

Abstract
Cardiovascular calcification is currently viewed as an active disease process similar to embryonic bone formation. Cardiovascular calcification mainly affects the aortic valve and arteries and is associated with increased mortality risk. Aortic valve and arterial calcification share similar risk factors, including age, gender, diabetes, chronic renal disease, and smoking. However, the exact cellular and molecular mechanism of cardiovascular calcification is unknown. Late-stage cardiovascular calcification can be visualized with conventional imaging modalities such as echocardiography and computed tomography. However, these modalities are limited in their ability to detect the development of early calcification and the progression of calcification until advanced tissue mineralization is apparent. Due to the subsequent late diagnosis of cardiovascular calcification, treatment is usually comprised of invasive interventions such as surgery. The need to understand the process of calcification is therefore warranted and requires new imaging modalities which are able to visualize early cardiovascular calcification. This review focuses on the use of new imaging techniques to visualize novel concepts of cardiovascular calcification.
Introduction
Calcification, a progressive disease of dysregulated mineral metabolism, has been observed in the cardiovascular system for many decades. Ectopic calcification of the cardiovascular system predominantly affects the aorta, coronary arteries, peripheral arteries, and the aortic valve. Traditionally, cardiovascular calcification has been considered as a passive phenomenon associated with aging; however, it is currently viewed as an actively regulated disease process. More specifically, mounting evidence suggests that the underlying mechanisms of cardiovascular calcification are similar to embryonic bone formation (Demer and Tintut, 2008; Otto, 2008; Towler and Demer, 2011).
The pathological mineralization of the arteries is often observed in atherosclerotic plaques, which clinically translates to reduced compliance of the vessel wall, associated with hypertension (Abedin et al., 2004). In addition, studies demonstrate that microcalcification in the fibrous cap overlying the necrotic core of atherosclerotic plaques could lead to microfractures and plaque rupture, leading to acute thrombosis and possibly fatal myocardial infarctions (Vengrenyuk et al., 2006; Virmani et al., 2006). Furthermore, calcification poses significant challenges for the outcome of interventional strategies such as percutaneous coronary interventions for coronary artery disease (Moses et al., 2004). Calcification of the aortic valve, or calcific aortic valve disease (CAVD), progresses from mild thickening to severe calcification and leads to stiffening of the aortic valve leaflets, eventually causing left ventricular outflow obstruction and heart failure (Mohler, 2004; Rajamannan et al., 2007; Otto, 2008). Even mild aortic valve calcification is associated with increased mortality risk (Lloyd-Jones et al., 2009). There are no known therapies that slow disease progression, and in case of aortic valve stenosis, surgical valve replacement and evolving transcatheter valve implantation (TAVI) are the only current treatments. Therefore, effective anti-calcification therapies are warranted.
Aortic valve calcification and arterial calcification share similar risk factors, such as age, gender, smoking, hypercholesterolemia, metabolic syndrome, end-stage renal disease, and diabetes mellitus (Stewart et al., 1997). Pathologically, explanted human stenotic aortic valves demonstrate similar lesions as observed in atherosclerotic plaques consisting of inflammatory cells and calcific deposits (Otto et al., 1994). Patients with familial hypercholesterolemia are prone to develop atherosclerosis in addition to developing valve lesions that calcify with age (Rajamannan et al., 2001a, 2001b). Moreover, preclinical animal studies show atherosclerotic-like lesion in aortic valve leaflets of rabbits and mice with established atherosclerosis (Rajamannan et al., 2002). Since aortic valve calcification and atherosclerosis potentially share a similar pathological mechanism, statins (3-hydroxy-3methylglutaryl-coenzyme A [HMG-CoA] reductase inhibitors) emerged as a therapeutic agent. Although several retrospective studies demonstrate a reduction in aortic valve stenosis when treated with statins (Aronow et al., 2001; Novaro et al., 2001; Bellamy et al., 2002), large prospective randomized clinical trials, do not support these findings and show no reduction in aortic valve calcification when treated with high doses of statins (Cowell et al., 2005; Rossebo et al., 2008). Although, this may be due to the late implementation of statins, after aortic valve calcification has progressed to an irreversible stage, these studies underscore our lack of understanding of underlying mechanisms of cardiovascular calcification. An increasing aging population necessitate further investigation of the pathways that contribute to cardiovascular calcification. The need to develop new therapeutic targets to prevent or reverse cardiovascular calcification warrants the use of novel imaging methods, as recently highlighted by the Working Group on Calcific Aortic Stenosis of the National heart, Lung, and Blood Institute (NHLBI) (Rajamannan et al., 2011).
Currently cardiovascular calcification is visualized by noninvasive conventional imaging modalities such as echocardiography, computed tomography (CT), and magnetic resonance imaging (MRI). Even though, CT and echocardiography are vital diagnostic tools because of their high sensitivity and ability to quantify calcium content of soft tissues, clinical imaging is usually only performed when patients are becoming symptomatic. It should be mentioned that coronary artery calcium (CAC) scoring is used in asymptomatic patients for risk stratification for CAD. Clinical symptoms arise only at advanced stages of calcification, leaving physicians too late to reduce or prevent calcification. In addition, CT and echocardiography have a relatively low spatial resolution and cannot detect early stages of cardiovascular calcification. Early detection methods are needed to not only elucidate early mechanisms of cardiovascular calcification, but also to establish whether the disease is reversible. Imaging modalities that can visualize molecular targets can contribute to understand important molecular and cellular aspects of cardiovascular calcification. The development of molecular imaging has enabled us to visualize early stages of calcification and piece together with the mechanism of disease progression leading to calcium deposition (Fig. 1). This review discusses how innovative imaging techniques have helped visualizing novel concepts of cardiovascular calcification.
Fig. 1.

Molecular imaging of cardiovascular calcification. Molecular imaging visualizes inflammatory activity, defined as macrophage accumulation, and osteogenesis in the aortic valves and aortas of apoE−/− mice. Magnetofluorescent nanoparticles were injected into mice to visualize macrophage accumulation (left). Spectrally distinct bisphosphonate-imaging agent that binds to hydroxyapatite were injected to detect osteogenic activity (right). Inflammatory and osteogenic activity colocalized in the aortic valve (top) and in the aorta (bottom), specifically in the areas of highest mechanical stresses at the aortic valve attachment (arrowheads) and at the atherosclerosis-prone areas, such as the innominate artery, aortic arch, and abdominal aorta (arrows). High signal intensities are shown in red/yellow/green (New and Aikawa, 2011).
Initiation of calcification
The mechanistic pathways involved in the development of cardiovascular calcification remain largely unknown. In calcification research much focus is shifting toward mapping the entire process of the disease and as such determining the initiation, progression and end-stage of the disease process. The current pathological concept of cardiovascular calcification, albeit arterial or valvular in nature, is that cardiovascular risk factors lead to endothelial dysfunction, followed by the deposition of lipoprotein (LDL) particles and other compounds that trigger inflammatory response (Aikawa et al., 2007b; Otto, 2008; Towler, 2008; Rajamannan, 2010). The inflammatory state leads to the activation of inflammatory signaling pathways, macrophage infiltration and T-lymphocyte activation. Activation of inflammatory pathways contributes to the disease process, which in turn activates smooth muscle cells (SMC) in the arterial wall and valvular interstitial cells (VIC) in the aortic valve to differentiate to osteoblast-like cells and deposit calcium. Although the initiation of cardiovascular calcification is still a matter of investigation, evidence suggests that endothelial dysfunction leads to osteoblastic differentiation of underlying SMC and VIC in the arterial wall and aortic valve, respectively.
Endothelial activation
The endothelium maintains normal homeostasis at the vasculature–blood interface, however dysfunctional endothelial cells promote an inflammatory response and the onset of atherosclerosis/intimal calcification (Gryglewski et al., 1988). Noninvasive ultrasound molecular imaging, via targeted imaging agents, can detect lesion-prone vasculature before the appearance of advanced calcified atherosclerotic plaques (Kaufmann et al., 2010). Sites of atherosclerotic lesion formation are associated with an increased surface expression of endothelial cell adhesion molecules, such as VCAM-1 and P-selectin (Cybulsky and Gimbrone, 1991; Nahrendorf et al., 2006; Kaufmann et al., 2010). This expression of VCAM-1 by endothelial cells precedes fatty streak formation (Nakashima et al., 1998) and is up-regulated in response to oxidized LDL cholesterol – a hypercholesterolemic milieu (Marui et al., 1993). VCAM-1 participates in the initiation and progression of atherosclerotic plaques can be visualized in vivo by noninvasive molecular imaging (Fig. 2) (Cybulsky and Gimbrone, 1991; Nahrendorf et al., 2006; Kaufmann et al., 2010).
Fig. 2.
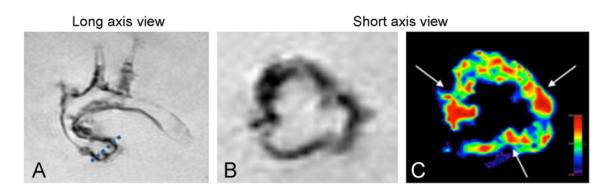
Visualization of VCAM-1 targeted agents. MRI visualizes endothelial activation detected by VCAM-1-targeted agent, in the commissures of aortic valves, which are regions of high flexure and mechanical stress: (A) dotted line = slice position of short axis view, (B) negative signal enhancement caused by uptake of VCAM-1-targeted nanoparticles, and (C) color-coded signal (red) show focused uptake of VCAM-1 in commissures (arrows) (Aikawa et al., 2007b).
The mechanism of initiation of vascular calcification is proposed to be similar to that which we have suggested for calcific aortic valve disease (Fig. 3) (Aikawa et al., 2007a, 2007b; Aikawa and Otto, 2012) Activated endothelial cells recruit inflammatory cells, mainly in the form of monocytes/macrophages, which contribute to the progression of this pathological disorder. Our recent work used molecular imaging to detect early changes in aortic valve disease. Utilizing a similar approach as imaging arterial endothelial cells, VCAM-1 targeted agents are distributed in the commissures of the aortic valve (Nahrendorf et al., 2006), which was validated by immunohistochemical analysis of VCAM-1 expression (Aikawa et al., 2007b). It has been confirmed that valvular endothelial dysfunction or injury leads to increased expression of adhesion molecules VCAM-1, ICAM-1 and E-selectin (Ghaisas et al., 2000; Muller et al., 2000). Mechanically the commissures endure the greatest amount of stress during the cardiac cycle, making these areas more prone to endothelial injury than other parts of the leaflets. In addition, high pulsatile shear stress has demonstrated the upregulation of inflammatory receptors by valvular endothelial cells for circulating cytokines and inflammatory monocytes, leukocytes and T-lymphocytes (Muller et al., 2000; Shavelle et al., 2008; Sucosky et al., 2009).
Fig. 3.

Mechanisms underlying arterial and aortic valve calcification. This figure depicts the theory that cardiovascular calcification follows similar pathways in both the artery and the aortic valve and summarizes how molecular imaging can visualize concepts of the calcification process. Cardiovascular calcification is an active process, initiated by endothelial dysfunction/activation and/or by inflammation and resulting in mineralization. Pro-inflammatory monocytes (1) are recruited to a site via activated/injured endothelial cells (ECs). (2) The activation of ECs causes increased expression of adhesion molecules, such as VCAM-1, which can be visualized by NIRF imaging VCAM-1 agent. Subsequent macrophage (3) accumulation follows, which can be analyzed with NIRF macrophage-targeted nanoparticles (AminoSPARK). The release of proteolytic enzymes, including matrix metalloproteases and cathepsins, by macrophages, which stimulates the differentiation of myofibroblasts (4) and smooth muscle cells (5) into osteoblasts, can be visualized by molecular imaging with activatable imaging agents (MMPSense, ProSense). Osteogenic activity in the form of osteoblast (6) formation and microcalcifications associated with generation of calcified matrix vesicles (7) can be identified by a bisphosphonate-conjugated imaging agent (OsteoSense) whereas apoptosis (8) can be detected by a fluorescently tagged Annexin A5 imaging probe. Calcification (9) can be readily detected by molecular imaging and conventional imaging techniques such as CT and Echocardiography (New and Aikawa, 2011).
Another endothelial mechanism suggested a role of reactive oxygen species (ROS) as a possible initiating factor of calcification (Sorescu et al., 2004; Butcher and Nerem, 2006; Sucosky et al., 2009). ROS, including oxidized lipids, can cause endothelial cell injury, leading to loss of cell alignment and upregulation of cell adhesion molecules for circulating inflammatory cells. To this end, a recent study demonstrated imaging ROS with allylhydrazine, a liquid compound that converts to nitrogen and propylene gas after reacting with ROS, in mice using a clinical echocardiography system. Allylhydrazine can be encapsulated within liposomes and is able to detect micromolar concentrations of radical oxidants (Perng et al., 2012).
Progression of calcification – An inflammatory concept
Evidence suggests that arterial and valvular calcification follows a pathway similar to that of endochondral bone formation (Rattazzi et al., 2005); osteoblastic differentiation of vascular SMC is via the Runx2 pathway (Steitz et al., 2001) similar to VIC. A series of in vitro studies (Watson et al., 1994; Parhami et al., 2002; Tintut et al., 2002; Radcliff et al., 2005) demonstrate that monocytes and macrophages release inflammatory cytokines that promote cardiovascular calcification by regulating the differentiation of calcifying vascular cells. These calcifying vascular cells express a number of phenotypic markers synonymous with osteoblasts and chondrocytes (Aikawa et al., 2007a, 2007b; El-Abbadi and Giachelli, 2007; Peacock et al., 2010). Our molecular imaging studies on arterial intimal calcification have provided the first in vivo evidence of the role that inflammation plays in initiating calcification (Aikawa et al., 2007a). Nanoparticle technology can be used to image not only the accumulation of macrophages within the intima of the atherosclerotic plaque, but also the co-localization of macrophages with osteogenic activity or active mineralization. Intravital microscopy has been performed on the carotid arteries of untreated and statin-treated cohort of apoE−/− mice at 20 and 30 weeks of age. Macrophage number, as visualized by macrophage-targeted iron-oxide nanoparticles, increased in association with osteogenic signal at 30 weeks. In this study of early calcification, anti-inflammatory statin therapy prevented both macrophage burden and osteogenic activity. Osteogenic differentiation of SMC and areas of active mineralization are effectively imaged via the use of a bisphosphonate-conjugated imaging agent (OsteoSense680) (Aikawa et al., 2007a; Hjortnaes et al., 2010a, 2010b). This imaging agent binds to calcium and will accumulate where there is increased osteogenesis, as confirmed by alkaline phophatase activity (Kozloff et al., 2007).
The progression of arterial calcification has shown to largely depend on an inflammatory process, which in turn associates with proteolysis and tissue remodeling. Early calcifying atherosclerotic plaques, often termed “spotty” calcifications, are associated with microcalcification and numerous macrophages undergoing proteolysis (Ehara et al., 2004). These vulnerable early atherosclerotic plaques are at risk of fatal rupture (Vengrenyuk et al., 2006; Virmani et al., 2006; Demer and Tintut, 2008; Li et al., 2012). During the progression of atherosclerosis, macrophages elaborate proteolytic activity, in the form of elastases (e.g., cathepsin S) and metalloproteinases (e.g., MMP-1, MMP-2, MMP-9). Our study utilized a novel protease-activatable imaging agent specific for cathepsin S and provided the first direct in vivo evidence for the role of this elastase in cardiovascular calcification (Aikawa et al., 2009). Molecular imaging detected increased cathepsin S and osteogenic activities in CRD mice, induced by 5/6 nephrectomy, compared to control apoE−/− mice. Furthermore, calcification was decreased in atherosclerotic plaques and aortic valves in mice lacking cathepsin S. These results were corroborated using optical projection tomography and quantitative histology.
In the early stages of cardiovascular calcification, macrophage-derived elastolytic and proteolytic enzymes degrade elastin and collagen, respectively, major components of the extracellular matrix (ECM) in both the aortic valve and arteries (New and Aikawa, 2011). Inflammation-induced elastolysis in the atherosclerotic plaques results in the release of biologically active, soluble elastin-derived peptides (Karnik et al., 2003; Simionescu et al., 2007). These biologically active peptides may regulate cell processes such as proliferation, as well as promote osteogenic differentiation of SMC and subsequent microcalcification formation (Saulnier et al., 1991; Karnik et al., 2003; Simionescu et al., 2007). Microcalcifications provoke a pro-inflammatory response in macrophages via mechanisms involving protein kinase C-α and ERK1/2 MAP kinase (Nadra et al., 2005, 2008). Therefore a positive feedback loop of inflammation and calcification drives progression of arterial calcification (Bostrom, 2005; Nadra et al., 2008), corroborated in vivo via the use of molecular imaging in our study (Aikawa et al., 2007a). Molecular imaging is capable of characterizing/diagnosing plaque activity, in the form of proteolytic activity, to identify high-risk patients. Thus the visualization of proteolytic activity, via the use of near-infrared fluorescence (NIRF) beacons/probes, may prove a valuable tool in the diagnosis of vulnerable rupture-prone plaques and onset of aortic stenosis.
The inflammatory cascade, that is initiated by the valvular endothelium, first results in excessive remodeling of the aortic valve extracellular matrix, which follows the clinical observation of aortic fibrosis preceding aortic valve stenosis (Otto et al., 1994). Similar to the arterial wall, proteolytic enzymes and their inhibitors maintain a balance that can degrade ECM compounds such as collagen and elastin during valvular remodeling. Using macrophage-targeted NIRF-conjugated iron nanoparticles and protease-activatable NIRF probes, we have demonstrated that macrophages correlate with increased levels of matrix metalloproteinases (MMP-1, MMP-2, MMP-9, MMP13,), and cysteine proteases (cathepsin S, cathepsin K) in early stage calcification animal models (Aikawa et al., 2007a, 2007b, 2009). In turn, we have also showed that inflammatory activity correlated with osteogenic activity using the same previously described calcification sensitive imaging probe (Hjortnaes et al., 2010). This imaging agent uses different NIRF wavelengths than other imaging agents, allowing for the correlation of osteogenic activity with other pathological processes of the valve. Using this approach, we have correlated inflammation with early osteogenic activity in the valve, and confirmed by alkaline phosphatase specific activity and immunohistochemistry for osteocalcin, osteopontin, and the osteogenic transcription factor Runx2 (Aikawa et al., 2007b).
Furthermore, it is also possible to use these NIRF agents for MRI visualization, making the translation to clinical practice very feasible. Recently developed iron-oxide based magnetic nanoparticles, which are engulfed by tissue macrophages by phagocytosis, can be used for T2 weighted contrast MRI and PET (Jaffer et al., 2006). This technology allows clinicians to discover a potential reversibility of the inflammatory response that leads to cardiovascular calcification by visualizing molecular, anatomic and physiological changes of the cardiovascular system. Another optical imaging technology, microoptical coherence tomography (micro-CT), has recently been developed and is able to visualize microcalcifications, cholesterol crystals, macrophages and proteins with a 1-μm resolution. It does not require the injection of molecular imaging agents and can thus be more easily translated from bench to bed. Nevertheless both NIRF imaging and micro-OCT have limited tissue penetration.
Recent evidence suggests that positron emission tomography (PET) imaging could overcome this limitation by using PET traces that target inflammation and calcification (Beheshti et al., 2011; Folco et al., 2011; Aikawa and Otto, 2012; Hyafil et al., 2012). Glucose analogue 18F-fluorodeoxyglucose (18F-FDG) is an imaging agent taken up by cells by glucose transport proteins, upon which the agent becomes trapped in macrophages. 18F-sodium fluoride (18F-NaF) binds to hydroxyapatite and is a marker for mineralization. A recent study that used this imaging approach confirmed, in patients with the full spectrum of aortic valve calcification, that inflammation and mineralization are present in early stages of aortic valve calcification (Dweck et al., 2012). It is the first clinical study to visualize early calcification processes in patients that confirms our current understanding of aortic valve calcification.
Although the exact role of inflammation remains to be elucidated, it is clear that specific imaging agents can be used to visualize mechanisms related to cardiovascular calcification. Moreover, specific targeting imaging agents, for particular molecules of interest, may prove beneficial as a means to visualize potential biomarkers or therapeutic targets.
Alternative mechanisms of arterial calcification
Although we are fully in support of the widely accepted concept that calcifying vascular cells play an important role in cardiovascular calcification, we also fervently believe that there are alternative mechanisms of mineralization. The apoptosis of macrophages has been proposed to contribute to plaque vulnerability (Virmani et al., 2006). In addition, macrophages induce the apoptosis of vascular SMC by direct interaction (Boyle et al., 2001, 2002). The release of apoptotic bodies from macrophages and SMC undergoing apoptosis may provide a nidus for calcification and provoke the generation of microcalcification (Proudfoot et al., 2000; Shanahan, 2007). Apoptosis can be monitored in the vasculature in realtime via the use of molecular imaging approaches with fluorescently tagged annexin V imaging agents (van Tilborg et al., 2010; Corsten and Bennaghmouch, 2011). The clinical translation of imaging approaches such as these may prove beneficial in assessing the effectiveness of specific therapies, as well as a means to visualize disease progression.
Osteoblasts and chondrocytes release hydroxyapatite-nucleating matrix vesicles during biological skeletal tissue mineralization (Anderson, 1981; Anderson et al., 1990). Matrix vesicles (30–300 nm), precursors of microcalcifications, bud from the plasma membrane of living cells and also believed to serve as a nidus for mineral nucleation in ectopic calcification (Kim, 1976; Anderson, 2003; Bobryshev et al., 2007). Evidence suggests that these matrix vesicles contain inhibitors of mineralization, such as Matrix Gla Protein and Fetuin-A, and a reduction in these inhibitors enhances their calcific potential (Reynolds et al., 2004; Chen et al., 2008; Kapustin et al., 2011). Calcifying matrix vesicles have been stated to possess exposed phosphatidylserine (Kapustin et al., 2011), therefore some of the current molecular imaging modalities used to image apoptosis and calcification can also detect matrix vesicle accumulation. The exact role that these vesicles play in cardiovascular calcification and the mechanism by which they are released still require further investigation.
Late-stage calcification
Late-stage cardiovascular calcification is characterized by advanced tissue mineralization and diminished inflammation. It is classically reviewed as irreversible due to the development of advanced calcified material and is readily detected by conventional imaging approaches (e.g., echocardiography, CT) (Johnson et al., 2006). All in all, cardiovascular calcification, albeit arterial or valvular, seems to follow similar pathways towards mineralization (Fig. 3). Specifically, calcification starts with endothelial dysfunction or activation due to inflammation, leading to an increased uptake of inflammatory cells. Inflammation therefore can be viewed as a propagation process eventually leading to the differentiation of SMC or VIC to actively deposit calcium in the arterial wall and aortic valve, respectively. Where conventional imaging modalities are limited, this early calcification process can be visualized by molecular imaging. Table 1 depicts molecular agents used in detecting various components of cardiovascular inflammation and calcification.
Table 1.
Molecular imaging agents in cardiovascular calcification
| Cardiovascular calcification | Target | Imaging agent | Imaging modality |
|---|---|---|---|
| Processes associated with early calcification | |||
| Endothelial dysfunction | VCAM-1 | VINP-28 Microbubbles |
Optical imaginga/MRI US |
| ICAM-1 | NIRF Microbubbles |
Optical imaging US/MRI |
|
| ROS | Microbubbles MPOb probe oxLDL-targeted nanoparticle |
US Optical imaging MRI |
|
| Inflammation | Macrophages | Nanoparticles (e.g., CLIOb) | Optical Imaging/MRI/PET |
| MMP | MMPSense | Optical imaging | |
| Cathepsin | ProSense | Optical imaging | |
| Progression of calcification | |||
| Inflammation | Macrophages | Nanoparticles (e.g., CLIOb) | Optical imaging/MRI/PET |
| Apoptosis | Annexin V | Nanoparticle | Optical imaging/MRI |
| Microcalcification | Caspase | 18F-FDG | PET |
| Hydroxyapatite | Activatable probes OsteoSense 18F-NaF |
Optical imaging/PET Optical imaging PET |
|
| Late-stage calcification | |||
| Advanced calcification | Hydroxyapatite | OsteoSense 18F-NaF |
Optical imaging PET |
| Calcium | – | Echocardiography CT |
Optical imaging indicates one or more of the following modalities: Optical projection tomography (OPT), micro-optical coherence tomography (micro-OCT), fluorescence molecular tomography (FMT) and intravital microscopy (IVM).
NIRF, near-infrared fluorescence; MRI, magnetic resonance imaging; US, ultrasound; ROS, reactive oxygen species; MPO, myeloperoxidase; CLIO, cross-linked iron oxide; PET, positron emission tomography; FDG, fluorodeoxyglucose; NaF, sodium fluoride; CT, computed tomography.
Conclusion
Cardiovascular calcification is currently viewed as an active disease process. Understanding the pathological pathways from initiation to late-stage mineralization is key to identifying therapeutic targets that can potentially treat cardiovascular calcification. Especially mapping early calcification before the disease process has passed a point of no return provides the window for potential treatment. Therapeutic agents have thus far not proved beneficial in the clinical setting, mainly because of late clinical diagnosis. In addition, because histopathological examination is not possible during longitudinal noninvasive imaging studies (e.g., echocardiography, CT, PET), validated high-resolution invasive imaging modalities (e.g., molecular imaging, micro-OCT) may be required to understand the mechanisms at various stages of CAVD.
The molecular imaging approach can unmask/resolve early pathophysiological changes in the cardiovascular system and may prove beneficial diagnostically in a clinical setting. Moreover, considering the amount of evidence demonstrating the role of inflammation in early stages of calcification (e.g., initiation, propagation), targeting inflammation could be beneficial in reducing and even halting subsequent mineralization. Although current clinical imaging approaches, such as echocardiography, remain essential for clinical management and for following CAVD progression once valve obstruction is present, PET may be the method of choice for detection of early calcification and inflammation in clinical trials. Molecular imaging modalities possess great potential in visualizing in vivo cardiovascular calcification and will be vital in the development of therapeutic targets. Large, prospective, event-driven studies of noninvasive imaging techniques will be required to determine the place of these modalities in future clinical practice.
REFERENCES
- Abedin M, Tintut Y, Demer LL. Vascular calcification: mechanisms and clinical ramifications. Arteriosclerosis, Thrombosis, and Vascular Biology. 2004;24:1161–70. doi: 10.1161/01.ATV.0000133194.94939.42. [DOI] [PubMed] [Google Scholar]
- Aikawa E, Aikawa M, Libby P, et al. Arterial and aortic valve calcification abolished by elastolytic cathepsin S deficiency in chronic renal disease. Circulation. 2009;119:1785–94. doi: 10.1161/CIRCULATIONAHA.108.827972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aikawa E, Nahrendorf M, Figueiredo JL, et al. Osteogenesis associates with inflammation in early-stage atherosclerosis evaluated by molecular imaging in vivo. Circulation. 2007;116:2841–50. doi: 10.1161/CIRCULATIONAHA.107.732867. [DOI] [PubMed] [Google Scholar]
- Aikawa E, Nahrendorf M, Sosnovik D, et al. Multimodality molecular imaging identifies proteolytic and osteogenic activities in early aortic valve disease. Circulation. 2007b;115:377–86. doi: 10.1161/CIRCULATIONAHA.106.654913. [DOI] [PubMed] [Google Scholar]
- Aikawa E, Otto CM. Look more closely at the valve: imaging calcific aortic valve disease. Circulation. 2012;125:9–11. doi: 10.1161/CIRCULATIONAHA.111.073452. [DOI] [PubMed] [Google Scholar]
- Anderson HC. Normal and abnormal mineralization in mammals. Transactions – American Society for Artificial Internal Organs. 1981;27:702–8. [PubMed] [Google Scholar]
- Anderson HC. Matrix vesicles and calcification. Current Rheumatology Reports. 2003;5:222–6. doi: 10.1007/s11926-003-0071-z. [DOI] [PubMed] [Google Scholar]
- Anderson HC, Stechschulte DJ, Jr., Collins DE, et al. Matrix vesicle biogenesis in vitro by rachitic and normal rat chondrocytes. American Journal of Pathology. 1990;136:391–8. [PMC free article] [PubMed] [Google Scholar]
- Aronow WS, Ahn C, Kronzon I, Goldman ME. Association of coronary risk factors and use of statins with progression of mild valvular aortic stenosis in older persons. The American Journal of Cardiology. 2001;88(6):693–5. doi: 10.1016/s0002-9149(01)01821-5. [DOI] [PubMed] [Google Scholar]
- Beheshti M, Saboury B, Mehta NN, et al. Detection and global quantification of cardiovascular molecular calcification by fluoro18-fluoride positron emission tomography/computed tomography – a novel concept. Hellenic Journal of Nuclear Medicine. 2011;14:114–20. [PubMed] [Google Scholar]
- Bellamy MF, Pellikka PA, Klarich KW, Tajik AJ, Enriquez-Sarano M. Association of cholesterol levels, hydroxymethylglutaryl coenzyme-A reductase inhibitor treatment, and progression of aortic stenosis in the community. Journal of the American College of Cardiology. 2002;40(10):1723–30. doi: 10.1016/s0735-1097(02)02496-8. [DOI] [PubMed] [Google Scholar]
- Bobryshev YV, Killingsworth MC, Huynh TG, Lord RS, Grabs AJ, Valenzuela SM. Are calcifying matrix vesicles in atherosclerotic lesions of cellular origin? Basic Research in Cardiology. 2007;102:133–43. doi: 10.1007/s00395-006-0637-9. [DOI] [PubMed] [Google Scholar]
- Bostrom K. Proinflammatory vascular calcification. Circulation Research. 2005;96:1219–20. doi: 10.1161/01.RES.0000172407.20974.e5. [DOI] [PubMed] [Google Scholar]
- Boyle JJ, Bowyer DE, Weissberg PL, Bennett MR. Human blood-derived macrophages induce apoptosis in human plaque-derived vascular smooth muscle cells by Fas-ligand/Fas interactions. Arteriosclerosis, Thrombosis, and Vascular Biology. 2001;21:1402–7. doi: 10.1161/hq0901.094279. [DOI] [PubMed] [Google Scholar]
- Boyle JJ, Weissberg PL, Bennett MR. Human macrophage-induced vascular smooth muscle cell apoptosis requires NO enhancement of Fas/Fas-L interactions. Arteriosclerosis, Thrombosis, and Vascular Biology. 2002;22:1624–30. doi: 10.1161/01.atv.0000033517.48444.1a. [DOI] [PubMed] [Google Scholar]
- Butcher JT, Nerem RM. Valvular endothelial cells regulate the phenotype of interstitial cells in co-culture: effects of steady shear stress. Tissue Engineering. 2006;12:905–15. doi: 10.1089/ten.2006.12.905. [DOI] [PubMed] [Google Scholar]
- Chen NX, O’Neill KD, Chen X, Moe SM. Annexin-mediated matrix vesicle calcification in vascular smooth muscle cells. Journal of Bone and Mineral Research. 2008;23:1798–805. doi: 10.1359/JBMR.080604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsten MF, Bennaghmouch A. Optical characterization of arterial apoptosis. Methods in Molecular Biology. 2011;680:117–129. doi: 10.1007/978-1-60761-901-7_8. [DOI] [PubMed] [Google Scholar]
- Cowell SJ, Newby DE, Prescott RJ, et al. A randomized trial of intensive lipid-lowering therapy in calcific aortic stenosis. New England Journal of Medicine. 2005;352:2389–97. doi: 10.1056/NEJMoa043876. [DOI] [PubMed] [Google Scholar]
- Cybulsky MI, Gimbrone MA., Jr Endothelial expression of a mononuclear leukocyte adhesion molecule during atherogenesis. Science. 1991;251:788–91. doi: 10.1126/science.1990440. [DOI] [PubMed] [Google Scholar]
- Demer LL, Tintut Y. Vascular calcification: pathobiology of a multifaceted disease. Circulation. 2008;117:2938–48. doi: 10.1161/CIRCULATIONAHA.107.743161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dweck MR, Jones C, Joshi NV, et al. Assessment of valvular calcification and inflammation by positron emission tomography in patients with aortic stenosis. Circulation. 2012;125:76–86. doi: 10.1161/CIRCULATIONAHA.111.051052. [DOI] [PubMed] [Google Scholar]
- Ehara S, Kobayashi Y, Yoshiyama M, et al. Spotty calcification typifies the culprit plaque in patients with acute myocardial infarction: an intravascular ultrasound study. Circulation. 2004;110:3424–9. doi: 10.1161/01.CIR.0000148131.41425.E9. [DOI] [PubMed] [Google Scholar]
- El-Abbadi M, Giachelli CM. Mechanisms of vascular calcification. Advances in Chronic Kidney Disease. 2007;14:54–66. doi: 10.1053/j.ackd.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Folco EJ, Sheikine Y, Rocha VZ, et al. Hypoxia but not inflammation augments glucose uptake in human macrophages: Implications for imaging atherosclerosis with 18fluorine-labeled 2-deoxy-d-glucose positron emission tomography. Journal of the American College of Cardiology. 2011;58:603–14. doi: 10.1016/j.jacc.2011.03.044. [DOI] [PubMed] [Google Scholar]
- Ghaisas NK, Foley JB, O’Briain DS, Crean P, Kelleher D, Walsh M. Adhesion molecules in nonrheumatic aortic valve disease: endothelial expression, serum levels and effects of valve replacement. Journal of the American College of Cardiology. 2000;36:2257–62. doi: 10.1016/s0735-1097(00)00998-0. [DOI] [PubMed] [Google Scholar]
- Gryglewski RJ, Botting RM, Vane JR. Mediators produced by the endothelial cell. Hypertension. 1988;12:530–48. doi: 10.1161/01.hyp.12.6.530. [DOI] [PubMed] [Google Scholar]
- Hjortnaes J, Butcher J, Figueiredo JL, et al. Arterial and aortic valve calcification inversely correlates with osteoporotic bone remodelling: a role for inflammation. European Heart Journal. 2010;31:1975–84. doi: 10.1093/eurheartj/ehq237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjortnaes J, Gottlieb D, Figueiredo JL, et al. Intravital molecular imaging of small-diameter tissue-engineered vascular grafts in mice: a feasibility study. Tissue Engineering Part C: Methods. 2010;16:597–607. doi: 10.1089/ten.tec.2009.0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyafil F, Messika-Zeitoun D, Burg S, et al. Detection of 18fluoride sodium accumulation by positron emission tomography in calcified stenotic aortic valves. The American Journal of Cardiology. 2012;109:1194–6. doi: 10.1016/j.amjcard.2011.11.060. [DOI] [PubMed] [Google Scholar]
- Jaffer FA, Nahrendorf M, Sosnovik D, Kelly KA, Aikawa E, Weissleder R. Cellular imaging of inflammation in atherosclerosis using magnetofluorescent nanomaterials. Molecular Imaging. 2006;5:85–92. [PubMed] [Google Scholar]
- Johnson RC, Leopold JA, Loscalzo J. Vascular calcification: pathobiological mechanisms and clinical implications. Circulation Research. 2006;99:1044–59. doi: 10.1161/01.RES.0000249379.55535.21. [DOI] [PubMed] [Google Scholar]
- Kapustin AN, Davies JD, Reynolds JL, et al. Calcium regulates key components of vascular smooth muscle cell-derived matrix vesicles to enhance mineralization. Circulation Research. 2011;109:e1–12. doi: 10.1161/CIRCRESAHA.110.238808. [DOI] [PubMed] [Google Scholar]
- Karnik SK, Brooke BS, Bayes-Genis A, et al. A critical role for elastin signaling in vascular morphogenesis and disease. Development. 2003;130:411–23. doi: 10.1242/dev.00223. [DOI] [PubMed] [Google Scholar]
- Kaufmann BA, Carr CL, Belcik JT, et al. Molecular imaging of the initial inflammatory response in atherosclerosis: implications for early detection of disease. Arteriosclerosis, Thrombosis, and Vascular Biology. 2010;30:54–9. doi: 10.1161/ATVBAHA.109.196386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KM. Calcification of matrix vesicles in human aortic valve and aortic media. Federal Proceedings. 1976;35:156–62. [PubMed] [Google Scholar]
- Kozloff KM, Weissleder R, Mahmood U. Noninvasive optical detection of bone mineral. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2007;22:1208–16. doi: 10.1359/jbmr.070504. [DOI] [PubMed] [Google Scholar]
- Li R, Mittelstein D, Lee J, et al. A dynamic model of calcific nodule destabilization in response to monocyte- and oxidized lipid-induced matrix metalloproteinases. American Journal of Physiology – Cell Physiology. 2012;302:C658–65. doi: 10.1152/ajpcell.00313.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd-Jones D, Adams R, Carnethon M, et al. Heart disease and stroke statistics – 2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2009;119:480–6. doi: 10.1161/CIRCULATIONAHA.108.191259. [DOI] [PubMed] [Google Scholar]
- Marui N, Offermann MK, Swerlick R, et al. Vascular cell adhesion molecule-1 (VCAM-1) gene transcription and expression are regulated through an antioxidant-sensitive mechanism in human vascular endothelial cells. Journal of Clinical Investigation. 1993;92:1866–74. doi: 10.1172/JCI116778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohler ER., 3rd Mechanisms of aortic valve calcification. The American Journal of Cardiology. 2004;94:1396–402. A1396. doi: 10.1016/j.amjcard.2004.08.013. [DOI] [PubMed] [Google Scholar]
- Moses JW, Carlier S, Moussa I. Lesion preparation prior to stenting. Reviews in Cardiovascular Medicine. 2004;5(Suppl. 2):S16–21. [PubMed] [Google Scholar]
- Muller AM, Cronen C, Kupferwasser LI, Oelert H, Muller KM, Kirkpatrick CJ. Expression of endothelial cell adhesion molecules on heart valves: up-regulation in degeneration as well as acute endocarditis. The Journal of Pathology. 2000;191:54–60. doi: 10.1002/(SICI)1096-9896(200005)191:1<54::AID-PATH568>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Nadra I, Boccaccini AR, Philippidis P, et al. Effect of particle size on hydroxyapatite crystal-induced tumor necrosis factor alpha secretion by macrophages. Atherosclerosis. 2008;196:98–105. doi: 10.1016/j.atherosclerosis.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Nadra I, Mason JC, Philippidis P, et al. Proinflammatory activation of macrophages by basic calcium phosphate crystals via protein kinase C and MAP kinase pathways: a vicious cycle of inflammation and arterial calcification? Circulation Research. 2005;96:1248–56. doi: 10.1161/01.RES.0000171451.88616.c2. [DOI] [PubMed] [Google Scholar]
- Nahrendorf M, Jaffer FA, Kelly KA, et al. Noninvasive vascular cell adhesion molecule-1 imaging identifies inflammatory activation of cells in atherosclerosis. Circulation. 2006;114:1504–11. doi: 10.1161/CIRCULATIONAHA.106.646380. [DOI] [PubMed] [Google Scholar]
- Nakashima Y, Raines EW, Plump AS, Breslow JL, Ross R. Upregulation of VCAM-1 and ICAM-1 at atherosclerosis-prone sites on the endothelium in the ApoE-deficient mouse. Arteriosclerosis, Thrombosis, and Vascular Biology. 1998;18:842–51. doi: 10.1161/01.atv.18.5.842. [DOI] [PubMed] [Google Scholar]
- New SE, Aikawa E. Molecular imaging insights into early inflammatory stages of arterial and aortic valve calcification. Circulation Research. 2011;108:1381–91. doi: 10.1161/CIRCRESAHA.110.234146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novaro GM, Tiong IY, Pearce GL, Lauer MS, Sprecher DL, Griffin BP. Effect of hydroxymethylglutaryl coenzyme a reductase inhibitors on the progression of calcific aortic stenosis. Circulation. 2001;104:2205–9. doi: 10.1161/hc4301.098249. [DOI] [PubMed] [Google Scholar]
- Otto CM. Calcific aortic stenosis – time to look more closely at the valve. New England Journal of Medicine. 2008;359:1395–8. doi: 10.1056/NEJMe0807001. [DOI] [PubMed] [Google Scholar]
- Otto CM, Kuusisto J, Reichenbach DD, Gown AM, O’Brien KD. Characterization of the early lesion of ‘degenerative’ valvular aortic stenosis. Histological and immunohistochemical studies. Circulation. 1994;90:844–53. doi: 10.1161/01.cir.90.2.844. [DOI] [PubMed] [Google Scholar]
- Parhami F, Basseri B, Hwang J, Tintut Y, Demer LL. High-density lipoprotein regulates calcification of vascular cells. Circulation Research. 2002;91:570–6. doi: 10.1161/01.res.0000036607.05037.da. [DOI] [PubMed] [Google Scholar]
- Peacock JD, Levay AK, Gillaspie DB, Tao G, Lincoln J. Reduced sox9 function promotes heart valve calcification phenotypes in vivo. Circulation Research. 2010;106:712–9. doi: 10.1161/CIRCRESAHA.109.213702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perng JK, Lee S, Kundu K, et al. Ultrasound imaging of oxidative stress in vivo with chemically-generated gas microbubbles. Annals of Biomedical Engineering. 2012 doi: 10.1007/s10439-012-0573-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proudfoot D, Skepper JN, Hegyi L, Bennett MR, Shanahan CM, Weissberg PL. Apoptosis regulates human vascular calcification in vitro: evidence for initiation of vascular calcification by apoptotic bodies. Circulation Research. 2000;87:1055–62. doi: 10.1161/01.res.87.11.1055. [DOI] [PubMed] [Google Scholar]
- Radcliff K, Tang TB, Lim J, Zhang Z, Abedin M, Demer LL, et al. Insulin-like growth factor-I regulates proliferation and osteoblastic differentiation of calcifying vascular cells via extracellular signal-regulated protein kinase and phosphatidylinositol 3-kinase pathways. Circulation Research. 2005;96:398–400. doi: 10.1161/01.RES.0000157671.47477.71. [DOI] [PubMed] [Google Scholar]
- Rajamannan NM. Mechanisms of aortic valve calcification: the LDL-density-radius theory: a translation from cell signaling to physiology. American Journal of Physiology – Heart and Circulatory Physiology. 2010;298:H5–15. doi: 10.1152/ajpheart.00824.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajamannan NM, Bonow RO, Rahimtoola SH. Calcific aortic stenosis: an update. Nature Clinical Practice Cardiovascular Medicine. 2007;4:254–62. doi: 10.1038/ncpcardio0827. [DOI] [PubMed] [Google Scholar]
- Rajamannan NM, Evans FJ, Aikawa E, et al. Calcific aortic valve disease: not simply a degenerative process: a review and agenda for research from the National Heart and Lung and Blood Institute Aortic Stenosis Working Group. Executive summary: Calcific aortic valve disease – 2011 update. Circulation. 2011;124:1783–91. doi: 10.1161/CIRCULATIONAHA.110.006767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajamannan NM, Sangiorgi G, Springett M, et al. Experimental hypercholesterolemia induces apoptosis in the aortic valve. The Journal of Heart Valve Disease. 2001;10:371–4. [PubMed] [Google Scholar]
- Rajamannan NM, Subramaniam M, Springett M, Sebo TC, Niekrasz M, McConnell JP, et al. Atorvastatin inhibits hypercholesterolemia-induced cellular proliferation and bone matrix production in the rabbit aortic valve. Circulation. 2002;105:2660–5. doi: 10.1161/01.cir.0000017435.87463.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattazzi M, Bennett BJ, Bea F, et al. Calcification of advanced atherosclerotic lesions in the innominate arteries of ApoE-deficient mice: potential role of chondrocyte-like cells. Arteriosclerosis Thrombosis and Vascular Biology. 2005;25:1420–5. doi: 10.1161/01.ATV.0000166600.58468.1b. [DOI] [PubMed] [Google Scholar]
- Reynolds JL, Joannides AJ, Skepper JN, et al. Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: a potential mechanism for accelerated vascular calcification in ESRD. Journal of the American Society of Nephrology. 2004;15:2857–67. doi: 10.1097/01.ASN.0000141960.01035.28. [DOI] [PubMed] [Google Scholar]
- Rossebo AB, Pedersen TR, Boman K, et al. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. New England Journal of Medicine. 2008;359:1343–56. doi: 10.1056/NEJMoa0804602. [DOI] [PubMed] [Google Scholar]
- Saulnier JM, Hauck M, Fulop T, Jr., Wallach JM. Human aortic elastin from normal individuals and atherosclerotic patients: lipid and cation contents; susceptibility to elastolysis. Clinica Chimica Acta. 1991;200:129–36. doi: 10.1016/0009-8981(91)90084-p. [DOI] [PubMed] [Google Scholar]
- Shanahan CM. Inflammation ushers in calcification: a cycle of damage and protection? Circulation. 2007;116:2782–5. doi: 10.1161/CIRCULATIONAHA.107.749655. [DOI] [PubMed] [Google Scholar]
- Shavelle DM, Katz R, Takasu J, Lima JA, Jenny NS, Budoff MJ, et al. Soluble intercellular adhesion molecule-1 (sICAM-1) and aortic valve calcification in the multi-ethnic study of atherosclerosis (MESA) The Journal of Heart Valve Disease. 2008;17:388–95. [PubMed] [Google Scholar]
- Simionescu A, Simionescu DT, Vyavahare NR. Osteogenic responses in fibroblasts activated by elastin degradation products and transforming growth factor-beta1: role of myofibroblasts in vascular calcification. American Journal of Pathology. 2007;171:116–23. doi: 10.2353/ajpath.2007.060930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorescu GP, Song H, Tressel SL, et al. Bone morphogenic protein 4 produced in endothelial cells by oscillatory shear stress induces monocyte adhesion by stimulating reactive oxygen species production from a nox1-based NADPH oxidase. Circulation Research. 2004;95:773–9. doi: 10.1161/01.RES.0000145728.22878.45. [DOI] [PubMed] [Google Scholar]
- Steitz SA, Speer MY, Curinga G, et al. Smooth muscle cell phenotypic transition associated with calcification: upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circulation Research. 2001;89:1147–54. doi: 10.1161/hh2401.101070. [DOI] [PubMed] [Google Scholar]
- Stewart BF, Siscovick D, Lind BK, et al. Clinical factors associated with calcific aortic valve disease. Cardiovascular Health Study. Journal of the American College of Cardiology. 1997;29:630–4. doi: 10.1016/s0735-1097(96)00563-3. [DOI] [PubMed] [Google Scholar]
- Sucosky P, Balachandran K, Elhammali A, Jo H, Yoganathan AP. Altered shear stress stimulates upregulation of endothelial VCAM-1 and ICAM-1 in a BMP-4- and TGF-beta1-dependent pathway. Arteriosclerosis, Thrombosis, and Vascular Biology. 2009;29:254–60. doi: 10.1161/ATVBAHA.108.176347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tintut Y, Patel J, Territo M, Saini T, Parhami F, Demer LL. Monocyte/macrophage regulation of vascular calcification in vitro. Circulation. 2002;105:650–5. doi: 10.1161/hc0502.102969. [DOI] [PubMed] [Google Scholar]
- Towler DA. Oxidation, inflammation, and aortic valve calcification peroxide paves an osteogenic path. Journal of the American College of Cardiology. 2008;52:851–4. doi: 10.1016/j.jacc.2008.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towler DA, Demer LL. Thematic series on the pathobiology of vascular calcification: an introduction. Circulation Research. 2011;108:1378–80. doi: 10.1161/CIRCRESAHA.110.234419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Tilborg GA, Vucic E, Strijkers GJ, et al. Annexin A5-functionalized bimodal nanoparticles for MRI and fluorescence imaging of atherosclerotic plaques. Bioconjugate Chemistry. 2010;21:1794–803. doi: 10.1021/bc100091q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vengrenyuk Y, Carlier S, Xanthos S, et al. A hypothesis for vulnerable plaque rupture due to stress-induced debonding around cellular microcalcifications in thin fibrous caps. Proceedings of the National Academy of Sciences, USA. 2006;103:14678–83. doi: 10.1073/pnas.0606310103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virmani R, Burke AP, Farb A, Kolodgie FD. Pathology of the vulnerable plaque. Journal of the American College of Cardiology. 2006;47(8 Suppl.):C13–8. doi: 10.1016/j.jacc.2005.10.065. [DOI] [PubMed] [Google Scholar]
- Watson KE, Bostrom K, Ravindranath R, Lam T, Norton B, Demer TGF-beta 1 and 25-hydroxycholesterol stimulate osteoblast-like vascular cells to calcify. Journal of Clinical Investigation. 1994;93:2106–13. doi: 10.1172/JCI117205. [DOI] [PMC free article] [PubMed] [Google Scholar]