

Abstract
Cognitive functions supported by neurons in the prefrontal cortex (PFC) are disrupted by acute and chronic exposure to alcohol, yet little is known about the mechanisms that underlie these effects. In the present study, in vivo and in vitro electrophysiology was used to determine the effects of ethanol on neuronal firing and network patterns of persistent activity in PFC neurons. In vivo, ethanol (0.375–3.5 g/kg) dose-dependently reduced spike activity in the PFC measured with multielectrode extracellular recording in the anesthetized rat. In an in vitro coculture system containing slices of PFC, hippocampus, and ventral tegmental area (VTA), ethanol (25–100 mm) decreased persistent activity of PFC neurons, but had little effect on firing evoked by direct current injection. Persistent activity was often enhanced after ethanol washout and this effect was maintained in cultures lacking the VTA. A low concentration of the NMDA antagonist APV (5 μm) mimicked the inhibition of ethanol of persistent activity with no change in activity after washout. Ethanol inhibition of spontaneous and VTA-evoked persistent activity was enhanced by the D1 dopamine receptor antagonist SCH23390 [R(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrochloride]. The results of this study show that ethanol inhibits persistent activity and spike firing of PFC neurons and that the degree of ethanol inhibition may be influenced by D1 receptor tone. Ethanol-induced alterations in the activity of deep-layer cortical neurons may underlie some of the behavioral effects associated with ethanol intake.
Keywords: alcohol, bistability, electrophysiology, slice culture, addiction, NMDA
Introduction
Alcohol disrupts behaviors and responses that require neurons in the prefrontal cortex (for review, see Moselhy et al., 2001). For example, ethanol reduces the detection of performance errors as monitored by scalp electrodes and decreases EEG activity evoked by stimulation of the prefrontal cortex (Ridderinkhof et al., 2002; Kahkonen et al., 2003). Long-term alcohol abuse causes changes in gray and white matter volume in the PFC that are correlated with the appearance of neurocognitive deficits (for review, see Sullivan and Pfefferbaum, 2005). In functional studies, human alcoholics show significant reductions in event-related oscillations in the frontal cortex during tests that require prefrontal processing (Kamarajan et al., 2004). Decreases in both δ and theta power in these subjects are greater during no-go trials, suggesting impairments in response inhibition. These findings suggest that alcohol disruption of PFC activity may contribute to the development and persistence of alcohol addiction (Goldman-Rakic, 1999; Kalivas et al., 2005). Despite evidence linking alcohol to impaired cortical function, little is known about the direct effects of ethanol on the behavior of individual PFC neurons.
Neurons within deep layers of the PFC display dynamic patterns of electrical activity during wakefulness that reflect their critical role in integrating inputs from cortical and subcortical structures. Untangling the mechanisms that regulate this activity is complicated by the number and diversity of these inputs and the presence of short and long neuromodulatory circuits that influence activity. During sleep or anesthesia when sensory input is diminished, PFC neurons undergo slow oscillations (0.5–1 Hz) in membrane potential between hyperpolarized down states and depolarized up states. Down states are periods of neuronal quiescence whereas up states reflect depolarization driven by synaptic activity (Steriade et al., 1993; O'Donnell and Grace, 1995; Lewis and O'Donnell, 2000). Activity during these up states is one source of the neocortical δ frequency observed in human EEG studies and is distinct from the slow oscillation (Steriade, 2001). Although these forms of activity are most often associated with loss of consciousness, slow oscillations and persistent activity may be important in strengthening and consolidating changes in synaptic plasticity that underlie various behaviors including certain forms of memory (Steriade, 2001; Marshall et al., 2006). Neuromodulators such as dopamine (DA) released by neurons located in the ventral tegmental area (VTA) influence the activity and excitability of PFC neurons (Lewis and O'Donnell, 2000; Seamans et al., 2003). These signals may allow prefrontal networks to attend to inputs that convey motivational or reward information, including those from alcohol and other drugs of abuse (Goldman-Rakic, 1999; Durstewitz et al., 2000).
In the present study, the effects of ethanol on neuronal activity within the PFC was investigated using in vivo and in vitro recording techniques. Because acute slices of cortex rarely show persistent activity, we used a slice coculture preparation that reproduces activity patterns similar to those observed in vivo (Seamans et al., 2003). The results of these studies demonstrate that acute ethanol alters persistent activity of deep-layer pyramidal neurons in the PFC at concentrations that are known to impair behavior.
Materials and Methods
Animals
All procedures involving animals were conducted under protocols approved by the Institutional Animal Care and Use Committee of the Medical University of South Carolina.
Organotypic slice cocultures
Slice cocultures were prepared as described previously (Seamans et al., 2003). Briefly, postnatal day 1–3 rat or mouse pups were killed and brains were placed in ice-cold, sucrose-substituted Ringer's solution containing (in mm) 200 sucrose, 1.9 KCl, 1.2 Na2HPO4, 33 NaHCO3, 6 MgCl2, 0.5 CaCl2, 10 glucose, and 0.4 ascorbic acid bubbled with 95% O2/5% CO2. Individual coronal sections containing the prelimbic and infralimbic regions of the prefrontal cortex, the midbrain containing the ventral tegmental area, and the hippocampus were cut at a thickness of 350 μm using a Leica (Nussloch, Germany) VT-1000 vibratome. Slices were oriented adjacent to one another on a Millipore (Temecula, CA) millicell insert in a six-well culture dish. Each well contained 1 ml of serum-containing media consisting of 50% basal medium Eagle, 25% Earle's balanced salt solution, 25% horse serum with 37 mg/ml glucose, 25 mm HEPES, 100 μg/ml streptomycin, and 0.235 mm Glutamax. After 3 d in culture, the media was changed to one containing reduced amounts of horse serum (5%). After 15 d in culture, 5-fluoro-2-deoxyuridine (0.08 mm) was added to prevent glial overgrowth. Cell culture dishes were kept in a humidified incubator equilibrated with 5% CO2 and were maintained for 15–40 d with media changes every 2–3 d. In some experiments examining the effects of ethanol on VTA dopamine neuron activity, cultures were prepared from transgenic mice that express green fluorescent protein (GFP) under the control of a tyrosine hydroxylase promoter (Matsushita et al., 2002).
Electrophysiology
In vivo recordings. Spontaneous firing of PFC neurons was monitored in vivo using a multielectrode recording system. Sprague Dawley rats (250–350 g) were anesthetized with urethane (1.5 g/kg, i.p.) and a 32-channel probe (8 × 1 tetrode assembly, NeuroNexus Technologies, Ann Arbor, MI) was lowered into the PFC using a stereotactic guide. The final coordinates relative to the center of the multiarray probe were (in millimeters from bregma) +2.7 anteroposterior, +0.7 mediolateral, and −3.0 dorsoventral. Each of the eight probe arms (15 μm thick, 2 mm long, 200 μm separation) contained four electrodes (electrode area, 312 μm2) that were separated by 25 μm (see Fig. 1A). Data were acquired at 10 kHz using a Multichannel Systems (Reutlingen, Germany) extracellular recording system and were stored off-line for additional processing. Spike sorting and clustering were performed using Off-line Sorter software (Plexon, Dallas, TX) and records were imported into NeuroExplorer (Nex Technologies, Littleton, MA) for spike-train analysis.
Figure 1.
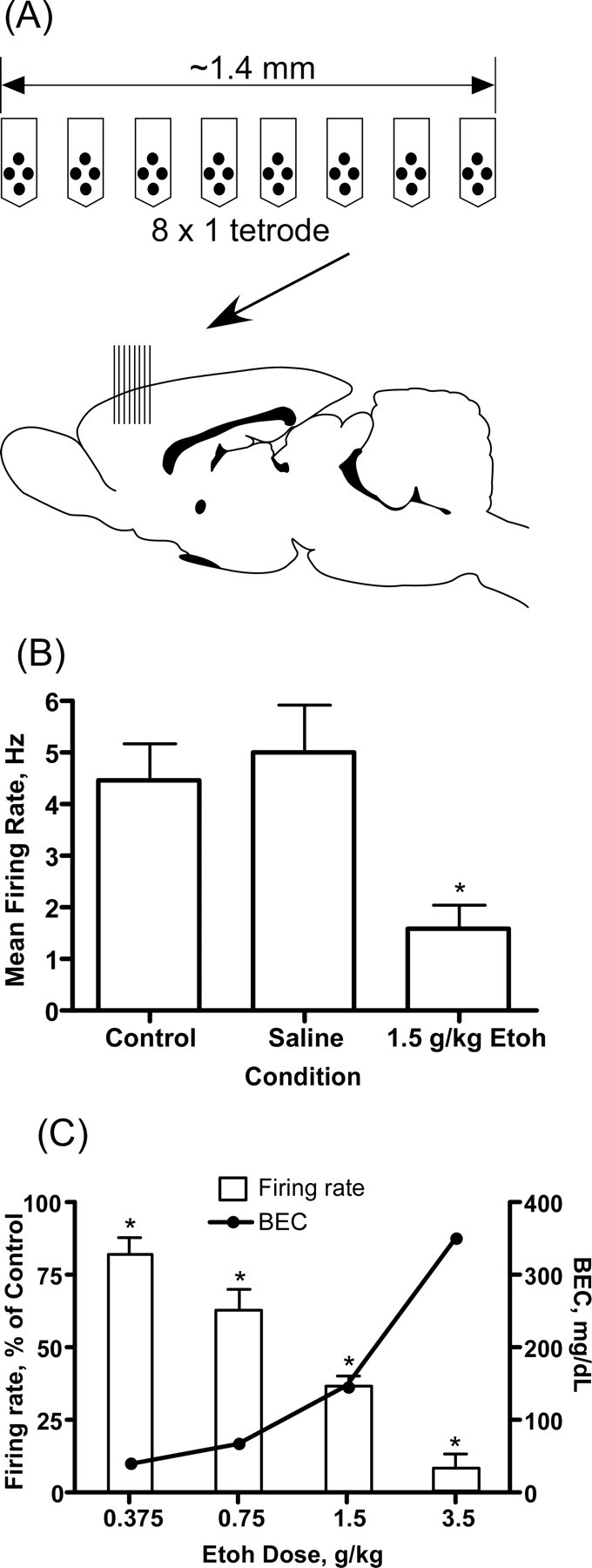
Effects of ethanol on PFC neuron firing in vivo. A, Top, Schematic showing orientation of the electrodes used to record spike activity in the PFC. Each of the eight arms contains four electrodes separated by 25 μm. Bottom, Sagittal section of the rat brain with the approximate location of the recording electrode (arrow and vertical bars). B, Representative exper-iment showing the effect of ethanol on the mean firing rate of PFC neurons. These results are from a single animal and each bar represents the mean (±SEM; n = 13 neurons per group) firing rate of all neurons measured during a 5 min period 30 min after either saline or ethanol (1.5 g/kg, i.p.). C, Dose dependence of the ethanol inhibition of PFC firing. Error bars represent the mean (±SEM; n = 15–17 neurons per concentration; 1 animal for each concentration) percent inhibition (left y-axis) of firing by ethanol relative to the control period. Closed circles show the blood ethanol concentration (right y-axis) 30 min after an intraperitoneal injection of ethanol. *Significantly (p ≤ 0.05, paired t test) different from corresponding control response.
In vitro recordings. For in vitro recordings, the section of the Millipore membrane containing a slice coculture was carefully removed from the insert and was submerged in a recording chamber. The slice coculture was perfused at ∼2 ml/min with artificial CSF (ACSF) containing (in mm) 125 NaCl, 2.5 KCl, 25 NaHCO3, 1.25 NaH2PO4, 1.3 MgCl2, 2 CaCl2, 0.4 ascorbic acid, and 10 dextrose. ACSF solutions were maintained at a temperature of 32–33°C using a flow-through solution heater and heated recording chamber under thermistor control of a TC-324B temperature controller (Warner Instruments, Hampden, CT). All solutions were continuously bubbled with a mixture of 95% O2/5% CO2. Patch pipettes (1.5 × 1.1 mm; 4–7 mΩ) were filled with an internal recording solution containing (in mm) 120 K-gluconate, 20 KCl, 0.1 EGTA, 10 HEPES, 2 Na2ATP, and 0.3 NaGTP, pH 7.2. In some experiments, biocytin was added to the internal solution to label the patched cell. Liquid junction potential errors were calculated to be ∼12 mV and were corrected unless otherwise stated. Whole-cell patch-clamp recordings were made from visually identified deep-layer pyramidal cells from the PFC using a Zeiss (Oberkochen, Germany) FS2 microscope with infrared video/differential interference contrast microscopy. Gigaohm seals were obtained in voltage-clamp mode using an Axoclamp 700A or 700B amplifier (Molecular Devices, Sunnyvale, CA) and the amplifier was switched to current-clamp mode after breakthrough. Pipette access resistance (15–35 MΩ) was monitored throughout the experiment and cells showing a significant deviation in access resistance (>25%) were not used for analysis. Data were acquired using an ITC-16 interface (Instrutech, Port Washington, NY) controlled by Axograph 4.9 software running on a Macintosh G4 computer (Apple, Cupertino, CA). For stimulus-evoked responses, square-wave pulses (0.2 ms) of variable intensity were delivered through a stimulus-isolation unit to a concentric bipolar stimulating electrode visually placed in the slice coculture. At the end of some experiments, slices were processed for tyrosine hydroxylase immunoreactivity as described previously (Seamans et al., 2003) to verify the location of the VTA and the extent of dopaminergic innervation of the PFC. Spontaneous firing of midbrain dopamine neurons was measured in voltage-clamp mode using cell-attached recordings with ACSF in the patch pipette. In some of these experiments, Alexa 594 (Invitrogen) was added to the pipette filling solution and cells were labeled after seal breakthrough after cell-attached recordings. Confocal images were acquired using a Zeiss LSM510 system as described previously (Carpenter-Hyland et al., 2004).
Data analysis
Unless otherwise noted, data were analyzed for statistical significance using repeated measures one-way ANOVA with post hoc testing where appropriate (Prism 4 software; GraphPad Software, San Diego, CA). A significance level was set at p < 0.05 for all analyses.
Results
Ethanol and PFC activity in vivo
The effect of ethanol on the firing activity of neurons in the prefrontal cortex was determined using multielectrode extracellular recording in urethane-anesthetized rats. Urethane was chosen because it has been reported to have minimal effects on excitatory and inhibitory synaptic responses (Sceniak and Maciver, 2006). In these experiments, each animal tested was given a single dose of ethanol and firing rates of individual neurons were compared before and after ethanol challenge. To quantify the effect of ethanol, mean firing rates were calculated from 5 min recording periods collected just before and 30 min after administration of ethanol. This period corresponds to the approximate peak of the blood ethanol concentrations after intraperitoneal injection of ethanol. After electrode implantation and a period of recovery (1–2 h), firing rates were relatively constant over time and were not significantly altered after an injection of saline (Fig. 1). In contrast, administration of ethanol to the same animal caused a sustained reduction in spike-firing. The magnitude of ethanol's inhibition of PFC spike-firing was dose dependent. As shown in Figure 1D, 0.375 g/kg ethanol reduced firing by ∼20% whereas firing was inhibited by ∼40% after 0.75 g/kg ethanol. Increasing the ethanol dose to 1.5 g/kg inhibited firing by ∼65% whereas the 3.5 g/kg dose nearly eliminated firing. The blood ethanol concentrations measured 30 min postinjection ranged from 41.3 mg/dl (0.0413%) for the 0.375 g/kg dose to 352.3 mg/dl (0.3523%) for the 3.5 g/kg dose (Fig. 1D). These blood levels of alcohol range from subintoxicating (0.04%) to sedating (0.35%) in the adult rat.
Persistent activity in slice cocultures
To examine the effects of ethanol on PFC neuron activity in more detail, we performed patch-clamp recordings in slice cocultures containing the PFC, VTA, and hippocampus (Fig. 2). Standard immunohistochemistry for tyrosine hydroxylase (TH) (Seamans et al., 2003), was performed in fixed tissue to verify innervation of the PFC from TH-positive cells in the midbrain slice (Fig. 2A). Similar results with VTA containing cultures have been obtained by other investigators (Franke et al., 2003) and suggest that brain slices maintained in vitro form appropriate synaptic connections and show responses that are similar to those observed in vivo.
Figure 2.

Ethanol inhibits persistent activity in brain-slice cocultures. A, Details of the slice coculture system. The diagram shows the orientation of brain slices in the coculture system used in the present study (HP, hippocampus). The middle panels show tyrosine-hydroxylase immunoreactivity (TH staining) with the bottom showing VTA cell bodies and top showing TH-positive fibers in the PFC portion of slice. The right panels show a biocytin-filled pyramidal neuron in the PFC portion of the slice coculture. B, Representative trace (and expanded section showing one up-state period) showing example of bistability and persistent activity recorded from a single PFC pyramidal neuron under current-clamp mode. C–E, Representative traces showing the effects of ethanol (10–50 mm) on spontaneous persistent activity of single PFC pyramidal neurons. In each set of traces, the top trace is the control, the middle trace is in the presence of ethanol, and the bottom trace is after washout. F, Effect of 100 mm ethanol on spike firing of PFC neurons induced by a series of current injections. The summary figures show the effects of 100 mm ethanol on resting membrane potential (left) and number of spikes (right) and are the mean ± SEM from four neurons.
Under baseline recording conditions, deep-layer pyramidal neurons displayed membrane bistability reflected as spontaneous transitions from hyperpolarized down states to a depolarized up state (Fig. 2B). Action potential-dependent spiking normally occurred only during up state periods and firing (but not up state transitions) could be suppressed by injection of negative current to bring membrane potentials below threshold for action potential generation (data not shown). The mean (±SEM) frequency of up states recorded from prefrontal neurons in all cultures showing spontaneous activity was 0.11 Hz (±0.01). The duration of up states and spike-firing during up states varied between slice cocultures, but were relatively constant within an individual neuron during recordings. For 49 control neurons recorded from the PFC area of the slice coculture that showed spontaneous persistent activity, the average membrane potential was −73.1 mV (±1.1) for the down state and −57.4 (±1.3) for the up state. The mean amplitude of depolarization from down state to up state in these neurons was 15.7 mV (±1.1) and the average duration of an up state was 3.7 s (±0.5). An average of 10.9 spikes per up state (±1.9) was observed with a mean firing rate of 4.7 Hz (±0.9).
Ethanol and persistent activity
To examine the effects of ethanol on persistent activity and bistability, a within-subjects experimental design was used. This consisted of recording spontaneous activity from a single deep-layer pyramidal neuron for ∼5–10 min in the absence of ethanol and then switching the perfusion to a solution containing ethanol. The time required for bath exchange between solutions was ∼2 min. Each coculture was exposed to a single concentration of ethanol for ∼10 min before returning to control recording solution. As shown in Figure 2C, 10 mm ethanol had little effect on persistent activity or spike activity. In contrast, concentrations of ethanol of 25 mm and higher significantly inhibited persistent activity (Fig. 2D,E). At a concentration that is the legal limit for driving in most US states (0.08% or 17 mm), ethanol produced noticeable inhibition of spontaneous persistent activity in three of four cells tested (data not shown). To determine whether ethanol affected the intrinsic excitability of deep-layer pyramidal neurons, spike-firing was induced by direct current injections through the patch pipette in the absence and presence of ethanol. As shown in the sample traces in Figure 2F, current injection induced regular spiking of deep-layer pyramidal neurons and firing immediately ceased with cessation of the depolarizing current. Ethanol, at concentrations as high as 100 mm, had little significant effect on the current-voltage relationship or on the rate of firing of deep-layer prefrontal neurons. After washout of the ethanol-containing solution, firing returned to pre-ethanol control levels.
To quantify the effects of ethanol on spontaneous persistent activity, the amplitude, duration, and number of spikes of individual up states in single neurons were measured before, during, and after exposure to ethanol. For each neuron, these data were expressed as a percentage of the pre-ethanol control value to account for baseline differences in activity between cultures. Figure 3 summarizes the effects of different concentrations of ethanol on up-state amplitude, duration, and the number of spikes per up state. Although ethanol reduced all three parameters in a dose-dependent manner, up-state duration appeared to be more sensitive to inhibition than up-state amplitude, especially at the lower concentrations of ethanol tested. For example, at a concentration of 25 mm, ethanol significantly depressed up-state duration by ∼40%, but did not affect up-state amplitude. As the ethanol concentration was increased to 50 mm, up-state amplitude and duration were reduced to ∼60 and 45%, respectively, of the control value and spike activity was inhibited by nearly 80%. A concentration of ethanol of 50 mm is equivalent to a blood alcohol level of ∼0.2% and is associated with moderate to severe intoxication in nontolerant humans. Increasing the ethanol concentration to 100 mm caused a slightly greater reduction in average up-state amplitude and duration and eliminated spike activity during up states in most neurons tested. The decrease in up-state amplitude by ethanol was caused by a decrease in the membrane potential achieved during the up state rather than a depolarized down state. For example, in 10 neurons tested with 50 mm ethanol, the mean down-state membrane potential before ethanol exposure was −71.1 mV (±2.4) whereas during ethanol exposure it was −69.2 mV (±2.2). Although the effect of ethanol on the number of spontaneous events (up states with or without firing) was not statistically significant, the number of events trended toward an increase at lower concentrations of ethanol (10–50 mm) and a decrease at 100 mm (data not shown). In addition, there was no significant correlation between the initial up-state amplitude, duration, or spiking during the control period and the degree of inhibition during ethanol exposure.
Figure 3.
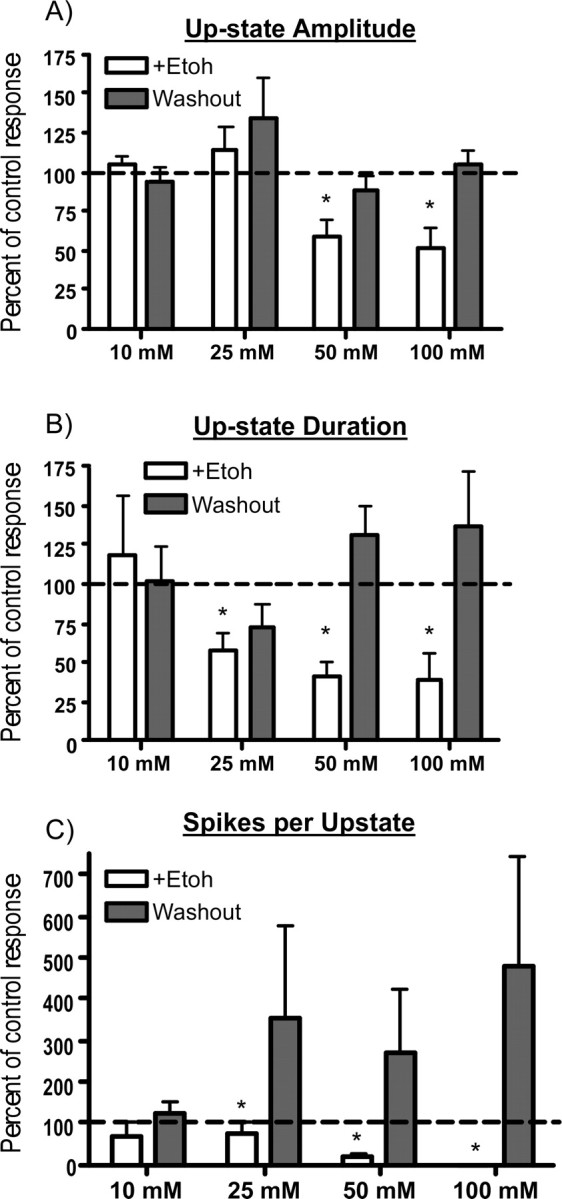
Effects of ethanol on individual components of spontaneous persistent activity of PFC neurons. A–C, Summary graphs show changes in up-state amplitude (A), up-state duration (B), and spike activity (C) during (open bars) and after (shaded bars) exposure to ethanol (Etoh; 10–100 mm). Data represent the mean (±SEM; n = 4–10 neurons per concentration) and are expressed as a percentage of the predrug control value. *Significantly different from corresponding control value (p < 0.05, ANOVA with repeated measures and post hoc testing).
After washout of ethanol, persistent activity was occasionally enhanced relative to the pre-ethanol control period (Fig. 2, washout traces). This rebound increase in activity was rapid in onset and, when present, was usually associated with increases in up-state duration or spike firing rather than changes in up-state amplitude (Fig. 3, shaded bars). In addition, although the magnitude of the increase in up-state duration and spiking after ethanol washout was cell-dependent, it was most often observed after exposure to ethanol concentrations of 25 mm and higher.
Effects of the NMDA antagonist APV
In a previous study by Seamans et al. (2003), saturating concentrations of the NMDA receptor antagonist APV mostly eliminated up-state transitions and persistent activity. Ethanol also reduces NMDA receptor activity although at the concentrations used in the present study (10–100 mm) this inhibition would be expected to be incomplete (Jin and Woodward, 2006). To further investigate the involvement of NMDA receptors in persistent activity of PFC neurons, slice cocultures were treated with a low concentration of APV (5 μm). This concentration only partially blocks NMDA receptor currents evoked by saturating concentrations of agonist and, thus, should more accurately reflect the low efficacy of ethanol as an NMDA antagonist (Blevins et al., 1995). As shown in Figure 4, 5 μm APV significantly reduced up-state amplitude, duration, and spike activity. For comparison, the effects of 50 mm ethanol on these parameters (taken from the data shown Fig. 3) are also shown. Although APV produced a slightly greater inhibition of persistent activity than 50 mm ethanol, no obvious rebound excitation was observed after washout.
Figure 4.

The NMDA antagonist APV reduces spontaneous persistent activity of PFC neurons in slice cocultures. A, Representative traces showing persistent activity of a single PFC pyramidal neuron before, during, and after exposure to 5 μm APV. B, C, Summary graphs show up-state amplitude, duration, and spike activity during (B) and after (C) exposure to APV (shaded bars). In B and C, the effects of 50 mm ethanol (Etoh) on persistent activity are shown for comparison (open bars; data taken from Fig. 3). Data represent the mean (±SEM; n = 5 for APV and 10 for 50 mm Etoh) and are expressed as a percentage of the corresponding predrug control value. *Significantly different from corresponding control value (p < 0.05, ANOVA with repeated measures and post hoc testing).
Figure 5.
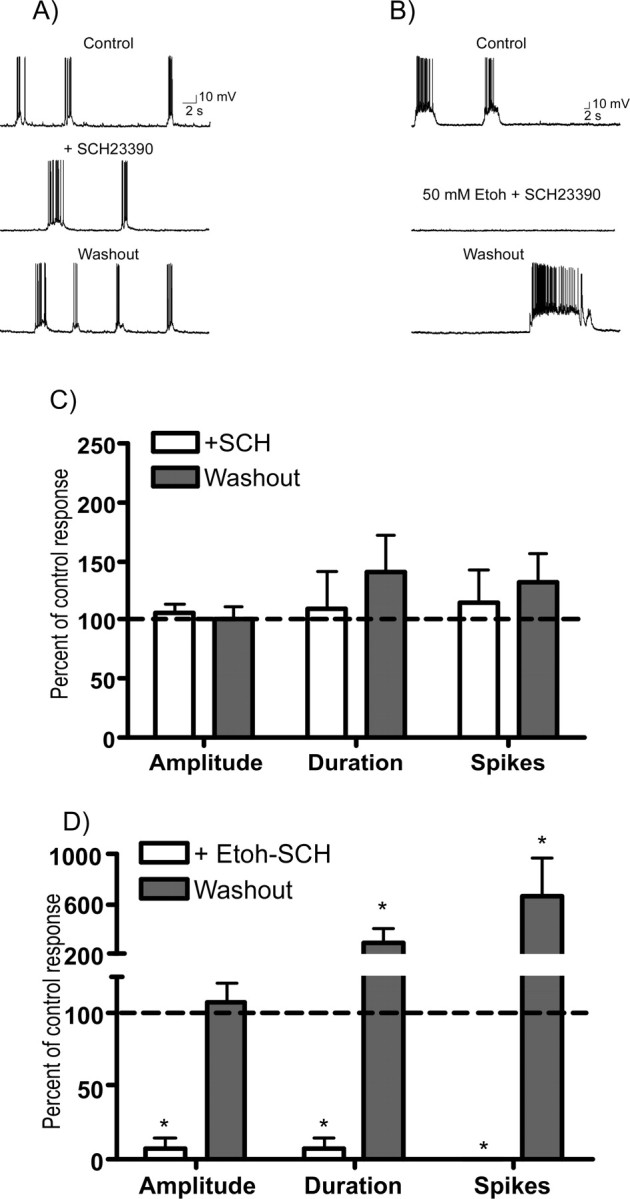
The D1 receptor antagonist SCH23390 enhances ethanol inhibition of spontaneous persistent activity. A, B, Representative traces of spontaneous persistent activity recorded from single PFC neurons in the presence of SCH23390 (SCH) alone (10 μm; A) or SCH23390 (10 μm) plus 50 mm ethanol (Etoh; B). C, D, Summary graphs show up-state amplitude, duration, and spike activity during (open bars) and after (shaded bars) exposure to SCH23990 (C) or SCH23390 plus ethanol (D). Data represent the mean (±SEM; n = 12 for SCH alone and 5 for SCH plus Etoh) and are expressed as a percentage of the corresponding predrug control value. *Significantly different from corresponding control value (p < 0.05, ANOVA with repeated measures with post hoc testing).
Ethanol, persistent activity, and dopamine
D1 antagonist studies
In vivo, the mesocorticolimbic dopamine system is a crucial component of the addiction neurocircuitry and dopaminergic fibers from the VTA make synaptic contact with deep-layer cortical pyramidal neurons (Berger et al., 1991). Dopamine has complex actions on both glutamatergic and GABAergic neurons within the prefrontal cortex and acts via multiple signaling pathways to modulate their excitability. D1 receptors are thought to be especially important in regulating PFC output and are highly expressed on deep-layer pyramidal neurons (for review, see Seamans and Yang, 2004). To investigate the potential involvement of D1 dopamine receptors in modulating the effects of ethanol on persistent activity, slices were exposed to the D1 antagonist R(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrochloride (SCH23390) alone or in combination with 50 mm ethanol. When administered alone, SCH23390 (10 μm) did not significantly alter persistent activity (Fig. 5A,C) suggesting that at this concentration, SCH23390 had no direct effect on NMDA receptors (Cui et al., 2006). In a separate group of neurons, coperfusion of slice cocultures with SCH23390 and 50 mm ethanol produced a marked inhibition of persistent activity that was greater than that seen in neurons exposed to 50 mm ethanol alone (compare Figs. 3, 5B,D). After washout of ethanol and SCH23390, average up-state duration and spike activity were greater than predrug control values whereas up-state amplitude showed little change (Fig. 5D).
VTA stimulation
In vivo, VTA dopamine neurons normally fire at a low frequency (1–5 Hz) whereas burst firing often accompanies presentation of relevant or novel stimuli (Schultz et al., 1997; Hyland et al., 2002). VTA stimulation evokes a glutamatergic EPSP in the PFC both in vitro and in vivo and can evoke up states and persistent activity in PFC neurons (Seamans et al., 2003; Lavin et al., 2005) In the present study, a stimulating electrode was placed in the VTA portion of the slice coculture and trains of stimuli (5 pulses at 20 Hz) were applied at regular intervals while recording from single PFC neurons. The amount of current delivered to the stimulating electrode was initially adjusted to yield reproducible responses in each recorded PFC neuron. Burst stimulation of the VTA induced up-state transitions and spike firing in PFC neurons (Fig. 6A). VTA-induced persistent activity was measured in 63 neurons in the absence and presence of ethanol. The average membrane potential during down states in these neurons was −74.2 mV (±0.9) whereas during up states it was −57.6 mV (±1.4). VTA-evoked up states had a mean amplitude of 16.6 mV (±1.1) and an average duration of 4.8 s (±0.3). The average number of spikes during each VTA-evoked up state was 19.4 (±3.5), with a mean frequency of firing of 3.9 Hz (±0.6).
Figure 6.
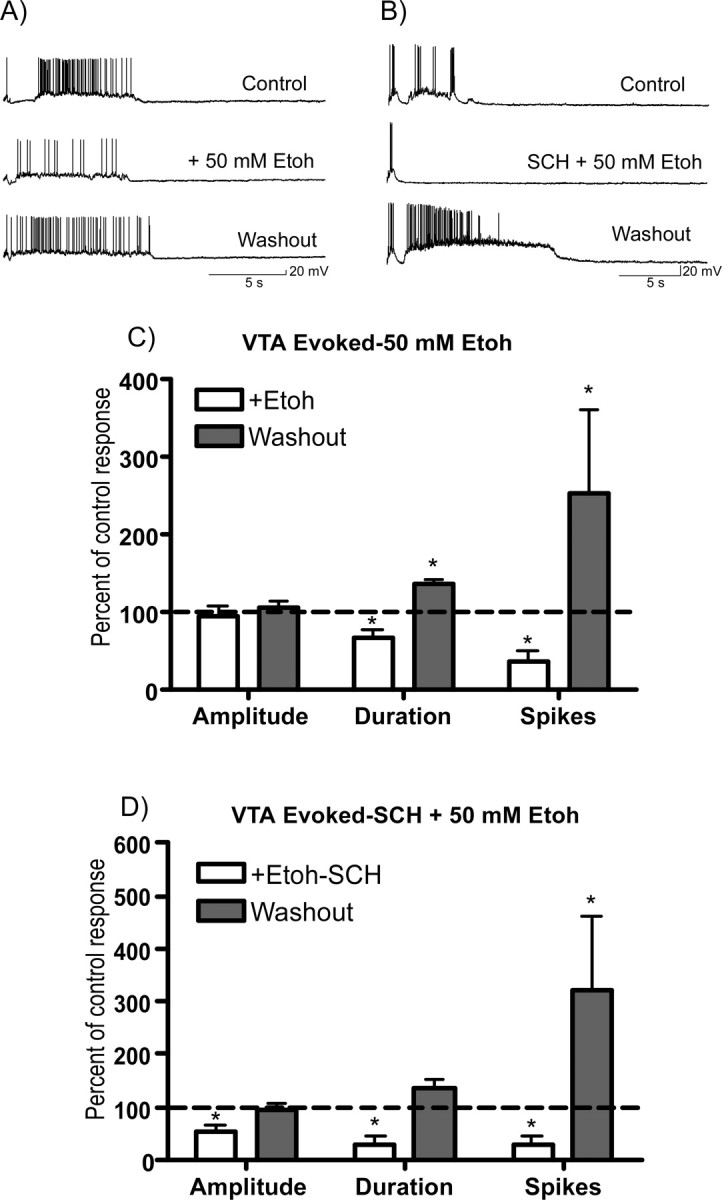
Effects of ethanol on persistent activity induced by VTA stimulation. A, B, Traces show VTA-evoked (5 pulse train, 20 Hz) persistent activity of single PFC neurons before, during, and after exposure to 50 mm ethanol (A) or the combination of 50 mm ethanol and 10 μm SCH23390 (B). C, D, Summary graphs below show effects of ethanol (C) or ethanol plus SCH23390 (D) on up-state amplitude, duration, and spike activity during (open bars) or after (shaded bars) washout of the test solution. Data represent the mean (±SEM; n = 8 VTA plus Etoh and 9 for VTA plus Etoh plus SCH) and are expressed as a percentage of the corresponding predrug control value. *Significantly different from corresponding control value (p < 0.05, ANOVA with repeated measures with post hoc testing).
Ethanol also reduced persistent activity of PFC neurons after VTA stimulation (Fig. 6A,C), although the magnitude of this inhibition was generally smaller than that observed for spontaneous activity. As shown in Figure 3, 50 mm ethanol inhibited the amplitude and duration of spontaneous up states by ∼40 and 60% of their corresponding control values. However, as shown in Figure 6C, during VTA stimulation, this same concentration of ethanol reduced up-state amplitude by ∼10% and up-state duration by 34%. A similar attenuation in the ethanol inhibition of VTA-stimulated activity was observed with other concentrations of ethanol (data not shown). After washout of the ethanol-containing solution, persistent activity evoked by VTA stimulation was often enhanced, although again the magnitude of this effect was cell-dependent (Fig. 6C, shaded bars). To determine the effect of D1 receptors on the ethanol inhibition of persistent activity induced by VTA stimulation, additional recordings were performed in the presence of the D1 antagonist SCH23390. In control studies, exposure of slice cultures to SCH23390 (10 μm) alone produced no significant effect on the mean (±SEM; n = 8) amplitude (104.5 ± 7.4%), duration (117.7 ± 23.7%), or spike-firing (109.7 ± 24.2%) of PFC neurons after VTA stimulation. In a separate group of neurons, persistent activity induced by VTA stimulation was markedly reduced during simultaneous application of SCH23390 (10 μm) and 50 mm ethanol (Fig. 6B,D). Under these conditions, up-state amplitude was reduced by ∼45% whereas up-state duration and number of spikes were inhibited by nearly 70%. After washout of ethanol and SCH23390, there was again evidence of a variable increase in persistent activity similar to that observed in the absence of SHC23390 (Fig. 6D).
Ethanol and VTA dopamine neuron firing
Ethanol enhances the spontaneous firing of VTA dopamine neurons from both rats and mice (Brodie et al., 1990; Okamoto et al., 2006). To determine whether ethanol causes a similar effect in slice cocultures, the spontaneous firing of VTA dopamine neurons was monitored by cell-attached recordings during application of ethanol. In these experiments, cultures were prepared from transgenic mice that express GFP under the control of a tyrosine hydroxylase promoter (Matsushita et al., 2002). Clusters of GFP-positive neurons were easily identified in the midbrain section of the coculture and PFC neurons in these cultures showed both spontaneous and evoked up states (Fig. 7A,B). The mean (± SEM; n = 17) firing rate of GFP-positive midbrain neurons under control conditions was 0.88 ± 0.13 Hz. Ethanol, at concentrations of 25 or 100 mm appeared to slightly enhance the spontaneous firing rate of midbrain dopamine neurons but this effect was variable and overall was not statistically significant (Fig. 7C,D).
Figure 7.
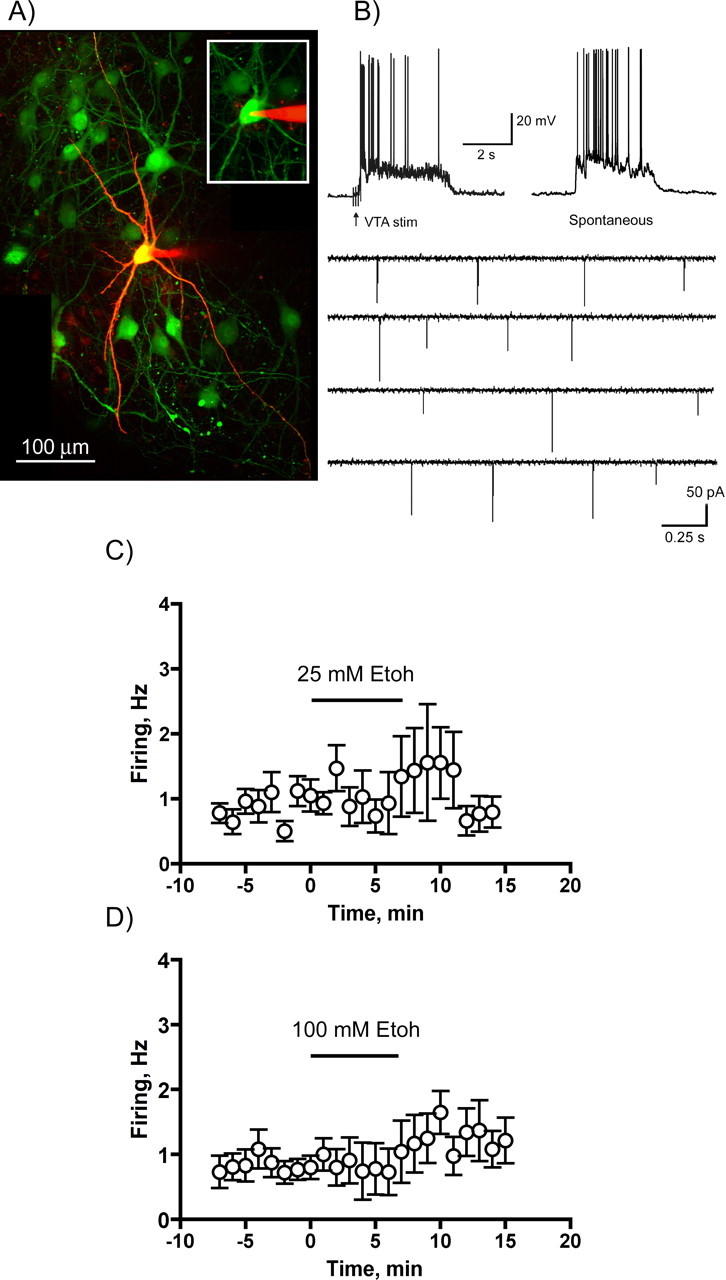
Effects of ethanol on the spontaneous firing of midbrain dopamine neurons in the slice cocultures. A, Midbrain portion of a slice coculture prepared from transgenic mice in which GFP expression is driven by a tyrosine-hydroxylase promoter. The neuron in the center was filled with Alexa 594 dye after recording of spontaneous firing activity. The inset shows the same neuron during cell-attached recording and before obtaining whole-cell access. B, Top two traces show VTA-evoked (left) and spontaneous (right) up states in PFC neurons from TH-GFP slice cocultures. The bottom traces show spontaneous firing of a single midbrain dopamine neuron. C, D, Summary figures show the average firing rate of dopamine neurons in the absence and presence (indicated by the horizontal bar) of 25 mm (C) or 100 mm (D) ethanol. Data are the mean (±SEM) from eight to nine different neurons for each ethanol concentration.
Persistent activity in PFC/hippocampal cultures
To further investigate the role of dopamine in the ethanol modulation of persistent activity, a series of recordings were performed in cultures containing only the PFC and hippocampal slices. Similar to triple-slice cultures, PFC neurons in cultures lacking DA neurons showed spontaneous episodes of persistent activity (Fig. 8A). The mean (±SEM, n = 5) amplitude, duration, and number of spikes per up state in these neurons were 17.4 ± 3.1 mV, 2.0 ± 0.4 s, and 15.3 ± 5.8; respectively. Ethanol (50 mm) inhibited up-state amplitude by ∼30% and up-state duration and spiking were reduced by 70–75%; respectively. After washout of the ethanol containing solution, up-state amplitude was similar to that of control values whereas up-state duration and number of spikes per up state were significantly increased relative to the control values.
Figure 8.

Effects of ethanol on spontaneous persistent activity of PFC neurons in slice cultures lacking midbrain dopamine neurons. A, Representative traces of persistent activity in a single PFC neuron before, during, and after exposure to 50 mm ethanol. B, Summary graph showing effect of ethanol on up-state amplitude, duration, and spike activity during (open bars) or after (shaded bars) washout of the ethanol containing solution. Data represent the mean (±SEM; n = 5) and are expressed as a percentage of the corresponding predrug control value. *Significantly different from corresponding control value (p < 0.05, ANOVA with repeated measures with post hoc testing).
Discussion
The results of this study demonstrate that ethanol markedly affects the activity of PFC neurons. Ethanol inhibited persistent activity and spike firing in the slice cocultures and reduced PFC firing in vivo. The concentrations of ethanol that produced these effects (17–100 mm; ∼0.08–0.4% blood alcohol concentration) are similar to those that alter PFC-mediated behaviors including working memory, error detection (Melchior et al., 1993; Givens and McMahon, 1997; Ridderinkhof et al., 2002; Rossetti et al., 2002), and EEG responses after stimulation of the frontal cortex in humans (Kahkonen et al., 2003). Ethanol's inhibition of persistent activity in the slice cocultures appeared to involve synaptic sites of action, as even high concentrations of ethanol had little effect on firing evoked by direct current injection.
Ethanol modulation of PFC persistent activity
The cellular mechanisms that regulate persistent activity of PFC neurons are complex and require coordinated glutamatergic and GABAergic synaptic inputs. Perfusion of slice cocultures with antagonists of non-NMDA or NMDA receptors inhibits persistent activity whereas direct activation of GABAA receptors clamps the membrane potential below the threshold needed for firing (Seamans et al., 2003). Inhibition of persistent activity could thus arise if ethanol inhibited excitatory glutamatergic activity or enhanced GABAergic tone or both. To our knowledge, the direct effect of ethanol on synaptic glutamatergic and GABAergic transmission in deep-layer neurons of the prefrontal cortex has not been reported. However, results from studies from our laboratory show that, in acute slices, synaptic NMDA currents from deep-layer PFC neurons are reliably inhibited by moderate (44 mm) concentrations of ethanol whereas those generated by AMPA or GABAA receptors are mostly unaffected (our unpublished observations). These findings are generally consistent with other studies showing that ethanol has little effect on most AMPA-mediated EPSCs (Lovinger et al., 1989; Weiner et al., 1999) (but see Mameli et al., 2005; Carta et al., 2006) and has variable effects on GABA-mediated currents (for review, see Weiner and Valenzuela, 2006). Importantly, NMDA-dependent response processes are consistently reported to be inhibited by ethanol including those from hippocampus (Lovinger et al., 1990), posterior cingulate (Li et al., 2002), nucleus accumbens (Nie et al., 1994), and amygdala (Roberto et al., 2004). It should be noted that the ethanol inhibition of NMDA receptors is incomplete even at concentrations associated with severe behavioral impairment. For example, in a study of 32 different recombinant NMDA receptors, 100 mm ethanol inhibited currents by 18–55%, depending on the subunits expressed (Jin and Woodward, 2006). Whether this range of inhibition is sufficient to reduce persistent activity of PFC neurons is not completely known. However, computational modeling studies show that NMDA currents are critical in maintaining persistent activity and that even modest reductions in NMDA receptor input significantly inhibit up states (Wang, 1999; Durstewitz et al., 2000; Wolf et al., 2005). This was verified in the present study as activity was significantly reduced by the NMDA antagonist APV at a concentration predicted to only partially inhibit synaptic NMDA receptor activity. Although these findings do not rule out an important role for other processes, they suggest that NMDA receptors are likely targets for ethanol's effect of persistent activity.
Dopamine modulation of ethanol inhibition of persistent activity
The degree of ethanol inhibition of persistent activity was affected by conditions designed to alter dopamine receptor-mediated signaling. For example, inhibition was enhanced in the presence of the D1 antagonist SCH23390 whereas VTA stimulated activity was less affected. The mechanism(s) underlying these effects are likely to be complex and involve multiple pathways given the wide range of action of dopamine on neuronal function (Seamans and Yang, 2004). Interestingly, D1 receptor agonists have been reported to decrease the ethanol inhibition of NMDA EPSCs in nucleus accumbens neurons (Maldve et al., 2002; Zhang et al., 2005). This effect was correlated with increased phosphorylation of the NR1 PKA (protein kinase A) site as measured by a phosphospecific antibody, suggesting that this site may directly regulate the ethanol sensitivity of NMDA receptors. Although this hypothesis was not supported by results of a subsequent study (Xu and Woodward, 2006), D1-mediated changes in network excitability could reduce ethanol inhibition of persistent activity without altering the acute ethanol sensitivity of NMDA receptors. For example, AMPA and NMDA receptor function are enhanced after D1 receptor activation through changes in channel kinetics and/or increases in forward trafficking and membrane expression of these receptors (Flores-Hernandez et al., 2002; Mangiavacchi and Wolf, 2004; Wirkner et al., 2004; Sun et al., 2005). D1-mediated potentiation of ionotropic glutamate receptor signaling may enhance excitatory drive of PFC networks to levels greater than that needed to initiate and maintain persistent activity. Pharmacological blockade of this signaling pathway would remove this modulatory influence and make the network more susceptible to ethanol-induced disruption.
Ethanol and rebound excitation after washout
A rebound increase in persistent activity of PFC neurons was often observed after washout of ethanol and was associated with changes in up-state duration and spiking, as up-state amplitude was not affected. Although the mechanism underlying this effect is unknown, NMDA receptors may be involved as these receptors are critical in maintaining the sustained depolarization underlying up states (Durstewitz et al., 2000; Seamans et al., 2003). Two pathways that could lead to enhanced NMDA receptor drive include ethanol-induced increases in D1 signaling and increased tyrosine phosphorylation of NMDA receptors. Ethanol may increase dopamine signaling through an enhancement of VTA dopamine neuron firing, leading to increased excitability of PFC neurons. Two observations reduce the chances that this action alone drives increased activity after ethanol washout. First, although ethanol does enhance DA neuron firing (Gessa et al., 1985; Appel et al., 2003; Okamoto et al., 2006), the magnitude of this change is relatively small, on the order of 15–20%. We found evidence for similar small changes in DA neuron firing in the present study, although these did not reach statistical significance. Second, and perhaps more importantly, slice cultures containing only the PFC and hippocampus still showed increases in up-state duration and spike firing after ethanol exposure, suggesting that dopamine is not required for this action. Similar ethanol-induced increases in NMDA-dependent responses have been reported in preparations of locus ceruleus, hippocampus, or spinal cord neurons, where dopamine cell bodies are also absent (Morrisett, 1994; Poelchen et al., 1997; Li and Kendig, 2003; Yaka et al., 2003). The rebound effect in these neurons was associated with increases in the tyrosine phosphorylation of NR2 NMDA receptor subunits and was blocked by inhibitors of Src-family tyrosine kinases (Li and Kendig, 2003; Yaka et al., 2003). These kinases increase the activity of both recombinant and native NMDA receptors via enhanced insertion and reduced internalization of surface receptors (Wang and Salter, 1994; Chen and Leonard, 1996; Skeberdis et al., 2001; Vissel et al., 2001; Dunah et al., 2004). The pathway linking ethanol to enhanced tyrosine phosphorylation of NMDA receptors is not completely known, but may involve dissociation of regulatory proteins such as Rack-1 that bind to and reduce the activity of Fyn and other Src-family tyrosine kinases (Yaka et al., 2002).
Whether PFC neurons in vivo show a similar rebound in activity is currently not known. Unlike the situation with the slice-culture system in which solution exchange is rapid, removal of ethanol in vivo is slow because of the zero-order kinetics of alcohol metabolism. Results from the in vivo recordings of PFC spike activity reflect this difference in pharmacokinetics because firing rates after ethanol administration were suppressed for prolonged periods. However, in nonanesthetized animals and human subjects, rapid tolerance to some effects of ethanol does occur, suggesting adaptation of neurons in certain brain regions during sustained exposure to behaviorally relevant concentrations of ethanol (Schwarz et al., 1981; Miyakawa et al., 1997; Chandler et al., 1998; Fadda and Rossetti, 1998). Although highly speculative, a similar recovery in the activity of PFC neurons during in vivo ethanol exposure could represent a form of acute functional tolerance that might limit ethanol-induced impairment of processes subserved by these neurons. Differences in the extent and/or rate of this compensation could contribute to differences in the susceptibility of individuals to ethanol-induced changes in prefrontal function that may contribute to the development of alcohol addiction.
Functional implications
The results of this study provide the first direct evidence that ethanol disrupts complex patterns of persistent activity in deep-layer pyramidal neurons of the PFC. Given the importance of this area in planning and decision-making, these effects may underlie some of the deficits in cognition, working memory, and executive function observed after ethanol intake. This impairment in prefrontal processing may also reduce performance and error monitoring during complex tasks and diminish one's ability to inhibit deleterious behaviors including control over alcohol drinking. Sustained alterations or adaptations in prefrontal function resulting from chronic drug and alcohol use could contribute to the loss of inhibitory control over drug taking that characterizes alcoholism and drug addiction. These results thus provide an important foundation from which to explore the mechanisms by which alcohol leads to these changes.
Footnotes
This work was supported by National Institutes of Health Grants P50AA10761 (Charleston Alcohol Research Center), R01AA010983 (L.J.C.), and R01AA09986 and K02AA00238 (J.J.W.).
References
- Appel SB, Liu Z, McElvain MA, Brodie MS. Ethanol excitation of dopaminergic ventral tegmental area neurons is blocked by quinidine. J Pharmacol Exp Ther. 2003;306:437–446. doi: 10.1124/jpet.103.050963. [DOI] [PubMed] [Google Scholar]
- Berger B, Gaspar P, Verney C. Dopaminergic innervation of the cerebral cortex: unexpected differences between rodents and primates. Trends Neurosci. 1991;14:21–27. doi: 10.1016/0166-2236(91)90179-x. [DOI] [PubMed] [Google Scholar]
- Blevins T, Mirshahi T, Woodward JJ. Increased agonist and antagonist sensitivity of N-methyl-d-aspartate stimulated calcium flux in cultured neurons following chronic ethanol exposure. Neurosci Lett. 1995;200:214–218. doi: 10.1016/0304-3940(95)12086-j. [DOI] [PubMed] [Google Scholar]
- Brodie MS, Shefner SA, Dunwiddie TV. Ethanol increases the firing rate of dopamine neurons of the rat ventral tegmental area in vitro. Brain Res. 1990;508:65–69. doi: 10.1016/0006-8993(90)91118-z. [DOI] [PubMed] [Google Scholar]
- Carpenter-Hyland EP, Woodward JJ, Chandler LJ. Chronic ethanol induces synaptic but not extrasynaptic targeting of NMDA receptors. J Neurosci. 2004;24:7859–7868. doi: 10.1523/JNEUROSCI.1902-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carta M, Mameli M, Valenzuela CF. Alcohol potently modulates climbing fiber→Purkinje neuron synapses: role of metabotropic glutamate receptors. J Neurosci. 2006;26:1906–1912. doi: 10.1523/JNEUROSCI.4430-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler LJ, Harris RA, Crews FT. Ethanol tolerance and synaptic plasticity. Trends Pharmacol Sci. 1998;19:491–495. doi: 10.1016/s0165-6147(98)01268-1. [DOI] [PubMed] [Google Scholar]
- Chen SJ, Leonard JP. Protein tyrosine kinase-mediated potentiation of currents from cloned NMDA receptors. J Neurochem. 1996;67:194–200. doi: 10.1046/j.1471-4159.1996.67010194.x. [DOI] [PubMed] [Google Scholar]
- Cui C, Xu M, Atzori M. Voltage-dependent block of N-methyl-D-aspartate receptors by dopamine D1 receptor ligands. Mol Pharmacol. 2006;70:1761–1770. doi: 10.1124/mol.106.028332. [DOI] [PubMed] [Google Scholar]
- Dunah AW, Sirianni AC, Fienberg AA, Bastia E, Schwarzschild MA, Standaert DG. Dopamine D1-dependent trafficking of striatal N-methyl-D-aspartate glutamate receptors requires Fyn protein tyrosine kinase but not DARPP-32. Mol Pharmacol. 2004;65:121–129. doi: 10.1124/mol.65.1.121. [DOI] [PubMed] [Google Scholar]
- Durstewitz D, Seamans JK, Sejnowski TJ. Neurocomputational models of working memory. Nat Neurosci. 2000;3(Suppl):1184–1191. doi: 10.1038/81460. [DOI] [PubMed] [Google Scholar]
- Fadda F, Rossetti ZL. Chronic ethanol consumption: from neuroadaptation to neurodegeneration. Prog Neurobiol. 1998;56:385–431. doi: 10.1016/s0301-0082(98)00032-x. [DOI] [PubMed] [Google Scholar]
- Flores-Hernandez J, Cepeda C, Hernandez-Echeagaray E, Calvert CR, Jokel ES, Fienberg AA, Greengard P, Levine MS. Dopamine enhancement of NMDA currents in dissociated medium-sized striatal neurons: role of D1 receptors and DARPP-32. J Neurophysiol. 2002;88:3010–3020. doi: 10.1152/jn.00361.2002. [DOI] [PubMed] [Google Scholar]
- Franke H, Schelhorn N, Illes P. Dopaminergic neurons develop axonal projections to their target areas in organotypic co-cultures of the ventral mesencephalon and the striatum/prefrontal cortex. Neurochem Int. 2003;42:431–439. doi: 10.1016/s0197-0186(02)00134-1. [DOI] [PubMed] [Google Scholar]
- Gessa GL, Muntoni F, Collu M, Vargiu L, Mereu G. Low doses of ethanol activate dopaminergic neurons in the ventral tegmental area. Brain Res. 1985;348:201–203. doi: 10.1016/0006-8993(85)90381-6. [DOI] [PubMed] [Google Scholar]
- Givens B, McMahon K. Effects of ethanol on nonspatial working memory and attention in rats. Behav Neurosci. 1997;111:275–282. doi: 10.1037//0735-7044.111.2.275. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS. The physiological approach: functional architecture of working memory and disordered cognition in schizophrenia. Biol Psychiatry. 1999;46:650–661. doi: 10.1016/s0006-3223(99)00130-4. [DOI] [PubMed] [Google Scholar]
- Hyland BI, Reynolds JN, Hay J, Perk CG, Miller R. Firing modes of midbrain dopamine cells in the freely moving rat. Neuroscience. 2002;114:475–492. doi: 10.1016/s0306-4522(02)00267-1. [DOI] [PubMed] [Google Scholar]
- Jin C, Woodward JJ. Effects of 8 different NR1 splice variants on the ethanol inhibition of recombinant NMDA receptors. Alcohol Clin Exp Res. 2006;30:673–679. doi: 10.1111/j.1530-0277.2006.00079.x. [DOI] [PubMed] [Google Scholar]
- Kahkonen S, Wilenius J, Nikulin VV, Ollikainen M, Ilmoniemi RJ. Alcohol reduces prefrontal cortical excitability in humans: a combined TMS and EEG study. Neuropsychopharmacology. 2003;28:747–754. doi: 10.1038/sj.npp.1300099. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Volkow N, Seamans J. Unmanageable motivation in addiction: a pathology in prefrontal-accumbens glutamate transmission. Neuron. 2005;45:647–650. doi: 10.1016/j.neuron.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Kamarajan C, Porjesz B, Jones KA, Choi K, Chorlian DB, Padmanabhapillai A, Rangaswamy M, Stimus AT, Begleiter H. The role of brain oscillations as functional correlates of cognitive systems: a study of frontal inhibitory control in alcoholism. Int J Psychophysiol. 2004;51:155–180. doi: 10.1016/j.ijpsycho.2003.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin A, Nogueira L, Lapish CC, Wightman RM, Phillips PE, Seamans JK. Mesocortical dopamine neurons operate in distinct temporal domains using multimodal signaling. J Neurosci. 2005;25:5013–5023. doi: 10.1523/JNEUROSCI.0557-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BL, O'Donnell P. Ventral tegmental area afferents to the prefrontal cortex maintain membrane potential “up” states in pyramidal neurons via D(1) dopamine receptors. Cereb Cortex. 2000;10:1168–1175. doi: 10.1093/cercor/10.12.1168. [DOI] [PubMed] [Google Scholar]
- Li HF, Kendig JJ. Ethanol withdrawal hyper-responsiveness mediated by NMDA receptors in spinal cord motor neurons. Br J Pharmacol. 2003;139:73–80. doi: 10.1038/sj.bjp.0705198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Wilson WA, Swartzwelder HS. Differential effect of ethanol on NMDA EPSCs in pyramidal cells in the posterior cingulate cortex of juvenile and adult rats. J Neurophysiol. 2002;87:705–711. doi: 10.1152/jn.00433.2001. [DOI] [PubMed] [Google Scholar]
- Lovinger DM, White G, Weight FF. Ethanol inhibits NMDA-activated ion current in hippocampal neurons. Science. 1989;243:1721–1724. doi: 10.1126/science.2467382. [DOI] [PubMed] [Google Scholar]
- Lovinger DM, White G, Weight FF. NMDA receptor-mediated synaptic excitation selectively inhibited by ethanol in hippocampal slice from adult rat. J Neurosci. 1990;10:1372–1379. doi: 10.1523/JNEUROSCI.10-04-01372.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldve RE, Zhang TA, Ferrani-Kile K, Schreiber SS, Lippmann MJ, Snyder GL, Fienberg AA, Leslie SW, Gonzales RA, Morrisett RA. DARPP-32 and regulation of the ethanol sensitivity of NMDA receptors in the nucleus accumbens. Nat Neurosci. 2002;5:641–648. doi: 10.1038/nn877. [DOI] [PubMed] [Google Scholar]
- Mameli M, Zamudio PA, Carta M, Valenzuela CF. Developmentally regulated actions of alcohol on hippocampal glutamatergic transmission. J Neurosci. 2005;25:8027–8036. doi: 10.1523/JNEUROSCI.2434-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangiavacchi S, Wolf ME. D1 dopamine receptor stimulation increases the rate of AMPA receptor insertion onto the surface of cultured nucleus accumbens neurons through a pathway dependent on protein kinase A. J Neurochem. 2004;88:1261–1271. doi: 10.1046/j.1471-4159.2003.02248.x. [DOI] [PubMed] [Google Scholar]
- Marshall L, Helgadottir H, Molle M, Born J. Boosting slow oscillations during sleep potentiates memory. Nature. 2006;444:610–613. doi: 10.1038/nature05278. [DOI] [PubMed] [Google Scholar]
- Matsushita N, Okada H, Yasoshima Y, Takahashi K, Kiuchi K, Kobayashi K. Dynamics of tyrosine hydroxylase promoter activity during midbrain dopaminergic neuron development. J Neurochem. 2002;82:295–304. doi: 10.1046/j.1471-4159.2002.00972.x. [DOI] [PubMed] [Google Scholar]
- Melchior CL, Glasky AJ, Ritzmann RF. A low dose of ethanol impairs working memory in mice in a win-shift foraging paradigm. Alcohol. 1993;10:491–493. doi: 10.1016/0741-8329(93)90071-u. [DOI] [PubMed] [Google Scholar]
- Miyakawa T, Yagi T, Kitazawa H, Yasuda M, Kawai N, Tsuboi K, Niki H. Fyn-kinase as a determinant of ethanol sensitivity: relation to NMDA-receptor function. Science. 1997;278:698–701. doi: 10.1126/science.278.5338.698. [DOI] [PubMed] [Google Scholar]
- Morrisett RA. Potentiation of N-methyl-D-aspartate receptor-dependent afterdischarges in rat dentate gyrus following in vitro ethanol withdrawal. Neurosci Lett. 1994;167:175–178. doi: 10.1016/0304-3940(94)91055-3. [DOI] [PubMed] [Google Scholar]
- Moselhy HF, Georgiou G, Kahn A. Frontal lobe changes in alcoholism: a review of the literature. Alcohol Alcohol. 2001;36:357–368. doi: 10.1093/alcalc/36.5.357. [DOI] [PubMed] [Google Scholar]
- Nie Z, Madamba SG, Siggins GR. Ethanol inhibits glutamatergic neurotransmission in nucleus accumbens neurons by multiple mechanisms. J Pharmacol Exp Ther. 1994;271:1566–1573. [PubMed] [Google Scholar]
- O'Donnell P, Grace AA. Synaptic interactions among excitatory afferents to nucleus accumbens neurons: hippocampal gating of prefrontal cortical input. J Neurosci. 1995;15:3622–3639. doi: 10.1523/JNEUROSCI.15-05-03622.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto T, Harnett MT, Morikawa H. Hyperpolarization-activated cation current (Ih) is an ethanol target in midbrain dopamine neurons of mice. J Neurophysiol. 2006;95:619–626. doi: 10.1152/jn.00682.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poelchen W, Nieber K, Illes P. Tolerance to inhibition by ethanol of N-methyl-D-aspartate-induced depolarization in rat locus coeruleus neurons in vitro. Eur J Pharmacol. 1997;332:267–271. doi: 10.1016/s0014-2999(97)01113-8. [DOI] [PubMed] [Google Scholar]
- Ridderinkhof KR, Vlugt Y, Bramlage A, Spaan M, Elton M, Snel J, Band GP. Alcohol consumption impairs detection of performance errors in mediofrontal cortex. Science. 2002;298:2209–2211. doi: 10.1126/science.1076929. [DOI] [PubMed] [Google Scholar]
- Roberto M, Schweitzer P, Madamba SG, Stouffer DG, Parsons LH, Siggins GR. Acute and chronic ethanol alter glutamatergic transmission in rat central amygdala: an in vitro and in vivo analysis. J Neurosci. 2004;24:1594–1603. doi: 10.1523/JNEUROSCI.5077-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossetti ZL, Carboni S, Stancampiano R, Sori P, Pepeu G, Fadda F. Bidirectional modulation of spatial working memory by ethanol. Alcohol Clin Exp Res. 2002;26:181–185. [PubMed] [Google Scholar]
- Sceniak MP, Maciver MB. Cellular actions of urethane on rat visual cortical neurons in vitro. J Neurophysiol. 2006;95:3865–3874. doi: 10.1152/jn.01196.2005. [DOI] [PubMed] [Google Scholar]
- Schultz W, Dayan P, Montague PR. A neural substrate of prediction and reward. Science. 1997;275:1593–1599. doi: 10.1126/science.275.5306.1593. [DOI] [PubMed] [Google Scholar]
- Schwarz E, Kielholz P, Hobi V, Goldberg L, Gilsdorf U, Hofstetter M, Ladewig D, Miest PC, Reggiani G, Richter R. Alcohol-induced biphasic background and stimulus-elicited EEG changes in relation to blood alcohol levels. Int J Clin Pharmacol Ther Toxicol. 1981;19:102–111. [PubMed] [Google Scholar]
- Seamans JK, Yang CR. The principal features and mechanisms of dopamine modulation in the prefrontal cortex. Prog Neurobiol. 2004;74:1–58. doi: 10.1016/j.pneurobio.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Seamans JK, Nogueira L, Lavin A. Synaptic basis of persistent activity in prefrontal cortex in vivo and in organotypic cultures. Cereb Cortex. 2003;13:1242–1250. doi: 10.1093/cercor/bhg094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skeberdis VA, Lan Jy, Zheng X, Zukin RS, Bennett MV. Insulin promotes rapid delivery of N-methyl-D-aspartate receptors to the cell surface by exocytosis. Proc Natl Acad Sci USA. 2001;98:3561–3566. doi: 10.1073/pnas.051634698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M. Impact of network activities on neuronal properties in corticothalamic systems. J Neurophysiol. 2001;86:1–39. doi: 10.1152/jn.2001.86.1.1. [DOI] [PubMed] [Google Scholar]
- Steriade M, Nunez A, Amzica F. A novel slow (<1 Hz) oscillation of neocortical neurons in vivo: depolarizing and hyperpolarizing components. J Neurosci. 1993;13:3252–3265. doi: 10.1523/JNEUROSCI.13-08-03252.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan EV, Pfefferbaum A. Neurocircuitry in alcoholism: a substrate of disruption and repair. Psychopharmacology (Berl) 2005;180:583–594. doi: 10.1007/s00213-005-2267-6. [DOI] [PubMed] [Google Scholar]
- Sun X, Zhao Y, Wolf ME. Dopamine receptor stimulation modulates AMPA receptor synaptic insertion in prefrontal cortex neurons. J Neurosci. 2005;25:7342–7351. doi: 10.1523/JNEUROSCI.4603-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vissel B, Krupp JJ, Heinemann SF, Westbrook GL. A use-dependent tyrosine dephosphorylation of NMDA receptors is independent of ion flux. Nat Neurosci. 2001;4:587–596. doi: 10.1038/88404. [DOI] [PubMed] [Google Scholar]
- Wang XJ. Synaptic basis of cortical persistent activity: the importance of NMDA receptors to working memory. J Neurosci. 1999;19:9587–9603. doi: 10.1523/JNEUROSCI.19-21-09587.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YT, Salter MW. Regulation of NMDA receptors by tyrosine kinases and phosphatases. Nature. 1994;369:233–235. doi: 10.1038/369233a0. [DOI] [PubMed] [Google Scholar]
- Weiner JL, Valenzuela CF. Ethanol modulation of GABAergic transmission: the view from the slice. Pharmacol Ther. 2006;111:533–554. doi: 10.1016/j.pharmthera.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Weiner JL, Dunwiddie TV, Valenzuela CF. Ethanol inhibition of synaptically evoked kainate responses in rat hippocampal CA3 pyramidal neurons. 1999;56:85–90. doi: 10.1124/mol.56.1.85. [DOI] [PubMed] [Google Scholar]
- Wirkner K, Krause T, Koles L, Thummler S, Al-Khrasani M, Illes P. D1 but not D2 dopamine receptors or adrenoceptors mediate dopamine-induced potentiation of N-methyl-D-aspartate currents in the rat prefrontal cortex. Neurosci Lett. 2004;372:89–93. doi: 10.1016/j.neulet.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Wolf JA, Moyer JT, Lazarewicz MT, Contreras D, Benoit-Marand M, O'Donnell P, Finkel LH. NMDA/AMPA ratio impacts state transitions and entrainment to oscillations in a computational model of the nucleus accumbens medium spiny projection neuron. J Neurosci. 2005;25:9080–9095. doi: 10.1523/JNEUROSCI.2220-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Woodward JJ. Ethanol inhibition of NMDA receptors under conditions of altered protein kinase A activity. J Neurochem. 2006;96:1760–1767. doi: 10.1111/j.1471-4159.2006.03703.x. [DOI] [PubMed] [Google Scholar]
- Yaka R, Thornton C, Vagts AJ, Phamluong K, Bonci A, Ron D. NMDA receptor function is regulated by the inhibitory scaffolding protein, RACK1. Proc Natl Acad Sci USA. 2002;99:5710–5715. doi: 10.1073/pnas.062046299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaka R, Phamluong K, Ron D. Scaffolding of Fyn kinase to the NMDA receptor determines brain region sensitivity to ethanol. J Neurosci. 2003;23:3623–3632. doi: 10.1523/JNEUROSCI.23-09-03623.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang TA, Hendricson AW, Morrisett RA. Dual synaptic sites of D(1)-dopaminergic regulation of ethanol sensitivity of NMDA receptors in nucleus accumbens. Synapse. 2005;58:30–44. doi: 10.1002/syn.20181. [DOI] [PubMed] [Google Scholar]