

Abstract
A role for annexin A7 (A7) is postulated in the obligatory fusion between lamellar bodies and the plasma membrane during surfactant secretion in alveolar type II cells. This study investigated if surfactant secretagogues increase cell surface A7, which could support A7 insertion into plasma membrane as annexin proteins reportedly lack membrane penetration ability. In vivo trafficking of A7 to cell surface was determined by immuno-staining after non-permeabilizing fixation of alveolar type II cells. Stimulation with various secretagogues increased protein kinase-dependent staining for A7 and ABCA3 in comparison to control cells. Biotin-labeling of surface proteins showed ~4% of total A7 in control cells, which increased ~3 – 4 folds in stimulated type II cells. Increased cell surface A7 was also observed by protein cross-linking studies showing ~70 kDa A7-adduct in the membranes but not in the cytosol fraction of PMA- or A23187-stimulated cells. In vitro phosphorylation increased the Ca2+-dependent binding of recombinant A7 to lung plasma membranes; and subsequent cross-linking showed increased levels of ~70 kDa A7-adduct. PMA-stimulation of type II cells increased A7 trafficking to lipid rafts suggesting that the latter are involved in A7 trafficking to the cell surface. However, in vitro membrane insertion of recombinant A7 and its tryptophan mutants as determined by fluorescence quenching with doxylPC suggested only shallow membrane insertion by A7. Together, our studies support in vivo association between surfactant secretion and cell surface A7 occurring by insertion into plasma membrane and by fusion of A7 containing lamellar bodies.
Keywords: Lung surfactant secretion, protein cross-linking, fluorescence quenching, biotin-labeling of proteins, immuno-fluorescence
INTRODUCTION
Lung surfactant is vital for lowering surface tension at the alveolar surface to prevent lung collapse during end-expiration. The principal surface-active component phosphatidylcholine (PC) together with surfactant proteins B and C, required for rapid recruitment and surface-spreading of lipids, lowers the surface tension (reviewed in [1]). All these components are synthesized in alveolar type II cells and stored in the lamellar bodies for secretion into air space, which occurs following lamellar body fusion with the plasma membrane. Although several agents increase surfactant secretion through Ca2+ and protein kinase-regulated mechanisms (reviewed in [2–4]), the regulatory processes for membrane fusion during surfactant secretion remain relatively poorly investigated.
Several proteins are suggested to function during membrane fusion between secretion vesicle and the plasma membrane. The classical and most-investigated system involves proteins of the SNARE-complex postulated to function in vesicle secretion during synaptic transmission [5, 6], a relatively rapid event in contrast with surfactant secretion [7]. Some of these proteins are also present in specialized cholesterol and gangliosides-rich domains called lipid rafts in plasma membrane of several cell types including type II cells [8, 9]. The lipid rafts are enriched in flotillins [10] that are palmitoylated and myristoylated plasma membrane-anchored proteins and are markers for lipid rafts [8, 10, 11]. The lipid rafts are postulated to function in the endocytosis and exocytosis functions of cells [8].
Several other proteins also function in membrane fusion during exocytosis [12, 13]. The membrane aggregation and membrane fusion properties of annexin A7 (A7) and other annexin proteins play a role in membrane fusion during exocytosis (reviewed in [14, 15]). In alveolar type II cells, A7 and A2 proteins are involved in membrane fusion for surfactant secretion [16–19]. We have previously suggested a role for A7 in membrane fusion since it promotes surfactant secretion in permeabilized type II cells [18] and shows in vitro interactions with lipid vesicles [20] and cell membranes including lamellar bodies and plasma membrane [21]. Stimulation of alveolar type II cells with several secretagogues of lung surfactant increases membrane-association of A7 [22]. The target membranes for A7 trafficking in stimulated type II cells include lamellar bodies and plasma membranes, as demonstrated by in vitro binding [21] and by immuno-fluorescence co-localization with ABCA3 [22] and SNAP23 [23]. Such trafficking is regulated by protein kinase-dependent phosphorylation of A7 [22, 23]. Together, these studies suggest a role for A7 in membrane fusion during surfactant secretion.
Although trafficking to lamellar bodies and SNAP23 domains suggests a role for A7 in membrane fusion, further studies are required to define if A7 could function as part of the fusion complex. Atomic force and electron microscopy studies of lipid-associated annexin A5 or A2 proteins suggesting a planar nature for annexin proteins [24–27] led to the conclusion that these proteins cannot insert into membranes and, therefore, can have only supportive role(s) in membrane fusion [28]. However, protein interactions with the specific membrane lipids, which can regulate protein molecule organization of A7 [20, 29, 30] or annexin A1 [31], could affect protein insertion into membranes. Alternately, association with proteins in biological membranes or post-translational modifications of A7 could affect the planar conformation and thereby facilitate membrane insertion. Therefore, we investigated if cell stimulation with surfactant secretagogues would increase A7 at the cell surface, which could be suggestive of its ability to penetrate plasma membrane. Our studies employing various techniques suggest that surfactant secretagogues increase A7 trafficking to the cell surface through protein kinase-dependent mechanisms. Phorbol myristate acetate (PMA) also increased A7 trafficking to lipid rafts in plasma membrane suggesting their involvement in A7 appearance at the cell surface. However, in agreement with others, in vitro biophysical studies suggested only shallow membrane insertion by recombinant A7 proteins. Thus, we speculate that stimulation with surfactant secretagogues increases A7 phosphorylation to facilitate its association with the plasma membrane and lamellar body proteins and appearance at the cell surface. The latter is possibly mediated through A7 insertion into plasma membrane or delivery during lamellar body fusion.
MATERIALS AND METHODS
Material
All cell culture supplies were obtained from Beckton Dickinson (Sparks, MD) or from Cell Culture and Hybridoma facility at the Stony Brook University (Stony Brook, NY). All fine chemicals and reagents were obtained from Sigma-Aldrich (St. Louis, MO). Lipids for vesicle preparations were obtained from Avanti Polar Lipids (Alabaster, AL) and were used without further purification. The membrane-permeable homobifunctional ethylene glycolbis(succinimidylsuccinate) (EGS) and impermeable sulfo-EGS (S-EGS) protein cross-linking reagents were used according to manufacturer’s instructions (Pierce). Purified brain PKC was obtained from Calbiochem (San Diego, CA). Actin polyclonal antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA) and ABCA3 monoclonal antibodies were from Covance (Gaithersburg, MD). Alexa-Fluor conjugated secondary antibodies were obtained from Invitrogen (Grand Island, NY). Bacterially expressed A7 full length protein was used for obtaining polyclonal antibodies (rabbit) through commercial services and have been described previously [22].
Isolation of Alveolar Type II Cells
The use of rats for alveolar type II cells isolation [32] was according to protocol approved by the Institutional Animal Care and Use Committee. Briefly, male Sprague-Dawley rats (180–200g) were anesthetized with Nembutal (50 mg/kg, ip), tracheostomized and ventilated with a rodent ventilator. The animal was sacrificed by exsanguinations without cessation of ventilation. Lungs were perfused through pulmonary artery until visibly clear of residual blood, excised and incubated at 37°C following intra-tracheal instillation of porcine elastase (3units/ml). The lung lobes were separated and chopped into 1 mm3 fragments on a tissue chopper. The lung fragments were vigorously shaken in buffer containing Ca2+, fetal bovine serum (FBS) and DNase I to release free cells that were sequentially filtered (×3) through nylon cloth of decreasing mesh-size. The cell suspension was enriched with type II cells by “panning” on rat IgG-coated bacteriological culture dishes to remove macrophages. Thus enriched cells (>85% type II cells by quinacrine staining [33]) were cultured for 20–22 h in minimum essential medium (MEM) containing 10% FBS, during which period the cells adherent to tissue culture plastic were routinely >90% alveolar type II cells.
Immuno-Fluorescence Studies
The cell surface and intracellular localization of target proteins was performed by immuno-fluorescence studies in type II cells fixed without or with permeabilization. Routinely, cells were plated and cultured for 20–22 h on poly-lysine coated glass cover slips. The adherent cells were washed and treated with agents as indicated. Thereafter, the cells were washed (3× 5 min) in phosphate-buffered saline (PBS) at room temperature and fixed as indicated. For non-permeabilization protocol, the cells were fixed for 10 min in 2% paraformaldehyde (PFA) at room temperature, and washed (3× 5 min) with PBS at 4°C. The non-specific sites were blocked by equilibration for 30 min at 4°C with detergent-free blocking buffer containing 10% donkey/goat serum and 1% BSA. For permeabilizing protocol [23], the cells fixed in 4% PFA for 10 min at room temperature, washed (×3) and stored in PBS at 4°C until further processing. Before staining, the cells were simultaneously blocked and permeabilized for 30 min in blocking buffer supplemented with 0.1% Triton X-100 and 0.05% Tween 20 [23]. Following fixation and blocking, the cells were washed once in PBS and then sequentially stained with primary (1:250) and secondary (1:500) antibodies, 1 h each, for the target proteins using polyclonal anti-rat A7 antibody and monoclonal anti-ABCA3 antibody. The respective secondary antibodies were anti rabbit Alexa568 anti mouse Alexa468 antibody (1:500).
Biotin labeling of Cell Surface Proteins
Adherent 20–22h cultured type II cells were washed, equilibrated for 30 min in fresh serum-free MEM and incubated for 2 h without or with indicated agents. The cell surface proteins were labeled using the membrane-impermeable biotin labeling kit according to manufacturer’s instructions (Pierce, Rockford, IL) with slight modifications. The cells were washed (×3) with PBS containing 0.1mM CaCl2 and 1mM MgCl2. All subsequent steps were performed at 4°C unless indicated. The cells were incubated for 2 h with sulfo-NHS-SS-biotin (1.25 mg/ml) in 50 mM triethanolamine buffer (pH 7.5) containing 2 mM MgCl2 and 150 mM NaCl. The cells were washed (×3) with PBS and the reaction stopped by washing and equilibration for 30 min on a rocker in 100 mM glycine in PBS. The cells were washed (×2) with PBS and harvested by scarping in lysis buffer (50 mM Tris buffer, pH 7.5, containing 150 mM NaCl, 5 mM EGTA, 1% Triton X-100) containing 1 mM PMSF and leupeptin, pepstatin and aprotinin (1 μg/ml each). The cell lysate was disrupted with a probe sonicator (10 sec ×4, at 20% of maximum output) and incubated for 1 h on ice with frequent mixing for further lysis. Equal aliquots of cell lysates were centrifuged for 10 min at 16,100g. The biotin-labeled proteins in the supernatant were bound to NeutrAvidin™ beads (100 μl) that were pre-washed (×4) and equilibrated in PBS. Equal volume of cell lysate proteins (~500 μg) and beads were equilibrated overnight at 4°C with end-over-end mixing. The biotin-labeled proteins bound to the beads were centrifuged, separated from the supernatant (the residual fraction), washed (×6) with PBS containing protease inhibitors and equilibrated for 2 h at 4°C in the elution buffer (62.5 mM Tris buffer, pH 7.5 containing 50 mM DTT and 10% glycerol). Equivalent proportions of the cell lysate, the residual fraction and the eluted biotin-labeled proteins were analyzed for A7 and actin by western analysis. The membranes were stained for 5 min in PonceauS (0.5% in 1% acetic acid) to assess the levels of transferred proteins.
In order to distinguish cell surface adherent A7 (possibly following secretion and reattachment), type II cells treated without or with PMA as indicated were incubated on ice for 30 min in 100 mM bicarbonate buffer (pH 11.5) as described previously [10]. The bicarbonate soluble proteins in the supernatant were separated following centrifugation at 16,100g for 60 min. The pellet was mixed with the cells on the plate that were harvested by scraping in the lysis buffer (see above). The relative levels of A7 in the cells and bicarbonate extract were analyzed by western blotting and densitometry.
Cell surface protein cross-linking
Following incubation for 2 h without (control) or with 100 nM PMA or 250nM A23187, the cells were equilibrated for 2 h on ice in the absence or presence of 2 mM S-EGS. The reaction was stopped by equilibration for 30 min on ice with TEE buffer (50 mM Tris-HCl, pH 7.4 containing 0.5 mM EGTA and 0.5 mM EDTA). The cells were washed and scraped either in TEE buffer without (for cell fractionation) or with 0.1% Triton X-100 (for cell lysis). Equal amounts of proteins in the cell lysate or in the membrane and the cytosol fractions were analyzed by western blotting for detection of actin- or A7-adducts.
A7 binding and cross-linking with lung plasma membrane proteins
Rat lungs visibly cleared of residual blood (see above) and dissected free of airways and other tissues were used for isolation of plasma membranes as described [34]. The Percoll® gradient purification was omitted because it reduces the yields. Briefly, the lung lobes were homogenized in ice-cold homogenization buffer (10 mM phosphate buffer, pH 7.4, containing 0.32 M sucrose, 1 mM MgCl2, 30 mM NaCl, 1 mM PMSF, 0.02% NaN3 and 10 μg/ml DNase) using a tissue disrupter (Model 5000, Omni International, Westbury, CT) and Potter-Elvejahm type homogenizer. The homogenate was filtered through gauze and centrifuged for 1 h at 95,000g through a discontinuous gradient of 0.5, 0.7, 0.9 and 1.2 M sucrose in the homogenizing buffer. The interface between 0.9 and 1.2 M sucrose was diluted to 0.2 M sucrose and centrifuged for 30 min at 95, 000g to isolate the plasma membrane enriched pellet, which was used for binding and cross-linking studies.
The binding of recombinant A7 to plasma membrane was performed as described for A7 binding to GST-SNAP23 beads in the binding buffer [23], but omitting the NP-40 detergent. The plasma membranes were washed and suspended in the binding buffer (40 mM Hepes, pH 7.0, 100 mM KCl, 1 mM EGTA, 2 mM MgCl2, 1 mM Na3VO4, 1 mM PMSF, and 1 μg/ml each of leupeptin, aprotinin and pepstatin). An aliquot of plasma membrane fraction (100 μg protein) was equilibrated with A7 (1 μg), phosphorylated previously for 2 h in the absence or presence of PKC [22]. The binding was conducted at 4°C for overnight in the absence or presence of 1 μM Ca2+ buffered with EGTA by end-over-end mixing [35]. Subsequently, the plasma membranes were centrifuged for 10 min at 16,100g at 4°C. The pellet was washed (×2) with the binding buffer containing respective Ca2+ concentrations. The membranes were further washed (×3), suspended in PBS and incubated for 30 min at room temperature with 10 mM EGS for cross-linking of proteins. The reaction was stopped with ice-cold TEE buffer as described above and proteins were analyzed for A7 by western blotting.
A7 in lamellar bodies
Since secretagogues increase A7 trafficking to lamellar bodies, we investigated the luminal and membrane-associated A7 in these organelles. The membrane-associated A7 was further distinguished into the adherent and integral components. Lamellar bodies were isolated from lung homogenate by upwards flotation on a discontinuous sucrose gradient [36], disrupted in hypotonic sucrose (0.05 M) and the lamellar body membrane fraction isolated by centrifugation (100,000g for 60 min) through 0.5 M sucrose [37]. The membranes were incubated for 30 min on ice with 100 mM NaHCO3 buffer (pH 11.5) and centrifuged for 60 min at 16,100g to separate the bicarbonate extract and the residual membranes. Equal aliquots of lamellar bodies, bicarbonate extract and the residual membranes were analyzed for A7 content by western blotting.
Lipid Rafts Isolation
Isolated type II cells were stimulated without or with 100 nM PMA and harvested in 1% Triton X-100-containing buffer as previously described for PC12 cells [8] with some modifications. Briefly, the cells were washed with ice-cold PBS for further processing at 4°C. The cells were washed once with 20 mM Hepes buffer, pH 7.0 containing 255 mM sucrose, 1 mM EDTA, incubated for 20 min and harvested in the same buffer supplemented with 1% Triton X-100 and the protease inhibitors (leupeptin, pepstatin and aprotinin, each 1 μg/ml, and 1 mM PMSF). The cells were further disrupted by passage (8 – 10×) through 27½G needle. Thus disrupted cells (0.5 ml) were mixed with appropriate volume of 80% sucrose in Hepes/EDTA buffer containing 1% Triton X-100 to yield 40% sucrose. The final suspension (1 ml) was placed at the bottom of the tube and overlaid with 1 ml each of 30% and 15% and 1.5 ml of 5% sucrose in the same buffer. After centrifugation for 18 h at 240,000 g, the pellet and 9 gradient fractions (0.5 ml each) were collected, and 20% of each fraction was analyzed for the target proteins by western blot analysis.
Bacterial expression of recombinant proteins
All recombinant proteins were expressed in E. coli and purified as described [20]. The single tryptophan residue (W239) in the second homologous repeat of the COOH-terminus of A7 was substituted with phenylalanine to obtain tryptophan-free mutant, which formed template for other DNA constructs for the expression of mutants with selected phenylalanine residue (176, 339 or 443) in repeats I, III and IV substituted with tryptophan. Thus, each mutant contained only one tryptophan residue and acted as reporter for molecular changes in the selected repeat during interaction with membranes.
Protein Fluorescence Measurements
The tryptophan fluorescence for the purified recombinant proteins was measured in spectrofluorometer as previously described [20]. Briefly, the fluorescence of ~0.5 to 1 μM protein solution (25 – 50 μg/ml) in 50mM Tris-acetate 1mM EGTA (TAE) buffer (pH 8.3) was measured (Exc. = 292 nm, Em = 310 to 400 nm, slit = 4 nm each). The solution was stirred at a low speed to ensure continuous mixing without air bubble formation. All other additions were at indicated final concentrations. The fluorescence spectra were corrected for the volume and emission changes due to buffer or other additions. All spectra were smoothed by 5 point averaging and expressed as 5th polynomial fit without or with normalization of emission intensities.
Preparation of Lipid Vesicles
The lipid mixture with indicated composition (mol:mol) were prepared using appropriate proportions of stock solutions in chloroform:methanol (20:1). The lipid mixture was dried under a stream of N2 and suspended in 50 mM Tris-Acetate-1 mM EGTA (TAE) buffer, pH 8.3, by vigorous mixing. The lipid suspension was sonicated briefly (3x10 sec) at room temperature and passed (7 – 10) through membrane filters (200 nm) in a membrane extruder. The vesicles were stored overnight at 4°C and centrifuged for 30 min at 16,100g to sediment large aggregates. The small vesicles in the supernatant were quantified for phospholipids by phosphorous analysis [38].
Lipid vesicle binding
Protein-lipid binding studies were carried out at room temperature. The binding was performed in TAE buffer in a total volume of 200 μl and contained 50 nmoles of phospholipids vesicles (PC:PE:PS:Cholesterol:PIP2, 42.5, 17.5, 25, 10, 5, mol%), 10 μg of recombinant protein and indicated Ca2+ concentration buffered with 1 mM EGTA. The mixture was equilibrated for 10 min at room temperature followed by centrifugation for 10 min at 16,100g. The pellet was washed with TAE buffer without or with indicated Ca2+. The final pellet was suspended in 45 μl of TAE buffer and used for quantification of phospholipids phosphorous and the protein.
Other Analyses
Purified recombinant proteins were quantified by protein absorbance (A280, 1mg/ml = 1 OD). The proteins in the cell lysate or cellular fractions were quantified according to Bradford [39] using protein dye binding reagent (Bio-Rad, Hercules, CA) and bovine γ-globulin as standard. The purified recombinant proteins were characterized by SDS-PAGE and stained with Coomassie Blue. The cell proteins were analyzed for target proteins by western blotting using rabbit antibodies to recombinant A7, actin or flotillin-1, and images obtained with a photoimager were analyzed by densitometry to compare relative levels of target protein(s).
The confocal images were analyzed for co-localization of two proteins using the software for the confocal microscope (Zeiss LSM510, version 4.2 Program, Carl Zeiss Inc., Chester, VA), which allows calculations of co-localizing pixels above the threshold level in each channel and calculation of the co-localization coefficients (CC). The CC is weighted for the total pixels for each fluorophore (weighted CC), which is independent of the pixel intensity variations between samples.
All statistical analyses were performed by GraphPad Prism (Version 5.0 for Windows, GraphPad Software, San Diego, CA). The weighted CCs from several fluorescence microscopy experiments were compared to determine the effects of various agents. All results (mean ± SE of indicated number of determinations) were analyzed for statistical significance by ANOVA followed by Tukey’s post-hoc test for evaluation of differences between different groups. P values < 0.05 were considered significant.
RESULTS
Immuno-fluorescence studies show A7 at cell surface
Preliminary studies showed lack of actin staining in cells fixed by non-permeabilization protocol in contrast to those fixed by permeabilization protocol (Figure 1A). Also, a comparison of A7 and ABCA3 staining showed that the staining for ABCA3 was higher in control cells fixed by permeabilization protocol (Figure 1B). As we recently reported [22], control type II cells showed low co-localization of the two proteins. PMA-stimulation for 30 min increased the co-localization of A7 with ABCA3. In comparison, non-permeabilized control cell showed lower staining for these two proteins indicating that only small amounts of these proteins were present at the cell surface. The PMA-stimulated cells, however, showed increased cell-surface staining and co-localization for the two proteins. Nevertheless, the increase was lower in comparison with PMA-stimulated permeabilization-fixed cells. Thus, the immuno-staining studies demonstrated increased cell surface levels of A7 and ABCA3 in PMA-stimulated type II cells.
Figure 1.

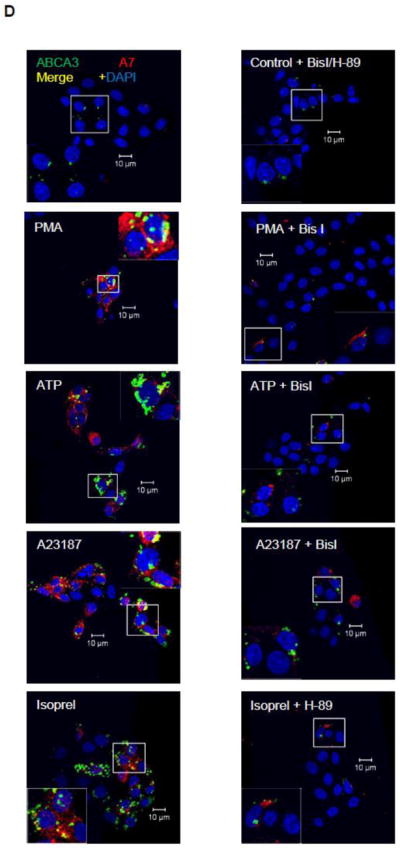

Cell surface levels of A7 and ABCA3 are increased in secretagogue-stimulated type II cells in a protein kinase-dependent manner. A. Type II cells fixed with non-permeabilization (non-perm) do not immune-stain for intracellular protein actin, while those fixed by permeabilization (perm) show actin staining. B. A comparison of A7 and ABCA3 staining in control and PMA-stimulated (30 min) type II cells fixed by permeabilzation and non-permeabilization protocols. The PMA-stimulation increased the staining for ABCA3 and A7 in type II cells, but the permeabilized cells showed higher staining. C. PMA increases A7 and ABCA3 co-localization in a time-dependent manner. Isolated type II cells were stimulated with 100 nM PMA for indicated periods and stained after fixation by non-permeabilization protocol. There was negligible co-localization of two proteins for up to 15 min. The co-localization was markedly increased after 30 min and was similar to that seen after 2 h of treatment with PMA. The lower panels show enlarged images of outlined areas in the respective upper panels. D. Protein kinase-dependence of increased cell surface staining for A7 and ABCA3. Isolated type II cells were stimulated for 2 h with 100 nM PMA, 1 mM ATP, 10 μM isoproterenol (Isoprel) or 250 nM A23187 without or with 30 min-preincubation with 100 nM BisI or H-89. The cells were fixed by non-permeabilization protocol. Representative confocal images show staining intensity under each condition. Enlarged view of outlined area in each image is shown as inset, which demonstrates inhibition of trafficking by indicated protein kinase inhibitor. E. The weighted co-localization coefficients for ABCA3 and A7 are expressed as mean ± SE, of indicated determinations in parentheses. Results were compared by ANOVA followed by post-hoc Tukey’s test. * P < 0.05 in comparison to control, ** P < 0.05 in comparison with protein kinase inhibitor (H-89 for isorproterenol and BisI for all others).
The staining and co-localization of the two proteins was also followed as a function of duration of cell stimulation with 100 nM PMA (Figure 1C). Although staining for A7 and ABCA3 increased after 5 and 15 min of treatment, a large increase in staining and co-localization of the two proteins was observed only at 30 min, which was similar to that after 2 h of stimulation. Selected areas of co-localization are presented as higher magnification of areas marked in upper panels. Similar increases were also observed in cells stimulated for 2 h with 250 nM A23187, 1 mM ATP or 10 μM isoproterenol (Figure 1D, inset – marked area magnified to show better view of co-localization). Preincubation of cells for 30 min with respective protein kinase inhibitor 100 nM Bis I (for PMA, ATP and A23187) or 100 nM H-89 (for isoproterenol) diminished the increase in cell surface levels of A7 and ABCA3 in secretagogue-stimulated type II cells. The quantitative data for weighted co-localization coefficients for ABCA3 and A7 from several analyses showed that protein kinase inhibitors diminished the co-localization of the two proteins (Figure 1E).
Biotin labeling of surface proteins
Biotin labeling of cell surface proteins with membrane-impermeable sulfo-NHS-SS-Biotin showed small amounts of A7 at the cell surface in the control cells, which was increased in cells stimulated for 2 h with 1 mM ATP or 10 μM isoproterenol (Figure 2Ai). Although equivalent volumes of relevant fractions (8% for cell lyate and residual fractions and 80% of the cell surface fraction) were used for analysis, equivalent loading of proteins in the cell surface fraction (notably of proteins at ~40 and ~90 kDa, arrows) was also assessed from PonceauS staining (Figure 2Aii) because of lack of actin in this fraction. Similarly increased levels of cell surface A7 were also observed in cells stimulated for 2 h with 100 nM PMA or 250 nM A23187 (Figure 2Bi), although PonceauS staining showed equivalent proteins levels in the cell surface fractions (Figure 2Bii, arrows). Densitometry analysis of western blots from several experiments showed that approximately 4% of total cellular A7 was present at the cell surface, which increased to almost 4-folds and 3-folds of the control levels in PMA or A23187-treated cells, respectively (Figure 2C). A7 trafficking to cell surface was mediated through PKC activation, since 30 min preincubation of cells with 100 nM BisI blocked the increase in biotin-labeled A7 in type II cells treated for 2 h with 100 nM PMA (Figure 2D).
Figure 2.

Biotin labeling shows increased levels of cell surface-exposed A7 in secretagogue-stimulated type II cells. Isolated type II cells were stimulated for 2 h without or with 1 mM ATP, 10 μM isoproterenol, 100 nM PMA or 250 nM A23187. The relative levels of A7 and actin were analyzed by western blotting of proteins from biotin-labeled (surface proteins), the residual fraction and the cell lysates. A. ATP and isoproterenol increase A7 levels at the cell surface. i) Western blots for actin and A7 showing relative levels of two proteins in each fraction. ii) PonceauS staining of membranes showing equivalent protein loading (notice proteins at ~40 and ~90 kDa, arrows) in the cell surface fraction for each condition. Densitometry showed that the cell surface A7 levels in the control, ATP and isoproterenol-treated cells were 6.6, 15.7 and 14.4% of the total cell A7. Although actin levels were similar in all groups, none was detected at the cell surface in any condition. B. Cell stimulation for 2 h with PMA and A23187 increases cell surface levels of A7. i) Western blot showing A7 and actin levels in the residual and surface fractions. ii) Western membrane were stained with PonceauS to demonstrate equivalent protein loading (notice proteins at ~40 and ~90 kDa, arrows) in the cell surface fraction, since actin could not be detected. C. Densitometry analysis of western blots from PMA and A23187-treated cells showed almost ~4-folds increase in the cell surface levels of A7 in comparison to the controls. Results are mean ± SE of experiments in parentheses. * P < 0.05 in comparison to PMA or A23187 by ANOVA and post-hoc Tukey’s test. D. Cells were incubated for 30 min with 100 nM BisI before incubation for 2 h with 100 nM PMA and the cell surface proteins were labeled with biotin. The presence of BisI blocked the PMA-stimulated increase in A7 at the cell surface. For comparison, the A23187-mediated increase in cell surface A7 is also shown. E. Luminal A7 in lung lamellar bodies was determined by analyzing the relative distribution in the limiting membrane (LBM) and the total A7 in lamellar bodies (LB) isolated from lung homogenate by upwards flotation on discontinuous sucrose. The isolated membranes were further treated with bicarbonate buffer to dissociate adherent proteins. Western blot analysis of equivalent proportions of the three fractions shows that most of the A7 was luminal in lamellar bodies and that only a part of membrane-associated A7 was inserted in the limiting membranes. F. Most cell surface A7 is not loosely attached to the cells. Control and PMA-stimulated (2 h) type II cells were treated with bicarbonate buffer for 30 min on ice, centrifuged for 30 min at 16,100g and the supernatant (wash) recovered. The pellet was pooled with the harvested cells from the plate. Western blot analysis of equivalent proportions of cell lysate and the wash followed by densitometry indicated that only 0.8% and 1.7% of the total cell A7 could be recovered in the wash. A comparison with the results from biotin-labeling of cell surface proteins, the levels in bicarbonate wash were less than 10% of the biotin-labeled A7.
Lamellar body A7
Since cell stimulation with secretagogue increases A7 association with the lamellar bodies, we analyzed the total and membrane-associated A7 in isolated lung lamellar bodies, as these organelles are unique to type II cells in the lung. Further, the yield from lung is greater in comparison to that from isolated type II cells. We have previously carried out extensive characterization of lamellar bodies and lamellar body membranes [33, 36, 37]. Equivalent proportions of various fractions were used for analysis and served as loading control. Thus, the image reflects the relative distribution of A7 in each fraction (Figure 2E). The A7 in lamellar bodies was mostly luminal and only a small amount was associated with the limiting membrane. Approximately, half of the membrane-associated A7 was adherent to membranes as it could be extracted in the bicarbonate buffer.
Because surfactant secretagogues increase the secretion of lamellar body contents (surfactant and possibly A7), we tested if reattachment of secreted A7 to the cell surface could account for the cell surface A7 (as determined by biotin-labeling) in control and stimulated cells (Figure 2A and B). Bicarbonate extraction of cell surface adherent proteins, previously used to demonstrate integral nature of flotillin [10], accounted for only 0.8% and 1.7% of total cellular A7 in control and PMA-stimulated type II cells, respectively (Figure 2F). Since these levels were about 80 – 90% lower than for the biotin-labeled cell surface proteins (Figure 2C), we suggest that the contribution of A7 to the cell surface pool by lamellar body secretion is relatively small in comparison to that by other mechanisms including A7 insertion into plasma membrane.
Cross-linking of cell surface proteins
The cell surface levels of A7 were also studied by cross-linking of cell surface protein using S-EGS, a membrane-impermeable cross-linking agent with a wide spacer arm (16.1 Å). In initial studies (Figure 3A), cross-linking was performed in control and PMA-stimulated cells and the cell lysates were evaluated for A7- and actin-adducts (including dimers or oligo-mers). Although the control cells did not show any adducts, the cells stimulated for 2 h with 100 nM PMA showed an A7-adduct at ~70 kDa but none for actin (Figure 3 A). In subsequent experiments, the surface proteins were cross-linked with S-EGS and the cells were fractionated into the membrane and the cytosol fractions. Western blot analysis showed ~70 kDa A7-adduct in the membrane, but not in the cytosol fraction, of the PMA or A23187-treated cells (Figure 3B). Thus, A7 at the cell surface interacts with a protein (~20 kDa) present in close proximity.
Figure 3.
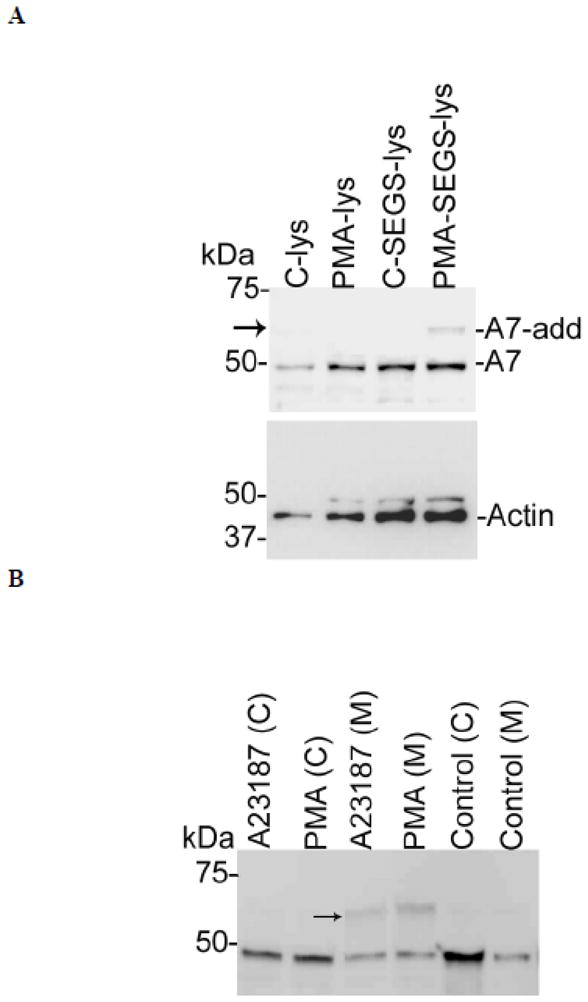
PMA andA23187 increase A7 interactions with cell surface proteins in type II cells. A. Type II cells were stimulated for 2 h without (Control) or with 100 nM PMA and the cell surface proteins were incubated in the absence or presence of membrane-impermeable cross-linker sulfo-ethylene glycol bis(succinimidyl succinate) (S-EGS). Western analysis of cell lysate (lys) shows A7-adduct (A7-add, arrow) in PMA-stimulated cells. The control cells without or with cross-linking did not show A7-adduct. The lower panel shows actin levels but no actin-adduct in these samples. B. Type II cells stimulated for 2 h without or with 100 nM PMA or 250 nM A23187 were cross-linked with S-EGS and fractionated into the membrane (M) and the cytosol (C) fractions. Western analysis showed that the membranes, but not the cytosol, fraction contained higher levels of ~70 kDa A7-adduct (arrow) in the PMA- or A23187-stimulated cells in comparison with the controls. This experiment was conducted at least two times with both agents.
Lung plasma membrane binding and cross-linking of A7
Protein phosphorylation and Ca2+ increase A7 binding to type II cell plasma membrane or SNAP23 [21, 23]. In current study, binding of recombinant A7 to lung plasma membrane was evaluated in the absence or presence of 1 μM Ca2+ following in vitro A7 phosphorylation with PKC (Figure 4). Additionally, the plasma membranes with the bound protein were cross-linked to determine direct interaction of A7 with other proteins in the plasma membrane fraction. The binding was higher for PKC-phosphorylated A7 in the absence and presence of 1 μM Ca2+, in comparison to the non-phosphorylated A7. The cross-linking of membrane bound A7 resulted in formation of several adducts, one of which was about the same size as observed by cross-linking of type II cells surface protein in the PMA- or A23187-stimulated type II cells (Figure 3A and B). These results suggest that the ~20 kDa A7-interacting protein is not recruited to the plasma membrane on cell-stimulation (Figure 3).
Figure 4.
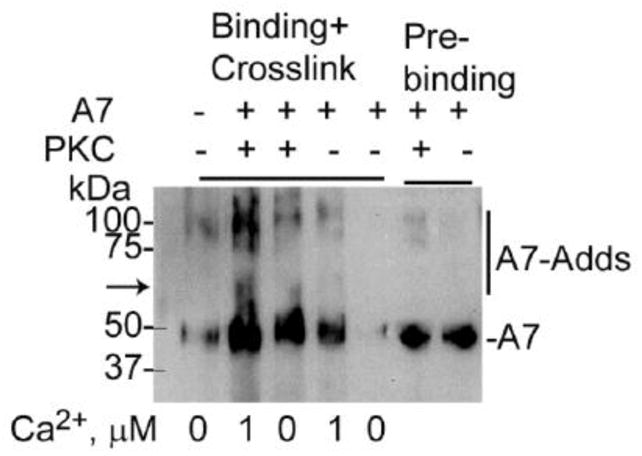
PKC phosphorylation of A7 increases binding and interaction with lung plasma membrane proteins. Recombinant A7 was phosphorylated in the absence or presence of PKC and incubated for binding to lung plasma membrane fraction without or with 1 μM Ca2+ followed by protein cross-linking. Phosphorylation with PKC increased A7 binding to plasma membrane, which was further increased in presence of 1 μM Ca2+. The presence of A7-adducts (highlighted by vertical line in the right margin) shows the interaction of A7 with several proteins. The A7-adduct at ~70 kDa (arrow) is possibly the same as observed with type II cells. The two right lanes show equivalent levels of A7 in the binding mixture.
PMA increases A7 trafficking to lipid rafts
The endocytosis and exocytosis functions are postulated to involve lipid rafts in plasma membrane that are enriched in cholesterol, sphingolipids and gangliosides [8] and proteins like flotillin and t-SNARE proteins [8, 10]. The lipid rafts, as judged by the marker flotillin-1, were present at the 30% and 15% sucrose inter-phase (fractions 5 and 6) in control cells, while in PMA-stimulated cells flotillin-1 was distributed in fractions 5 – 7. PMA-stimulation of type II cells also affected A7 distribution on the gradient showing increased protein in fraction 5 in comparison to the control cells (Figure 5). This change in distribution was detected possibly because of the extra layer of 15% sucrose between the 30% and 5% sucrose layers. Thus, PMA-stimulation increases trafficking of A7 to flotillin-1 containing domains in type II cells.
Figure 5.
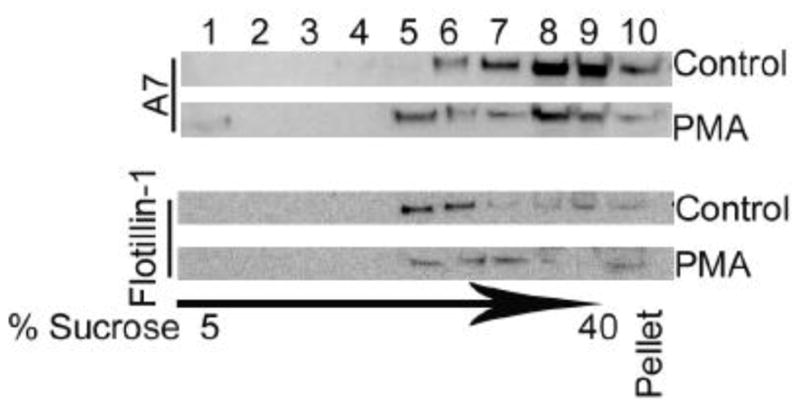
PMA stimulation increases A7 trafficking to lipid rafts in type II cells. Equal aliquots of fractions and the pellet from lipid rafts gradient from cells incubated for 2 h without or with 100 nM PMA were analyzed for A7 and flotillin-1 (lipid rafts marker). Most of flotillin-1 was present in fractions 5 and 6 in the control cells. PMA stimulation causes redistribution of flotillin-1 into fractions 5 – 7. The arrow indicates gradient of 5% to 40% sucrose with fractions 1 – 3, 4 and 5, 6 and 7, and 8 and 9 belonging to pre-centrifugation sucrose concentrations of 5, 15, 30 and 40%, respectively. In comparison to control cells, A7 was enriched in fraction 5 in PMA-stimulated cells, suggesting increased trafficking of A7 to lipid rafts.
Tryptophan protein fluorescence of recombinant proteins
The crystal structure of mammalian A7 is not known. Previous attempts with Dictyostelium discoideum A7 (later classified as annexin C1 [40]) were successful in obtaining only a partial structure [41], which showed homology with annexin A5, a protein with a short NH2-terminus [42]. Therefore, we have shown relative location of the tryptophan and neighboring residues as linear peptide (Figure 6A) for each homologous repeat (I, F176W; II, W239 wild type; III, F339W; and IV, F433W) [20]. Tryptophan in all repeats was present in one of the helical regions (underlined), except in repeat I [20]. All purified protein preparations showed a single ~47 kDa protein by SDS-PAGE analysis (Figure 6B). The tryptophan-free A7W239F and A7 showed similar calcium-dependent binding to phospholipids vesicles (Figure 6C). The binding of lipids increased with increasing Ca2+ reaching a maximum at about 10 μM. Thus, tryptophan residue is not critical for membrane binding of A7.
Figure 6.



Protein fluorescence quenching studies show lack of membrane insertion by recombinant A7. A. Amino acid sequence for relevant regions in the repeats I – IV showing the position of the tryptophan (W) residue in each repeat. The underlined sequences denote helical regions. Tryptophan at 239 is present in the wild type protein. The mutant F176W, F339W or F433W containing only a single tryptophan was expressed using the tryptophan-free mutant A7W239F (not shown) as the template. B. Coomassie brilliant blue®-stained SDS gel of purified proteins shows the presence of a major protein at ~47 kDa. C. Ca2+-dependence of recombinant A7 and tryptophan-free mutant A7W239F binding to lipid vesicles was similar. D. Protein tryptophan fluorescence of recombinant proteins shows lack of fluorescence in A7W239F and that the fluorescence emission characteristics are influenced by tryptophan location within the molecule as suggested by differences in emission peaks and intensities. E. Fluorescence emission spectra of each recombinant protein in the absence and presence of 1 mM Ca2+ without or with 50 mM acrylamide. The spectra were corrected for buffer and dilution and are presented as 5th polynomial fit after 5-point smoothing. Acrylamide quenching of fluorescence indicates partial exposure of the tryptophan residue to the aqueous phase. F. Fluorescence spectra (5th polynomial fit) in presence of 1 mM Ca2+ without or with 20 μM of indicated vesicles. Fluorescence quenching with 7-doxylPC containing lipid vesicles (PC:PS:7-doxylPC, 4:5:1) in comparison to PC:PS (1:1) suggest shallow insertion by A7 through repeat II (W239), since no quenching was observed with PC:PS:12-doxylPC (4:5:1) vesicles (not shown).
A comparison of fluorescence scans (20–25 μg protein) showed that the emission was due largely to the tryptophan residue in A7 and its three other mutants (A7F176W, A7F339W and A7F433W), since very low fluorescence was observed for mutant A7W239F (Figure 6D). The emission intensity and the emission peak differed for each protein possibly due to the relative position of the tryptophan residue and the quenching and enhancing effects of the neighboring amino acids. The presence of 1mM Ca2+ caused minor change in the total fluorescence for each protein (Figure 6E). For all proteins, the tryptophan residue was partially exposed to the aqueous phase as suggested by acrylamide quenching of total fluorescence - 15% for the wild type (35.43 to 30.25), 27% for A7F176W (19.42 to 14.23) and A7F443W (34.78 vs. 25.66) and 43% for A7F339W (59.3 to 33.7).
Our previous studies on protein tryptophan fluorescence suggested NH2-terminus modulation of molecular organization of A7 and its interactions with phospholipid membranes [20, 29, 30]. We also demonstrated the ability of A7 to interact with trans-membranes as determined by fluorescence quenching of membrane-bound A7 with dansyl-PE vesicles [29]. The fluorescence quenching by dansyl moiety in the polar head-group of PE, however, does not distinguish membrane association from insertion. The present study determined membrane insertion of A7 by measuring fluorescence quenching with doxylPC, a spin-labeled lipid where the nitroxide quencher group, doxyl, is located in the fatty acid chain of the lipid [43]. The fluorescence emission of A7 was increased in presence of 20 μM PC:PS (1:1) vesicles and 1 mM Ca2+. In comparison, the fluorescence was unchanged in presence of 1 mM Ca2+ and PC:PS vesicles containing 10% 7-doxylPC. Similar studies with tryptophan mutants did not show any differences in fluorescence intensity with vesicles containing 0% or 10% 7-doxylPC (Figure 6F). Although these results suggested A7 insertion into membranes, it did not appear deep since vesicles with 10% 12-doxylPC (doxyl embedded deeper in the bilayer) did not cause fluorescence quenching for A7 or its mutants (not shown).
DISCUSSION
In this study, we demonstrate trafficking of A7 to the cell surface in type II cells during stimulated secretion of lung surfactant and suggest possible insertion of A7 into the plasma membrane in vivo. The cell surface A7 was increased with several agents that increase surfactant secretion through different signaling mechanisms. However, in vitro protein tryptophan fluorescence studies with recombinant A7 and its tryptophan mutants did not provide a conclusive evidence for A7 insertion into artificial membrane except for possible shallow penetration. Thus, we suggest that in vivo membrane insertion occurs possibly due to interaction with other proteins or post-translational modifications like phosphorylation of A7, which facilitate protein insertion into membranes. These observations support an involved role of A7 in membrane fusion during surfactant secretion.
In vitro A7 interactions with artificial membranes
A7, like other annexin proteins, can bind to membranes in a calcium-dependent manner and promotes membrane fusion during catecholamines secretion in charomaffin cells and lung surfactant secretion in type II cells. The hypothetical scheme of membrane fusion suggested membrane binding of A7 followed by reorganization of membrane phospholipids around the protein molecules to form the fusion pore [44] implying membrane insertion of A7. This important phase of membrane fusion is supported by the ability of A7 to form calcium channels in lipid membranes [45]. However, purified annexin A5 and A2 show only peripheral association with lipid bilayers as demonstrated by ultrastructural and atomic force microscopy studies of the lipid-protein complex [24, 25]. Also, the thermodynamic aspects of conformational change and the lack of seven trans-membrane domains argue against A7 function as conventional channel [28]. In agreement, our in vitro studies with doxylPC vesicles show only shallow membrane insertion by A7 (Figure 6). Nevertheless, our in vivo studies suggest that part of A7 trafficking to cell surface could occur through penetration into plasma membrane, possibly in cooperation with other proteins or following phosphorylation during stimulation of surfactant secretion. Thus, A7 may have a more involved role in membrane fusion in vivo.
Annexin A7 insertion into membranes in vivo
Despite physico-chemical studies with artificial membranes suggesting inability of annexin proteins (including A7) to insert into artificial membranes (Figure 6 and [24, 25]), our in vivo studies show that endogenous A7 likely inserts and traverses the plasma membrane as revealed by immuno-fluorescence (Figure 1) and biotin-labeling of cell surface proteins (Figure 2). Some of the cell surface A7 could arise from its association with the lamellar bodies (this study and [22, 23]) during surfactant secretion. Although most of the A7 in lamellar bodies was luminal (Figure 2E), its release and reattachment to the cell surface does not appear significant because A7 concentrations following release in the cell culture medium will be highly diluted. This is supported by recovery of only a small amount of cellular A7 in the bicarbonate wash (Figure 2F) in comparison to that recovered as biotin-labeled A7 in control and stimulated cells (Figure 2B and C). Thus, only a small proportion of cell surface A7 could be derived from lamellar body secretion, while the remaining is likely through mechanisms including protein insertion into plasma membrane. Also, the presence of A7 in lamellar body lumen (Figure 2E) suggests that A7 can insert into the lamellar body membrane. Finally, the presence of A7 in detergent insoluble lipid rafts argues against mere association with the membranes (Figure 5). Thus, contrary to the findings using artificial membrane and physical analysis showing lack of membrane insertion (Figure 6 and [24, 25]), in vivo studies suggest that cellular A7 can likely insert and traverse biological membranes under specific conditions.
Mechanisms of annexin A7 insertion into membranes
Stimulation of type II cells causing A7 trafficking to lamellar bodies and the presence of A7 in lamellar body lumen would suggest that secretion of lung surfactant, at best, could only partly function as a mechanism for A7 trafficking to the cell surface. The presence of luminal A7 in lamellar bodies also supports that A7 association with lamellar bodies is followed by insertion into the membranes and transfer to the lipid (including anionic phospholipids)-rich interior that bind A7 with high affinity. The mechanisms regulating A7 insertion into various membranes are not clear, but two possibilities – phosphorylation and interaction with other proteins – are considered. Our studies thus far show that protein kinase-dependent phosphorylation of A7 may be predominant for its trafficking either directly or indirectly (through lamellar bodies) into the plasma membrane (Figures 1D and E, and 2D). The immuno-staining studies showing A7 trafficking to non-lamellar body compartment(s) suggest that all of the A7 in the plasma membrane is not directed through the lamellar bodies. Direct phosphorylation-dependent trafficking of A7 to plasma membrane can also occur as supported by in vitro and in vivo interactions of A7 with SNAP23 [23] and by A7 binding to plasma membrane (Figure 4). Additional mechanism(s) for membrane insertion may involve interaction with other membrane proteins (Figure 3) or trafficking to specific domains like lipid rafts (Figure 5) that may regulate membrane insertion following phosphorylation of A7.
The increased A7 levels in lipid rafts (Figure 5) in PMA-stimulated cells suggests that other secretagogues may also promote A7 trafficking to lipid rafts, which may be accompanied by its insertion into membranes and exposure at the cells surface, since all secretagogues showed increased immuno-staining and biotin-labeling of A7 (Figures 1 and 2). We propose that such trafficking requires protein kinase mediated phosphorylation, which alters A7 affinity for membranes [21, 22, 46]. Our in vitro binding and cross-linking studies also suggest a role for A7 phosphorylation and Ca2+ for binding and interactions with proteins in the plasma membrane as supported by formation of several adducts including the ~70 kDa adduct seen during cross-linking of cell surface proteins (Figures 3). Although the in vivo cross-linking studies do not quantify A7 levels at the cell surface, the relative increase in ~70 kDa protein implies increased A7 at the cell surface.
In conclusion, our studies show that the in vivo function of annexin A7, and possibly other annexin proteins, is regulated by protein phosphorylation and, possibly, by interaction with other membrane proteins. Identification of interacting proteins and in vitro interactions using recombinant proteins without and with phosphorylation may help further understand the precise mechanism and function of A7 and other annexin proteins.
Highlights.
Suggested planar configuration can limit membrane insertion by annexin proteins.
Stimulation of surfactant secretion increases annexin A7 on type II cell surface.
Protein kinase activation increases cell surface annexin A7 possibly via lipid rafts.
Recombinant annexin A7, however, failed to show insertion into artificial membranes.
Phosphorylation could promote in vivo membrane penetration by annexin A7.
Acknowledgments
This study was supported by a grant (HL 049959 to AC) from the National Heart, Lung and Blood Institute, National Institutes of Health, Bethesda, MD. We are thankful for the expert advice and the use of confocal microscope facility at the core facility at the Stony Brook University, NY.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rooney SA. Regulation of surfactant secretion. Comp Biochem Physiol A Mol Integr Physiol. 2001;129:233–243. doi: 10.1016/s1095-6433(01)00320-8. [DOI] [PubMed] [Google Scholar]
- 2.Chander A, Fisher AB. Regulation of lung surfactant secretion. The American journal of physiology. 1990;258:L241–253. doi: 10.1152/ajplung.1990.258.6.L241. [DOI] [PubMed] [Google Scholar]
- 3.Andreeva AV, Kutuzov MA, Voyno-Yasenetskaya TA. Regulation of surfactant secretion in alveolar type II cells. American journal of physiology. 2007;293:L259–271. doi: 10.1152/ajplung.00112.2007. [DOI] [PubMed] [Google Scholar]
- 4.Dietl P, Haller T. Exocytosis of lung surfactant: from the secretory vesicle to the air-liquid interface. Annu Rev Physiol. 2005;67:595–621. doi: 10.1146/annurev.physiol.67.040403.102553. [DOI] [PubMed] [Google Scholar]
- 5.Rothman JE. Mechanisms of intracellular protein transport. Nature. 1994;372:55–63. doi: 10.1038/372055a0. [DOI] [PubMed] [Google Scholar]
- 6.Dietrich LE, Boeddinghaus C, LaGrassa TJ, Ungermann C. Control of eukaryotic membrane fusion by N-terminal domains of SNARE proteins. Biochimica et biophysica acta. 2003;1641:111–119. doi: 10.1016/s0167-4889(03)00094-6. [DOI] [PubMed] [Google Scholar]
- 7.Dietl P, Liss B, Felder E, Miklavc P, Wirtz H. Lamellar body exocytosis by cell stretch or purinergic stimulation: possible physiological roles, messengers and mechanisms. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology. 2010;25:1–12. doi: 10.1159/000272046. [DOI] [PubMed] [Google Scholar]
- 8.Chamberlain LH, Burgoyne RD, Gould GW. SNARE proteins are highly enriched in lipid rafts in PC12 cells: implications for the spatial control of exocytosis. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:5619–5624. doi: 10.1073/pnas.091502398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chintagari NR, Jin N, Wang P, Narasaraju TA, Chen J, Liu L. Effect of cholesterol depletion on exocytosis of alveolar type II cells. American journal of respiratory cell and molecular biology. 2006;34:677–687. doi: 10.1165/rcmb.2005-0418OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bickel PE, Scherer PE, Schnitzer JE, Oh P, Lisanti MP, Lodish HF. Flotillin and epidermal surface antigen define a new family of caveolae-associated integral membrane proteins. The Journal of biological chemistry. 1997;272:13793–13802. doi: 10.1074/jbc.272.21.13793. [DOI] [PubMed] [Google Scholar]
- 11.Schneider A, Rajendran L, Honsho M, Gralle M, Donnert G, Wouters F, Hell SW, Simons M. Flotillin-dependent clustering of the amyloid precursor protein regulates its endocytosis and amyloidogenic processing in neurons. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2008;28:2874–2882. doi: 10.1523/JNEUROSCI.5345-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gerst JE. SNARE regulators: matchmakers and matchbreakers. Biochimica et biophysica acta. 2003;1641:99–110. doi: 10.1016/s0167-4889(03)00096-x. [DOI] [PubMed] [Google Scholar]
- 13.Duman JG, Forte JG. What is the role of SNARE proteins in membrane fusion? Am J Physiol Cell Physiol. 2003;285:C237–249. doi: 10.1152/ajpcell.00091.2003. [DOI] [PubMed] [Google Scholar]
- 14.Creutz CE. The annexins and exocytosis. Science (New York, NY. 1992;258:924–931. doi: 10.1126/science.1439804. [DOI] [PubMed] [Google Scholar]
- 15.Pollard HB, Guy HR, Arispe N, de la Fuente M, Lee G, Rojas EM, Pollard JR, Srivastava M, Zhang-Keck ZY, Merezhinskaya N, et al. Calcium channel and membrane fusion activity of synexin and other members of the Annexin gene family. Biophysical journal. 1992;62:15–18. doi: 10.1016/S0006-3495(92)81764-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chattopadhyay S, Sun P, Wang P, Abonyo B, Cross NL, Liu L. Fusion of lamellar body with plasma membrane is driven by the dual action of annexin II tetramer and arachidonic acid. The Journal of biological chemistry. 2003;278:39675–39683. doi: 10.1074/jbc.M212594200. [DOI] [PubMed] [Google Scholar]
- 17.Liu L, Wang M, Fisher AB, Zimmerman UJ. Involvement of annexin II in exocytosis of lamellar bodies from alveolar epithelial type II cells. The American journal of physiology. 1996;270:L668–676. doi: 10.1152/ajplung.1996.270.4.L668. [DOI] [PubMed] [Google Scholar]
- 18.Chander A, Sen N, Spitzer AR. Synexin and GTP increase surfactant secretion in permeabilized alveolar type II cells. American journal of physiology. 2001;280:L991–998. doi: 10.1152/ajplung.2001.280.5.L991. [DOI] [PubMed] [Google Scholar]
- 19.Chander A, Wu RD. In vitro fusion of lung lamellar bodies and plasma membrane is augmented by lung synexin. Biochimica et biophysica acta. 1991;1086:157–166. doi: 10.1016/0005-2760(91)90003-z. [DOI] [PubMed] [Google Scholar]
- 20.Naidu DG, Raha A, Chen XL, Spitzer AR, Chander A. Partial truncation of the NH2-terminus affects physical characteristics and membrane binding, aggregation, and fusion properties of annexin A7. Biochimica et biophysica acta. 2005;1734:152–168. doi: 10.1016/j.bbalip.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 21.Chander A, Sen N, Naidu DG, Spitzer AR. Calcium ionophore and phorbol ester increase membrane binding of annexin a7 in alveolar type II cells. Cell calcium. 2003;33:11–17. doi: 10.1016/s0143-4160(02)00177-x. [DOI] [PubMed] [Google Scholar]
- 22.Gerelsaikhan T, Chen XL, Chander A. Secretagogues of lung surfactant increase annexin A7 localization with ABCA3 in alveolar type II cells. Biochimica et biophysica acta. 2011;1813:2017–2025. doi: 10.1016/j.bbamcr.2011.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gerelsaikhan T, Vasa PK, Chander A. Annexin A7 and SNAP23 interactions in alveolar type II cells and in vitro: A role for Ca(2+) and PKC. Biochimica et biophysica acta. 2012;1823:1796–1806. doi: 10.1016/j.bbamcr.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oling F, Santos JS, Govorukhina N, Mazeres-Dubut C, Bergsma-Schutter W, Oostergetel G, Keegstra W, Lambert O, Lewit-Bentley A, Brisson A. Structure of membrane-bound annexin A5 trimers: a hybrid cryo-EM - X-ray crystallography study. Journal of molecular biology. 2000;304:561–573. doi: 10.1006/jmbi.2000.4183. [DOI] [PubMed] [Google Scholar]
- 25.Pigault C, Follenius-Wund A, Schmutz M, Freyssinet JM, Brisson A. Formation of two-dimensional arrays of annexin V on phosphatidylserine-containing liposomes. Journal of molecular biology. 1994;236:199–208. doi: 10.1006/jmbi.1994.1129. [DOI] [PubMed] [Google Scholar]
- 26.Reviakine II, Bergsma-Schutter W, Brisson A. Growth of Protein 2-D Crystals on Supported Planar Lipid Bilayers Imaged in Situ by AFM. Journal of structural biology. 1998;121:356–361. doi: 10.1006/jsbi.1998.4003. [DOI] [PubMed] [Google Scholar]
- 27.Voges D, Berendes R, Burger A, Demange P, Baumeister W, Huber R. Three-dimensional structure of membrane-bound annexin V. A correlative electron microscopy-X-ray crystallography study. Journal of molecular biology. 1994;238:199–213. doi: 10.1006/jmbi.1994.1281. [DOI] [PubMed] [Google Scholar]
- 28.Gerke V, Creutz CE, Moss SE. Annexins: linking Ca2+ signalling to membrane dynamics. Nature reviews Molecular cell biology. 2005;6:449–461. doi: 10.1038/nrm1661. [DOI] [PubMed] [Google Scholar]
- 29.Chander A, Chen XL, Naidu DG. A role for diacylglycerol in annexin A7-mediated fusion of lung lamellar bodies. Biochimica et biophysica acta. 2007;1771:1308–1318. doi: 10.1016/j.bbalip.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chander A, Naidu DG, Chen XL. A ten-residue domain (Y11-A20) in the NH2-terminus modulates membrane association of annexin A7. Biochimica et biophysica acta. 2006;1761:775–784. doi: 10.1016/j.bbalip.2006.05.018. [DOI] [PubMed] [Google Scholar]
- 31.Rosengarth A, Gerke V, Luecke H. X-ray structure of full-length annexin 1 and implications for membrane aggregation. Journal of molecular biology. 2001;306:489–498. doi: 10.1006/jmbi.2000.4423. [DOI] [PubMed] [Google Scholar]
- 32.Chander A, Sen N. Inhibition of phosphatidylcholine secretion by stilbene disulfonates in alveolar type II cells. Biochemical pharmacology. 1993;45:1905–1912. doi: 10.1016/0006-2952(93)90450-b. [DOI] [PubMed] [Google Scholar]
- 33.Chander A, Johnson RG, Reicherter J, Fisher AB. Lung lamellar bodies maintain an acidic internal pH. The Journal of biological chemistry. 1986;261:6126–6131. [PubMed] [Google Scholar]
- 34.Sen N, Spitzer AR, Chander A. Calcium-dependence of synexin binding may determine aggregation and fusion of lamellar bodies. The Biochemical journal. 1997;322(Pt 1):103–109. doi: 10.1042/bj3220103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bers DM. A simple method for the accurate determination of free [Ca] in Ca-EGTA solutions. The American journal of physiology. 1982;242:C404–408. doi: 10.1152/ajpcell.1982.242.5.C404. [DOI] [PubMed] [Google Scholar]
- 36.Chander A, Dodia CR, Gil J, Fisher AB. Isolation of lamellar bodies from rat granular pneumocytes in primary culture. Biochimica et biophysica acta. 1983;753:119–129. doi: 10.1016/0005-2760(83)90105-4. [DOI] [PubMed] [Google Scholar]
- 37.Chander A. Dicyclohexylcarbodiimide and vanadate sensitive ATPase of lung lamellar bodies. Biochimica et biophysica acta. 1992;1123:198–206. doi: 10.1016/0005-2760(92)90112-9. [DOI] [PubMed] [Google Scholar]
- 38.Chander A, Fisher AB, Strauss JF., 3rd Role of an acidic compartment in synthesis of disaturated phosphatidylcholine by rat granular pneumocytes. The Biochemical journal. 1982;208:651–658. doi: 10.1042/bj2080651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 40.Gerke V, Moss SE. Annexins: from structure to function. Physiological reviews. 2002;82:331–371. doi: 10.1152/physrev.00030.2001. [DOI] [PubMed] [Google Scholar]
- 41.Liemann S, Bringemeier I, Benz J, Gottig P, Hofmann A, Huber R, Noegel AA, Jacob U. Crystal structure of the C-terminal tetrad repeat from synexin (annexin VII) of Dictyostelium discoideum. Journal of molecular biology. 1997;270:79–88. doi: 10.1006/jmbi.1997.1091. [DOI] [PubMed] [Google Scholar]
- 42.Huber R, Romisch J, Paques EP. The crystal and molecular structure of human annexin V, an anticoagulant protein that binds to calcium and membranes. The EMBO journal. 1990;9:3867–3874. doi: 10.1002/j.1460-2075.1990.tb07605.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chapman ER, Davis AF. Direct interaction of a Ca2+-binding loop of synaptotagmin with lipid bilayers. The Journal of biological chemistry. 1998;273:13995–14001. doi: 10.1074/jbc.273.22.13995. [DOI] [PubMed] [Google Scholar]
- 44.Pollard HB, Burns AL, Rojas E. A molecular basis for synexin-driven, calcium-dependent membrane fusion. The Journal of experimental biology. 1988;139:267–286. doi: 10.1242/jeb.139.1.267. [DOI] [PubMed] [Google Scholar]
- 45.Burns AL, Magendzo K, Shirvan A, Srivastava M, Rojas E, Alijani MR, Pollard HB. Calcium channel activity of purified human synexin and structure of the human synexin gene. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:3798–3802. doi: 10.1073/pnas.86.10.3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Caohuy H, Pollard HB. Activation of annexin 7 by protein kinase C in vitro and in vivo. The Journal of biological chemistry. 2001;276:12813–12821. doi: 10.1074/jbc.M008482200. [DOI] [PubMed] [Google Scholar]