

Abstract
Organophosphorus pesticides (OPs) are implicated in human asthma. We previously demonstrated that, at concentrations that do not inhibit acetylcholinesterase activity, the OP parathion causes airway hyperreactivity in guinea pigs as a result of functional loss of inhibitory M2 muscarinic receptors on parasympathetic nerves. Because macrophages are associated with asthma, we investigated whether macrophages mediate parathion-induced M2 receptor dysfunction and airway hyperreactivity. Airway physiology was measured in guinea pigs 24 h after a subcutaneous injection of parathion. Pretreatment with liposome-encapsulated clodronate induced alveolar macrophage apoptosis and prevented parathion-induced airway hyperreactivity in response to electrical stimulation of the vagus nerves. As determined by qPCR, TNF-α and IL-1β mRNA levels were increased in alveolar macrophages isolated from parathion-treated guinea pigs. Parathion treatment of alveolar macrophages ex vivo did not significantly increase IL-1β and TNF-α mRNA but did significantly increase TNF-α protein release. Consistent with these data, pretreatment with the TNF-α inhibitor etanercept but not the IL-1β receptor inhibitor anakinra prevented parathion-induced airway hyperreactivity and protected M2 receptor function. These data suggest a novel mechanism of OP-induced airway hyperreactivity in which low-level parathion activates macrophages to release TNF-α-causing M2 receptor dysfunction and airway hyperreactivity. These observations have important implications regarding therapeutic approaches for treating respiratory disease associated with OP exposures.
Keywords: airway hyperreactivity, alveolar macrophages, organophosphorus pesticides, parathion, tumor necrosis factor-α
organophosphorus pesticides (OPs) are the most widely used insecticides in the United States and throughout the world (45), both in agriculture (30) and in residential and urban communities (33). An early study reported that OP metabolites were detected in the urine of 99% of a cohort of Seattle children (33), and subsequent studies have reported that a mixture of pesticide residues are detected in the blood and/or urine of nearly all persons sampled in the United States (3). Increased use of OPs over the last two decades correlates with increased incidence of asthma, particularly in children (20). Both epidemiological and clinical studies have shown an association between OP exposure and symptoms of asthma, including wheezing and increased airway responsiveness (9, 22, 23). OPs also increase airway reactivity in animal models (48). In guinea pigs, we have shown that a single subcutaneous injection of phosphorothionate OPs, specifically diazinon, chlorpyrifos, or parathion, causes airway hyperreactivity 24 h later, and, at least for chlorpyrifos, this effect persists for at least 7 days (14, 32, 41).
The primary mechanism of acute OP intoxication is inhibition of acetylcholinesterase (AChE) activity, the enzyme responsible for hydrolyzing acetylcholine (ACh). Significant inhibition of AChE activity causes airway smooth muscle contraction and excessive mucus secretion. However, we have demonstrated that diazinon, chlorpyrifos, and parathion cause airway hyperreactivity in guinea pigs at doses too low to inhibit AChE activity (14, 32, 41). OPs target enzymes and receptors other than AChE, including cholinergic receptors (39, 47, 51). In lungs, postganglionic parasympathetic nerves release ACh onto M3 muscarinic receptors on airway smooth muscle, causing contraction, resulting in bronchoconstriction. ACh activation of inhibitory M2 muscarinic receptors located prejunctionally on parasympathetic nerves decreases ACh release and limits vagally induced bronchoconstriction (15). M2 muscarinic receptors are dysfunctional in some humans with asthma (34) and in animal models of antigen-, virus-, and ozone-induced airway hyperreactivity (12, 16, 18). Although OPs are reported to inhibit muscarinic receptor function in brain (28), we were unable to show a direct interaction between parathion or its oxon metabolite with M2 muscarinic receptor expression or function in autonomic neurons (42). These data suggest that parathion causes neuronal M2 muscarinic receptor dysfunction and airway hyperreactivity via an intermediary cell type, which then in turn impacts parasympathetic nerve function in the airways.
Alveolar and interstitial macrophages are the main immune cell type throughout the lung. In asthmatics, numbers of macrophages, as well as eosinophils and T cells (40), are increased, and monocyte chemoattractant protein is elevated (46). Macrophages release TNF-α and IL-1β, both of which are increased in the lungs of asthmatics and in animal models of hyperreactivity (5, 7). In experimental models, administration of TNF-α or IL-1β enhances airway reactivity (29, 57). Conversely, blocking TNF-α or IL-1β with specific antagonists prevents airway hyperreactivity caused by antigen and other environmental triggers, such as ozone (35, 49, 56). TNF-α and IL-1β and other macrophage factors can decrease M2 muscarinic receptor function or expression (35). OPs and related organophosphate nerve agents, given to animals at low doses, affect isolated monocytes and macrophages, increasing respiratory burst capacity (44) and decreasing phagocytic activity (27). However, whether OPs similarly influence macrophage function in vivo has not been reported. OPs and related organophosphate nerve agents have been demonstrated to increase mRNA of interleukin-6, IL-1β, and TNF-α and increase NF-κB activation in whole lung (38), suggesting the possibility of macrophage involvement. Thus we investigated whether macrophages and these inflammatory pathways contribute to OP-induced airway hyperreactivity.
Here we report that inducing macrophage apoptosis with clodronate prevents parathion-induced airway hyperreactivity. Furthermore, parathion increases TNF-α and IL-1β expression in macrophages isolated from the lungs, and blocking TNF-α, but not IL-1β, prevents parathion-induced airway hyperreactivity and neuronal M2 muscarinic receptor dysfunction.
MATERIALS AND METHODS
Animals.
Young adult, pathogen-free female Hartley guinea pigs (300–375 g) were obtained from Elm Hill Laboratory (Chelmsford, MA). Guinea pigs were shipped in filtered crates, housed in rooms with air filtered using high-efficiency particulate absorbers, and fed standard guinea pig diet (Prolab; Agway, Syracuse, NY). Guinea pigs were treated humanely in accordance with standards established by the U.S. Animal Welfare Act set forth by National Institutes of Health guidelines. All protocols were approved by the Animal Care and Use Committee at Oregon Health and Science University.
Animal pretreatments.
Clodronate, a gift of Roche Diagnostics (Mannheim, Germany), was used to induce alveolar macrophage apoptosis (53). Liposome-encapsulated clodronate (7 mg clodronate/ml) and liposome-encapsulated PBS were prepared as previously described (54). On day 1, liposome-encapsulated clodronate or liposome-encapsulated PBS was administered intranasally (i.n.; 0.3 ml) and i.p. (1 ml), and, on day 3, liposomes were administered again i.n. (0.3 ml). On day 4, guinea pigs were treated with 1.0 mg/kg s.c. parathion (o,o-diethyl-o-p-nitrophenyl phosphorothioate, 99.5% pure; Chem Service, West Chester, PA) in peanut oil or an equivalent volume (300 μl) of peanut oil (vehicle controls) injected into the subscapular region (32). Subcutaneous dosing results in slow sustained pesticide release into the systemic circulation (50), approximating human dermal exposure (17). Animals were monitored 1, 4, and 24 h after pesticide administration for signs of cholinergic intoxication (tremors, altered gait, and excessive excretions), none of which were observed. Recombinant human IL-1 receptor antagonist anakinra (Kineret; Amgen, Thousand Oaks, CA) was diluted in PBS, and either 30 mg/kg anakinra or PBS (control) was administered i.p. 30 min before s.c. parathion or peanut oil. TNF-α inhibitor, etanercept (Enbrel; Amgen), was diluted in PBS, and either 3 mg/kg etanercept or PBS (control) was administered i.p. 24 h before s.c. parathion or peanut oil. Doses and timing of anakinra (49, 56) and etanercept (35) were chosen based on their effectiveness in preventing airway hyperreactivity in other guinea pig asthma models.
Measurement of pulmonary inflation pressure and bradycardia.
Physiological measurements of lung function were performed 24 h after parathion administration as previously described (13). Guinea pigs were anesthetized with urethane (1.7–1.9 g/kg, i.p.; Sigma-Aldrich, St. Louis, MO). Depth of anesthesia was determined by a foot pinch and a touch to the inner corner of the eye. Both jugular veins were cannulated for i.v. drug administration. A carotid artery was cannulated and attached to a pressure transducer (BD Biosciences, San Jose, CA) to measure heart rate and blood pressure. These recordings were also used to monitor the depth of anesthesia after neuromuscular blockade. Guinea pigs were tracheostomized, cannulated, paralyzed with succinylcholine (10 μg·kg−1·min−1, i.v.; Sigma-Aldrich), and ventilated with positive pressure and constant volume (1 ml/100 g body wt and 100 breaths/min). Guanethidine (2 mg/kg, i.v.; Sigma-Aldrich) was used to deplete noradrenaline. Both vagus nerves were cut and distal ends placed on platinum electrodes. Pulmonary inflation pressure (mmH2O) was measured with a pressure transducer off a side arm of the tracheal cannula. Electrical stimulation of the vagus nerves (1–15 Hz, 10 V, 0.2 ms, for 5 s at 40–60-s intervals) induced bradycardia and bronchoconstriction that was measured as a decrease in beats per minute and an increase in pulmonary inflation pressure, respectively, as previously described (13). Treatment with parathion, liposome-encapsulated clodronate, liposome-encapsulated PBS, etanercept, or anakinra independently or in combination had no effect on baseline pulmonary inflation pressure, baseline heart rate, or baseline blood pressure in age-matched guinea pigs (Table 1).
Table 1.
Baseline physiological values in age-matched guinea pigs
| Baseline Values | Weight, g | Ppi, mmH2O | Heart Rate, bpm | Systolic BP, mmHg | Diastolic BP, mmHg |
|---|---|---|---|---|---|
| Clodronate (n = 4–7) [Figure 1] | |||||
| PBSlip | 358.6 ± 6.4 | 101.4 ± 2.6 | 279.3 ± 10.6 | 44.6 ± 2.1 | 23.7 ± 2.1 |
| Cldlip | 370.5 ± 11.0 | 97.5 ± 5.3 | 294.4 ± 7.7 | 42.3 ± 2.8 | 22.0 ± 2.1 |
| PBSlip + Pth | 366.0 ± 9.6 | 101.1 ± 4.2 | 297.2 ± 8.1 | 49.8 ± 2.2 | 25.6 ± 2.4 |
| Cldlip + Pth | 354.6 ± 6.8 | 103.0 ± 5.8 | 285.5 ± 8.1 | 40.0 ± 1.4 | 19.2 ± 1.1 |
| Anakinra (n = 6–7) [Figure 6] | |||||
| Control | 360.4 ± 11.0 | 98.6 ± 1.4 | 283.6 ± 16.5 | 50.6 ± 2.1 | 18.3 ± 1.7 |
| Ank | 366.3 ± 5.6 | 93.3 ± 5.6 | 241.7 ± 6.1 | 40.7 ± 3.0 | 12.3 ± 2.0 |
| Pth | 366.1 ± 5.6 | 91.4 ± 3.4 | 269.3 ± 15.4 | 52.6 ± 4.8 | 16.0 ± 1.6 |
| Ank + Pth | 359.5 ± 8.9 | 101.7 ± 4.8 | 284.2 ± 12.3 | 52.0 ± 1.9 | 17.0 ± 1.7 |
| Etanercept (n = 5–7) [Figure 7] | |||||
| Control | 369.5 ± 11.7 | 95.0 ± 3.4 | 287.5 ± 6.4 | 47.7 ± 3.4 | 21.0 ± 2.2 |
| Etn | 387.3 ± 4.1 | 90.0 ± 2.2 | 267.9 ± 15.8 | 48.6 ± 1.0 | 22.9 ± 3.1 |
| Pth | 365.1 ± 9.8 | 100.0 ± 4.4 | 280.7 ± 8.0 | 50.6 ± 2.6 | 22.3 ± 1.8 |
| Etn + Pth | 380.3 ± 11.1 | 92.5 ± 2.5 | 283.1 ± 15.5 | 44.0 ± 4.2 | 24.5 ± 2.9 |
| M2 function Etanercept (n = 5–6) [Figure 10] | |||||
| Control | 356.0 ± 14.1 | 91.7 ± 3.1 | 282.5 ± 17.3 | 45.3 ± 4.4 | 17.3 ± 2.2 |
| Etn | 346.2 ± 3.4 | 94.0 ± 6.0 | 254.0 ± 12.0 | 45.2 ± 3.0 | 14.8 ± 1.6 |
| Pth | 352.8 ± 5.6 | 88.3 ± 7.0 | 291.7 ± 10.0 | 42.0 ± 0.9 | 16.7 ± 1.8 |
| Etn + Pth | 340.3 ± 7.5 | 95.5 ± 6.7 | 261.7 ± 17.5 | 48.0 ± 2.7 | 16.0 ± 1.5 |
Data are presented as means ± SE. Ank, anakinra; Cldlip, liposome-encapsulated clodronate; Etn, etanercept; PBSlip, liposome-encapsulated PBS (control); Ppi, pulmonary inflation pressure; Pth, parathion.
Measurement of M2 receptor function.
M2 muscarinic receptor function was measured using the selective M2 receptor antagonist gallamine in animals separate from those used to measure vagally induced and ACh-induced airway reactivity. The distal ends of both vagus nerves were stimulated at 15 Hz, 2–20 V, 0.2-ms pulse duration, for 3 s at 40-s intervals until 10 consistent bronchoconstrictions of 10–25 mmH2O were measured (≤5 mmH2O difference) above baseline inflation pressure. The last five bronchoconstrictions were averaged to obtain a baseline. Gallamine (0.1, 0.3, 1.0, 3.0, and 10 mg/kg) was administered i.v., and four bronchoconstrictions were measured after each gallamine dose and averaged. The effect of gallamine on M2 receptor function was assessed as the ratio of mean bronchoconstriction after each dose of gallamine to the mean bronchoconstriction before gallamine. Voltages were not significantly different between groups (data not shown).
AChE activity assay.
PBS-perfused lung and brain tissue and heparinized blood were collected and measured for AChE activity using the Ellman assay (10) as previously described (32).
Bronchoalveolar lavage.
In experiments that did not involve culturing bronchoalveolar lavage (BAL) macrophages, BAL fluid was collected immediately following physiological experiments using five aliquots of 10 ml PBS (room temperature). Cells were centrifuged for 10 min at 300 g and resuspended in 20 ml PBS. Cells were counted on a hemocytometer to obtain total cell counts and cytospun onto slides and stained with Hemacolor (EMD Chemicals, Philadelphia, PA) to obtain differential cell counts.
Culturing alveolar macrophages.
BAL was performed as described above with the exception that antibiotics (100 I.U. penicillin/ml and 100 μg/ml streptomycin; Mediatech, Manassas, VA) were added to sterile PBS (4°C). BAL fluid was centrifuged for 10 min at 300 g, and cells were resuspended in RPMI-1640 media (Cellgro 15-040-CV; Mediatech) containing glutamine (2 μM; Invitrogen, Carlsbad, CA), antibiotics, and 10% heat-inactivated fetal bovine serum (Hyclone 30071.63; Thermo Scientific, Waltham, MA). All BAL cells were plated into tissue culture dishes and incubated for 1 h at 37°C at 95% O2-5% CO2. After 1 h, nonadherent cells were gently dislodged and removed, leaving adherent macrophages (>95% purity as identified by Hemacolor stain; EMD Chemicals). Alveolar macrophages used for mRNA analysis were isolated and cultured from parathion- or control-treated treated guinea pigs following physiological experiments and frozen (−80°C) for RNA isolation (see below).
Caspase-3 activity assay.
Alveolar macrophages were cultured from the BAL of animals treated with liposome-encapsulated PBS or liposome-encapsulated clodronate. Macrophages were rinsed with PBS, lysed with 0.4 ml lysis buffer [10 mM Tris·HCl, 10 mM NaH2PO4/Na2HPO4 (pH 7.5), 130 mM NaCl, 1% Triton X-100, 10 mM Na pyrophosphate in sterile H2O], and stored at −80°C until used. Lysed macrophages were thawed and centrifuged. For both control (no cell lysate) and macrophage cell lysate samples, a duplicate sample was treated with caspase inhibitor (10 μM AC-DEVD-CHO; Sigma-Aldrich) for 30 min on ice. A caspase-3 fluorogenic substrate (20 μM AC-DEVD-AMC; BD Biosciences) was added to each sample. Samples were loaded in a 96-well plate in duplicate and incubated for 1 h at 37°C. The 7-amino-4-methylcoumarin standard (Sigma-Aldrich) was serially diluted in cell lysis buffer and loaded in duplicate in the same 96-well plate. The plate was read on a spectrofluorometer with an excitation wavelength of 380 nm and an emission wavelength of 430–460 nm. Background fluorescence (averaged from the control sample) was subtracted from each sample.
Alveolar macrophages exposed to parathion and paraoxon ex vivo.
Macrophages were isolated from BAL (as described above) collected from control anesthetized guinea pigs not used in physiological experiments. Stock parathion and paraoxon (Chem Service) were diluted first in DMSO and subsequently in phenol-free RPMI media. Parathion (100 μM), paraoxon (100 nM), or 0.1% DMSO (control) was added to the macrophages for 24 h before the conditioned medium was collected for ELISA assay (see below) and mRNA was isolated from the cells. All experiments were run in triplicate.
qPCR.
Alveolar macrophage RNA was isolated using the RNeasy Mini Kit (Qiagen, Valencia, CA). RNA was reverse transcribed with SuperScript III (Invitrogen) using random hexamers. cDNA (1 μl of 1:100 dilution for 18S and 2 μl of 1:10 dilution for IL-1β and TNF-α) was amplified for 40 cycles at an annealing temperature of 59°C using QuantiTect SYBR Green (Qiagen) in duplicate using the 7500 Fast Real-Time PCR System (Applied Biosystems, Carlsbad, CA). PCR products were quantified on the Mx3000P real-time PCR System (Stratagene, Wilmington, DE). Specific PCR primers for 18S, IL-1β, and TNF-α were synthesized (Integrated DNA Technologies, Coralville, IA) as follows: 18S rRNA 5′ GTAACCCGTTGAACCCCATT and 18S rRNA 3′ CCATCCAATCGGTAGTAGCG; guinea pig IL-1β 5′ CTTTGAAGAAGAGCCCATCG and guinea pig IL-1β 3′ CGAAGCTCATGGAGAACACC; and guinea pig TNF-α 5′ CCTACCTGCTTCTCACCCATACC and guinea pig TNF-α 3′ TTGATGGCAGAGAAGGTTGA. For each gene product, a linear regression formula was generated by plotting the log nanogram of input RNA vs. the critical threshold (CT) value obtained from a serial dilution of a random cDNA sample (6). Using this standard curve, the CT values for each sample were converted to relative mRNA concentrations. The relative amount of mRNA for each gene product was normalized to the relative amount of 18S mRNA expression for that particular sample.
ELISA.
Guinea pig IL-1β and TNF-α were measured using commercially available ELISA kits (R&D Systems, Minneapolis, MN). Detection limits were 31.25 pg TNF-α/ml and 62.5 pg IL-1β/ml.
Macrophage immunohistochemistry and analysis.
Guinea pig lungs were removed, fixed in zinc formalin, and embedded in paraffin. Staining of paraffin sections required using Antigen Unmasking Solution (Vector Laboratories, Burlingame, CA), followed by quenching of endogenous peroxidase with 3% hydrogen peroxide in methanol. Nonspecific binding was blocked with 2.5% normal horse serum. Macrophages in lung sections were identified using a primary antibody for calprotectin (1:1,000 MAC 387 antibody; AbD Serotec, Raleigh, NC) and secondary anti-mouse ImmPRESS Reagent kit (Vector Laboratories) with ImmPACT DAB (Vector Laboratories). In each of four lung sections from each animal, 19–25 randomly selected views of lung parenchyma encompassing the entire section were photographed at ×10 (CoolSNAP; PhotoMetrics, Huntington Beach, CA) using the Metamorph Imaging System (Universal Imaging, Downingtown, PA). For each picture, MAC 387-positive cells were manually counted (avoiding labeled monocytes in blood vessels) by experimenters blinded to the experimental condition. The number of MAC 387-positive cells per field of view in the four lung sections was averaged for each animal.
Data analysis.
Frequency and dose response curves of vagally induced and ACh-induced bronchoconstriction and bradycardia and gallamine dose response curves were analyzed by repeated-measures two-way ANOVA. BAL and blood differential counts, AChE activity, and macrophage immunohistochemistry were analyzed by one-way ANOVA with post hoc Bonferroni correction. Real-time quantification of BAL macrophages treated with parathion ex vivo and TNF-α ELISA were analyzed by one-way ANOVA with post hoc Dunnett's multiple-comparison test. Caspase-3 activity and real-time PCR quantification of BAL macrophages treated with parathion in vivo were analyzed using an unpaired, one-tailed Student's t-test. Statistical probability of P ≤ 0.05 was considered significant. Data are represented as means ± SE.
RESULTS
Clodronate pretreatment blocks parathion-induced airway hyperreactivity.
Electrical stimulation of the vagus nerves caused frequency-dependent bronchoconstriction measured as an increase in pulmonary inflation pressure (Fig. 1A) and frequency-dependent decrease in heart rate (Fig. 1B) in animals pretreated with PBS encapsulated in liposomes (controls). Pretreatment with clodronate encapsulated in liposomes did not change either vagally induced bronchoconstriction or vagally induced bradycardia relative to the controls. Consistent with our previous studies (14, 32, 41), parathion significantly increased vagally mediated bronchoconstriction in animals pretreated with PBS liposomes. In contrast, this parathion-induced hyperresponsiveness was completely prevented by pretreating guinea pigs with clodronate liposomes (Fig. 1A). Neither parathion nor clodronate, alone or in combination, had any effect on vagally induced bradycardia (Fig. 1B).
Fig. 1.

Pretreatment with liposome-encapsulated clodronate (Cldlip) prevented parathion (Pth)-induced airway hyperreactivity. A: electrical stimulation of the vagus nerves caused frequency-dependent bronchoconstriction that was similar in guinea pigs pretreated with either liposome-encapsulated PBS (PBSlip) or Cldlip. Parathion (1.0 mg/kg, s.c.) significantly potentiated vagally induced bronchoconstriction in animals pretreated with PBSlip. Cldlip pretreatment prevented parathion potentiation of vagally induced bronchoconstriction (n = 4–7). B: electrical stimulation of the vagus nerves caused frequency-dependent bradycardia that was not affected by parathion or Cldlip, administered independently or in combination (n = 4–5). Data are presented as means ± SE (*P < 0.05). Ppi, pulmonary inflation pressure; HR, heart rate.
Neither parathion nor clodronate affected airway smooth muscle responsiveness.
Direct stimulation of M3 muscarinic receptors on airway smooth muscle via intravenous ACh caused dose-dependent bronchoconstriction in all treatment groups. ACh-induced bronchoconstriction was not different between control and parathion-treated guinea pigs in the absence or presence of clodronate (Fig. 2A). Neither liposome treatment nor parathion inhibited AChE activity in blood or lung relative to controls (Fig. 2B).
Fig. 2.

Pretreatment with Cldlip had no effect on acetylcholine-induced bronchoconstriction or acetylcholinesterase (AChE) activity. A: ACh (1–10 μg/kg, i.v.) induced dose-dependent bronchoconstriction. Treatment with parathion or Cldlip independently or in combination had no effect on ACh-induced bronchoconstriction relative to animals treated with PBSlip (n = 4–7). B: treatment with Cldlip or parathion independently or in combination did not inhibit AChE activity in lung or blood relative to levels observed in the same tissues from guinea pigs treated with PBSlip (n = 3). Data are presented as means ± SE.
Effects of clodronate and parathion on macrophages in the lung.
To confirm that clodronate was targeting macrophages, caspase-3 activity was measured in alveolar macrophages isolated from guinea pig BAL. As shown in Fig. 3A, caspase-3 activity was significantly increased in macrophages obtained from guinea pigs treated with clodronate liposomes compared with guinea pigs treated with PBS liposomes. Clodronate increased caspase-3 activity independent of parathion treatment (Fig. 3A, ◇ and △). Addition of a caspase-3 inhibitor to the isolated macrophages completely inhibited the signal, confirming that the assay was measuring caspase-3 activity (Fig. 3A, ●, ◆, ▲, and ■). Neither parathion nor clodronate affected inflammatory cell numbers in the BAL (Fig. 3B). Clodronate also did not change the number of lung interstitial macrophages as identified by calprotectin immunoreactivity (Fig. 3, C–D).
Fig. 3.
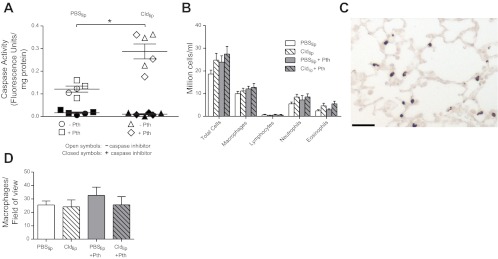
Pretreatment with Cldlip increased caspase-3 activity in alveolar macrophages but did not deplete the number of alveolar or interstitial macrophages. A: caspase-3 activity was measured in alveolar macrophages using a fluorogenic substrate and normalized to protein concentration. Pretreatment with Cldlip (n = 6) significantly increased caspase-3 activity in alveolar macrophages isolated from control or parathion-treated guinea pigs relative to alveolar macrophages isolated from guinea pigs that received PBSlip (n = 5). To confirm that the fluorescent signal was due to caspase activity, fluorescence was also quantified in duplicate samples to which the caspase-3 inhibitor Ac-DEVD-CHO had been added. B: the number of macrophages and other inflammatory cells in the bronchoalveolar lavage (BAL) was not changed by treatment with either parathion or Cldlip (n = 7–9). As shown in a representative photomicrograph from a guinea pig that received Cldlip (C), interstitial lung macrophages were identified by calprotectin immunoreactivity (bar = 100 μm). D: the number of interstitial macrophages per unit area was not changed by either parathion or Cldlip treatment (n = 4–7). Data are presented as means ± SE (*P ≤ 0.05).
Effects of parathion and paraoxon on proinflammatory cytokine expression in alveolar macrophages.
Both TNF-α and IL-1β mRNA were significantly increased in alveolar macrophages isolated from guinea pigs treated with parathion compared with peanut oil (vehicle) controls (Fig. 4). When alveolar macrophages were isolated from untreated guinea pigs and exposed ex vivo to parathion for 24 h, no significant changes were detected in transcript levels of either TNF-α (Fig. 5A) or IL-1β (Fig. 5B). In contrast, TNF-α protein was significantly increased in conditioned media from these same cells (Fig. 5C). IL-1β protein was not detected in conditioned media, and neither IL-1β nor TNF-α proteins were detected in the BAL fluid by ELISA (data not shown). Ex vivo exposure to the oxon metabolite of parathion, paraoxon, had no effect on alveolar macrophage levels of TNF-α mRNA (Fig. 5A), IL-1β mRNA (Fig. 5B), or TNF-α protein (Fig. 5C). Thus production of TNF-α protein was increased by parathion but not by the more active metabolite paraoxon, consistent with inhibition of cholinesterase activity not being central to this effect.
Fig. 4.

TNF-α and IL-1β mRNA expression in BAL macrophages isolated from guinea pigs treated with parathion. RNA was isolated from BAL macrophages collected from guinea pigs 24 h after administration of parathion (1.0 mg/kg, s.c.) or an equal volume of peanut oil (vehicle control). As determined by qPCR, parathion significantly increased TNF-α (A) and IL-1β (B) mRNA expression in BAL macrophages (n = 5–7). Data are presented as means ± SE (*P ≤ 0.05).
Fig. 5.
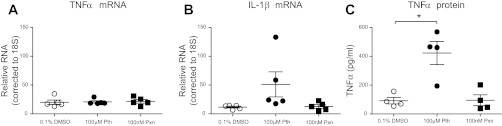
TNF-α and IL-1β expression in guinea pig BAL macrophages treated ex vivo with parathion. BAL macrophages were cultured from naïve guinea pigs and treated ex vivo with 100 μM parathion or 100 nM paraoxon (Pxn) for 24 h. Both mRNA and cell-conditioned media were collected. As determined by qPCR, neither parathion nor paraoxon had a significant effect on TNF-α (A) or IL-1β mRNA expression (B). Exposure to parathion but not paraoxon significantly increased TNF-α protein levels as determined by ELISA (C). IL-1β protein was undetectable by ELISA in control and treated macrophages (data not shown). (A–C; n = 4–5). Data are presented as means ± SE (*P ≤ 0.05).
The IL-1β receptor inhibitor anakinra did not prevent parathion-induced airway hyperreactivity.
Electrical stimulation of the vagus nerves caused frequency-dependent bronchoconstriction that was not changed by pretreatment with 30 mg/kg anakinra (Fig. 6A). Parathion significantly potentiated vagally induced bronchoconstriction, and this parathion-induced airway hyperreactivity was not prevented by anakinra pretreatment (Fig. 6A). Intravenous ACh caused dose-dependent bronchoconstriction (Fig. 6B). Neither anakinra nor parathion, alone or in combination, affected airway smooth muscle responsiveness to ACh (Fig. 6B). Neither parathion nor anakinra, alone or in combination, affected total and differential counts of leukocytes in the peripheral blood and BAL (data not shown). None of these treatments altered blood, brain, and lung AChE relative to controls (data not shown).
Fig. 6.

Pretreatment with the IL-1β antagonist anakinra (Ank) had no effect on parathion-induced airway hyperreactivity. A: electrical stimulation of the vagus nerves caused frequency-dependent bronchoconstriction in control animals that was not changed with anakinra (30 mg/kg, i.p.) treatment. Parathion (1.0 mg/kg, s.c.) significantly potentiated vagally induced bronchoconstriction, and this effect was not blocked by pretreatment with anakinra (n = 6–7). B: intravenous administration of ACh caused a dose-dependent increase in bronchoconstriction in control animals. Neither parathion, anakinra, nor the combination of these compounds had a significant effect on ACh-induced bronchoconstriction (n = 6–7). Data are presented as means ± SE (*P < 0.05).
The TNF-α inhibitor etanercept prevented parathion-induced airway hyperreactivity at the level of the parasympathetic nerves.
Electrical stimulation of the vagus nerves caused frequency-dependent bronchoconstriction (Fig. 7A) and frequency-dependent bradycardia (Fig. 7B). The potentiation of vagally induced bronchoconstriction by parathion was prevented by pretreatment with etanercept (Fig. 7A). Etanercept had no effect on vagally induced bronchoconstriction in controls. Vagally induced bradycardia was not significantly increased by parathion (Fig. 7B). Intravenous ACh caused dose-dependent bronchoconstriction and bradycardia that was not altered by parathion or etanercept, either alone or in combination (Fig. 7, C–D). Neither parathion, etanercept, nor the combination affected AChE activity in lung, blood, or brain that was collected following physiological experiments (Fig. 8).
Fig. 7.
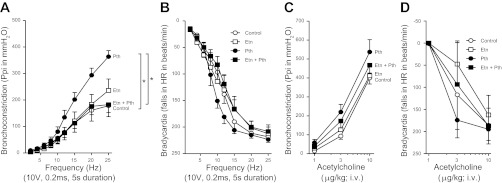
Pretreatment with the TNF-α antagonist etanercept (Etn) prevented parathion-induced airway hyperreactivity. A: electrical stimulation of the vagus nerves caused frequency-dependent bronchoconstriction in control animals that was not changed by etanercept (3 mg/kg, i.p.). The significant potentiation of vagally induced bronchoconstriction caused by parathion (1.0 mg/kg, s.c.) was prevented by pretreatment with etanercept. B: electrical stimulation of the vagus nerves also caused a frequency-dependent increase in bradycardia in control animals. Neither parathion nor etanercept alone or in combination had a significant effect on frequency-dependent bradycardia. C–D: intravenous administration of ACh caused a dose-dependent increase in bronchoconstriction (C) and bradycardia (D) in control animals. Parathion did not potentiate ACh-induced bronchoconstriction (C) or bradycardia (D). Etanercept in the presence or absence of parathion did not increase ACh-induced bronchoconstrictions (C) or bradycardia (D) above control responses (n = 5–7). Data are presented as means ± SE (*P < 0.05).
Fig. 8.
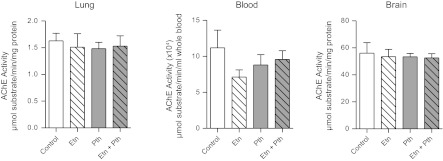
Neither parathion nor etanercept affected AChE activity. Lung, blood, and brain were collected after physiological experiments to measure AChE activity. Neither parathion nor etanercept, alone or in combination, affected AChE activity compared with comparable tissues from vehicle control animals (n = 5–8). Data are presented as means ± SE.
The effect of parathion and etanercept on BAL and blood cells.
Neither parathion, etanercept, nor the combination affected the number of macrophages, lymphocytes, neutrophils, eosinophils, or total number of cells in the BAL (Fig. 9A). In the blood, monocytes, lymphocytes, neutrophils, and eosinophils were not affected by any treatment (Fig. 9B).
Fig. 9.

Macrophage and monocyte cell numbers were not affected by etanercept or parathion in BAL or peripheral blood, respectively. BAL and blood were collected following physiological experiments for total cell and differential cell counts. A: neither parathion nor etanercept, alone or in combination, affected the number of total cells, macrophages, lymphocytes, neutrophils, or eosinophils in the BAL. B: neither parathion nor etanercept, alone or in combination, affected the total number of cells, monocytes, lymphocytes, neutrophils, or eosinophils in the blood (n = 5–8.) Data are presented as means ± SE.
Etanercept inhibits parathion-induced M2 muscarinic dysfunction.
The M2 muscarinic antagonist gallamine increased vagally induced bronchoconstriction in a dose-related manner in control animals, demonstrating that M2 receptors were functioning normally to inhibit ACh release (Fig. 10). In contrast, the ability of gallamine to potentiate bronchoconstriction was significantly inhibited in parathion-treated guinea pigs, indicating M2 receptor dysfunction (Fig. 10). The TNF-α inhibitor etanercept had no effect on gallamine-induced potentiation in controls, but, in the presence of etanercept, parathion no longer inhibited M2 receptor function (Fig. 10).
Fig. 10.

Etanercept partially prevented parathion-induced M2 muscarinic receptor dysfunction. Guinea pigs were administered increasing doses of the M2 receptor antagonist gallamine (i.v.) to test M2 receptor function. In control animals, gallamine dose-dependently increased bronchoconstrictions compared with bronchoconstrictions measured before gallamine administration, indicating the M2 receptor is functional. Pretreatment with etanercept alone did not affect M2 receptor function. The gallamine response in parathion-treated guinea pigs was significantly lower than control animals, indicating M2 receptor dysfunction. In the presence of etanercept, parathion no longer inhibited M2 receptor function (n = 5–6). Data are presented as means ± SE (*P < 0.05).
DISCUSSION
Consistent with our previous studies (14, 32, 41), parathion potentiated vagally induced bronchoconstriction and caused M2 receptor dysfunction on airway nerves in guinea pigs at a dose that did not inhibit AChE. This same dose of parathion caused significant M2 receptor dysfunction in airway nerves but had no effect on any other physiological parameters measured, including animal weight, baseline pulmonary inflation pressure, heart rate, and blood pressure. Also consistent with previous data showing that airway hyperreactivity is independent of BAL inflammatory cell number (11), parathion did not increase the number of cells in the BAL. Parathion also had no effect on bronchoconstriction induced by intravenous ACh. Collectively, these data demonstrate that parathion-induced airway hyperreactivity is mediated at the vagus nerves and is not due to a postjunctional increase in airway smooth muscle contraction.
M2 receptors are expressed prejunctionally on the vagus nerve innervating the heart and postjunctionally on cardiac myocytes. Parathion did not significantly increase bradycardia in response to electrical stimulation or i.v. ACh, indicating the M2 receptors in the heart were not affected by parathion. This dissociation between a loss of M2 receptor function on lung parasympathetic nerves but not heart parasympathetic nerves has also been observed with virus- and antigen challenge-induced airway hyperreactivity in guinea pigs (16, 36). These data are consistent with other data indicating that M2 receptors are differentially regulated in different tissues (19).
Whereas parathion caused a loss of M2 receptor function on parasympathetic nerves, our previous studies indicated that neither parathion nor its oxon metabolite, paraoxon, interact directly with parasympathetic nerves to alter M2 receptor expression, subcellular localization, or function (42). Therefore, we hypothesized that parathion acts on an intermediate cell type in the lung that subsequently influences the function of airway nerves to cause airway hyperreactivity. The data reported here identify alveolar macrophages as an intermediary cell targeted by parathion. The most direct evidence supporting a key role for alveolar macrophages is that parathion-induced airway hyperreactivity was completely prevented by pretreating guinea pigs with liposome-encapsulated clodronate. Macrophages phagocytize the liposomes containing clodronate, and lysosomal phospholipases release clodronate into the cell, inducing apoptosis (55). In rats and mice, intratracheal or intranasal instillation of clodronate is effective at depleting alveolar macrophages by 80–95% within 24 h (21, 52), and this effect persists for at least 72 h (4, 54). Whereas pretreatment with liposome-encapsulated clodronate clearly prevented parathion-induced airway hyperreactivity in guinea pigs, we were unable to demonstrate macrophage depletion in the BAL or lung, even though it was administered by simultaneous intranasal instillation and intraperitoneal injection. This is consistent with our previous report of clodronate use in guinea pigs (31). Clodronate did, however, induce apoptosis of alveolar macrophages. Caspase-3 activity, a marker of apoptosis, was significantly increased in alveolar macrophages isolated from clodronate-treated guinea pigs regardless of parathion treatment. Thus, although clodronate did not decrease macrophage cell numbers, it is likely that, at the time when airway hyperreactivity was measured, alveolar macrophages were apoptotic, suggesting that loss of macrophage function is the mechanism underlying the protective effect of clodronate against parathion-induced airway hyperreactivity.
Alveolar macrophages potentially modulate parathion-induced airway hyperreactivity via release of soluble mediators that cause neuronal M2 receptor dysfunction. Activated macrophages release IL-1β and TNF-α, both of which are associated with asthma and airway hyperresponsiveness (2, 7, 29, 57) and have been shown to decrease M2 receptor expression (35). This study supports the hypothesis that release of TNF-α from alveolar macrophages mediates parathion-induced airway hyperreactivity. Transcript levels of TNF-α were significantly elevated in BAL macrophages from guinea pigs exposed to parathion, and ex vivo exposure to parathion increased release of TNF-α from alveolar macrophages isolated from untreated guinea pigs. Lastly, pretreatment with the TNF-α inhibitor etanercept blocked parathion-induced airway hyperreactivity. Whereas IL-1β mRNA was also increased in alveolar macrophages exposed to parathion in vivo, pretreatment with the IL-1β receptor inhibitor anakinra did not prevent parathion-induced airway hyperreactivity. This is likely not due to insufficient levels of anakinra because we previously showed that this dose of anakinra prevented vagally mediated airway hyperreactivity in guinea pigs after ozone exposure (56). Thus parathion-induced airway hyperresponsiveness is not mediated by a general upregulation of inflammatory mediators but is mediated selectively by TNF-α from macrophages.
Here we show that parathion has direct effects on macrophages as evidenced by increased release of TNF-α from alveolar macrophages exposed to parathion ex vivo. The observation that parathion increased TNF-α mRNA in alveolar macrophages exposed in vivo, whereas ex vivo exposure increased TNF-α release but not mRNA expression, suggests that parathion influences macrophages via direct and indirect effects. Other inflammatory cells may contribute to parathion-induced airway hyperreactivity in vivo. For example, our preliminary observations (P. Lein and A. Grodzki, unpublished observations) and other data (44) show that OPs activate mast cells. Mast cells release several mediators that modulate macrophage function (43) and airway smooth muscle contractility (37). Determining whether mast cells contribute to parathion-induced airway hyperreactivity is a focus of ongoing research in our laboratories.
Loss of M2 receptor function contributes to airway hyperreactivity in some patients with asthma (34) and mediates hyperreactivity in animals that are antigen challenged (16), exposed to ozone (18), virus infected (12), or exposed to OPs (14, 32). In all these models, treatments that protect or restore M2 receptor function also prevent or reverse airway hyperreactivity. Consistent with our previous reports (14, 32), parathion caused M2 muscarinic receptor dysfunction, measured as the inability of gallamine to potentiate vagally induced bronchoconstriction. Pretreatment with etanercept, not only prevented airway hyperreactivity, but also protected neuronal M2 receptor function. These data, together with the finding that neither parathion nor etanercept alter intravenous ACh-induced bronchoconstriction, suggest that TNF-α is mediating parathion-induced airway hyperreactivity at the level of the nerve.
Etanercept is used clinically to treat arthritis and plaque psoriasis, which are disorders characterized by inflammation. Inflammation is also a characteristic of asthma, and anti-TNF-α therapy reduces airway hyperreactivity and frequency exacerbation in patients with severe steroid refractory asthma (5, 24), particularly in patients with increased TNF-α (5). Our data predict that etanercept could be a potential therapy for OP-induced airway hyperreactivity and asthma.
There is an increasing recognition that OPs cause toxic effects independent of AChE inhibition via mechanism(s) that include immunomodulation (1, 26, 39, 44). In this study, our data support other studies showing that OPs, specifically parathion, modulate macrophages. However, unlike other studies, we show that this interaction has a significant deleterious effect on airway function, specifically through the release of macrophage TNF-α but not macrophage IL-1β. Furthermore, we show that it is the parent compound, parathion, but not the metabolite that inhibits AChE, paraoxon, that increases TNF-α release from alveolar macrophages exposed in culture. This concurs with our previous studies (32, 41) that show parathion causes airway hyperreactivity at doses lower than those required to inhibit AChE activity in the blood, brain, or lung. That the parent compound may be responsible for non-AChE effects is consistent with a recent paper demonstrating that parathion was more potent than paraoxon in producing genotoxicity (25). Understanding how OP parent compounds and metabolites affect non-AChE targets will improve regulatory policies regarding the safe use of these pesticides because current exposure limits are based on AChE activity in the blood.
There is widespread human exposure to OPs, both in agricultural and urban settings (30, 33). The data presented here may have important implications for the role of OPs in asthma, especially in agricultural workers (23) and inner city children (8). Our study presents a mechanism whereby OPs activate macrophages to release TNF-α, resulting in loss of inhibitory M2 muscarinic receptor function on airway parasympathetic nerves. Macrophages and muscarinic receptors control important physiological functions in the brain, heart, gastrointestinal tract, and urogenital system. Although we have not examined M2 receptor function in the central nervous system or other parasympathetic target organs, there is no reason to suspect that OP-induced activation of macrophages and subsequent loss of neuronal M2 function is limited to the lungs.
GRANTS
This work was supported by the National Institutes of Health (ES017592 awarded to P. Lein and A. Fryer, ES014521 awarded to B. Proskocil, ES014601 awarded to A. Fryer and P. Lein, and HL113023 and AR061567 awarded to D. Jacoby).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: B.J.P., D.B.J., P.J.L., and A.D.F. conception and design of research; B.J.P. and D.A.B. performed experiments; B.J.P. analyzed data; B.J.P., P.J.L., and A.D.F. interpreted results of experiments; B.J.P. prepared figures; B.J.P. and A.D.F. drafted manuscript; B.J.P., D.B.J., N.v.R., P.J.L., and A.D.F. edited and revised manuscript; B.J.P., D.A.B., D.B.J., N.v.R., P.J.L., and A.D.F. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Ana Cristina G. Grodzki (UC Davis) for comments on an earlier version of the manuscript.
REFERENCES
- 1. Banks CN, Lein PJ. A review of experimental evidence linking neurotoxic organophosphorus compounds and inflammation. Neurotoxicology 33: 575–584, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barnes PJ. Cytokines as mediators of chronic asthma. Am J Respir Crit Care Med 150: S42–S49, 1994 [DOI] [PubMed] [Google Scholar]
- 3. Barr DB, Allen R, Olsson AO, Bravo R, Caltabiano LM, Montesano A, Nguyen J, Udunka S, Walden D, Walker RD, Weerasekera G, Whitehead RD, Jr, Schober SE, Needham LL. Concentrations of selective metabolites of organophosphorus pesticides in the United States population. Environ Res 99: 314–326, 2005 [DOI] [PubMed] [Google Scholar]
- 4. Berg JT, Lee ST, Thepen T, Lee CY, Tsan MF. Depletion of alveolar macrophages by liposome-encapsulated dichloromethylene diphosphonate. J Appl Physiol 74: 2812–2819, 1993 [DOI] [PubMed] [Google Scholar]
- 5. Berry MA, Hargadon B, Shelley M, Parker D, Shaw DE, Green RH, Bradding P, Brightling CE, Wardlaw AJ, Pavord ID. Evidence of a role of tumor necrosis factor alpha in refractory asthma. N Engl J Med 354: 697–708, 2006 [DOI] [PubMed] [Google Scholar]
- 6. Bookout AL, Cummins CL, Mangelsdorf DJ, Pesola JM, Kramer MF. High-throughput real-time quantitative reverse transcription PCR. Curr Protoc Mol Biol 15: Unit 15.8, 2006 [DOI] [PubMed] [Google Scholar]
- 7. Borish L, Mascali JJ, Dishuck J, Beam WR, Martin RJ, Rosenwasser LJ. Detection of alveolar macrophage-derived IL-1 beta in asthma. Inhibition with corticosteroids. J Immunol 149: 3078–3082, 1992 [PubMed] [Google Scholar]
- 8. Crain EF, Weiss KB, Bijur PE, Hersh M, Westbrook L, Stein RE. An estimate of the prevalence of asthma and wheezing among inner-city children. Pediatrics 94: 356–362, 1994 [PubMed] [Google Scholar]
- 9. Crinnion WJ. Do environmental toxicants contribute to allergy and asthma? Altern Med Rev 17: 6–18, 2012 [PubMed] [Google Scholar]
- 10. Ellman GL, Courtney KD, Andres V, Featherstone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 7: 88–95, 1961 [DOI] [PubMed] [Google Scholar]
- 11. Evans CM, Jacoby DB, Fryer AD. Effects of dexamethasone on antigen-induced airway eosinophilia and M(2) receptor dysfunction. Am J Respir Crit Care Med 163: 1484–1492, 2001 [DOI] [PubMed] [Google Scholar]
- 12. Fryer AD, Jacoby DB. Parainfluenza virus infection damages inhibitory M2 muscarinic receptors on pulmonary parasympathetic nerves in the guinea-pig. Br J Pharmacol 102: 267–271, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fryer AD, Jacoby DB. Function of pulmonary M2 muscarinic receptors in antigen-challenged guinea pigs is restored by heparin and poly-L-glutamate. J Clin Invest 90: 2292–2298, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fryer AD, Lein PJ, Howard AS, Yost BL, Beckles RA, Jett DA. Mechanisms of organophosphate insecticide-induced airway hyperreactivity. Am J Physiol Lung Cell Mol Physiol 286: L963–L969, 2004 [DOI] [PubMed] [Google Scholar]
- 15. Fryer AD, Maclagan J. Muscarinic inhibitory receptors in pulmonary parasympathetic nerves in the guinea-pig. Br J Pharmacol 83: 973–978, 1984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fryer AD, Wills-Karp M. Dysfunction of M2-muscarinic receptors in pulmonary parasympathetic nerves after antigen challenge. J Appl Physiol 71: 2255–2261, 1991 [DOI] [PubMed] [Google Scholar]
- 17. Gallo MA, Lawryk NJ. Organic phosphorus pesticides. In: Handbook of Pesticide Toxicology, edited by Hayes WJ, Laws ER. New York, NY: Academic Press, 1991 [Google Scholar]
- 18. Gambone LM, Elbon CL, Fryer AD. Ozone-induced loss of neuronal M2 muscarinic receptor function is prevented by cyclophosphamide. J Appl Physiol 77: 1492–1499, 1994 [DOI] [PubMed] [Google Scholar]
- 19. Grodzki AC, Ghogha A, Mangini L, Fryer AD, Lein PJ. IFNgamma increases M2 muscarinic receptor expression in cultured sympathetic neurons. Curr Neurobiol 2: 23–29, 2011 [PMC free article] [PubMed] [Google Scholar]
- 20. Hartert TV, Peebles RS., Jr Epidemiology of asthma: the year in review. Curr Opin Pulm Med 6: 4–9, 2000 [DOI] [PubMed] [Google Scholar]
- 21. Hashimoto S, Pittet JF, Hong K, Folkesson H, Bagby G, Kobzik L, Frevert C, Watanabe K, Tsurufuji S, Wiener-Kronish J. Depletion of alveolar macrophages decreases neutrophil chemotaxis to Pseudomonas airspace infections. Am J Physiol Lung Cell Mol Physiol 270: L819–L828, 1996 [DOI] [PubMed] [Google Scholar]
- 22. Hernandez AF, Parron T, Alarcon R. Pesticides and asthma. Curr Opin Allergy Clin Immunol 11: 90–96, 2011 [DOI] [PubMed] [Google Scholar]
- 23. Hoppin JA, Umbach DM, London SJ, Henneberger PK, Kullman GJ, Coble J, Alavanja MC, Beane Freeman LE, Sandler DP. Pesticide use and adult-onset asthma among male farmers in the Agricultural Health Study. Eur Respir J 34: 1296–1303, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Howarth PH, Babu KS, Arshad HS, Lau L, Buckley M, McConnell W, Beckett P, Al AM, Chauhan A, Wilson SJ, Reynolds A, Davies DE, Holgate ST. Tumour necrosis factor (TNFalpha) as a novel therapeutic target in symptomatic corticosteroid dependent asthma. Thorax 60: 1012–1018, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hreljac I, Filipic M. Organophosphorus pesticides enhance the genotoxicity of benzo(a)pyrene by modulating its metabolism. Mutat Res 671: 84–92, 2009 [DOI] [PubMed] [Google Scholar]
- 26. Johnson VJ, Rosenberg AM, Lee K, Blakley BR. Increased T-lymphocyte dependent antibody production in female SJL/J mice following exposure to commercial grade malathion. Toxicology 170: 119–129, 2002 [DOI] [PubMed] [Google Scholar]
- 27. Kassa J, Krocova Z, Sevelova L, Sheshko V, Kasalova I, Neubauerova V. The influence of single or repeated low-level sarin exposure on immune functions of inbred BALB/c mice. Basic Clin Pharmacol Toxicol 94: 139–143, 2004 [DOI] [PubMed] [Google Scholar]
- 28. Katz LS, Marquis JK. Modulation of central muscarinic receptor binding in vitro by ultralow levels of the organophosphate paraoxon. Toxicol Appl Pharmacol 101: 114–123, 1989 [DOI] [PubMed] [Google Scholar]
- 29. Kips JC, Tavernier J, Pauwels RA. Tumor necrosis factor causes bronchial hyperresponsiveness in rats. Am Rev Respir Dis 145: 332–336, 1992 [DOI] [PubMed] [Google Scholar]
- 30. Koch D, Lu C, Fisker-Andersen J, Jolley L, Fenske RA. Temporal association of children's pesticide exposure and agricultural spraying: report of a longitudinal biological monitoring study. Environ Health Perspect 110: 829–833, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lee AM, Fryer AD, van Rooijen N, Jacoby DB. Role of macrophages in virus-induced airway hyperresponsiveness and neuronal M2 muscarinic receptor dysfunction. Am J Physiol Lung Cell Mol Physiol 286: L1255–L1259, 2004 [DOI] [PubMed] [Google Scholar]
- 32. Lein PJ, Fryer AD. Organophosphorus insecticides induce airway hyperreactivity by decreasing neuronal M2 muscarinic receptor function independent of acetylcholinesterase inhibition. Toxicol Sci 83: 166–176, 2005 [DOI] [PubMed] [Google Scholar]
- 33. Lu C, Knutson DE, Fisker-Andersen J, Fenske RA. Biological monitoring survey of organophosphorus pesticide exposure among pre-school children in the Seattle metropolitan area. Environ Health Perspect 109: 299–303, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Minette PA, Lammers JW, Dixon CM, McCusker MT, Barnes PJ. A muscarinic agonist inhibits reflex bronchoconstriction in normal but not in asthmatic subjects. J Appl Physiol 67: 2461–2465, 1989 [DOI] [PubMed] [Google Scholar]
- 35. Nie Z, Jacoby DB, Fryer AD. Etanercept prevents airway hyperresponsiveness by protecting neuronal M2 muscarinic receptors in antigen-challenged guinea pigs. Br J Pharmacol 156: 201–210, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nie Z, Scott GD, Weis PD, Itakura A, Fryer AD, Jacoby DB. Role of TNF-alpha in virus-induced airway hyperresponsiveness and neuronal M muscarinic receptor dysfunction. Br J Pharmacol 164: 444–452, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Page S, Ammit AJ, Black JL, Armour CL. Human mast cell and airway smooth muscle cell interactions: implications for asthma. Am J Physiol Lung Cell Mol Physiol 281: L1313–L1323, 2001 [DOI] [PubMed] [Google Scholar]
- 38. Pena-Philippides JC, Razani-Boroujerdi S, Singh SP, Langley RJ, Mishra NC, Henderson RF, Sopori ML. Long- and short-term changes in the neuroimmune-endocrine parameters following inhalation exposures of F344 rats to low-dose sarin. Toxicol Sci 97: 181–188, 2007 [DOI] [PubMed] [Google Scholar]
- 39. Pope CN. Organophosphorus pesticides: do they all have the same mechanism of toxicity? J Toxicol Environ Health B Crit Rev 2: 161–181, 1999 [DOI] [PubMed] [Google Scholar]
- 40. Poston RN, Chanez P, Lacoste JY, Litchfield T, Lee TH, Bousquet J. Immunohistochemical characterization of the cellular infiltration in asthmatic bronchi. Am Rev Respir Dis 145: 918–921, 1992 [DOI] [PubMed] [Google Scholar]
- 41. Proskocil BJ, Bruun DA, Lorton JK, Blensly KC, Jacoby DB, Lein PJ, Fryer AD. Antigen sensitization influences organophosphorus pesticide-induced airway hyperreactivity. Environ Health Perspect 116: 381–388, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Proskocil BJ, Bruun DA, Thompson CM, Fryer AD, Lein PJ. Organophosphorus pesticides decrease M2 muscarinic receptor function in guinea pig airway nerves via indirect mechanisms. PLoS One 5: e10562, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rodgers K, Xiong S. Contribution of mast cell mediators to alterations in macrophage function after malathion administration. Fundam Appl Toxicol 33: 100–108, 1997 [DOI] [PubMed] [Google Scholar]
- 44. Rodgers K, Xiong S. Effect of administration of malathion for 14 days on macrophage function and mast cell degranulation. Fundam Appl Toxicol 37: 95–99, 1997 [DOI] [PubMed] [Google Scholar]
- 45. Rohlman DS, Anger WK, Lein PJ. Correlating neurobehavioral performance with biomarkers of organophosphorous pesticide exposure. Neurotoxicology 32: 268–276, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rozyk KJ, Plusa T, Kuna P, Pirozynska E. Monocyte chemotactic and activating factor/monocyte chemoattractant protein in bronchoalveolar lavage fluid from patients with atopic asthma and chronic bronchitis. Relationship to lung function tests, bronchial hyper-responsiveness and cytology of bronchoalveolar lavage fluid. Immunol Lett 58: 47–52, 1997 [DOI] [PubMed] [Google Scholar]
- 47. Schuh RA, Lein PJ, Beckles RA, Jett DA. Noncholinesterase mechanisms of chlorpyrifos neurotoxicity: altered phosphorylation of Ca2+/cAMP response element binding protein in cultured neurons. Toxicol Appl Pharmacol 182: 176–185, 2002 [DOI] [PubMed] [Google Scholar]
- 48. Segura P, Chavez J, Montano LM, Vargas MH, Delaunois A, Carbajal V, Gustin P. Identification of mechanisms involved in the acute airway toxicity induced by parathion. Naunyn Schmiedebergs Arch Pharmacol 360: 699–710, 1999 [DOI] [PubMed] [Google Scholar]
- 49. Selig W, Tocker J. Effect of interleukin-1 receptor antagonist on antigen-induced pulmonary responses in guinea pigs. Eur J Pharmacol 213: 331–336, 1992 [DOI] [PubMed] [Google Scholar]
- 50. Smith JN, Campbell JA, Busby-Hjerpe AL, Lee S, Poet TS, Barr DB, Timchalk C. Comparative chlorpyrifos pharmacokinetics via multiple routes of exposure and vehicles of administration in the adult rat. Toxicology 261: 47–58, 2009 [DOI] [PubMed] [Google Scholar]
- 51. Sultatos LG. Mammalian toxicology of organophosphorus pesticides. J Toxicol Environ Health 43: 271–289, 1994 [DOI] [PubMed] [Google Scholar]
- 52. Thepen T, van Rooijen N, Kraal G. Alveolar macrophage elimination in vivo is associated with an increase in pulmonary immune response in mice. J Exp Med 170: 499–509, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. van Rooijen N, Hendrikx E. Liposomes for specific depletion of macrophages from organs and tissues. Methods Mol Biol 605: 189–203, 2010 [DOI] [PubMed] [Google Scholar]
- 54. van Rooijen N, Sanders A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J Immunol Methods 174: 83–93, 1994 [DOI] [PubMed] [Google Scholar]
- 55. van Rooijen N, Sanders A, van den Berg TK. Apoptosis of macrophages induced by liposome-mediated intracellular delivery of clodronate and propamidine. J Immunol Methods 193: 93–99, 1996 [DOI] [PubMed] [Google Scholar]
- 56. Verhein KC, Jacoby DB, Fryer AD. IL-1 receptors mediate persistent, but not acute, airway hyperreactivity to ozone in guinea pigs. Am J Respir Cell Mol Biol 39: 730–738, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhang Y, Cardell LO, Adner M. IL-1beta induces murine airway 5-HT2A receptor hyperresponsiveness via a non-transcriptional MAPK-dependent mechanism. Respir Res 8: 29, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]