

Abstract
Numerous cellular responses to hypoxia are mediated by the transcription factor hypoxia-inducible factor-1 (HIF-1). HIF-1 plays a central role in the pathogenesis of hypoxic pulmonary hypertension. Under certain conditions, HIF-1 may utilize feedforward mechanisms to amplify its activity. Since hypoxia increases endothelin-1 (ET-1) levels in the lung, we hypothesized that during moderate, prolonged hypoxia ET-1 might contribute to HIF-1 signaling in pulmonary arterial smooth muscle cells (PASMCs). Primary cultures of rat PASMCs were treated with ET-1 or exposed to moderate, prolonged hypoxia (4% O2 for 60 h). Levels of the oxygen-sensitive HIF-1α subunit and expression of HIF target genes were increased in both hypoxic cells and cells treated with ET-1. Both hypoxia and ET-1 also increased HIF-1α mRNA expression and decreased mRNA and protein expression of prolyl hydroxylase 2 (PHD2), which is the protein responsible for targeting HIF-1α for O2-dependent degradation. The induction of HIF-1α by moderate, prolonged hypoxia was blocked by BQ-123, an antagonist of ET-1 receptor subtype A. The effects of ET-1 were mediated by increased intracellular calcium, generation of reactive oxygen species, and ERK1/2 activation. Neither ET-1 nor moderate hypoxia induced the expression of HIF-1α or HIF target genes in aortic smooth muscle cells. These results suggest that ET-1 induces a PASMC-specific increase in HIF-1α levels by upregulation of HIF-1α synthesis and downregulation of PHD2-mediated degradation, thereby amplifying the induction of HIF-1α in PASMCs during moderate, prolonged hypoxia.
Keywords: calcium, prolyl hydroxylase, pulmonary hypertension
in a variety of chronic lung diseases, the pulmonary circulation is exposed to prolonged periods of hypoxia, often resulting in the development of pulmonary hypertension. Numerous studies have described the structural and functional changes that occur in the pulmonary circulation in response to chronic hypoxia (60). Structural remodeling, characterized by pulmonary arterial smooth muscle cell (PASMC) proliferation, intimal thickening, and extension of muscle into previously nonmuscular arterioles, is commonly observed with pulmonary hypertension (23, 40). Changes in the vascular wall are accompanied by active contraction of vessels, evidenced by acute reduction in pulmonary arterial pressure in response to vasodilatory agents (42, 44). These pulmonary vascular changes result, in large part, from altered expression of genes encoding ion channels and transporters that control PASMC ion homeostasis, including increased expression of Na+/H+ exchanger isoform 1 (NHE1) and the canonical transient receptor potential (TRPC) family members TRPC1 and TRPC6, as well as reduced levels of mRNAs encoding voltage-gated K+ (KV) channel family members KV1.5 and KV2.1 (22, 48, 61, 63).
We have demonstrated a key role for the transcription factor, hypoxia-inducible factor-1 (HIF-1), in the development of hypoxic pulmonary hypertension (1, 48, 49, 61, 70). HIF-1 exists as a heterodimer, consisting of HIF-1α and HIF-1β subunits. HIF-1β is ubiquitously expressed, whereas expression of HIF-1α is tightly regulated by oxygen tension, thus conferring sensitivity and specificity for hypoxic induction (26, 69). While HIF-1α mRNA transiently increased within 30 min of exposure to 7% O2 in mouse lung (64), HIF-1α is also subjected to posttranslational regulation. Under normoxic conditions, human HIF-1α protein is ubiquitinated by a mechanism requiring hydroxylation at proline residue 403 and/or 564, which is catalyzed by prolyl hydroxylase domain (PHD) proteins with molecular O2 as a substrate (25, 27). Although four PHD isoforms have been identified, only PHD1–3 hydroxylate HIF-1α, with data suggesting that PHD2 is the primary isoform responsible for in vivo hydroxylation of HIF-1α (2, 4, 39). At reduced O2 concentrations, PHD activity decreases, with almost complete inhibition below 2% O2 (16), resulting in rapid stabilization of HIF-1α, which then translocates into the nucleus, binds HIF-1β, and recruits coactivators to increase the transcription of hundreds of genes. PHD1–3 are expressed in a variety of tissues (58), although the isoforms expressed in PASMCs have not been determined. HIF-1 activity is also controlled by factor inhibiting HIF-1 (FIH-1), an asparaginyl hydroxylase that regulates the transactivation function of HIF-1α (34).
HIF-1 mediates its effects in the hypoxic pulmonary circulation, in part, via regulation of NHE1, TRPC1, TRPC6, KV1.5, and KV2.1 expression (48, 49, 61, 63). Recently, we reported that the HIF-1-dependent effects of hypoxia on KV channel expression were mediated by the vasoactive peptide, endothelin-1 (ET-1) (63), which is encoded by a HIF-1 target gene that is upregulated during hypoxia (8, 24, 35, 66). Hypoxia increases ET-1 synthesis and secretion from several cell types (24, 30), including PASMCs (63), and treatment with an ET-1 receptor antagonist prevents and reverses the development of hypoxic pulmonary hypertension in animal models (6, 11, 14) and has beneficial effects in patients (56). Although we initially speculated that the effect of ET-1 on KV channel expression would be downstream of HIF-1, recent publications describing activation of HIF-1 by ET-1 in tumors suggested that the reverse might also be true (53, 54). Thus, in this study, we tested the hypothesis that upregulation of HIF-1 in PASMCs during moderate hypoxia is augmented by ET-1 and determined the downstream signaling mechanisms involved.
METHODS
All protocols were reviewed and approved by the Johns Hopkins University Animal Care and Use Committee.
In vivo hypoxic exposure.
Adult male Wistar rats (250–350 g) were placed in a hypoxic chamber for 3 wk, as previously described (61). The chamber was constantly flushed with room air to achieve low CO2 concentrations (<0.5%) and a servo-control system (PRO-OX, RCI Hudson, Anaheim, CA) continuously monitored O2 levels inside the chamber and injected 100% N2 as needed to maintain 10 ± 0.5% O2. Animals were removed from the chamber twice a week (<5 min) to replenish food and water supplies and to clean the cages. Normoxic animals were kept in room air on a rack adjacent to the chamber. All animals were allowed free access to food and water. In a separate series of experiments, animals were subjected to normoxia (room air) or hypoxia [inspired O2 fraction (FiO2) = 10%] for 24 h. In these experiments, rats were injected with BQ-123 (1 mg/kg ip) or an equal volume of saline 15 min before being placed in the hypoxic chamber. Because of the short half-life of circulating BQ-123 (55), rats were given a second injection of BQ-123 or saline at 10 h. The concentration of drug and route of administration were chosen based on methods described in the literature (12).
Isolation of arterial smooth muscle cells.
The methods for obtaining primary cultures of rat PASMCs have been previously described (61, 63). Animals were anesthetized with pentobarbital sodium (130 mg/kg ip) before the heart and lungs were removed en bloc. Intralobar pulmonary arteries (200–600 μm OD) were dissected and cleaned of connective tissue in cold HEPES-buffered saline solution (HBSS) containing (in mM) 130 NaCl, 5 KCl, 1.2 MgCl2, 10 HEPES, and 10 glucose with pH adjusted to 7.2 with 5 M NaOH. The arteries were cut open longitudinally and the lumen was gently rubbed with a cotton swab to remove the endothelium. Cleaned arteries were allowed to recover for 30 min in cold (4°C) HBSS followed by 20 min in reduced-Ca2+ HBSS (20 μM CaCl2) at room temperature. Following recovery, the tissue was incubated for 20 min at 37°C in reduced-Ca2+ HBSS containing collagenase (type I; 1,750 U/ml), papain (9.5 U/ml), bovine serum albumin (2 mg/ml), and dithiothreitol (1 mM). Single smooth muscle cells were dispersed by gentle trituration of the tissue in Ca2+-free HBSS. For aortic smooth muscle cells (AoSMCs), a segment of thoracic aorta was cleaned and digested as described above except that the digestion time was increased to 40 min. For intracellular Ca2+ concentration ([Ca2+]i) and reactive oxygen species (ROS) measurements, the cell suspension was placed on glass coverslips. Cells were cultured in Ham's F-12 media supplemented with 0.5% fetal calf serum and 1% penicillin-streptomycin for 24–48 h. For all other experiments, cells were plated in tissue culture dishes containing Ham's F-12 media supplemented with 10% fetal calf serum and 1% penicillin-streptomycin until 80% confluent (3–5 days). Cells were then placed in low-serum media (0.5% FCS) for 24 h before beginning exposures.
Ex vivo exposure to hypoxia.
PASMCs were exposed to hypoxia ex vivo by placing cells in an airtight chamber (Billups-Rothberg) gassed with 4% O2-5% CO2. The chamber and the normoxic control plates were placed in an incubator at 37°C. Initial experiments were performed with a hand-held oxygen monitor (model 5577; Hudson RCI) to assure that the chamber was able to sustain the desired level of hypoxia for a minimum of 48 h. For experiments longer than 48 h in duration, the chamber was reflushed at 36–48 h.
Measurement of [Ca2+]i and ROS.
For [Ca2+]i measurements, PASMCs were incubated with 5 μM fura 2-AM, a membrane permeant form of fura 2, at 37°C for 60 min. Following incubation, PASMCs were placed in a cell chamber and perfused with modified Krebs solution (KRB) containing (in mM) 118.3 NaCl, 4.7 KCl, 1.2 MgSO4, 25 NaHCO3, 10 glucose, 1.2 KH2PO4, and 2.5 CaCl2, and gassed with 16% O2-5% CO2. Cells were washed for 15 min at 37°C to remove extracellular dye and allow complete deesterification of cytosolic dye. Fura 2 fluorescence was measured with a Nikon TES 100 Ellipse inverted microscope. The light beam from a xenon arc lamp was filtered at 340 and 380 nm, passed through a ×20 fluorescence objective (Super Fluor 20, Nikon), and focused onto the field of PASMCs under examination. Emitted fluorescence was returned through the objective and detected by a CCD imaging camera. An electronic shutter was used to minimize photobleaching of dye. Protocols were executed and data were collected online with InCyte software (Intracellular Imaging, Cincinnati, OH). For ROS measurements, cells were loaded with H2DCFDA by continuous perfusion with Krebs solution containing 5 μM H2DCFDA for at least 30 min before beginning measurements to ensure stable uptake of dye. Since dichlorofluorescein (DCF) fluorescence is excited (490 nm) and detected (510 nm) at a single wavelength, any change in the concentration of the dye in the perfusion medium (i.e., differing concentrations between perfusion reservoirs) could result in a change in fluorescence that is not due to a change in ROS production. To minimize the possibility of this type of error, all experiments were performed by perfusing from a single reservoir into which drugs were dissolved directly.
RNA isolation, reverse transcription, and real-time PCR.
Total RNA was prepared from samples by use of the RNeasy extraction kit with on-column DNase treatment (Qiagen), and 1 μg of total RNA was reverse transcribed by use of the iScript cDNA synthesis kit (Bio-Rad). Real-time PCR was performed on an iCyclerIQ thermocycler (Bio-Rad) with 50 ng of the first-strand cDNA mixture and 250 nM forward and reverse primers in a 25-μl reaction volume containing QuantiTect SYBR Green PCR Master Mix (Qiagen). Specific primers were designed from sequences of the coding regions corresponding to the genes of interest (Table 1). Transcripts of both the gene of interest and a housekeeping gene (cyclophilin B) were amplified for 45 cycles by annealing at 58°C for 20 s, extending at 72°C for 20 s and denaturing at 94°C for 15 s. At the end of the run, melt curves were performed to confirm amplification of a single product. Prior to measurements, specificity of the PCR products was also confirmed by running the products on an agarose gel, excising the band, and sequencing at the Johns Hopkins Sequencing facility. Comparisons between treatments were always run on the same 96-well plate. Quantification of each gene was calculated by the Pfaffl method (45). PCR detection threshold cycle (CT) values for each plate were calculated by using iCycler software and the efficiency of each primer pair was determined from a five-point standard curve for each gene of interest. Data were expressed as a ratio of gene of interest to housekeeping gene (cyclophilin B) within a sample.
Table 1.
List of PCR primers
| Gene | Accession Number | Sequence (sense/antisense) | Product Size |
|---|---|---|---|
| Real-time | |||
| HIF-1α | NM_024359 | 5′-TGCTCATCAGTTGCCACTTC-3′ | 128 |
| 5′-CCATCCAGGGCTTTCAGATA-3′ | |||
| PHD2 | NM_178334 | 5′-ACCGTTTGGTATTTCGATGC-3′ | 110 |
| 5′-GGCAACTGAGAGGCTGTAGG-3′ | |||
| GLUT1 | NM_138827.1 | 5′-GCCTGAGACCAGTTGAAAGCAC-3′ | 292 |
| 5′-CTGCTTAGGTAAAGTTACAGGAG-3′ | |||
| TRPC1 | NM_053558 | 5′-AGCCTCTTGACAAACGAGGA-3′ | 146 |
| 5′-ACCTGACATCTGTCCGAACC-3′ | |||
| KV1.5 | NM_012972 | 5′-ACTTCGCAGAGGCAGACAAT-3′ | 128 |
| 5′-GATCTTGCCCCCTACAGTGA-3′ | |||
| NHE1 | NM_053811 | 5′-CTCTGATGGAGCTGTGGTGA-3′ | 137 |
| 5′-GGGCTGCTACCTGTTCTCAG-3′ | |||
| Cyclophilin | NM_022536 | 5′-GGACGAGTGACCTTTGGACT-3′ | 118 |
| 5′-TGACACGATGGAACTTGCTG-3′ | |||
| Conventional | |||
| PHD1 | NM_001004083 | 5′-GGTGAAGCCAGCCTATGC-3′ | 299 |
| 5′-CTCTCCTTGTTGCTCCTCAG-3′ | |||
| PHD2 | NM_178334 | 5′-CAGACGAGCGAGCAAGAG-3′ | 237 |
| 5′-GATGACACCTGGCGAAGTAG-3′ | |||
| PHD3 | NM_019371 | 5′-GCCGCTGTATCACCTGTATC-3′ | 306 |
| 5′-CTTCACACCACCGTCAGTC-3′ | |||
| β-Actin | NM_031144 | 5′-AGTGTGACGTTGACATCCGT-3′ | 244 |
| 5′-GACTCATCGTACTCCTGCTT-3′ |
HIF-1α, hypoxia-inducible factor 1α; PHD, prolyl hydroxylase; TRPC1, canonical transient receptor potential 1; KV1.5, voltage-dependent potassium channel; NHE1, Na+/H+ exchanger isoform 1.
Immunoblotting.
Tissue was homogenized and cells were lysed in cold lysis buffer containing (in mM) 30 HEPES, 100 NaCl, 1 EGTA, 50 NaF, 1 benzamidine, and 1 phenylmethylsulfonyl fluoride with 1% Triton X-100 and 5 μg/ml each leupeptin and aprotinin. For analysis of HIF-1α, lysates were clarified by centrifugation at 15,000 g for 10 min at 4°C. The supernatant was removed, assayed for total protein content (Pierce), incubated with antibodies overnight at 4°C, and sedimented with protein A-Sepharose for 2 h. The nuclear pellet was obtained by centrifugation, washed, and resuspended in SDS-PAGE buffer. For all other proteins, total protein samples were homogenized by sonication before being assayed for total protein content. Proteins were fractionated by SDS-PAGE and transferred to polyvinylidene difluoride membranes, which were probed with appropriate primary and secondary antibodies and visualized by enhanced chemiluminescence (ECL). The primary antibodies used were PHD2 (Novus Biologicals), smooth muscle-specific α-actin (Sigma); phosphorylated and total ERK1/2 (Cell Signaling), and HIF1-α (Abcam). For all experiments, membranes were probed for the protein of interest, stripped, and reprobed for the housekeeping protein (actin; loading control).
EMSA.
A commercially available kit (Panomics) was used according to the manufacturer's instructions. Nuclear extracts were incubated with probe at room temperature for 30 min. Samples were then fractionated by 6% PAGE and transferred to nylon membranes. At the end of transfer, membranes were baked for 1 h and then placed in a UV cross-linker for 3 min to immobilize bound oligonucleotides. Membranes were blocked, incubated with streptavidin-horseradish peroxidase, and visualized by ECL.
Drugs and solutions.
Ca2+-free KBR contained (in mM) 118.3 NaCl, 4.7 KCl, 1.2 MgSO4, 25 NaHCO3, 10 glucose, 1.2 KH2PO4, and 1 EGTA. ET-1 and BQ-123 were obtained from American Peptides (Sunnyvale, CA). Nifedipine was obtained from Calbiochem (La Jolla, CA). Fura 2, H2DCFDA, and BAPTA-AM were obtained from Molecular Probes (Eugene, OR). All other reagents were obtained from Sigma Aldrich (St. Louis, MO). Stock solutions of BQ-123 (10 mM in deionized H2O), ET-1 (10−5 M in deionized H2O), and PD98059 (10 mM in DMSO) were prepared, divided into aliquots, and stored at −20°C until used. Nifedipine stock solution (10 mM in 2:10 ethanol-KRB) was prepared on the day of the experiment. Stock solution of H2DCFDA (20 mM in DMSO) was prepared, aliquoted, and stored under argon at −20°C. Stock solutions of BAPTA-AM (50 mM in DMSO), 4-hydroxy-2,2,6,6-tetramethylpiperidinyloxy (TEMPOL; 10 mM in ethanol), and diphenyleneiodonium (DPI; 10 mM in DMSO) were prepared fresh for each experiment. All stock solutions were diluted to working concentrations in perfusate or media.
Data analysis.
All values are expressed as means ± SE. For all experiments, cells isolated from different animals were used; thus n refers to both the number of independent experiments and the number of animals. For [Ca2+]i and ROS measurements, data were collected from up to 30 cells, and the values were averaged to obtain a single value for each experiment. Change in [Ca2+]i (Δ[Ca2+]i) was computed by subtracting the average basal [Ca2+]i, determined from 1 min of data collected immediately prior to beginning challenge from the average of five data points at the peak of the response. Change in ROS was measured as the change in DCF fluorescence emitted when cells were excited at 490 nm (ΔF490) computed by subtracting the average baseline F490 (average of 1 min before challenge) from the maximal F490 at the peak of the response. Data were compared by unpaired Student's t-test or groupwise by one-way ANOVA with a Holms-Sidak post hoc test to determine differences between groups. A P value <0.05 was accepted as statistically significant.
RESULTS
Effect of ET-1 on HIF-1 activity in PASMCs.
In nuclear extracts from PASMCs exposed to ET-1 (10−8 M), HIF-1α protein levels increased in a time-dependent manner. As previously described (69), a faint band for HIF-1α was observed under nonhypoxic conditions in untreated cells, with no significant increase in HIF-1α after 4 h of incubation with ET-1 (Fig. 1A). By 24 h, HIF-1α protein levels were increased almost twofold, with a further increase observed at 48 h. The increase in HIF-1α protein levels correlated with increased HIF-1 transcriptional activity, since EMSA showed increased HIF-1 DNA-binding activity in cells treated with ET-1 (Fig. 1B), as well as increased mRNA levels of several HIF-1 target genes (GLUT1, NHE1, and TRPC1) and decreased expression of KV1.5 mRNA (Fig. 1C), which we have previously demonstrated was inhibited in a HIF-1-dependent manner. Since the increase in HIF-1α protein levels in PASMCs exposed to ET-1 was not immediate, as occurs when PHD activity is acutely inhibited under hypoxic conditions, we investigated the effect of ET-1 treatment on HIF-1α mRNA expression. ET-1 caused a time-dependent increase in HIF-1α mRNA levels (Fig. 1D). Consistent with the effects of ET-1 on HIF-1α protein, no induction of mRNA was observed at 4 h. At 24 h, HIF-1α mRNA increased twofold, but the increase did not reach statistical significance (P = 0.08), whereas an almost 10-fold increase was observed at 48 h. Application of different concentrations revealed that ET-1 was a potent stimulator of HIF-1α mRNA levels, with a marked increase observed at concentrations as low as 10−10 M (Fig. 1E). These results indicate that ET-1 induces an accumulation of HIF-1α protein under nonhypoxic conditions in PASMCs by a mechanism that includes increased HIF-1α mRNA levels.
Fig. 1.
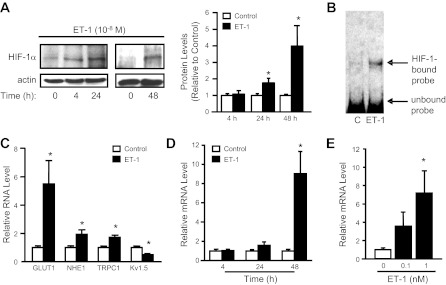
Endothelin-1 (ET-1) increases hypoxia inducible factor 1α (HIF-1α) expression. A: immunoblot assays were performed to assess the time dependence of HIF-1α protein accumulation in pulmonary arterial smooth muscle cells (PASMCs) treated with ET-1 (10−8 M). Bar graph shows values for 3–4 independent experiments. B: electrophoretic mobility shift assay of nuclear extracts isolated from rat PASMCs that were treated with vehicle control (C) or ET-1 (10−8 M) for 48 h. Similar results were observed in 3 independent experiments. C–E: real-time PCR was used to measure mRNA expression in PASMCs treated with ET-1. C: HIF-1 target gene expression in PASMCs treated with ET-1 (48 h; 10−8 M; n = 3–4). D: HIF-1α mRNA levels in PASMCs exposed to ET-1 (10−8 M) for the indicated periods of time relative to vehicle-treated cells (n = 3–4). E: HIF-1α mRNA levels in PASMCs exposed to varying concentrations of ET-1 for 48 h (n = 3). For all bar graphs, mean ± SE values are shown. Data are normalized to cyclophilin B or actin for mRNA and protein, respectively, and expressed relative to control. *P < 0.05 vs. control. NHE1, Na+/H+ exchanger isoform 1; TRPC1, canonical transient receptor potential 1; KV1.5, voltage-gated potassium channel.
Effect of ET-1 on PHD expression in PASMCs.
HIF-1α is also subjected to posttranslational regulation and increasing HIF-1α synthesis alone would not necessarily increase HIF-1α protein expression in the absence of reduced PHD activity. Since an acute reduction in PHD activity appeared unlikely, given the late onset of HIF-1α protein accumulation, we investigated the effects of ET-1 on PHD expression in PASMCs. An initial screen of PHD isoforms revealed that PASMCS express PHD1, PHD2, and PHD3 mRNA (Fig. 2A). Even with use of nonquantitative, conventional PCR, decreased expression of all three mRNAs was evident in response to ET-1. Since PHD2 has been suggested to be the primary regulator of HIF-1α stability under nonhypoxic conditions in vivo (4), we concentrated on defining the effects of ET-1 on PHD2 expression. We found that ET-1 caused a time- (Fig. 2B) and concentration- (Fig. 2C) dependent reduction in PHD2 mRNA expression, with inhibition of PHD2 expression occurring at concentrations of ET-1 greater than or equal to 10−9 M and with greater than 24 h of exposure. Similar time- and concentration-dependent effects of ET-1 were observed with respect to PHD2 protein expression (Fig. 2, D and E). ET-1 had no effect on FIH-1 mRNA levels (Fig. 2F).
Fig. 2.
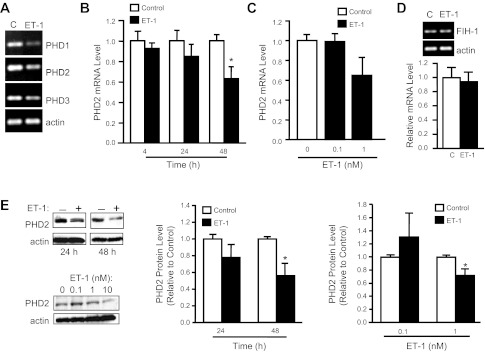
ET-1 decreases prolyl hydroxylase (PHD) expression in PASMCs. A: conventional RT-PCR was used to analyze PHD1, PHD2, and PHD3 mRNA levels in PASMCs exposed to ET-1 (48 h; 10−8 M). B and C: real-time RT-PCR was used to measure PHD2 mRNA levels in PASMCs exposed to ET-1 (10−8 M) at different time points (B; n = 3–4) and different concentrations of ET-1 for 48 h (C; n = 3). D and E: PASMCs were exposed to 10−8 M ET-1 at different time points (D; n = 3–4) or for 48 h at different concentrations (E; n = 4) and immunoblot assays for PHD2 protein were performed. F: representative images and bar graph (n = 3) showing factor inhibiting HIF-1 (FIH-1) mRNA levels measured in PASMCs exposed to ET-1 (10−8 M; 48 h). For all bar graphs, mean ± SE values are shown. Data are normalized to cyclophilin B or actin for mRNA and protein, respectively, and expressed relative to control. *P < 0.05 vs. control.
Role of ET-1 in activation of HIF-1 during moderate hypoxia.
The data exploring the effect of ET-1 on HIF-1α and PHD2 expression indicated that ET-1 was capable of inducing HIF-1 under nonhypoxic conditions in PASMCs by inducing HIF-1α synthesis and decreasing PHD2 levels. ET-1 secretion increases rapidly in response to hypoxia, likely due to release from preformed pools, and is maintained by HIF-1-dependent transcription of ET-1 mRNA (24, 63). Thus we next determined whether ET-1 was involved in the activation of HIF-1 in PASMCs during moderate hypoxia. We found that short-term (4 h) exposure of PASMCs to severe (1% O2), but not moderate (4% O2), hypoxia resulted in increased HIF-1α protein levels (Fig. 3A), suggesting that moderate hypoxia was not sufficient to inhibit PHD activity and induce HIF-1α stabilization in cultured PASMCs. However, when the challenge with moderate hypoxia was prolonged (60 h), HIF-1α mRNA and protein levels were significantly increased and were correlated with decreased PHD2 mRNA and protein levels (Fig. 3, B and C). To test whether in vivo exposure to hypoxia induced similar changes in the HIF system, distal pulmonary arteries were isolated from chronically hypoxic rats and the endothelium removed to provide a preparation that is mainly smooth muscle. In vivo exposure to hypoxia also increased HIF-1α and decreased PHD2 mRNA levels (Fig. 3D). Although the short half-life prevented the determination of HIF-1α protein levels in the pulmonary arteries of chronically hypoxic animals, PHD2 protein expression was decreased to 69.9 ± 9.8% of normoxic levels (Fig. 3E), indicating that similar changes are induced by exposure of PASMCs to physiological levels of hypoxia ex vivo and in vivo.
Fig. 3.
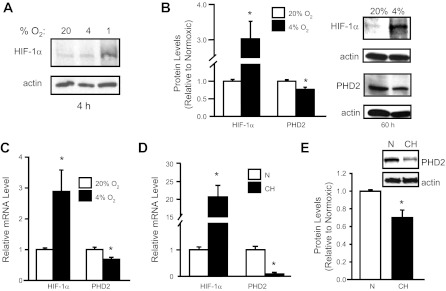
Hypoxia alters HIF-1α and PHD2 expression in PASMCs. PASMCs were exposed to a short (4 h) or long (60 h) duration of hypoxia (1 or 4% O2) ex vivo or pulmonary arteries were isolated from rats exposed to ambient hypoxia [3 wk; inspired O2 fraction (FiO2) = 10% O2]. Protein and mRNA levels were measured via immunoblot and real-time RT-PCR assays, respectively. A: HIF-1α protein in PASMCs after short-duration ex vivo hypoxic exposure at different O2 levels. B: HIF-1α and PHD2 protein expression in PASMCs after ex vivo exposure to prolonged, moderate hypoxia (60 h; 4% O2). Bar graphs show mean ± SE from 4 separate experiments. C: HIF-1α and PHD2 mRNA levels in PASMCs exposed to ex vivo hypoxia (60 h; 4% O2) relative to levels measured in nonhypoxic cells (n = 4). *P < 0.05 vs. 20% O2. D: HIF-1α and PHD2 mRNA levels in endothelium-denuded intralobar pulmonary arteries from rats exposed to chronic hypoxia (CH) relative to levels in pulmonary arteries from normoxic (N) rats (n = 3). *P < 0.05 vs. N. E: PHD2 protein expression in intralobar pulmonary arteries from N and CH rats. For all bar graphs, mean ± SE values are shown. Data are normalized to cyclophilin B or actin for mRNA and protein, respectively, and expressed relative to normoxic. *P < 0.05 vs. N.
We next investigated whether ET-1 was required for the induction of HIF-1α expression in PASMCs exposed to moderate hypoxia. Cells were pretreated with the well-characterized, highly selective ETA receptor antagonist, BQ-123 (10−6 M), for 30 min prior to exposure to hypoxia (4% O2; 60 h). BQ-123 had no effect on basal HIF-1α protein levels but prevented the induction of HIF-1α RNA and protein by hypoxia (Fig. 4, A and B), as well as the downregulation of PHD2. In addition, BQ-123 prevented the hypoxia-induced changes in HIF-1 target gene expression (Fig. 4C), indicating that activation of ETA receptors was critical for the activation of HIF-1 in isolated PASMCs exposed to moderate hypoxia. In contrast to results obtained with moderate hypoxia, BQ-123 had no effect on accumulation of HIF-1α protein during severe hypoxia (1% O2; 4 h) (Fig. 4D). To test whether ET-1 was involved in the hypoxia-induced changes in HIF-1α and PHD2 mRNA observed in vivo, rats were treated with saline or BQ-123 and exposed to hypoxia for 24 h. In saline treated animals, exposure to hypoxia increased HIF-1α and decreased PHD2 mRNA levels (Fig. 4, E and F). In animals treated with BQ-123, no significant differences in HIF-1α or PHD2 mRNA levels were observed with hypoxia.
Fig. 4.

Effect of BQ-123 treatment on the hypoxia-induced increase in HIF-1α and PHD2. PASMCs were exposed to control (20% O2) or hypoxic (4% O2) conditions in the absence or presence of BQ-123 (10 μM) for 60 h. Protein (n = 3–4; A) and mRNA (n = 3; B) levels of HIF-1α and PHD2 relative to cells treated with BQ-123 under normoxic conditions. C: HIF-1 target genes induced by hypoxia in PASMCs in the absence and presence of BQ-123 (n = 3–4). D: HIF-1α protein accumulation in response to short-term, severe hypoxia (4 h; 1% O2) in the absence and presence of BQ-123. Data are normalized to cyclophilin B or actin for mRNA and protein, respectively, and expressed relative to 20% O2. *P < 0.05 from 20% O2. E: HIF-1α mRNA levels in endothelium-denuded pulmonary arteries from rats treated with either saline (vehicle; n = 4 each) or BQ-123 (1 mg/kg; n = 3–4 each) and exposed to normoxia (room air) or hypoxia (FiO2 = 10%) for 24 h. F: PHD2 mRNA levels in endothelium-denuded pulmonary arteries from normoxic and hypoxic rats treated with either saline (n = 4 each) or BQ-123 (n = 3–4 each). mRNA data for HIF-1α and PHD2 are normalized to 18s and expressed relative to saline-treated normoxia. *P < 0.05 from saline-treated room air. For all bar graphs, mean ± SE values are shown.
Role of [Ca2+]i, ROS, and ERK in ET-1-induced HIF-1 activation.
We next defined the mechanism by which ET-1 modulated HIF-1α and PHD2 levels. One of the best characterized effects of ET-1 on PASMCs is an increase in [Ca2+]i. Consistent with our previous results (50), ET-1 caused a concentration-dependent increase in [Ca2+]i (Fig. 5, A and C) that could be prevented by pretreating cells with the voltage-gated Ca2+ channel (VGCC) antagonist nifedipine (10−6 M) or the intracellular Ca2+ chelator BAPTA-AM (50 μM), or by removal of extracellular Ca2+ by perfusing the cells with a Ca2+-free solution (Fig. 5D). In PASMCs loaded with the oxidant-sensitive dye H2DCF-AM, application of ET-1 also caused a concentration-dependent production of ROS (Fig. 5, B and C), which could be prevented by pretreating cells with the antioxidant TEMPOL (10 μM) or the oxidase inhibitor DPI (10 μM) (Fig. 5E). Inhibition of the ET-1-induced change in [Ca2+]i by treatment with BAPTA or nifedipine, or by removal of extracellular Ca2+ also blocked the increase in ROS levels in response to ET-1 (Fig. 5E). In contrast, reduction of ROS levels by treatment with TEMPOL or DPI had no significant effect on the ET-1-induced increase in [Ca2+]i (Fig. 5D), suggesting that ROS production occurs secondary to Ca2+ influx through VGCCs. Finally, we investigated the effect of ET-1 on ERK1/2 activation. Consistent with previous reports (57, 67), we found that ET-1 increased phosphorylation of ERK1/2 in PASMCs (Fig. 6A). The effect of ET-1 on ERK activation was blocked by pretreating cells with nifedipine to block VGCCs, with TEMPOL or DPI to reduce ROS levels, or by PD98059 (10 μM) to inhibit ERK1/2 activation. In PASMCs pretreated with PD98059, nifedipine, TEMPOL, or DPI, the ET-1-induced expression of HIF-1α mRNA and decreased expression of PHD2 mRNA was markedly inhibited (Fig. 6, B and C).
Fig. 5.
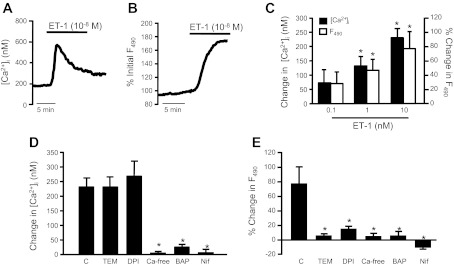
Effect of ET-1 on intracellular Ca2+ concentration ([Ca2+]i) and reactive oxygen species (ROS) generation. [Ca2+]i and ROS were measured via fluorescent microscopy with the intracellular dyes fura 2 and H2DCFDA, respectively, in PASMCs challenged with ET-1. A: change in [Ca2+]i in response to ET-1. B: change in ROS in response to ET-1. C: bar graphs show mean change in DCF fluorescence (F490) and [Ca2+]i in response to increasing concentrations of ET-1 and H2O2 (n = 3–7). D and E: PASMCs were pretreated with the antioxidants 4-hydroxy-2,2,6,6-tetramethylpiperidinyloxy (TEM; 10 mM), a membrane permeable superoxide scavenger, or diphenyleneiodonium (DPI; 10 mM), an oxidase inhibitor, the intracellular Ca2+ chelator BAPTA (BAP), the l-type Ca2+ channel inhibitor nifedipine (Nif), or the absence of extracellular calcium (Ca-free) before challenge with ET-1 (n = 3–7). For all bar graphs, mean ± SE values are shown. *P < 0.05 vs. control (C).
Fig. 6.

ERK1/2 mediates effects of ET-1 in PASMCs. A: immunoblots and mean data (bar graph) from PASMCs challenged with ET-1 (10−8 M; 30 min) in the absence (n = 4) or presence of nifedipine (Nifed; n = 3), PD98059 (PD; 10 mM), TEMPO (n = 3), or DPI (n = 3). ERK1/2 phosphorylation was measured by immunoblot using a phospho-specific anti-ERK1/2 antibody, normalized to total ERK, and expressed relative to control (Con; untreated cells). B and C: real-time RT-PCR data for HIF-1α and PHD2 mRNA levels in PASMCs challenged with ET-1 (48 h; 10−8 M) under control conditions, or in the presence of nifedipine (n = 4), PD98059 (n = 4), TEMPO (n = 3), or DPI (n = 3). Data are normalized to cyclophilin and expressed relative to untreated cells. For all bar graphs, mean ± SE values are shown. *P < 0.05 vs. control (Con).
Effect of ET-1 on AoSMCs.
Many of the effects of hypoxia on PASMCs are not found in smooth muscle from systemic arteries, including the aorta. To test whether this could be a function of differential regulation of HIF-1α between pulmonary and systemic vascular smooth muscle, we determined the effects of hypoxia and ET-1 on HIF-1α and PHD2 in AoSMCs. In contrast to the results obtained in PASMCs, exposing AoSMCs to prolonged moderate hypoxia (4% O2; 60 h) had no detectable effect on HIF-1α or PHD2 protein levels (Fig. 7A). In contrast, HIF-1α protein levels were increased after exposure to short-term (4 h), severe hypoxia (1% O2). In AoSMCs, exposure to prolonged, moderate hypoxia also had no effect on the expression of HIF-1α or PHD2 mRNA (Fig. 7B) or HIF-1 target genes (Fig. 7C).
Fig. 7.

Moderate hypoxia has no effect on aortic smooth muscle cells (AoSMCs). AoSMCs were exposed to short (4 h) or long (60 h) duration hypoxia (1 or 4% O2) or control (20% O2) conditions. A: immunoblot assays of HIF-1α and PHD2 protein in AoSMCs exposed to hypoxia. Data are normalized to actin and expressed relative to 20% O2. Bar graph shows means ± SE from 3–4 experiments. B: HIF-1α and PHD2 mRNA levels in AoSMCs exposed to hypoxia (60 h; 4% O2) (n = 3–5). C: HIF-1 target gene expression in AoSMCs exposed to hypoxia for 60 h (n = 3 each). For B and C, data are normalized to cyclophilin and expressed relative to 20% O2 values. *P < 0.05 vs. 20%.
We next determined whether the lack of effect of moderate hypoxia on AoSMCs might be due to a differential response of AoSMCs to ET-1. We found that ET-1 challenge had no effect on HIF-1α and PHD2 mRNA or protein levels in AoSMCs (Fig. 8, A and B). Moreover, HIF-1 target gene expression was not altered by ET-1 treatment of AoSMCs (Fig. 8C). To determine whether the lack of effect of ET-1 on HIF-1α levels in AoSMCs was due to differential ET-1 signaling between PASMCs and AoSMCs, we tested the effect of ET-1 on [Ca2+]i, ROS formation, and ERK1/2 activation. We found that the increase in both [Ca2+]i and ROS in response to ET-1 in AoSMCs (Fig. 8, D–F) was similar to that observed in PASMCs. In contrast to PASMCs, however, ET-1 did not cause activation of ERK1/2 in AoSMCs (Fig. 8G).
Fig. 8.
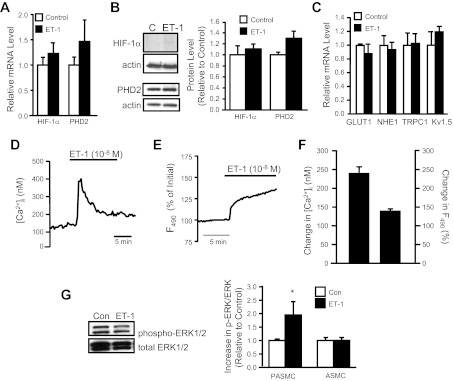
Effect of ET-1 in AoSMCs. A and B: AoSMCs were incubated with ET-1 (48 h; 10−8 M) and analyzed for HIF-1α and PHD2 mRNA and protein. Data were normalized to cyclophilin (mRNA) or actin (protein) and expressed relative to untreated cells (control). Bar graphs show mean ± SE from 3–4 experiments. C: HIF-1 target gene expression in AoSMCs incubated in the absence (control) or presence of ET-1 (n = 3). Data are normalized to cyclophilin and expressed relative to control. D and E: representative traces showing that ET-1 increases [Ca2+]i and ROS in AoSMCs. F: bar graph shows mean change in DCF fluorescence (F490) and [Ca2+]i in response to ET-1 (n = 4–5). G: AoSMCs were challenged with ET-1 (10−8 M; 30 min) and ERK1/2 phosphorylation was measured, normalized to total ERK, and expressed relative to control (untreated cells). For all bar graphs, mean ± SE values are shown. *P < 0.05 vs. control.
DISCUSSION
In this study, we demonstrated that ET-1 increased HIF-1α accumulation, enhanced expression of HIF-1 target genes under nonhypoxic conditions, and was required for the induction of HIF-1 in PASMCs exposed to moderate hypoxia. Our results indicate that the ET-1-induced increase in HIF-1α protein levels involves both increased synthesis and decreased degradation of HIF-1α as a result of increased [Ca2+]i and ROS, and subsequent ERK1/2 activation (Fig. 9). Moreover, the effects of ET-1 on HIF-1 appear to be specific for PASMCs, since similar effects were not observed in AoSMCs.
Fig. 9.

Regulation of HIF-1 by ET-1 in PASMCs. Under conditions of moderate, prolonged hypoxia, increased ET-1 production results in increased [Ca2+]i levels, ROS generation, and ERK1/2 activation. This leads to increased HIF-1α and decreased PHD2 mRNA and protein levels. HIF-1 activates Trpc1 and Nhe1 gene expression, whereas Kv1.5 gene expression is inhibited. These changes in gene expression lead to enhanced PASMC contraction, proliferation, and migration, which contribute to the pathogenesis of pulmonary hypertension.
That ET-1 induced HIF-1α protein accumulation in PASMCs is consistent with studies of human melanoma and ovarian cancer cells (52–54). ET-1 induced HIF-1α accumulation was also observed in lymphatic endothelial cells (51), suggesting that noncancer cells also possess the ability to respond to ET-1 with an upregulation of HIF-1. However, that PASMCs, but not AoSMCs, exhibited upregulation of HIF-1 when challenged with ET-1 indicates that not all cells possess the ability to respond to ET-1 in the same manner.
ET-1 increases [Ca2+]i in PASMCs (50) and induces generation of ROS and activation of ERK1/2 in a variety of vascular smooth muscle preparations (13, 29, 62, 67). We verified that ET-1 increased ROS levels and activated ERK1/2 in PASMCs and that increased [Ca2+]i was necessary for both responses. Ca2+-dependent ROS generation may be due to the action of NADPH oxidase (Nox) (18, 28), since ROS generation was inhibited by the Nox inhibitor DPI; however, DPI binds nonspecifically to flavins and also inhibits other oxidases. The activation of ERK1/2 depended on increased ROS levels, since antioxidants blocked ET-1-dependent ERK1/2 activation. ET-1 also increased [Ca2+]i and ROS levels in AoSMCs, consistent with previous reports (21, 31, 41), but did not activate ERK1/2 or induce HIF-1α protein accumulation. The mechanism by which ROS leads to ERK1/2 activation in PASMCs is unclear, as is the reason why ET-1-induced increases in ROS do not activate ERK1/2 in AoSMCs. Other laboratories have also failed to find activation of ERK1/2 by ET-1 in aortic smooth muscle (38), although some studies found that high concentrations of ET-1 (≥10−7 M) could activate ERK1/2 in AoSMCs (10, 17, 31, 38, 68). However, unlike PASMCs, in these studies the effects of ET-1 on ERK1/2 were independent of Ca2+ (10) and ROS (17).
ET-1-induced induction of HIF-1α in the absence of hypoxia does not appear to be due to direct inhibition of PHD activity, as HIF-1α protein accumulation in response to ET-1 required >24 h to become significant, whereas HIF-1α accumulation was observed with severe hypoxia by 4 h. Instead, our data indicate that ET-1 both increased synthesis of HIF-1α (as a result of increased HIF-1 mRNA levels) and decreased expression of PHD2, with the latter likely leading to decreased degradation of HIF-1α. Previous studies have shown that PHD2 and PHD3 mRNA expression can be regulated by hypoxia, via HIF-1-dependent transcriptional activation (3, 5, 36), and by physiological stimuli other than hypoxia, including estrogen, transforming growth factor-β, p53, platelet-derived growth factor, and angiotensin II (33, 37, 47, 65). Our data indicate that ET-1 also regulates PHD2 expression through a mechanism requiring activation of ETA receptors, Ca2+ influx, increased ROS levels, and activation of ERK1/2. In melanoma cells, the effects of ET-1 on PHD2 mRNA levels were mediated by the ETB receptor subtype and rely on activation of the phosphatidylinositol 3-kinase-AKT-mTOR pathway (52). Thus the effect of ET-1 on PHD2 expression, as well as the mechanism involved, appears to be cell type specific.
The accumulation of HIF-1α protein triggered by ET-1 results in the transcriptional activation of HIF-1 target genes, as indicated by increased HIF-1 DNA-binding activity and increased levels of mRNAs encoding HIF-1 target genes. HIF-1 transcriptional activity is negatively regulated by O2- and FIH-1-dependent hydroxylation of HIF-1α at an asparagine residue within the transactivation domain, which prevents binding of the coactivators CBP/p300 (34). That ET-1 induced the expression of HIF-1 target genes suggests that ET-1 also altered FIH-1 activity, although the mechanism by which this occurs does not appear to involve a reduction in FIH-1 mRNA levels. Instead, Ca2+- and ROS-dependent phosphorylation of p300 may stimulate HIF-1α-p300 interactions, as seen in cells subjected to intermittent hypoxia (71).
During hypoxia, PASMCs exhibit increased HIF-1α protein levels. When the hypoxic stimulus is severe (i.e., 1–2% O2), accumulation of HIF-1α protein occurs rapidly, as a consequence of inhibited PHD catalytic activity due to reduced substrate (O2) or generation of mitochondrial ROS (9). With more moderate hypoxia, as used in the present study, rapid accumulation of HIF-1α was not observed in PASMCs, suggesting that inhibition of PHD catalytic activity is minimal. With longer duration of moderate hypoxia, HIF-1α accumulation becomes evident, and is associated with increased HIF-1α and decreased PHD2 mRNA levels. We previously showed that PASMCs release ET-1 in response to hypoxia (63). Our results using a highly specific ETA receptor antagonist indicate that increased elaboration of ET-1 during moderate hypoxia was required for induction of HIF-1α, as well as the hypoxia-induced changes in HIF target gene expression. In contrast, rapid induction of HIF-1α accumulation in response to severe hypoxia was not altered by ET receptor inhibition, confirming that ET-1 does not play an essential role in activating HIF-1 under those conditions.
We previously demonstrated that hypoxia-induced pulmonary hypertension is critically dependent on the activation of HIF-1 (70). Moreover, ET-1 levels are elevated in chronically hypoxic animals and in patients with hypoxia-associated pulmonary hypertension (20) and ET receptor antagonists prevent and/or reduce pulmonary hypertension in animal models (6, 11, 14, 15) and have beneficial effects in patients (19). Previously, the effects of these drugs have been attributed to either alleviation of vasoconstriction or inhibition of ET-1-induced mitogenic effects. The finding of increased HIF-1α mRNA and decreased PHD2 expression in pulmonary vascular smooth muscle from chronically hypoxic rats, where we have previously demonstrated changes in HIF-1 target gene expression (48, 49, 61, 63) and where ET-1 levels are elevated (32, 43), is consistent with the possibility that ET-1 also contributes to pulmonary hypertension by activating HIF-1. Indeed, the increased HIF-1α, and decreased PHD2, mRNA levels observed in pulmonary vascular smooth muscle from rats exposed to hypoxia in vivo for only 24 h were completely prevented in rats treated with an ETA receptor antagonist, further supporting a role for ET-1 in regulating the HIF pathway in vivo. Whether ET-1 also mediates the rapid, transient increase in HIF-1α mRNA observed in lung tissue from mice exposed to more severe FiO2 remains to be determined; however, since HIF-1α mRNA levels returned to baseline within 4 h in those studies (64), the mechanisms involved may be different. Both increased ET-1 levels (in lungs and plasma) and HIF-1α levels (in lungs) have been reported in pulmonary hypertension patients without associated hypoxemic lung disease (7, 20, 46, 59), offering further supporting evidence that elevated ET-1 levels in the absence of hypoxia can be associated with increased HIF-1α levels, although whether ET-1 is responsible for the increased HIF-1 activation under these conditions has yet to be evaluated. The fact that ET receptor antagonists are less effective in patients might reflect the complexity of human disease, in which other factors may share the ability to induce HIF-1 in a manner similar to ET-1. HIF-1 inhibitors block the development of pulmonary hypertension in animal models (1), suggesting that clinical trials of these drugs are warranted.
In summary, we have demonstrated that ET-1 is essential for induction of HIF-1α in PASMCs during moderate hypoxia via a mechanism requiring Ca2+ influx through VGCCs, increased ROS levels, and activation of ERK1/2. This mechanism is not observed in AoSMCs and thus may explain the differential effects of hypoxia on these cell types and why chronic hypoxia causes vasoconstriction, vascular remodeling, and hypertension selectively in the pulmonary circulation.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants HL67191 and HL67919 and by the Johns Hopkins Institute for Cell Engineering. G. L. Semenza is the C. Michael Armstrong Professor at the Johns Hopkins University School of Medicine. X. Yun was supported by funds from the Overseas Study Project of Guangzhou Elite Project.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
S.P., J.M., W.L., X.Y., C.U., and L.A.S. performed experiments; S.P., X.Y., and L.A.S. analyzed data; S.P. and L.A.S. prepared figures; S.P., J.M., W.L., C.U., J.T.S., G.L.S., and L.A.S. edited and revised manuscript; S.P., J.M., W.L., X.Y., C.U., J.T.S., G.L.S., and L.A.S. approved final version of manuscript; J.T.S., G.L.S., and L.A.S. conception and design of research; J.T.S., G.L.S., and L.A.S. interpreted results of experiments; L.A.S. drafted manuscript.
REFERENCES
- 1. Abud EM, Maylor J, Undem C, Punjabi A, Zaiman AL, Myers AC, Sylvester JT, Semenza GL, Shimoda LA. Digoxin inhibits development of hypoxic pulmonary hypertension in mice. Proc Natl Acad Sci USA 109: 1239–1244, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Appelhoff RJ, Tian YM, Raval RR, Turley H, Harris AL, Pugh CW, Ratcliffe PJ, Gleadle JM. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem 279: 38458–38465, 2004 [DOI] [PubMed] [Google Scholar]
- 3. Berchner-Pfannschmidt U, Yamac H, Trinidad B, Fandrey J. Nitric oxide modulates oxygen sensing by hypoxia-inducible factor 1-dependent induction of prolyl hydroxylase 2. J Biol Chem 282: 1788–1796, 2007 [DOI] [PubMed] [Google Scholar]
- 4. Berra E, Benizri E, Ginouves A, Volmat V, Roux D, Pouyssegur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1α in normoxia. EMBO J 22: 4082–4090, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Berra E, Richard DE, Gothie E, Pouyssegur J. HIF-1-dependent transcriptional activity is required for oxygen-mediated HIF-1α degradation. FEBS Lett 491: 85–90, 2001 [DOI] [PubMed] [Google Scholar]
- 6. Bonvallet ST, Zamora MR, Hasunuma K, Sato K, Hanasato N, Anderson D, Sato K, Stelzner TJ. BQ123, an ETA-receptor antagonist, attenuates hypoxic pulmonary hypertension in rats. Am J Physiol Heart Circ Physiol 266: H1327–H1331, 1994 [DOI] [PubMed] [Google Scholar]
- 7. Cacoub P, Dorent R, Maistre G, Nataf P, Carayon A, Piette C, Godeau P, Cabrol C, Gandjbakhch I. Endothelin-1 in primary pulmonary hypertension and the Eisenmenger syndrome. Am J Cardiol 71: 448–450, 1993 [DOI] [PubMed] [Google Scholar]
- 8. Camenisch G, Stroka DM, Gassmann M, Wenger RH. Attenuation of HIF-1 DNA-binding activity limits hypoxia-inducible endothelin-1 expression. Pflügers Arch 443: 240–249, 2001 [DOI] [PubMed] [Google Scholar]
- 9. Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, Schumacker PT. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem 275: 25130–25138, 2000 [DOI] [PubMed] [Google Scholar]
- 10. Chen QW, Edvinsson L, Xu CB. Role of ERK/MAPK in endothelin receptor signaling in human aortic smooth muscle cells. BMC Cell Biol 10: 52, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen SJ, Chen YF, Opgenorth TJ, Wessale JL, Meng QC, Durand J, DiCarlo VS, Oparil S. The orally active nonpeptide endothelin A-receptor antagonist A-127722 prevents and reverses hypoxia-induced pulmonary hypertension and pulmonary vascular remodeling in Sprague-Dawley rats. J Cardiovasc Pharmacol 29: 713–725, 1997 [DOI] [PubMed] [Google Scholar]
- 12. Chen Y, Hanaoka M, Droma Y, Chen P, Voelkel NF, Kubo K. Endothelin-1 receptor antagonists prevent the development of pulmonary emphysema in rats. Eur Respir J 35: 904–912, 2010 [DOI] [PubMed] [Google Scholar]
- 13. Daou GB, Srivastava AK. Reactive oxygen species mediate endothelin-1-induced activation of ERK1/2, PKB, and Pyk2 signaling, as well as protein synthesis, in vascular smooth muscle cells. Free Radic Biol Med 37: 208–215, 2004 [DOI] [PubMed] [Google Scholar]
- 14. DiCarlo VS, Chen SJ, Meng QC, Durand J, Yano M, Chen YF, Oparil S. ETA-receptor antagonist prevents and reverses chronic hypoxia-induced pulmonary hypertension in rat. Am J Physiol Lung Cell Mol Physiol 269: L690–L697, 1995 [DOI] [PubMed] [Google Scholar]
- 15. Eddahibi S, Raffestin B, Clozel M, Levame M, Adnot S. Protection from pulmonary hypertension with an orally active endothelin receptor antagonist in hypoxic rats. Am J Physiol Heart Circ Physiol 268: H828–H835, 1995 [DOI] [PubMed] [Google Scholar]
- 16. Ehrismann D, Flashman E, Genn DN, Mathioudakis N, Hewitson KS, Ratcliffe PJ, Schofield CJ. Studies on the activity of the hypoxia-inducible-factor hydroxylases using an oxygen consumption assay. Biochem J 401: 227–234, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fei J, Viedt C, Soto U, Elsing C, Jahn L, Kreuzer J. Endothelin-1 and smooth muscle cells: induction of jun amino-terminal kinase through an oxygen radical-sensitive mechanism. Arterioscler Thromb Vasc Biol 20: 1244–1249, 2000 [DOI] [PubMed] [Google Scholar]
- 18. Fulton DJ. Nox5 and the regulation of cellular function. Antioxid Redox Signal 11: 2443–2452, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gabbay E, Fraser J, McNeil K. Review of bosentan in the management of pulmonary arterial hypertension. Vasc Health Risk Manag 3: 887–900, 2007 [PMC free article] [PubMed] [Google Scholar]
- 20. Giaid A, Yanagisawa M, Langleben D, Michel RP, Levy R, Shennib H, Kimura S, Masaki T, Duguid WP, Stewart DJ. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N Engl J Med 328: 1732–1739, 1993 [DOI] [PubMed] [Google Scholar]
- 21. Hirata Y, Yoshimi H, Emori T, Shichiri M, Marumo F, Watanabe TX, Kumagaye S, Nakajima K, Kimura T, Sakakibara S. Receptor binding activity and cytosolic free calcium response by synthetic endothelin analogs in cultured rat vascular smooth muscle cells. Biochem Biophys Res Commun 160: 228–234, 1989 [DOI] [PubMed] [Google Scholar]
- 22. Hong Z, Weir EK, Nelson DP, Olschewski A. Subacute hypoxia decreases voltage-activated potassium channel expression and function in pulmonary artery myocytes. Am J Respir Cell Mol Biol 31: 337–343, 2004 [DOI] [PubMed] [Google Scholar]
- 23. Howell K, Preston RJ, McLoughlin P. Chronic hypoxia causes angiogenesis in addition to remodelling in the adult rat pulmonary circulation. J Physiol 547: 133–145, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hu J, Discher DJ, Bishopric NH, Webster KA. Hypoxia regulates expression of the endothelin-1 gene through a proximal hypoxia-inducible factor-1 binding site on the antisense strand. Biochem Biophys Res Commun 245: 894–899, 1998 [DOI] [PubMed] [Google Scholar]
- 25. Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG., Jr HIFα targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292: 464–468, 2001 [DOI] [PubMed] [Google Scholar]
- 26. Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY, Semenza GL. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1α. Genes Dev 12: 149–162, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292: 468–472, 2001 [DOI] [PubMed] [Google Scholar]
- 28. Jagnandan D, Church JE, Banfi B, Stuehr DJ, Marrero MB, Fulton DJ. Novel mechanism of activation of NADPH oxidase 5: calcium sensitization via phosphorylation. J Biol Chem 282: 6494–6507, 2007 [DOI] [PubMed] [Google Scholar]
- 29. Kawanabe Y, Hashimoto N, Masaki T. Extracellular Ca2+ influx and endothelin-1-induced intracellular mitogenic cascades in rabbit internal carotid artery vascular smooth muscle cells. J Cardiovasc Pharmacol 40: 307–314, 2002 [DOI] [PubMed] [Google Scholar]
- 30. Kourembanas S, Marsden PA, McQuillan LP, Faller DV. Hypoxia induces endothelin gene expression and secretion in cultured human endothelium. J Clin Invest 88: 1054–1057, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kyaw M, Yoshizumi M, Tsuchiya K, Kirima K, Suzaki Y, Abe S, Hasegawa T, Tamaki T. Antioxidants inhibit endothelin-1 (1–31)-induced proliferation of vascular smooth muscle cells via the inhibition of mitogen-activated protein (MAP) kinase and activator protein-1 (AP-1). Biochem Pharmacol 64: 1521–1531, 2002 [DOI] [PubMed] [Google Scholar]
- 32. Li H, Chen SJ, Chen YF, Meng QC, Durand J, Oparil S, Elton TS. Enhanced endothelin-1 and endothelin receptor gene expression in chronic hypoxia. J Appl Physiol 77: 1451–1459, 1994 [DOI] [PubMed] [Google Scholar]
- 33. Lipscomb EA, Sarmiere PD, Freeman RS. SM-20 is a novel mitochondrial protein that causes caspase-dependent cell death in nerve growth factor-dependent neurons. J Biol Chem 276: 5085–5092, 2001 [DOI] [PubMed] [Google Scholar]
- 34. Mahon PC, Hirota K, Semenza GL. FIH-1: a novel protein that interacts with HIF-1α and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev 15: 2675–2686, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Manalo DJ, Rowan A, Lavoie T, Natarajan L, Kelly BD, Ye SQ, Garcia JG, Semenza GL. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 105: 659–669, 2005 [DOI] [PubMed] [Google Scholar]
- 36. Marxsen JH, Stengel P, Doege K, Heikkinen P, Jokilehto T, Wagner T, Jelkmann W, Jaakkola P, Metzen E. Hypoxia-inducible factor-1 (HIF-1) promotes its degradation by induction of HIF-α-prolyl-4-hydroxylases. Biochem J 381: 761–767, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McMahon S, Charbonneau M, Grandmont S, Richard DE, Dubois CM. Transforming growth factor β1 induces hypoxia-inducible factor-1 stabilization through selective inhibition of PHD2 expression. J Biol Chem 281: 24171–24181, 2006 [DOI] [PubMed] [Google Scholar]
- 38. Meoli DF, White RJ. Endothelin-1 induces pulmonary but not aortic smooth muscle cell migration by activating ERK1/2 MAP kinase. Can J Physiol Pharmacol 88: 830–839, 2010 [DOI] [PubMed] [Google Scholar]
- 39. Metzen E, Berchner-Pfannschmidt U, Stengel P, Marxsen JH, Stolze I, Klinger M, Huang WQ, Wotzlaw C, Hellwig-Burgel T, Jelkmann W, Acker H, Fandrey J. Intracellular localisation of human HIF-1α hydroxylases: implications for oxygen sensing. J Cell Sci 116: 1319–1326, 2003 [DOI] [PubMed] [Google Scholar]
- 40. Meyrick B, Reid L. The effect of continued hypoxia on rat pulmonary arterial circulation. An ultrastructural study. Lab Invest 38: 188–200, 1978 [PubMed] [Google Scholar]
- 41. Minowa T, Miwa S, Kobayashi S, Enoki T, Zhang XF, Komuro T, Iwamuro Y, Masaki T. Inhibitory effect of nitrovasodilators and cyclic GMP on ET-1-activated Ca2+-permeable nonselective cation channel in rat aortic smooth muscle cells. Br J Pharmacol 120: 1536–1544, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nagaoka T, Fagan KA, Gebb SA, Morris KG, Suzuki T, Shimokawa H, McMurtry IF, Oka M. Inhaled Rho kinase inhibitors are potent and selective vasodilators in rat pulmonary hypertension. Am J Respir Crit Care Med 171: 494–499, 2005 [DOI] [PubMed] [Google Scholar]
- 43. Nakanishi K, Tajima F, Nakata Y, Osada H, Tachibana S, Kawai T, Torikata C, Suga T, Takishima K, Aurues T, Ikeda T. Expression of endothelin-1 in rats developing hypobaric hypoxia-induced pulmonary hypertension. Lab Invest 79: 1347–1357, 1999 [PubMed] [Google Scholar]
- 44. Oka M, Morris KG, McMurtry IF. NIP-121 is more effective than nifedipine in acutely reversing chronic pulmonary hypertension. J Appl Physiol 75: 1075–1080, 1993 [DOI] [PubMed] [Google Scholar]
- 45. Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29: e45, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rubens C, Ewert R, Halank M, Wensel R, Orzechowski HD, Schultheiss HP, Hoeffken G. Big endothelin-1 and endothelin-1 plasma levels are correlated with the severity of primary pulmonary hypertension. Chest 120: 1562–1569, 2001 [DOI] [PubMed] [Google Scholar]
- 47. Seth P, Krop I, Porter D, Polyak K. Novel estrogen and tamoxifen induced genes identified by SAGE (Serial Analysis of Gene Expression). Oncogene 21: 836–843, 2002 [DOI] [PubMed] [Google Scholar]
- 48. Shimoda LA, Fallon M, Pisarcik S, Wang J, Semenza GL. HIF-1 regulates hypoxic induction of NHE1 expression and alkalinization of intracellular pH in pulmonary arterial myocytes. Am J Physiol Lung Cell Mol Physiol 291: L941–L949, 2006 [DOI] [PubMed] [Google Scholar]
- 49. Shimoda LA, Manalo DJ, Sham JS, Semenza GL, Sylvester JT. Partial HIF-1α deficiency impairs pulmonary arterial myocyte electrophysiological responses to hypoxia. Am J Physiol Lung Cell Mol Physiol 281: L202–L208, 2001 [DOI] [PubMed] [Google Scholar]
- 50. Shimoda LA, Sylvester JT, Sham JS. Mobilization of intracellular Ca2+ by endothelin-1 in rat intrapulmonary arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 278: L157–L164, 2000 [DOI] [PubMed] [Google Scholar]
- 51. Spinella F, Garrafa E, Di Castro V, Rosano L, Nicotra MR, Caruso A, Natali PG, Bagnato A. Endothelin-1 stimulates lymphatic endothelial cells and lymphatic vessels to grow and invade. Cancer Res 69: 2669–2676, 2009 [DOI] [PubMed] [Google Scholar]
- 52. Spinella F, Rosano L, Del Duca M, Di Castro V, Nicotra MR, Natali PG, Bagnato A. Endothelin-1 inhibits prolyl hydroxylase domain 2 to activate hypoxia-inducible factor-1α in melanoma cells. PLoS One 5: e11241, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 53. Spinella F, Rosano L, Di Castro V, Decandia S, Nicotra MR, Natali PG, Bagnato A. Endothelin-1 and endothelin-3 promote invasive behavior via hypoxia-inducible factor-1α in human melanoma cells. Cancer Res 67: 1725–1734, 2007 [DOI] [PubMed] [Google Scholar]
- 54. Spinella F, Rosano L, Di Castro V, Natali PG, Bagnato A. Endothelin-1 induces vascular endothelial growth factor by increasing hypoxia-inducible factor-1α in ovarian carcinoma cells. J Biol Chem 277: 27850–27855, 2002 [DOI] [PubMed] [Google Scholar]
- 55. Spratt JC, Goddard J, Patel N, Strachan FE, Rankin AJ, Webb DJ. Systemic ETA receptor antagonism with BQ-123 blocks ET-1 induced forearm vasoconstriction and decreases peripheral vascular resistance in healthy men. Br J Pharmacol 134: 648–654, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Stenmark KR, Rabinovitch M. Emerging therapies for the treatment of pulmonary hypertension. Pediatr Crit Care Med 11: S85–S90, 2010 [DOI] [PubMed] [Google Scholar]
- 57. Suzuki YJ, Day RM, Tan CC, Sandven TH, Liang Q, Molkentin JD, Fanburg BL. Activation of GATA-4 by serotonin in pulmonary artery smooth muscle cells. J Biol Chem 278: 17525–17531, 2003 [DOI] [PubMed] [Google Scholar]
- 58. Takeda K, Aguila HL, Parikh NS, Li X, Lamothe K, Duan LJ, Takeda H, Lee FS, Fong GH. Regulation of adult erythropoiesis by prolyl hydroxylase domain proteins. Blood 111: 3229–3235, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tuder RM, Chacon M, Alger L, Wang J, Taraseviciene-Stewart L, Kasahara Y, Cool CD, Bishop AE, Geraci M, Semenza GL, Yacoub M, Polak JM, Voelkel NF. Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: evidence for a process of disordered angiogenesis. J Pathol 195: 367–374, 2001 [DOI] [PubMed] [Google Scholar]
- 60. Tuder RM, Yun JH, Bhunia A, Fijalkowska I. Hypoxia and chronic lung disease. J Mol Med 85: 1317–1324, 2007 [DOI] [PubMed] [Google Scholar]
- 61. Wang J, Weigand L, Lu W, Sylvester JT, Semenza GL, Shimoda LA. Hypoxia inducible factor 1 mediates hypoxia-induced TRPC expression and elevated intracellular Ca2+ in pulmonary arterial smooth muscle cells. Circ Res 98: 1528–1537, 2006 [DOI] [PubMed] [Google Scholar]
- 62. Wedgwood S, Dettman RW, Black SM. ET-1 stimulates pulmonary arterial smooth muscle cell proliferation via induction of reactive oxygen species. Am J Physiol Lung Cell Mol Physiol 281: L1058–L1067, 2001 [DOI] [PubMed] [Google Scholar]
- 63. Whitman EM, Pisarcik S, Luke T, Fallon M, Wang J, Sylvester JT, Semenza GL, Shimoda LA. Endothelin-1 mediates hypoxia-induced inhibition of voltage-gated K+ channel expression in pulmonary arterial myocytes. Am J Physiol Lung Cell Mol Physiol 294: L309–L318, 2008 [DOI] [PubMed] [Google Scholar]
- 64. Wiener CM, Booth G, Semenza GL. In vivo expression of mRNAs encoding hypoxia-inducible factor 1. Biochem Biophys Res Commun 225: 485–488, 1996 [DOI] [PubMed] [Google Scholar]
- 65. Wolf G, Harendza S, Schroeder R, Wenzel U, Zahner G, Butzmann U, Freeman RS, Stahl RA. Angiotensin II's antiproliferative effects mediated through AT2-receptors depend on down-regulation of SM-20. Lab Invest 82: 1305–1317, 2002 [DOI] [PubMed] [Google Scholar]
- 66. Yamashita K, Discher DJ, Hu J, Bishopric NH, Webster KA. Molecular regulation of the endothelin-1 gene by hypoxia. Contributions of hypoxia-inducible factor-1, activator protein-1, GATA-2, and p300/CBP. J Biol Chem 276: 12645–12653, 2001 [DOI] [PubMed] [Google Scholar]
- 67. Yamboliev IA, Hruby A, Gerthoffer WT. Endothelin-1 activates MAP kinases and c-Jun in pulmonary artery smooth muscle. Pulm Pharmacol Ther 11: 205–208, 1998 [DOI] [PubMed] [Google Scholar]
- 68. Yang Z, Krasnici N, Luscher TF. Endothelin-1 potentiates human smooth muscle cell growth to PDGF: effects of ETA and ETB receptor blockade. Circulation 100: 5–8, 1999 [DOI] [PubMed] [Google Scholar]
- 69. Yu AY, Frid MG, Shimoda LA, Wiener CM, Stenmark K, Semenza GL. Temporal, spatial, and oxygen-regulated expression of hypoxia-inducible factor-1 in the lung. Am J Physiol Lung Cell Mol Physiol 275: L818–L826, 1998 [DOI] [PubMed] [Google Scholar]
- 70. Yu AY, Shimoda LA, Iyer NV, Huso DL, Sun X, McWilliams R, Beaty T, Sham JS, Wiener CM, Sylvester JT, Semenza GL. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1α. J Clin Invest 103: 691–696, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Yuan G, Nanduri J, Bhasker CR, Semenza GL, Prabhakar NR. Ca2+/calmodulin kinase-dependent activation of hypoxia inducible factor 1 transcriptional activity in cells subjected to intermittent hypoxia. J Biol Chem 280: 4321–4328, 2005 [DOI] [PubMed] [Google Scholar]