H3K27 (14-17). Phosphorylation levels at both S21 and T492 were elevated in abl cells compared with LNCaP, whereas phosphorylation at T350 was equivalent (Fig. 4A). We replaced the endogenous EZH2 with phosphorylation site mutants to determine the potential role of site-specific phosphorylation in EZH2-mediated gene activation, and found that S21A mutant failed to rescue the down-regulation of EZH2-activated genes upon EZH2 silencing in abl (fig. S16A). In addition, only the antibody specific for phospho-S21 could detect EZH2 enrichment preferentially at the solo peaks (Fig. 4B and fig. S16B). Furthermore, replacement of endogenous EZH2 by wild-type EZH2 or a phosphomimetic mutant of S21 (S21D), but not the S21A mutant, could support the androgen-independent growth of CRPC cells (Fig. 4C and fig. S17, A and B). This same dependence on S21 phosphorylation was also found for the ability of EZH2 to induce the androgen-independent growth of LNCaP cells (fig. S17C). These results suggest the importance of phosphorylation at S21 in both EZH2-mediated gene activation and androgen-independent growth. We further confirmed the up-regulation of EZH2 phosphorylation at S21 in two additional hormone-refractory prostate cancer cell lines, C4-2B and CWR22Rv1 (fig. S18A). Consistent with the report that AKT is the kinase for EZH2 phosphorylation at S21 (16), we found higher levels of the active AKT in abl cells (Fig. 4A). Although both LNCaP and abl cells are PTEN-null, PHLPP-1, an AKT phosphatase (18), was decreased in abl cells (fig. S18B), which may contribute to the activation of the PI3K/AKT signaling in abl. To gain further insight into the involvement of EZH2 phosphorylation in the EZH2 interaction with AR, we investigated the co-purification of phosphorylated forms of EZH2 and AR by gel filtration, and found that pS21 EZH2 predominantly co-eluted with AR in a large molecular weight complex (Fig. 4D). These results suggest a potential role for EZH2 phosphorylation at S21 to promote its association with an AR containing complex. The importance of EZH2 phosphorylation at S21 in prostate cancer progression was further analyzed by immunohistochemistry in tissue microarrays containing early stage prostate tumors from a neoadjuvant androgen deprivation therapy trial and metastatic, hormone-refractory tumors (Fig. 4E and fig. S19). As previously reported (1), the level of EZH2 is increased in CRPC as compared with early stage disease. Strikingly, pS21 EZH2 was even more significantly increased in CRPC. Intriguingly, H3K27me3 levels significantly decrease with prostate cancer progression, consistent with our observation that the global level of H3K27me3 in abl is considerably lower than in LNCaP (Fig. 1A). This result further supports our conclusion that the oncogenic activity of EZH2 in CRPC is independent of its Polycomb repressive function.
Fig. 4. Critical role of EZH2 phosphorylation at S21 for its functional switch.
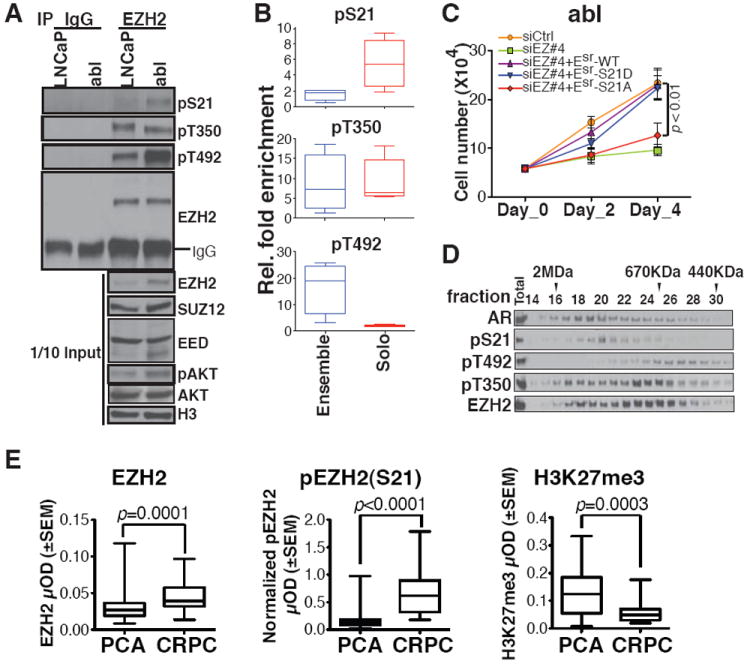
(A) Immunoprecipitation in LNCaP and abl cells using IgG or EZH2 antibodies, followed by immunoblotting with indicated antibodies. (B) Box plots of ChIP-qPCR values for EZH2 recruitment to selected ensemble or solo sites using phosphorylation-specific antibodies. (C) Growth of abl cells in androgen-depleted medium after replacement with wild-type or mutant EZH2 as indicated. (D) Immunoblot of nuclear extracts from abl cells after gel filtration fractionation. Molecular mass standards are indicated. (E) Analysis of EZH2, S21 phosphorylated EZH2 [pEZH2(S21)] and H3K27me3 protein levels by quantitative immunohistochemistry in neoadjuvant prostate tumors (PCA) and CRPC.
This study demonstrates that phosphorylation of EZH2 at S21 mediated directly or indirectly by PI3K/AKT pathway can switch its function from a Polycomb repressor to a transcriptional co-activator of AR and potentially other factors. Rescue experiments and the lack of correlation with H3K27me3 levels support a role for EZH2 directed methylation of substrates other than H3K27 including potential non-histone proteins. The current rationale for EZH2 inhibitor design is based primarily on targeting its Polycomb repressive activity and uses H3K27me3 as the pharmacodynamic readout (19). However, the observed loss-of-function mutations of EZH2 in myelodysplastic syndrome and acute leukemia raise concerns that such inhibitors might exhibit significant hematologic side effects (20, 21). Our finding of an altered function for EZH2 in CRPC raises the potential to develop inhibitors that specifically target the EZH2 activation function while sparing its PRC2 repressive function. In addition, our finding that EZH2 cooperates with AR-associated complexes and requires phosphorylation to support CRPC growth suggests novel combination therapies for the treatment of metastatic, hormone-refractory prostate cancer (fig. S20).
Supplementary Material
Acknowledgments
We thank H. Huang, A. Chinnaiyan, Y. Shang and D. Reinberg for sharing their reagents. We thank Y. Qiu, X. Yang, J. Xu and M. Ni for technical support and advice. This work was supported by the Prostate Cancer Foundation (PCF) Young Investigator Award (KX), Swiss National Science Foundation Prospective Researcher Fellowship (ACG), NSF Graduate Research Fellowship (JET), an A. David Mazzone Project Development Award (MB), PCF Challenge Awards (SPB and MB), DOD Idea Development Award PC100950 (SPB), NCI grants CA131945, CA89021, and CA90381 (ML), NIH grants CA166507 (CC), CA111803 (SPB), CA090381 (MB, SPB, PWK), GM99409 (XSL), CA85859 and CA097186 (CM, RLV). All data sets have been deposited in the Gene Expression Omnibus database (GSE39461). MB serves as a consultant to Novartis Pharmaceuticals.
References and Notes
- 1.Varambally S, et al. Nature. 2002;419:624. doi: 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- 2.Cao R, et al. Science. 2002;298:1039. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 3.Chen H, Tu SW, Hsieh JT. J Biol Chem. 2005;280:22437. doi: 10.1074/jbc.M501379200. [DOI] [PubMed] [Google Scholar]
- 4.Lee ST, et al. Mol Cell. 2011;43:798. doi: 10.1016/j.molcel.2011.08.011. [DOI] [PubMed] [Google Scholar]
- 5.LaJeunesse D, Shearn A. Development. 1996;122:2189. doi: 10.1242/dev.122.7.2189. [DOI] [PubMed] [Google Scholar]
- 6.Strutt H, Cavalli G, Paro R. Embo J. 1997;16:3621. doi: 10.1093/emboj/16.12.3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Culig Z, et al. Br J Cancer. 1999;81:242. doi: 10.1038/sj.bjc.6690684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu YP, et al. J Clin Oncol. 2004;22:2790. doi: 10.1200/JCO.2004.05.158. [DOI] [PubMed] [Google Scholar]
- 9.Varambally S, et al. Cancer Cell. 2005;8:393. doi: 10.1016/j.ccr.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 10.Glinsky GV, Glinskii AB, Stephenson AJ, Hoffman RM, Gerald WL. J Clin Invest. 2004;113:913. doi: 10.1172/JCI20032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cao R, Zhang Y. Curr Opin Genet Dev. 2004;14:155. doi: 10.1016/j.gde.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 12.Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. Genes Dev. 2002;16:2893. doi: 10.1101/gad.1035902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Joshi P, et al. J Biol Chem. 2008;283:27757. doi: 10.1074/jbc.M804442200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen S, et al. Nat Cell Biol. 2010;12:1108. doi: 10.1038/ncb2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaneko S, et al. Genes Dev. 2010;24:2615. doi: 10.1101/gad.1983810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cha TL, et al. Science. 2005;310:306. doi: 10.1126/science.1118947. [DOI] [PubMed] [Google Scholar]
- 17.Wei Y, et al. Nat Cell Biol. 2011;13:87. doi: 10.1038/ncb2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao T, Furnari F, Newton AC. Mol Cell. 2005;18:13. doi: 10.1016/j.molcel.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 19.Sparmann A, van Lohuizen M. Nat Rev Cancer. 2006;6:846. doi: 10.1038/nrc1991. [DOI] [PubMed] [Google Scholar]
- 20.Nikoloski G, et al. Nat Genet. 2010;42:665. doi: 10.1038/ng.620. [DOI] [PubMed] [Google Scholar]
- 21.Ntziachristos P, et al. Nat Med. 2012;18:298. doi: 10.1038/nm.2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuzmichev A, Jenuwein T, Tempst P, Reinberg D. Mol Cell. 2004;14:183. doi: 10.1016/s1097-2765(04)00185-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.