

SUMMARY
RIG-I is a cytosolic sensor of viral RNA, comprised of two N-terminal CARDs followed by helicase and C-terminal regulatory domains (helicase-CTD). Viral RNA binds to the helicase-CTD and “exposes” the CARDs for downstream signaling. The role of the second CARD (CARD2) is essential as RIG-I activation requires dephosphorylation of Thr170 followed by ubiquitination at Lys172. Here, we present the solution structure and dynamics of human RIG-I CARD2. Surprisingly, we find that Thr170 is mostly buried. Parallel studies on the phosphomimetic T170E mutant suggest that the loss of function upon Thr170 phosphorylation is likely associated with changes in the CARD1–CARD2 interface that may prevent Lys172 ubiquitination and/or binding to free K63-linked polyubiquitin. We also demonstrate a strong interaction between CARD2 and the helicase-CTD, and show that mutations at the interface result in constitutive activation of RIG-I. Collectively, our data suggests a close interplay between phosphorylation, ubiquitination, and activation of human RIG-I, all mediated by CARD2.
INTRODUCTION
RIG-I (retinoic acid inducible gene I) is a key protein in the cellular chordates' sentinel system to detect viral infection (Foy et al., 2005; Yoneyama et al., 2004). The protein contains three characteristic functional domains. The amino terminal part of RIG-I contains two caspase recruitment domains (CARDs) so named for their homology with described CARDs (Qin et al., 1999; Zhou et al., 1999). In addition, RIG-I contains an internal helicase-translocase domain and a C-terminal regulatory domain (CTD). RIG-I binds to several types of RNAs (Schlee and Hartmann, 2010); in particular, to viral RNAs containing double stranded regions with a 5′ end triphosphate group. The binding of RNA switches RIG-I from a passive to an active conformation, wherein the CARDs are “exposed” for interactions with downstream signaling proteins including the adaptor protein MAVS (mitochondrial-associated viral signaling protein), also known as VISA, IPS1, or Cardif (Belgnaoui et al., 2011). This, in turn, triggers a signaling cascade leading to the production of interferon-β (IFN-β), an important factor in the activation of the antiviral cellular innate immune response. Intriguingly, the expression of the tandem CARDs (CARD12) alone is sufficient for signaling, leading to IFN-β production (Yoneyama et al., 2004).
A number of RIG-I crystal structures have been reported recently, including the CTDs, the helicase domains, and the helicase-CTD constructs in complex with RNA in a variety of organisms: humans (Jiang et al., 2011; Luo et al., 2011), mouse (Civril et al., 2011), and duck (Kowalinski et al., 2011). Together, these structures show the interplay between the CTD and the helicase domain in binding the 5′ triphosphate group and the double stranded portion of viral RNA, respectively. A crystal structure of full-length duck RIG-I (Kowalinski et al., 2011) in the absence of RNA has also been determined. The structure has revealed RIG-I in the repressed conformation, whereby CARD12 is sequestered by an “insertion” subdomain (Hel2i) within the two RecA-like subdomains (Hel1 and Hel2) of the helicase domain. The binding of RNA is postulated to induce a conformational change in RIG-I that releases CARD12 for downstream signaling (Jiang et al., 2011; Kowalinski et al., 2011; Saito et al., 2007; Takahasi et al., 2008).
The central role of CARD12 in RIG-I signaling is reflected in its intense regulation. Several proteins bind and modify CARD12, mainly by ubiquitination and phosphorylation. Ubiquitin ligase RNF125, for example, mediates lysine 48-linked polyubiquitination of the second CARD (CARD2), which leads to the degradation of RIG-I by the proteasome (Arimoto et al., 2007). Other ubiquitin ligases such as the E3 ligases TRIM25 (tripartite containing motif protein 25) (Gack et al., 2007) and REUL (RIG-I E3 ubiquitin ligase) (Gao et al., 2009) mediate the polyubiquitination with Lys63-linked polyubiquitin (K63) of CARD2 at Lys172. K63 polyubiquitination is essential for the interaction with MAVS and activation of downstream signaling. Both REUL and TRIM25 mediate the K63-linked polyubiquitination at residue lysine 172 of RIG-I's CARD2, a process that can be reversed by the deubiquitinating enzyme CYLD (Friedman et al., 2008). The activity of TRIM25 can also be modified by the HOIL-1L/HOIP complex, also known as LUBAC (linear ubiquitin assembly complex). LUBAC suppresses the ubiquitination of RIG-I CARD12 and the subsequent activation by inducing both the degradation of TRIM25 and the inhibition of the interaction of TRIM25 and RIG-I CARD12 (Inn et al., 2011). In addition, PKC-α and PKC-β phosphorylate the first CARD (CARD1) and CARD2 at the side-chain hydroxyls of serine 8 and threonine 170 respectively (Gack et al., 2010; Maharaj et al., 2012; Nistal-Villán et al., 2010). Both phosphorylation events lead to the inhibition of downstream signaling. In uninfected cells, CARD12 is phosphorylated and not ubiquitinated. Dephosphorylation occurs after the induction of RIG-I, by an unidentified phosphatase. Removal of phosphate groups allows TRIM25 binding and ubiquitination, suggesting that the two posttranslational modifications are mutually exclusive. An alternative scenario has been proposed, where CARD12 from both RIG-I and MDA5 are able, in the absence of conjugation, to bind free Lys63-linked polyubiquitin chains produced in situ by TRIM25 and contribute to the activation of downstream events (Jiang et al., 2012; Zeng et al., 2010).
Altogether, much has been learned about how RIG-I CARD12 is modified and regulated in cells but how these modifications modulate the structure and interactions of CARD12 remains uncertain. Attempts to crystallize human RIG-I CARDs in isolation or as part of larger construct have been unsuccessful. While a full-length duck RIG-I structure has recently being generated, the lack of lysine 172 and threonine 170 residues in this species as compared to humans makes it difficult to interpret the human RIG-I phosphorylation and ubiquitination regulatory events.
We present the structure, dynamics, and interactions of human CARD2 determined by nuclear magnetic resonance (NMR). We also determined the structure of a T170E mutant in which phosphorylation of threonine 170 is mimicked by a threonine to glutamic acid substitution. The structures (WT and T170E) show the possible impact of phosphorylation on CARD2 structure and shed light on the mutually exclusive nature of phosphorylation and ubiquitination. Surprisingly, we find that in the wild-type structure, threonine 170 is not surface-exposed but that conformational exchange is likely to make the side-chain of threonine 170 transiently accessible. Also, a comparison of the structures shows that the T170E mutation modifies the surface of the protein around the mutation site, in particular due to the repulsive interaction with the carboxylate group of glutamic acid 129. However, no structural change is detected around the ammonium group of lysine 172, the site for ubiquitination, which is accessible in both the wild-type and the phosphomimetic mutant. We have also studied an interaction in trans between the CARD2 and the helicase-CTD domains of human RIG-I and identified an interface on CARD2, which is in good agreement with the recently described cis interaction between these domains seen in the structure of duck RIG-I (Kowalinski et al., 2011). Mutations of key CARD2 amino acids interacting with the helicase-CTD relieve the inhibitory state of full length RIG-I and lead to ligand independent induction of IFN. Based on these observations, we propose a refined model of activation of human RIG-I upon viral RNA binding.
RESULTS
Overall Structure of RIG-I CARD2
We used NMR to solve the structure of the second CARD of human RIG-I (CARD2; residues 95–190). The [15N,1H] heteronu-clear single quantum coherence (HSQC) spectrum of CARD2 shows a good dispersion and a relative homogeneity of peak intensities (see Figure S1 available online), which indicate that the protein is well folded and not highly dynamic. A uniformly 13C- and 15N-labeled sample was employed to carry out the assignment of backbone and side-chain resonances as well as the collection of distance constraints. Figure 1A shows the backbone trace of the ensemble of the 20 lowest energy conformers while Figure 1B presents a ribbon diagram of the structure with lowest energy. The structural model is very precise, with a backbone root mean square deviation (RMSD) of 0.44Å. The overall structure of CARD2 consists of six helices, as typically seen in CARD structures, a group of the death domains (DD) superfamily (Park et al., 2007a). The first, long alpha helix (h1) goes from Ile97 to Arg117. It is kinked at residue Leu110, and the second part is referred to as helix h1′. The second helix (h2) comprises residues Thr121 to Glu129 but only the core four residues (Ile124 to Leu127) form a canonical alpha helix in all models. Helix 3 (h3) consists of residues Asn133 to Thr145, helix 4 (h4) from Met148 to Arg161, helix 5 (h5) from Asn166 to Glu178, while helix 6 (h6), which is the only 310 helix, is much shorter than seen in most CARD structures with only four residues (Phe182 to Leu185). Notably, helix 5 contains amino acid residues Thr170 and Lys172 known to undergo post-translational modification by phosphorylation and ubiquitination, respectively.
Figure 1. NMR Structure of CARD2.
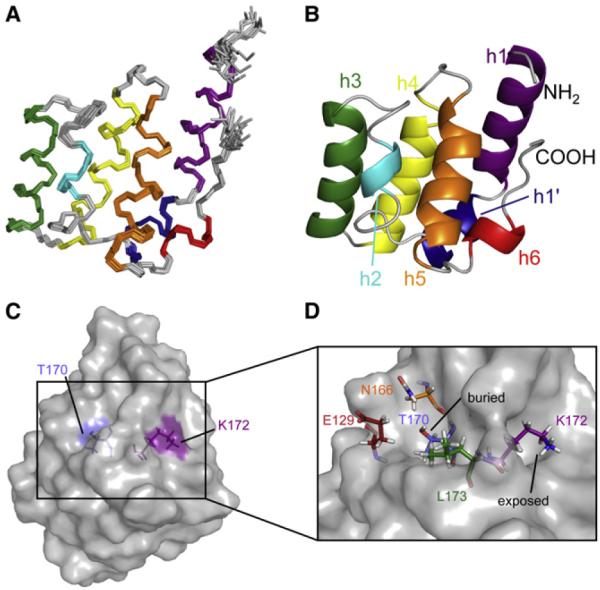
(A) Backbone traces for the ensemble of the 20 conformers with lowest energy.
(B) Ribbon representation of the lowest energy model. (A and B) Helices are color-coded in the following way: h1, purple; h1′, blue; h2, cyan; h3, green; h4, yellow; h5, orange; h6, red.
(C and D) Relative orientation and surface exposure of Thr170 and Lys172. Thr170 and Lys172 are shown as sticks as well as all residues covering Thr170. (C) The surface of CARD2 is shown in gray. The side chains of Thr170 and Lys172 point toward opposite sides of the domain. (D) The remainder of the protein is shown as a surface plot. Thr170 is not exposed at the surface. The side-chain ammonium group of Lys172 is surface accessible. Figures were generated with Pymol (http://www.pymol.org; Schrodinger, 2012).
See also the NMR spectrum of wt CARD2 in Figure S1.
The depiction of the surface of CARD2 in Figure 1C shows that the side-chain of Lys172 (purple) is well exposed at a site distant from the Thr170 phosphorylation site (the average distance between the hydroxyl oxygen of Thr170 and the nitrogen epsilon of Lys172 is 11Å). The ammonium group of Lys172 is surrounded by the C-terminal part of the protein with interactions in the ensemble of 20 structures with the backbone and side-chains of residues Glu176, Ser183, Trp186, and Val188. Surprisingly, Thr170 is mostly buried (blue) and its hydroxyl group is not well exposed at the surface, covered by the side-chains of Glu129, Asn166 and Leu173 (Figure 1D). The average surface area for Thr170 over the 20 conformers is only 1.4Å2 as calculated by MolMol (Koradi et al., 1996).
The structures of human and duck CARD2 (PDB code 4A2Q, molecule A) were aligned using the DALI server (Holm and Rosenström, 2010). The superimposed structures are shown in Figure 2A. The backbone structures of the two CARDs are very similar, with an RMSD of 1.6Å, using 91 residues for alignment. Small translations of helices 1′ and 6 can be observed, which may be due to contacts with the helicase domain in the structure of duck CARD2. The structures of human CARD2 and the CARD of human MAVS (PDB code 2VGQ) were also aligned (see Figure 2B). These two structures are similar, with an RMSD of 2.3Å, using 89 residues for alignment. The most notable difference is the length of helix h6, which is particularly short in RIG-I CARD2.
Figure 2. Alignment of the Structures of Human CARD2 with Duck CARD2 and the CARD Domain of MAVS.
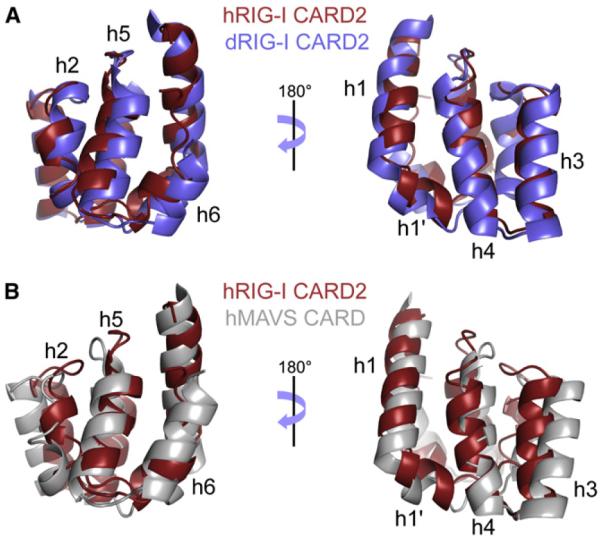
(A and B) The ribbon representation of the lowest energy structural model of CARD2, shown in red, is shown aligned to duck CARD2 (residues 95–186, from the PDB file 4A2Q, molecule A) in blue, and to the CARD of MAVS (residues 1–93, from the PDB file 2VGQ) in gray.
Multiple Timescale Dynamics of RIG-I CARD2
The atomic-level understanding of the function of a protein requires the characterization not only of its structure but also of its dynamics (Henzler-Wildman and Kern, 2007). NMR is a powerful tool to characterize site-specific dynamics in biomolecules over a wide range of timescales (Mittermaier and Kay, 2006; Palmer, 2004). A full series of 15N relaxation experiments was carried out to quantify backbone dynamics on sub-nanosecond as well as micro- and millisecond timescales on a uniformly 15N-labeled sample. Transverse, R2 and longitudinal, R1, relaxation rates were used along with NOEs to determine the features of overall rotational diffusion of CARD2 with the program Rotdif (Fushman et al., 2004). Rotational diffusion was found to be axially symmetric, with an anisotropy of 1.15 and an average correlation time τc = 7.15 ns. The value of τc is higher than expected for a protein of this size. In order to address the possibility of a monomer/dimer equilibrium, transverse relaxation rates were also measured on a low-concentration sample (70 μM compared to 300 μM for all other relaxation measurements). It was found that the low-concentration R2 rates were, on average 3% lower than in the high-concentration sample. This difference is too small to account for the higher value of τc and cannot be explained by a monomer/dimer (or monomer/oligomer) equilibrium. A model-free analysis (Lipari and Szabo, 1982) of the transverse, R2, longitudinal, R1, rates and NOEs was carried out to quantify fast (sub-nanosecond) internal motions with the program Dynamics (Hall and Fushman, 2006). Order parameters for fast motions of the backbone NHN vectors were found to be high for most of the protein: between residues 100 and 187, 72% (resp. 97%) of NHN vectors have an order parameter above 0.9 (resp. 0.8). In addition to the C terminus, a few NHN vectors showed order parameters below 0.85: at the center and at the extremities of helix h2, with residues Asp122, Leu127, and Leu131 as well as the loop residues Thr145, Lys146 and Gly147, present between helix h3 and h4 (see Figure S2).
The contribution of chemical exchange, Rex to the transverse relaxation rate R2 was quantified with the method of Kroenke et al. (Kroenke et al., 1998) adapted by Pelupessy et al. (Pelupessy et al., 2007). Results are shown in Figures 3A and 3B. The high accuracy of our approach allowed for the identification of small contributions of chemical exchange to transverse relaxation Rex, as low as 2 s−1. Important Rex contributions were found as evidence for conformational exchange in residues from helix h1′, the N-terminus of helix h2, loops h2–h3, and h4–h5, as well as residues from helix h6, which lies close to the ammonium group of Lys172. Dynamic residues in helices h1′ and h6 form a continuous surface. Significant values of Rex were also measured for residues Asn166, Lys169, and Thr170, which shows that, despite being buried in the major conformation of CARD2, the side chain of Thr170 may be solvent exposed in a minor conformation. To test this hypothesis, we used the Phase-Modulated CLEAN chemical EXchange (CLEANEX-PM) experiment (Hwang et al., 1998) to test for chemical exchange of backbone amide protons with water. Results are shown in Figure 3C and S2B. Several residues with high proton exchange contributions belong to the disordered N terminus of the protein. Another amide proton with efficient exchange with water is that of Lys181, which also displays an exceptionally high value of Rex such that fast nitrogen-15 relaxation prevents its accurate quantification. Most importantly, both amide protons of Glu129 and Asn166 show small but significant exchange with water, which supports the existence of an excited state with an open conformation where the hydroxyl of Thr170 is surface exposed to allow phosphorylation by PKC-α and PKC-β.
Figure 3. Slow Dynamics of the Second CARD of RIG-I.

(A) Contributions of chemical exchange Rex to 15N transverse relaxation. Error bars are obtained by the propagation of the spectral noise (versus peak intensity) in all spectra used for the relaxation rate analysis.
(B) These contributions are color-coded onto three orientations of the lowest energy structure of CARD2. The positions of Thr170 and Lys172 as well as residues covering Thr170 are shown in at least one of the orientations.
(C) Amide proton exchange with water derived from the CLEANEX-PM experiment. The color coding corresponds to the following CLEANEX efficiencies: ηCL > 0.02 (red); 0.02 > ηCL > 0.003 (orange); 0.003 > ηCL > 0.001 (yellow). The side-chains of Glu129, Asn166, Thr170, and Lys172 are shown.
See also Figure S2.
Effect of Phosphorylation
The inhibitory role of the phosphorylation of threonine 170 in the function of RIG-I prompted us to investigate the conformational changes associated with the modification of this residue. To address this, we expressed a CARD2 mutant where a glutamate residue was introduced at position 170, to mimic the change of charge upon phosphorylation of Thr170. Results are shown in Figures 4, S3, and S4. Overlay of the spectra of wild-type CARD2 and of the T170E mutant reveals significant chemical shift changes (Figure 4A). Backbone and side-chain assignments were obtained from triple-resonance experiments measured at 800 and 600 MHz, respectively. Changes in the structure were elucidated from the chemical shift perturbations of the peptide plane nuclei (15N, 1HN, and 13CO) (see Figure S4A) and were found to be limited to helices h2 and h5. In order to characterize the nature of these changes, we determined the structure of the T170E mutant of human CARD2 (see Table 1 and Figures S3A–S3D). The lowest energy models of the structure ensembles of the wild-type and mutant proteins were aligned and are shown in Figure 4B. The two structures are remarkably similar, with a RMSD of 1.3Å, using 94 residues for alignment. The changes between these structures are limited and, in most cases, likely comparable to their accuracies. A small translation of the h2–h5 helices can be observed, as well as a tilt of the N terminus of helix 1. This slight change in the N terminus of helix 1 is likely due to the small number of NOE restraints available for the structure calculations of the mutant compared to WT CARD2 (see Table 1). The small structural changes in helix h2 are likely transmitted to helix h5 through the strong repulsive interaction between the carboxylate groups of Glu129 in h2 and Glu170 in h5, as can be seen in the significantly different orientations of the sidechain of Glu129 in the two structures (Figure 4C). The surface electrostatics of wild-type and mutant CARD2 are shown in Figures 4D and 4E, with the same orientation as in Figure 4B. As expected, the main difference is a larger, negatively-charged surface around residues E129 and E170 in the mutant domain. Changes in dynamics upon the T170E mutation were evaluated from 15N-{1H} nuclear Overhauser effects for sub-ns motions and cross-correlated chemical shift modulation measurements with the method of Kloiber and Konrat (2000) for μs-ms timescales (see Figures S3E and S3F). Changes are limited both on sub-nanosecond and micro-millisecond timescales. In particular, the region around Glu129 is not disordered in the phosphomimetic mutant protein. Although previous studies (Gack et al., 2010) have demonstrated the absence of ubiquitin conjugation at Lys172 when Thr170 is phosphorylated, we did not observe significant chemical shift perturbations around the side-chain ammonium of Lys172. Therefore, the structural role of phosphorylation is likely to be the alteration of an interaction surface that overlaps with helices h2 and h5, vide infra. To test this hypothesis, we designed a series of single and double mutants that would modulate the key Glu170–Glu129 repulsion.
Figure 4. Comparison of NMR Spectra and Structures of Wild-type CARD2 and the T170E CARD2 Mutant.

(A) Changes of peak positions is shown on the two-dimensional HSQC spectra of the wild-type and T170E mutant proteins. Assignments are shown for residues with a chemical shift perturbation above an arbitrary threshold (see supporting information).
(B) Lowest-energy structures of the wild-type (red) and T170E mutant (green) CARD2.
(C–E) Focus on the conformations of Glu129 and Thr170/Glu170 sidechains (C). Surface electrostatics of WT CARD2 (D) and T170E CARD2 (E) are shown in the same orientation as in (B).
See full structural details and dynamics in Figure S3.
Table 1.
NMR Restraints and Structural Statistics for the Best 20 Structures of WT and T170E Mutant of Human CARD2
| Restraints and Statistics | WT | T170E Mutant |
|---|---|---|
| Total number of restraints | 2,900 | 2,212 |
| NOE restraints | 2,646 | 1,944 |
| Unambiguous | 2,173 | 1,636 |
| Intraresidue | 945 | 822 |
| Sequential | 445 | 345 |
| Short-range | 274 | 217 |
| Medium-range | 128 | 66 |
| Long-range | 381 | 186 |
| Ambiguous | 473 | 308 |
| Dihedral angle restraints | 174 | 182 |
| Hydrogen bond restraintsa | 80 | 86 |
| Structure statisticsb | ||
| NOE violations > 0.5 Å | 0% | 0% |
| Dihedral violations > 5° | 0% | 0% |
| RMSD from average structurec,d | ||
| Backbone (N, Cα, C) (Å) | 0.44 ± 0.08 | 0.44 ± 0.09 |
| Heavy atoms (Å) | 0.97 ± 0.08 | 0.93 ± 0.08 |
| Ramachandran Statisticse | ||
| Most favored region (%) | 90.7 | 91.1 |
| Additionally allowed (%) | 8.6 | 8.6 |
| Generously allowed (%) | 0.3 | 0.1 |
| Disallowed (%) | 0.3 | 0.2 |
Hydrogen bond restraints were HN-O distance of 1.8–2.3 Å and an N-O distance of 2.8–3.3 Å.
Structural characteristics for the final ensemble of 20 water-refined structures.
RMSD of the mean structure from individual structures in the ensemble.
RMSD for residues 95–190 shown.
Ramachandran plot data shown for residues 95–1190.
We generated a battery of Glu129 mutants in a mammalian expression vector containing WT or T170E RIG-I GST-CARD12. CARD12, lacking the repressing helicase-CTD domain, is known to constitutively induce IFN-β expression, and the T170E phosphomimetic mutation has previously been shown to reduce this induction by reducing K172 ubiquitination (Gack et al., 2010). Briefly, we mutated Glu129 to valine, alanine, or glutamine. We analyzed the ability of the different constructs to induce IFN-β reporter activity using a reporter assay (see Figure 5). Differences between WT and T170E GST-CARD12 constructs to induce IFN-β reporter activity were significant (p < 0.001) but not large (about 2-fold). Importantly, we were able to recover some WT phenotype in some of the 170E mutants. In particular, the T170E-E129A mutant was able to moderately increase IFN-β promoter activity in the context of the 170E mutation, while the same amino acid change, E129A, did not have a significant effect in the absence of T170E mutation. Mutation of Glu129 to valine in both E129V and T170E-E129V contexts showed a small but consistent negative effect on the ability to induce IFN-β. Similarly, a mutant with larger side-chains T170E-E129Q did not show any enhancement to the T170E phenotype in their ability to induce the IFN-β promoter despite similar protein levels (Figure 5B). Taken together, the mutational data supports the idea that E129 constrains the E170 side-chain and that the repulsion can be partially overcome by reducing amino acid size and/or removing the negative charge at position 129.
Figure 5. Comparison of RIG-I GST-CARD12 Mutant's Ability to Induce IFN-β Promoter.

(A) Data shows IFN-β reporter assay driven by overexpression of constitutively active GST-CARD12 RIG-I in 293T cells. Cells were cotransfected with IFN-β-luciferase plasmid reporter plasmid and pRL-TK plasmid to normalize firefly luciferase values, together with plasmids expressing RIG-I CARD12 E129 mutations in corresponding WT or phosphomimetic E170 background. The experiment was done in quadruplicate, and the error bars represent the standard deviation between sample values. *Represents significant differences (p < 0.001) using one-way ANOVA followed by a Bonferroni's comparison test between specified data sets.
(B) GST-CARD12 and actin protein level expression from (A) compared by western blot.
Interaction between RIG-I CARD2 and Helicase-CTD
The structure of the helicase and CTD domains of human RIG-I (helicase-CTD) bound to dsRNA (PDB: 3TMI and 2YKG) shows a closed conformation in which the helicase and CTD domains curl around the dsRNA (Jiang et al., 2011; Luo et al., 2011). In addition, SAXS data on full-length RIG-I in complex with RNA (Jiang et al., 2011) suggest that CARD12 is well exposed and available for intermolecular interactions. In the absence of RNA, CARD12 is sequestered by the helicase-CTD region of RIG-I. This interaction has been observed in the crystal structure of duck RIG-I (Kowalinski et al., 2011). In particular, CARD2 interacts with the insertion domain Hel2i in the crystal environment. In order to confirm this observation and its occurrence in solution, in trans, we investigated the interactions of CARD2 and helicase-CTD by monitoring the NMR signals of CARD2 in a titration with helicase-CTD (residues 230–925). A series of 15N-1H HSQC spectra of CARD2 (concentration 50 μM) were recorded with 0 to 1.7 equivalents of helicase-CTD. Figure 6A compares the HSQC spectra of CARD2 with 0 (apo) and 1 equivalent of helicase-CTD. Upon binding, there was no evidence of any chemical shift perturbation, however almost all peaks decrease in intensity, which suggests that the CARD2:helicase-CTD interaction is in the slow exchange regime. Upon binding to helicase-CTD, the effective molecular mass of CARD2 becomes about 90 kDa. Thus, even if a fraction of CARD2 is bound, transverse relaxation rates increase significantly, leading to reduced sensitivity of HSQC experiments. In addition to this effect, which is unambiguous evidence of interaction between the two proteins, slow exchange leads to additional reduction of the intensities of signals of most residues of CARD2 at the interface with helicase-CTD (those with different chemical shifts in the apo and holo forms; see for example the signal of Met149 in Figure 6B). The residues with the largest decrease in signal were identified and plotted onto the surface of CARD2 in Figure 6D. Most signals identified belong to residues from the N terminus of helix h1, helix h1′, loop h1′–h2, the N- and C termini of helix 3, the N terminus of helix h4, the C terminus of h5, and helix h6 as well as the C terminus of the domain and together they delineate a mostly continuous surface. This surface corresponds very well to the one identified in the crystal structure of duck RIG-I as interacting in cis with the Hel2i domain. As noted in the description of the structure of duck RIG-I (Kowalinski et al., 2011), the ammonium group of Lys172 in human RIG-I is likely in close contact with this interface. The global analysis of the decay of intensity upon binding of a group of peaks that belong to the interface, neglecting the effect of intermediate chemical exchange on relaxation rates, provides a dissociation constant in the low micromolar range (Kd = 1.66 ± 1.3 μM, see Figure 6C), which confirms the presence of a tight interaction for these domains of RIG-I in trans, likely reflecting their interactions in cis when in the context of full length RIG-I.
Figure 6. Interaction of CARD2 with Helicase-CTD.

(A) [1H, 15N] HSQC spectra recorded for two samples of CARD2 (uniformly 13C- and 15N- labeled, 50 μM) in the absence (blue) or with one equivalent (red) of unlabeled helicase-CTD.
(B) A one-dimensional slice at 118.8 ppm in the 15N dimension (dashed line) is shown with an additional spectrum in green for half an equivalent of unlabeled helicase-CTD. All peaks are attenuated upon the addition of helicase-CTD but the effect is more pronounced for some peaks, such as the correlation for Met149, corresponding to residues at the interface.
(C) The evolution of normalized peak intensities upon titration were used to fit the dissociation constant Kd. A global fit on the data obtained for four NHN pairs of residues located at the interface is shown here.
(D) Residues in CARD2 with the strongest attenuation are shown in red onto the surface of CARD2. For reference, the ammonium group of Lys172 is depicted in purple. Proline residues, for which no signal can be detected are rendered in black.
Mutations in CARD2 at the CARD2:Helicase Interface Render RIG-I Constitutively Active
In current models of RIG-I function, the CARDs are held in an inactive state by interaction with the helicase insertion domain and activation comes from the de-repression of CARDs in the presence of an activating RNA ligand. To study the functional significance of the structural data presented in this study and also in the recently published study (Kowalinski et al., 2011) on the interaction of CARD domains with the helicase domain, a series of alanine point mutations were generated in the CARD2 domain of full length (FL) RIG-I and CARD12. Mutations that disrupt the interaction between CARD2 and helicase insertion domain can be expected to dissociate the CARDs from the helicase, allowing interaction with TRIM25 and MAVS for downstream signaling, resulting in IFN-β induction. Residues R109, E113, R117, and L185 were selected from our mapped surface on CARD2 and their proximity to the helicase insertion domain. As previously shown (Nistal-Villán et al., 2010), overexpression of full length RIG-I induces minimal interferon production, while the N-terminal CARDs, when expressed alone, stimulate very high levels of interferon production, due to the absence of the inhibitory helicase domain. Although, point mutants of FL RIG-I, R109A, and L185A did not express as well as wild-type (WT) RIG-I, their expression induced significantly enhanced IFN-β promoter activity, suggesting de-repression of the CARDs (Figure 7A). The E113A-R117A mutant, however, did not display any constitutive activity. The non-interface mutation T145A was used as a negative control, which also did not show any enhancement in interferon promoter activity compared to WT RIG-I.
Figure 7. Interferon-β Promoter Induction by CARD2 Mutants.

(A and B) HA-tagged (A), full length, or CARD12 WT and CARD2 (B) mutants as indicated were transfected in HEK293T cells stably expressing IFN-β promoter driven firefly luciferase gene and luciferase activity (RLU) was measured 48 hr later. Protein expression levels were detected by western blotting using an HA-HRP antibody. β-actin was used as a loading control.
(C) Graph depicts arbitrary units of de-repression of CARD12 for each mutant calculated by the ratio of interferon induction (RLU) of full-length (FL) RIG-I to CARD12 RIG-I normalized to respective quantified band intensity ratios of HA to β-actin.
(D) Five-fold serially diluted empty plasmid, full-length or CARD12 WT or mutants were transfected in HEK293T cells stably expressing IFN-β promoter driven firefly luciferase gene and luciferase activity (RLU) was measured 48 hr later. Protein expression levels were detected by western blotting using an HA-HRP antibody. β-actin was used as a loading control. Dashed line indicates background luciferase expression and dotted line indicates the threshold of IFN-β induction by WT RIG-I. Data represents mean of three to four independent experiments with error bars showing standard deviation. Statistically significant differences are indicated with a p value.
As expected, WT CARD12 showed increased IFN-β reporter activity induction compared to FL WT RIG-I, but the levels did not change with the CARD12 R109A and E113A-R117A, even though the expression level of CARD12 R109A was lower than CARD12 WT. This suggests that these mutations did not affect the ability of the CARDs to activate downstream signaling (Figure 7B). Although there was a significant decrease in the induction level of IFN-β reporter activity by CARD12 L185A when compared to CARD12 WT or CARD12 R109A, its poor protein expression levels may partly explain its reduction in interferon promoter induction. The CARD12 T145A also showed a reduction in interferon activation; however, it was not a statistically significant result when compared to CARD12 WT.
De-repression of CARDs of each mutant was normalized by calculating the ratio of interferon induction (RLU) of FL to CARD12 RIG-I (Figure 7C). Both L185A and R109A mutations enhanced interferon promoter activation up to 10-fold as compared with WT RIG-I (Figure 7C). However, the mean de-repression values for both are similar when corrected for the apparent defect in L185A to activate interferon efficiently. In order to see if there is any cumulative effect of the two constitutive active mutants R109A and L185A, a double mutant R109A-L185A was constructed and a protein dose experiment was performed to study interferon activation. The double mutant did not show any enhanced interferon activation compared to the single mutant R109A, which suggests no increased disruption of the interaction between CARD2 and Hel2i by the L185A mutation in the context of the R109A mutation (Figure 7D).
DISCUSSION
Here, we show that the structure of human RIG-I CARD2 is very similar to the structures of the CARDs of both MAVS (Potter et al., 2008) and Apaf-1 (Qin et al., 1999; Zhou et al., 1999). The main difference, like in duck RIG-I, lies in a very short helix h6 in CARD2, with the C-terminal part of CARD2 curled around the ammonium group of Lys172 (absent in the crystal structure of duck RIG-I), the site of ubiquitination by TRIM25. This defines a very specific surface for the ubiquitination site in CARD2 of human RIG-I. In contrast, it is unclear whether duck RIG-I is similarly polyubiquitinated since in birds (duck, goose or finch) and mouse RIG-I, Lys172 is substituted by a glutamine. Note that both Lys172 and the C terminus of human CARD2 (in particular residues 187–188) are also essential for interaction with unanchored polyubiquitin chains (Jiang et al., 2012).
The sequence alignments of the CARDs of RIG-I across species are shown in Figure 8A. Several regions of CARD2 are highly conserved across species, in particular residues in helices h4, h5 as well as in loop h4–h5, which lies at the core of a highly conserved surface area (see Figure 8B) that comprises the interaction surface with CARD1, as demonstrated on the crystal structure of duck RIG-I, see Figure 8B. Interestingly, this conserved surface is broader than the interface with CARD1, expanding in particular along most of helix h4, and is contiguous to another conserved surface constituted by the second and third helices of CARD1. Together, these regions form a deep groove, which could open up to accommodate a downstream binding partner. We also note that the interface with CARD1 is also the locus of significant conformational exchange (Figure S4C), revealing a conformational flexibility in the free form of CARD2 that likely facilitates the binding of CARD1 (and possibly downstream cofactors).
Figure 8. Sequence Alignment of CARD12 across Species.

(A)The color coding reflects sequence conservation. Secondary structure elements are shown in gray for CARD1 with helices numbers corresponding to the structure of duck CARD12, while we used the same color coding as in Figure 1 for CARD2. Triangles are used to identify residues Glu129, Thr170 and Lys172. The figure was prepared with Jalview (Waterhouse et al., 2009).
(B and C) The structure of human CARD2 was aligned to CARD2 from the duck RIG-IΔCTD crystal structure (PDB code 4A2Q, molecule A). (B) Representation of the conservation of surface residues in CARD12. The color coding is identical as in (A) for CARD2 and similar, in shades of red for CARD1. (C) Surface electrostatics of human CARD2 and a model of CARD1 based on the structure of duck CARD1. See additional representations in Figure S4.
Phosphorylation and Ubiquitination
Surprisingly, Thr170, the site of phosphorylation, is mostly buried in the structure of CARD2, such that its hydroxyl group is not exposed to the surface in the dominant conformation in solution. The side-chain of Thr170 is occluded by the side-chains of Glu129, Asn166, and Leu173 (see Figure 1D). However, our site-specific studies of micro- and millisecond dynamics in CARD2 show that this region of the protein and, in particular, the backbone NHN groups of Asn166 and Thr170 are prone to conformational exchange (Figures 3A and 3B). This exchange likely leads to sampling an excited state with an open conformation around Thr170, as confirmed by the detection of enhanced chemical exchange for the amide protons of Glu129 and Asn166 with water (see Figures 3C and S2B) compared to most residues from other parts of the protein.
In spite of their proximity in the amino-acid sequence, the sidechains of Thr170 and Lys172 are distant (~ 11Å apart) and point toward different sides of CARD2. A direct interaction between the phosphate group of pThr170 and the ammonium group of Lys172 cannot thus account for the reported absence of ubiquitination in phosphorylated CARD2 (Gack et al., 2010). However, the determination of chemical shift perturbations upon the phosphomimetic mutation T170E shows that the addition of a negatively charged group at the side-chain of residue 170 leads to an alteration of the surface of CARD2 at helices h2 and h5, and that the perturbed surface overlaps with the interface with CARD1 (see Figure S4B). Both steric interactions with the side-chain of phosphorylated Thr170, and the electrostatic repulsion with the carboxylate of Glu129 are likely key in the propagation of chemical shift changes. The presence of a bulky phosphate group will likely enhance this structural modification and further deteriorate the CARD1–CARD2 interaction.
The interaction of TRIM25 with CARD1 has been shown to be essential for the ubiquitination of CARD2 at residue Lys172. The T55I mutation in CARD1, for example, suppresses both the interaction between CARD12 and TRIM25 and the ubiquitination of CARD2 (Gack et al., 2008). The modification of the CARD1-CARD2 interaction by Thr170 phosphorylation may alter the relative orientation of CARD1 and CARD2 for proper ubiquitination of CARD2 by TRIM25. Our measurements of IFN-β promoter activation in human cells transfected with mutants of the tandem CARD12 provides partial support to this hypothesis. Thus, whereas overexpression of the T170E phosphomimetic tandem CARD12 in 293T cells leads to an ~50% reduction in the ability to induce IFN-β compared to the WT CARD12, the E129A mutation restores the response to about 63% of WT. By contrast, mutations to amino acids with a side-chain of similar size to glutamate (E129Q) have very limited impact.
Recently, it has been suggested that ubiquitin conjugation of Lys172 might not be essential for human RIG-I activation. Rather, RIG-I CARD12 binds to unconjugated K63-linked polyubiquitin, and this results in RIG-I activation (Zeng et al., 2010). A following study, showed that mutations in CARD2 at the interface with CARD1 (E135A and K164A) could reduce or suppress binding to polyubiquitin (Jiang et al., 2012). The structural changes associated with Thr170 phosphorylation alter the same part of the interface with CARD1 and are also likely to reduce the ability of CARD12 to bind unanchored K63 polyubiquitin.
RIG-I Activation
In all current models of the function of RIG-I (Cui et al., 2008; Jiang et al., 2011; Luo et al., 2011; Saito et al., 2007; Kowalinski et al., 2011) CARD12 is sequestered in an inactive, pre-viral RNA binding state. CARD12 is believed to become exposed and available for interactions and signaling in the active, RNA bound form. The structure of RIG-IΔCTD (Kowalinski et al., 2011) shows that the interaction of CARD2 with the insertion domain Hel2i of the helicase domain likely locks RIG-I in its inactive state. Here, we show (see Figure 6) that there is a direct interaction between CARD2 and helicase-CTD that can be detected in trans in solution. As such, we anticipate even stronger binding in the full-length protein where the CARD2 and the helicase-CTD are in cis. Interestingly, as can be seen in Figure 8B, this interface is not very conserved across species, although two of the four highly conserved hydrophobic residues with solvent-exposed side-chains (Ala150 and Leu185) participate in the interaction. Notably, the interface of CARD2 with helicase-CTD does not overlap with the area perturbed by the T170E mutation. This suggests that Thr170 phosphorylation should not alter the interaction of CARD2 with helicase-CTD, but may affect the interaction with TRIM25 (and possibly REUL) that is required for RIG-I ubiquitination and full activation. Interestingly, residue Ser183, which is the locus of the single-nucleotide polymorphism (SNP) giving rise to S183I (Pothlichet et al., 2009; Shigemoto et al., 2009), is located at the CARD2:helicase-CTD interface and may thus alter the interaction of CARD12 and helicase-CTD. Also, the surface identified in our titration is larger than the interface in the crystal structure of duck RIG-I. As can be seen in Figure 9A, the region of CARD2 facing helix α28 of the Hel2 domain is also perturbed by the interaction with helicase-CTD. This could be because of the existence of a broader interface in human RIG-I. Alternatively, binding of CARD2 to Hel2i could lead to a broad conformational transition extending beyond the interface to the region facing helix α28.
Figure 9. Interaction between CARD2 and the Helicase Domain of RIG-I.
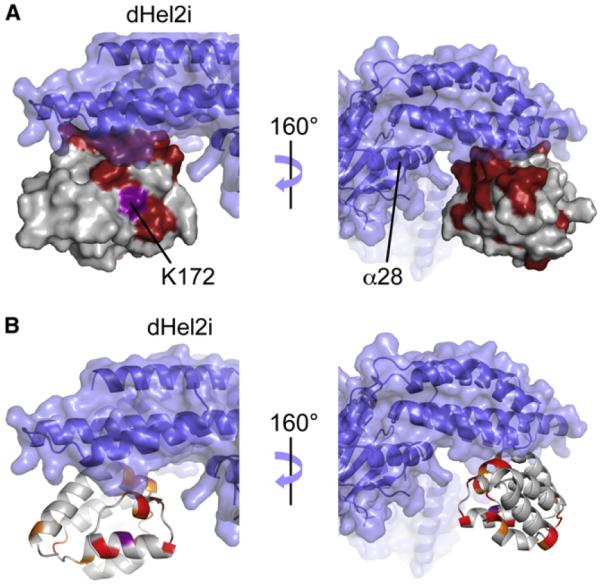
(A and B) The structure of human CARD2 was aligned to duck CARD2 in the structure of duck RIG-IΔCTD (PDB code 4A2Q). Part of the helicase domain is depicted in blue in both a ribbon representation and a transparent rendition of its surface. Helix α28 in the Hel2 domain is indicated in (A). (A) Surface of human CARD2 corresponding to the interface identified in the titration experiment (see Figure 6) is represented in red. (B) The residues presenting a contribution of micro-millisecond chemical exchange to relaxation, Rex are depicted on a ribbon representation of CARD2 in the same alignment as in (A). The color coding is the following: Rex > 3 s−1 (red); 3 > Rex > 2 s−1 (orange); Lys181, shown in brown, displays a large value of Rex that was not quantified. Ribbon representation of human CARD2.
A full model for RIG-I activation is shown in Figure S5.
As observed in the structure of duck RIG-I, the residue equivalent to Lys172 is in proximity to the binding interface with helicase-CTD (Figure 9). This proximity has two important consequences. First, the ubiquitination site is unlikely to be accessible in the inactive state of the protein. Second, the presence of a polyubiquitin chain bonded to the interface with the helicase-CTD would likely hinder this interaction. Therefore, in addition to its function for signaling (Gack et al., 2007), ubiquitination may be a mechanism to lock RIG-I in the active state preventing binding to helicase-CTD, where CARD12 would be exposed beyond the typical timescale of RNA binding. The CARD2:helicase-CTD interface is also characterized by a high level of conformational exchange (Figures 3 and 9B). This conformational flexibility may be a mechanism to adapt locally to more than one binding partner, which supports the hypothesis that CARD2 commits the same surface to the interactions with the helicase-CTD and polyubiquitin, covalently or noncovalently bound (Zeng et al., 2010). Polyubiquitination of CARD2 may promote the formation of oligomeric RIG-I structures (Jiang et al., 2012), possibly in conjunction with MAVS CARD, in analogy to structures formed by CARDs and other members of the death domain family such as the apoptosome (Shi, 2002), the Piddosome (Park et al., 2007b), or the Myddosome (Lin et al., 2010). Following ATP hydrolysis, RIG-I may translocate along the dsRNA (Myong et al., 2009) and leave the end accessible for another RIG-I molecule. The resulting accumulation of RIG-I oligomers onto dsRNA may provide the high local concentration for interaction with MAVS clusters on the mitochondria (Figure S5) as a step to induce IFN-β transcription.
EXPERIMENTAL PROCEDURES
A detailed description of plasmid construction, mutagenesis, western blotting and protein purification is described in Supplemental Information and Table S1. Dual-Luciferase Reporter (DLR) Assay (Promega) was performed following manufacturer instructions (details of assays and statistical analysis are also described in detail in the Supplemental Information). A group of reporter assays experiments were performed in a stable HEK293T cell line carrying an IFN-β promoter-driven firefly luciferase gene (Rodriguez-Madoz et al., 2010).
NMR Experiments
A detailed description of all NMR experiments and structure calculation is described in the Supplemental Information. Unless otherwise mentioned (see Supplemental Information), the concentration of both WT and T170E mutant of CARD2 in NMR samples was 300 μM. A series of double- and triple-resonance three-dimensional experiment was carried out to obtain backbone and sidechain assignments (Sattler et al., 1999) of WT and T170E CARD2. Aromatic side-chain resonances were assigned with (HB)CB(CGCD) HD; (HB)CB(CGCDCE)HE (Yamazaki et al., 1993) and aromatic 1H-13C NOESY-HSQC experiments. Distance constraints were obtained from aliphatic 1H-13C NOESY-HSQC, aromatic 1H-13C NOESY-HSQC and 1H-15N NOESY-HSQC. Dihedral angle constraints were derived from backbone chemical shifts using the program TALOS+ (Shen et al., 2009).
A full series of 15N relaxation experiments was carried out on a uniformly 15N-labeled sample to quantify backbone dynamics on sub-nanosecond as well as micro- and millisecond timescales (Kroenke et al., 1998). The following rates were measured on a uniformly 15N-labeled sample at 600 MHz: longitudinal and transverse relaxation rates; longitudinal (RL(N/NH) (Pelupessy et al., 2007) and transverse RT(N/NH) (Pelupessy et al., 2003) cross-correlated relaxation rates. 15N-{1H} nuclear Overhauser effects were measured at 500 and 600 MHz (Ferrage et al., 2008, 2009, 2010). To identify the fingerprint of conformational exchange, a full analysis of backbone 15N auto- and cross-correlated relaxation was carried out following the method of Kroenke et al. (Kroenke et al., 1998). Chemical exchange of amide protons with water was explored with the CLEANEX-PM scheme with a fast HSQC detection (Hwang et al., 1998).
The interaction of CARD2 and helicase-CTD was studied by recording a series of HSQC spectra with increasing concentration of unlabeled helicase-CTD into 15N-13C-labeled CARD2. All spectra were processed with NMRPipe (Delaglio et al., 1995) and analyzed using NMRView (Johnson and Blevins, 1994).
Structure Calculation
Structure calculation was carried out with partially assigned NOEs, dihedral and hydrogen-bonding constraints using the program ARIA/CNS (Linge et al., 2003). Structure refinement was performed using a protocol described in detail previously (Linge et al., 2003). The structural ensemble was then analyzed with PROCHECK-NMR (Laskowski et al., 1996). The secondary structure regions were defined using DSSP (Joosten et al., 2011; Kabsch and Sander, 1983).
All protein sequence alignments were carried out with ClustalW2 (Larkin et al., 2007). The structures of human CARD2 with duck CARD2 and the CARD of MAVS were aligned with the use of the DALI server (Holm and Rosenström, 2010).
Supplementary Material
ACKNOWLEDGMENTS
We thank Richard Cadagan and Osman Lizardo (MSSM) as well as Roberto Ferrero Laborda (CIMA) for excellent technical assistance. We thank David Cowburn (Albert Einstein College of Medicine) and Wolfgang Peti (Brown University) for fruitful discussions. This work was partly supported by grants from the UTE project CIMA, JCI-2011-09179 to E. N.-V., and SAF2009-08524 (Ministerio de Ciencia e Innovación) to G.G.-A. This work was also partly supported by NIAID Grant U19AI083025 to A.G.-S. A.K.A. is a member of the New York Structural Biology Center. The data collection at NYSBC was made possible by a grant from NYSTAR, ORIP/NIH facility improvement Grant CO6RR015495, NIH Grant P41GM066354, the Keck Foundation, New York State, and the NYC Economic Development Corporation.
Footnotes
ACCESSION NUMBERS The Protein Data Bank accession numbers for the coordinates and structural constraints reported in this paper are 2LWD (wild-type CARD2) and 2LWE (T170E CARD2). The BMRB accession numbers for the NMR resonance assignments reported in this paper are 18622 (WT) and 18623 (T170E).
SUPPLEMENTAL INFORMATION Supplemental Information includes five figures, one table, and Supplemental Experimental Procedures and can be found with this article online at http://dx.doi.org/10.1016/j.str.2012.09.003.
REFERENCES
- Arimoto KI, Takahashi H, Hishiki T, Konishi H, Fujita T, Shimotohno K. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc. Natl. Acad. Sci. USA. 2007;104:7500–7505. doi: 10.1073/pnas.0611551104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belgnaoui SM, Paz S, Hiscott J. Orchestrating the interferon antiviral response through the mitochondrial antiviral signaling (MAVS) adapter. Curr. Opin. Immunol. 2011;23:564–572. doi: 10.1016/j.coi.2011.08.001. [DOI] [PubMed] [Google Scholar]
- Civril F, Bennett M, Moldt M, Deimling T, Witte G, Schiesser S, Carell T, Hopfner KP. The RIG-I ATPase domain structure reveals insights into ATP-dependent antiviral signalling. EMBO Rep. 2011;12:1127–1134. doi: 10.1038/embor.2011.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui S, Eisenächer K, Kirchhofer A, Brzózka K, Lammens A, Lammens K, Fujita T, Conzelmann KK, Krug A, Hopfner KP. The C-terminal regulatory domain is the RNA 5′-triphosphate sensor of RIG-I. Mol. Cell. 2008;29:169–179. doi: 10.1016/j.molcel.2007.10.032. [DOI] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Ferrage F, Piserchio A, Cowburn D, Ghose R. On the measurement of 15N–1H nuclear Overhauser effects. J. Magn. Reson. 2008;192:302–313. doi: 10.1016/j.jmr.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrage F, Cowburn D, Ghose R. Accurate sampling of high-frequency motions in proteins by steady-state (15)N-(1)H nuclear Overhauser effect measurements in the presence of cross-correlated relaxation. J. Am. Chem. Soc. 2009;131:6048–6049. doi: 10.1021/ja809526q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrage F, Reichel A, Battacharya S, Cowburn D, Ghose R. On the measurement of 15N-1H nuclear Overhauser effects. 2. Effects of the saturation scheme and water signal suppression. J. Magn. Reson. 2010;207:294–303. doi: 10.1016/j.jmr.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foy E, Li K, Sumpter R, Jr., Loo YM, Johnson CL, Wang CF, Fish PM, Yoneyama M, Fujita T, Lemon SM, Gale M., Jr. Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc. Natl. Acad. Sci. USA. 2005;102:2986–2991. doi: 10.1073/pnas.0408707102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman CS, O'Donnell MA, Legarda-Addison D, Ng A, Cárdenas WB, Yount JS, Moran TM, Basler CF, Komuro A, Horvath CM, et al. The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep. 2008;9:930–936. doi: 10.1038/embor.2008.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fushman D, Varadan R, Assfalg M, Walker O. Determining domain orientation in macromolecules by using spin-relaxation and residual dipolar coupling measurements. Prog. Nucl. Magn. Reson. Spectrosc. 2004;44:189–214. [Google Scholar]
- Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun LJ, Takeuchi O, Akira S, Chen ZJ, Inoue SS, Jung JU. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature. 2007;446:916–920. doi: 10.1038/nature05732. [DOI] [PubMed] [Google Scholar]
- Gack MU, Kirchhofer A, Shin YC, Inn KS, Liang CY, Cui S, Myong S, Ha T, Hopfner KP, Jung JU. Roles of RIG-I N-terminal tandem CARD and splice variant in TRIM25-mediated antiviral signal transduction. Proc. Natl. Acad. Sci. USA. 2008;105:16743–16748. doi: 10.1073/pnas.0804947105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gack MU, Nistal-Villán E, Inn KS, García-Sastre A, Jung JU. Phosphorylation-mediated negative regulation of RIG-I antiviral activity. J. Virol. 2010;84:3220–3229. doi: 10.1128/JVI.02241-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Yang YK, Wang RP, Zhou X, Diao FC, Li MD, Zhai ZH, Jiang ZF, Chen DY. REUL is a novel E3 ubiquitin ligase and stimulator of retinoic-acid-inducible gene-I. PLoS ONE. 2009;4:e5760. doi: 10.1371/journal.pone.0005760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JB, Fushman D. Variability of the 15N chemical shielding tensors in the B3 domain of protein G from 15N relaxation measurements at several fields. Implications for backbone order parameters. J. Am. Chem. Soc. 2006;128:7855–7870. doi: 10.1021/ja060406x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henzler-Wildman K, Kern D. Dynamic personalities of proteins. Nature. 2007;450:964–972. doi: 10.1038/nature06522. [DOI] [PubMed] [Google Scholar]
- Holm L, Rosenström P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38(Web Server issue):W545–9. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang TL, van Zijl PCM, Mori S. Accurate quantitation of water-amide proton exchange rates using the phase-modulated CLEAN chemical EXchange (CLEANEX-PM) approach with a Fast-HSQC (FHSQC) detection scheme. J. Biomol. NMR. 1998;11:221–226. doi: 10.1023/a:1008276004875. [DOI] [PubMed] [Google Scholar]
- Inn K-S, Gack MU, Tokunaga F, Shi M, Wong L-Y, Iwai K, Jung JU. Linear ubiquitin assembly complex negatively regulates RIG-I- and TRIM25-mediated type I interferon induction. Mol. Cell. 2011;41:354–365. doi: 10.1016/j.molcel.2010.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang FG, Ramanathan A, Miller MT, Tang GQ, Gale M, Jr., Patel SS, Marcotrigiano J. Structural basis of RNA recognition and activation by innate immune receptor RIG-I. Nature. 2011;479:423–427. doi: 10.1038/nature10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Kinch LN, Brautigam CA, Chen X, Du F, Grishin NV, Chen ZJ. Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity. 2012;36:959–973. doi: 10.1016/j.immuni.2012.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BA, Blevins RA. NMR View: A computer program for the visualization and analysis of NMR data. J. Biomol. NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- Joosten RP, te Beek TA, Krieger E, Hekkelman ML, Hooft RWW, Schneider R, Sander C, Vriend G. A series of PDB related databases for everyday needs. Nucleic Acids Res. 2011;39(Database issue):D411–D419. doi: 10.1093/nar/gkq1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- Kloiber K, Konrat R. Differential multiple-quantum relaxation arising from cross-correlated time-modulation of isotropic chemical shifts. J. Biomol. NMR. 2000;18:33–42. doi: 10.1023/a:1008317212558. [DOI] [PubMed] [Google Scholar]
- Koradi R, Billeter M, Wüthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 1996;14:51–55. 29–32. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- Kowalinski E, Lunardi T, McCarthy AA, Louber J, Brunel J, Grigorov B, Gerlier D, Cusack S. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell. 2011;147:423–435. doi: 10.1016/j.cell.2011.09.039. [DOI] [PubMed] [Google Scholar]
- Kroenke CD, Loria JP, Lee LK, Rance M, Palmer AG., III Longitudinal and Transverse H-1-N-15 Dipolar N-15 Chemical Shift Anisotropy Relaxation Interference: Unambiguous Determination of Rotational Diffusion Tensors and Chemical Exchange Effects in Biological Macromolecules. J. Am. Chem. Soc. 1998;120:7905–7915. [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. Clustal W and clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- Lin SC, Lo YC, Wu H. Helical assembly in the MyD88-IRAK4–IRAK2 complex in TLR/IL-1R signalling. Nature. 2010;465:885–890. doi: 10.1038/nature09121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linge JP, Habeck M, Rieping W, Nilges M. ARIA: automated NOE assignment and NMR structure calculation. Bioinformatics. 2003;19:315–316. doi: 10.1093/bioinformatics/19.2.315. [DOI] [PubMed] [Google Scholar]
- Lipari G, Szabo A. Model-Free Approach to the Interpretation of Nuclear Magnetic Resonance Relaxation In Macromolecules 1. Theory and Range of Validity. J. Am. Chem. Soc. 1982;104:4546–4559. [Google Scholar]
- Luo DH, Ding SC, Vela A, Kohlway A, Lindenbach BD, Pyle AM. Structural insights into RNA recognition by RIG-I. Cell. 2011;147:409–422. doi: 10.1016/j.cell.2011.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maharaj NP, Wies E, Stoll A, Gack MU. Conventional protein kinase C-α (PKC-α) and PKC-β negatively regulate RIG-I antiviral signal transduction. J. Virol. 2012;86:1358–1371. doi: 10.1128/JVI.06543-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittermaier A, Kay LE. New tools provide new insights in NMR studies of protein dynamics. Science. 2006;312:224–228. doi: 10.1126/science.1124964. [DOI] [PubMed] [Google Scholar]
- Myong S, Cui S, Cornish PV, Kirchhofer A, Gack MU, Jung JU, Hopfner KP, Ha T. Cytosolic viral sensor RIG-I is a 5′-triphosphate-dependent translocase on double-stranded RNA. Science. 2009;323:1070–1074. doi: 10.1126/science.1168352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nistal-Villán E, Gack MU, Martínez-Delgado G, Maharaj NP, Inn KS, Yang HY, Wang R, Aggarwal AK, Jung JU, García-Sastre A. Negative role of RIG-I serine 8 phosphorylation in the regulation of inter-feron-beta production. J. Biol. Chem. 2010;285:20252–20261. doi: 10.1074/jbc.M109.089912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer AG., 3rd NMR characterization of the dynamics of bio-macromolecules. Chem. Rev. 2004;104:3623–3640. doi: 10.1021/cr030413t. [DOI] [PubMed] [Google Scholar]
- Park HH, Lo YC, Lin SC, Wang L, Yang JK, Wu H. Annu. Rev. Immunol. Annual Reviews; Palo Alto: 2007a. The death domain superfamily in intracellular signaling of apoptosis and inflammation; pp. 561–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HH, Logette E, Raunser S, Cuenin S, Walz T, Tschopp J, Wu H. Death domain assembly mechanism revealed by crystal structure of the oligomeric PIDDosome core complex. Cell. 2007b;128:533–546. doi: 10.1016/j.cell.2007.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelupessy P, Espallargas GM, Bodenhausen G. Symmetrical reconversion: measuring cross-correlation rates with enhanced accuracy. J. Magn. Reson. 2003;161:258–264. doi: 10.1016/s1090-7807(02)00190-8. [DOI] [PubMed] [Google Scholar]
- Pelupessy P, Ferrage F, Bodenhausen G. Accurate measurement of longitudinal cross-relaxation rates in nuclear magnetic resonance. J. Chem. Phys. 2007;126:134508. doi: 10.1063/1.2715583. [DOI] [PubMed] [Google Scholar]
- Pothlichet J, Burtey A, Kubarenko AV, Caignard G, Solhonne B, Tangy F, Ben-Ali M, Quintana-Murci L, Heinzmann A, Chiche JD, et al. Study of human RIG-I polymorphisms identifies two variants with an opposite impact on the antiviral immune response. PLoS ONE. 2009;4:e7582. doi: 10.1371/journal.pone.0007582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter JA, Randall RE, Taylor GL. Crystal structure of human IPS-1/MAVS/VISA/Cardif caspase activation recruitment domain. BMC Struct. Biol. 2008;8:8. doi: 10.1186/1472-6807-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin HX, Srinivasula SM, Wu G, Fernandes-Alnemri T, Alnemri ES, Shi YG. Structural basis of procaspase-9 recruitment by the apoptotic protease-activating factor 1. Nature. 1999;399:549–557. doi: 10.1038/21124. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Madoz JR, Belicha-Villanueva A, Bernal-Rubio D, Ashour J, Ayllon J, Fernandez-Sesma A. Inhibition of the type I interferon response in human dendritic cells by dengue virus infection requires a catalytically active NS2B3 complex. J. Virol. 2010;84:9760–9774. doi: 10.1128/JVI.01051-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Hirai R, Loo YM, Owen D, Johnson CL, Sinha SC, Akira S, Fujita T, Gale M., Jr. Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc. Natl. Acad. Sci. USA. 2007;104:582–587. doi: 10.1073/pnas.0606699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler M, Schleucher J, Griesinger C. Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog. Nucl. Magn. Reson. Spectrosc. 1999;34:93–158. [Google Scholar]
- Schlee M, Hartmann G. The chase for the RIG-I ligand—recent advances. Mol. Ther. 2010;18:1254–1262. doi: 10.1038/mt.2010.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrodinger L. The PyMOL Molecular Graphics System. Version 0.99rc6. Schrödinger, LLC; 2012. http://www.pymol.org. [Google Scholar]
- Shen Y, Delaglio F, Cornilescu G, Bax A. TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR. 2009;44:213–223. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi YG. Mechanisms of caspase activation and inhibition during apoptosis. Mol. Cell. 2002;9:459–470. doi: 10.1016/s1097-2765(02)00482-3. [DOI] [PubMed] [Google Scholar]
- Shigemoto T, Kageyama M, Hirai R, Zheng JP, Yoneyama M, Fujita T. Identification of loss of function mutations in human genes encoding RIG-I and MDA5: implications for resistance to type I diabetes. J. Biol. Chem. 2009;284:13348–13354. doi: 10.1074/jbc.M809449200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahasi K, Yoneyama M, Nishihori T, Hirai R, Kumeta H, Narita R, Gale M, Jr., Inagaki F, Fujita T. Nonself RNA-sensing mechanism of RIG-I helicase and activation of antiviral immune responses. Mol. Cell. 2008;29:428–440. doi: 10.1016/j.molcel.2007.11.028. [DOI] [PubMed] [Google Scholar]
- Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki T, Forman-Kay JD, Kay LE. J. Am. Chem. Soc. 1993;115:11054–11055. [Google Scholar]
- Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- Zeng WW, Sun LJ, Jiang XM, Chen X, Hou FJ, Adhikari A, Xu M, Chen ZJJ. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell. 2010;141:315–330. doi: 10.1016/j.cell.2010.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P, Chou J, Olea RS, Yuan JY, Wagner G. Solution structure of Apaf-1 CARD and its interaction with caspase-9 CARD: a structural basis for specific adaptor/caspase interaction. Proc. Natl. Acad. Sci. USA. 1999;96:11265–11270. doi: 10.1073/pnas.96.20.11265. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.